

Abstract
Using a preclinical model, we investigated whether excess estradiol (E2) or leptin during pregnancy affects maternal mammary tumorigenesis in rats initiated by administering carcinogen DMBA on day 50. Two weeks later, rats were mated, and pregnant dams were treated daily with 10 μg of 17β-estradiol, 15 μg of leptin or vehicle from gestation day 8 to 19. Tumor development was assessed separately during weeks 1–12 and 13–22 after DMBA administration, since pregnancy is known to induce a transient increase in breast cancer risk, followed by a persistent reduction. Parous rats developed less (32%) mammary tumors than nulliparous rats (59%, p<0.001), and the majority (93%) of tumors in the parous rats appeared before week 13 (versus 41% in nulliparous rats), indicating that pregnancy induced a transient increase in breast cancer risk. Parous rats exposed to leptin (final tumor incidence 65%) or E2 (45%) during pregnancy developed mammary tumors throughout the tumor monitoring period, similar to nulliparous control rats, and the incidence was significantly higher in both the leptin and E2 exposed dams after week 12 than in the vehicle exposed parous dams (p<0.001). The mammary glands of the exposed parous rats contained significantly more proliferating cells (p<0.001). In addition, the E2 or leptin treated parous rats did not exhibit the protective genomic signature induced by pregnancy and seen in the parous control rats. Specifically, these rats exhibited down-regulation of genes involved in differentiation and immune functions and up-regulation of genes involved in angiogenesis, growth, and epithelial to mesenchymal transition.
Keywords: Pregnancy, estradiol, leptin, genomic signature, breast cancer
Introduction
Pregnancy affects a woman’s breast cancer risk by first inducing a transient increase in risk, lasting for 5–7 years (1–4), and then either permanently reducing or increasing the risk, depending upon the age of the woman. Women who gave birth before age 20 decrease their breast cancer risk by half compared to women who were over 30 when they had their first child (5). The latter, in turn, have a significantly higher lifetime risk of breast cancer than nulliparous women (6, 7). The protective effect of early pregnancy is limited to estrogen and progesterone receptor positive (ER+ and PR+) breast cancers (8, 9), whilst late first pregnancy can increase the risk of developing either ER+ or ER negative cancers (9, 10).
Several theories have been offered to explain the protective effects of early pregnancy on breast cancer risk (11, 12). Importantly, parous women and animals exhibit permanent changes in gene expression patterns, resulting in a pregnancy-induced protective genomic signature. This signature involves genes that can prevent malignant transformation, including those that reduce mammary epithelial cell proliferation and increase differentiation (13–15). It is less clear why a late first pregnancy increases breast cancer risk, but it may be caused by an aging-related increase the presence of transformed mammary epithelial cells that can start proliferating when exposed to high pregnancy hormonal environment. Accumulating evidence indicates that women who had the highest circulating estrogen levels during pregnancy (16, 17) or were exposed to synthetic estrogen diethylstilbestrol (DES) (18, 19) are at highest risk of developing breast cancer. In addition, giving birth to an infant with high birth weight is associated with high maternal estriol/alpha-fetoprotein ratio and increased breast cancer risk (20).
The possibility that elevated leptin levels during pregnancy also may increase breast cancer risk has not been explored. Serum leptin concentrations increase during pregnancy, peaking during the second trimester (21, 22), although the increase is not nearly as dramatic as with estrogens. Pregnant women who gain an excessive amount of weight have high leptin levels (23–25) and are significantly more likely to develop breast cancer after menopause than women whose weight gain during pregnancy does not exceed the recommendations provided by the Institute of Medicine (IOM) (26). In preclinical studies, excessive weight gain induced by feeding pregnant dams an obesity-inducing high fat diet increases pregnancy leptin levels and subsequent mammary tumorigenesis (27). Importantly, leptin interacts with estradiol (E2) and the ER. Leptin has been shown to activate ER-α, likely through its ability to stimulate aromatase and/or mitogen activated protein kinases (MAPKs) (28, 29). Further leptin decreases ER-α ubiquitination and increases ER-α half-life, potentially leading to increased ER-α activity (30). E2, in turn, can interfere with leptin’s actions by regulating the expression of the leptin receptor (31). Similar to E2, leptin promotes the growth of ER+ human breast cancer cells in culture (32, 33), but it also induces proliferation of ER- breast cancer cells (34).
In this study, we sought to establish experimentally whether treating pregnant dams with excess E2 or leptin during pregnancy increases later mammary tumorigenesis in rats. Our results indicate that in the vehicle treated control rats pregnancy induced a transient increase in mammary cancer risk that lasted until mammary glands had undergone involution and returned to a non-pregnant and non-lactating stage. When back to this stage, the risk of developing breast cancer was dramatically reduced, resulting in a lower lifetime risk than what was seen in nulliparous rats. Rats exposed to an excess of either E2 or leptin during pregnancy exhibited a sustained increase in mammary tumorigenesis, similar to nulliparous rats. Higher breast cancer risk in the parous E2 or leptin rats than in vehicle treated parous control rats may be related to a persistent increase in cell proliferation in their mammary glands, and absence of parity-induced protective changes in the genome. Thus, our preclinical study suggests that an exposure to excess E2 or leptin during pregnancy prevents pregnancy-induced reduction in breast cancer risk and the protective changes in genomic signaling pathways seen in the parous mammary gland.
Materials and methods
Animals
Five-week-old Sprague Dawley rats were obtained from Charles River (Wilmington, MA) and fed AIN93G diet upon arrival. Animals were housed in a temperature- and humidity-controlled room at the Georgetown University Resource Animal Facility under a 12-hour light-dark cycle. All animal procedures were approved by the Georgetown University Animal Care and Use Committee, and the experiments were performed following the National Institutes of Health guidelines for the proper and humane use of animals in biomedical research.
Carcinogen exposure
At 50 days of age, a total of 223 female rats were administered 10 mg of the mammary carcinogen 7,12-dimethylbenz[a]anthracene (DMBA) (Sigma Chemical Co., St. Louis, MO) by oral gavage. Carcinogen was dissolved in peanut oil and given in a volume of 1 ml.
Mating and hormonal exposures
Two weeks after DMBA exposure, female rats were mated by housing two female rats and one male rat together. Positive vaginal plug was used to determine the first day of pregnancy. On gestation day 8, pregnant females were divided into three experimental groups: control dams receiving subcutaneous vehicle injections (n=43), E2 dams receiving subcutaneous injections of 10 μg of 17β-estradiol (Sigma Chemical Co., St. Louis, MO; n=42), or leptin dams receiving subcutaneous injections of 15 μg of leptin (R&D systems, Minneapolis, MN; n=40). Injections were given daily until gestation day 19. The doses were chosen based upon a pilot study that indicated that neither 10 μg E2 nor 15 μg leptin affected weight development during pregnancy. After giving birth, dams were allowed to nurse their offspring for three weeks, and then the pups were weaned.
Exposure of nulliparous rats to hormones
An additional set of 78 DMBA exposed female rats, three weeks after the carcinogen exposure (to match with day 8 of gestation), were divided to three groups and given subcutaneous injections of vehicle (n=29), 10 μg of E2 (n=41) or 15 μg of leptin (n=28). Injections were given daily for a total of two weeks.
Monitoring tumorigenesis
Four weeks post-DMBA administration, we began checking rats weekly for mammary tumors by palpation. Tumor growth was measured using a caliper, and the length, width, and height of each tumor were recorded. Animals were sacrificed if any tumor reached a size of 25–30 mm in diameter. The remaining animals, including those that did not develop tumors, were sacrificed 17 weeks after pregnancy ended/22 weeks after DMBA administration. End-points for this study were time to tumor appearance (tumor latency), the number of tumors per animal (tumor multiplicity), and the percentage of rats that developed tumors per experimental group (tumor incidence).
Pregnancy hormone measurements
Concentrations of circulating leptin and E2 were determined in serum collected by tail bleeding on gestation day 19 (n=5–7 per group), using a rodent leptin EIA kit from Assay Designs, Inc. (Ann Arbor, MI) and a rodent E2 EIA kit from Cayman Chemical Company (Ann Arbor, MI), respectively, following the manufacturers’ instructions.
Immunohistochemical detection of cell proliferation
At the end of the tumor monitoring period (22 weeks post-DMBA exposure) all rats were sacrificed and their mammary tissues and tumors were obtained. Cell proliferation in the mammary tissue was assessed by immunohistochemistry staining for PCNA in 6 rats per group. The 2nd–3rd glands were used and they were fixed in 10% buffered formalin, embedded in paraffin and sectioned (5 μm). Sections were deparaffinized in xylene, hydrated through graded alcohols, and incubated with 3% H2O2 for 10 min to block endogenous peroxidases. Non-specific binding was blocked with normal rabbit serum from the Vectastain Elite ABC Kit (Vector Laboratories, Inc.) for 20 min. Tissue sections were incubated overnight with the primary antibody against PCNA at a 1:500 dilution (Santa Cruz Biotechnology, Inc., CA). After several washes, sections were treated with the secondary antibody (biotinylated anti-goat IgG from the Vectastain Elite ABC Kit; Vector Laboratories, Inc.) for 30 minutes at room temperature, followed by treatment with an avidin and biotinylated horseradish peroxidase complex from the Vectastain Elite ABC Kit (Vector Laboratories, Inc.) for 30 min at room temperature. Sections were washed and stained with 3,3′-diaminobenzidine (DAB) (Vector Laboratories, Inc) for 1 min, washed, and counterstained with Vector’s Hematoxylin QS Nuclear Counterstain (Vector Laboratories, Inc.) for 45 sec. Proliferation index was determined by calculating the percentage of cells with positive PCNA staining in at least 1,000 cells per mammary gland section. Slides were evaluated using the Metamorph software, without knowledge of treatment group.
Detection of apoptosis
Apoptosis was assessed in the same mammary gland sections used to determine proliferation (n=6 per group) by in situ oligo ligation (ISOL) assay with an ApopTag Kit (Serologicals Corporation, Norcross, GA) following the manufacturer’s instructions. Briefly, sections were deparaffinized in xylene and hydrated in a series of graded alcohols. The sections were then treated with 20 μg/ml of Proteinase K for 15 min. Endogenous peroxidases were quenched with 3% H2O2 for 5 min. Sections were washed with equilibration buffer (ApopTag Kit) and incubated with the Ligase enzyme for 16 hours at 16–22 °C. The reaction was stopped and sections were incubated with a streptavidin-peroxidase conjugate at room temperature. Sections were again washed, incubated with the peroxidase substrate for 10 min, and counterstained with 0.5% methyl green (Vector Laboratories, Inc., Burlingame, CA) for 10 min. Apoptotic index was determined by calculating the percentage of cells that were apoptotic through both positive staining and histological evaluation amongst 1,000 cells per mammary gland section. All sections were evaluated using the Metamorph software, without knowledge of treatment group.
Microarray analysis
Array hybridization and scanning
The 4th mammary glands that contained no palpable growth or non-palpable microtumors were obtained from 5 rats per group (control, E2, and leptin exposed), sacrificed 22 weeks after DMBA exposure. Six micrograms of purified total RNA was used to synthesize cDNA and then generate cRNA, which was labeled with biotin according to techniques recommended by Affymetrix (Santa Clara, CA). Labeled cRNA was fragmented at 94 °C for 35 min in a fragmentation buffer and then hybridized to Affymetrix Rat U34 A GeneChips, which contained approximately 7,000 full-length sequences and 1,000 EST clusters. After washing, the chips were stained with strepavidin-phycoerythrin conjugate and then scanned using the Affymetrix GeneChip Scanner 3000 (Hewllet-Packard Co). Raw data were generated using Affimetrix GeneChip 3.1 software.
Data normalization
In Affimetrix GeneChip experiments, variations in the amount and quality of target hybridized to the array may contribute to an overall variability in hybridization intensities. To reliably compare data from multiple probe arrays, differences of non-biological origin must be minimized. We accomplished this by normalizing the data using the MicroArray Suite 5.0 (Affymetrix) software to average the intensities for each GeneChip and to calculate a normalization factor. The normalized intensities were obtained from each chip by multiplying raw intensities by the normalization factor.
Identification of gene expression profiles
Normalized results obtained from each group were used to calculate the ratio (control / treated) for each gene. Hybridization signal intensities of relative fold changes, which ranged from ≤ 0.5 for down-regulation or ≥ 2-fold for up-regulation, were considered to be significant and were reported. The level of significance was set at p<0.05. Dimensionality reduction (elimination of non-informative data) was performed by filtering out genes with low threshold (intensity < 0.1 in both groups) and low fold change (< 2.0). In addition, comparisons made had to be significantly different in at least one of three statistical tests (i.e., equal and unequal variance t tests, equal and unequal variance t tests on log transformed data, Wilcoxon test).
Data visualization
We calculated the 3-dimensional projections of multidimensional gene expression microarray data sets by using Principal Component Analyses (PCA) and Discriminant Component Analyses (DCA).
Generation and testing of a neural network
To determine whether the model could accurately predict the leptin/E2 exposure, a neural network was trained, independent of gene expression profile selection.
Quantitative Real Time PCR (qRT PCR)
qRT PCR was used to confirm the differential expression of selected genes between the control and high risk groups shown in the microarray analysis. The 4th mammary glands were obtained from a different set of rats (n=6–8 per group) than the ones used for microarray analysis. Briefly, cDNA was reverse transcribed from 50 μg/ml of total input RNA using Taqman Reverse Transcription Reagents (Applied Biosystems, Foster City, CA). The reverse transcription reaction was carried out in a Taqman master mix under the following conditions: 25°C for 10 minutes, 48°C for 30 minutes, and 95°C for 5 minutes. Next, PCR products were generated from the cDNA samples using the Taqman Universal PCR Master Mix (Applied Biosystems) and Assays-on-Demand (Applied Biosystems) for the appropriate target gene (VEGF, Pleiotrophin, Nras, Mapk 9 and Eif4e). The 18S Assay-on-Demand (Applied Biosystems) was used as an endogenous control in all assays. All assays were run on 384 well plates so that each cDNA sample was run in triplicate for the target gene and the endogenous control. qRT PCR was performed on an ABI Prism 7900 Sequence Detection System and the results assessed by relative quantification of gene expression using the ΔΔCT method.
Statistical analysis
Data for pregnancy hormone levels and gene expression were analyzed by one-way analysis of variance (ANOVA) (only assessed in parous rats). Some mammary tumor end-points (latency and multiplicity) were analyzed by two-way ANOVA, using nulliparous or parous, and treatments as independent variables. Cell proliferation and apoptosis were only assessed in parous rats, and because the estrous cycle may influence mammary cell proliferation and apoptosis in rats (low proliferation: pro-estrus, estrus and the second part of diestrus; and high proliferation: metestrus and the first part of diestrus), the proliferation and apoptosis indices data were analyzed by two-way ANOVA, using the stage of estrous cycle and E2/leptin exposure as independent variables. Where appropriate, comparisons between groups were done using Holm-Sidak method. Kaplan-Meier curves were used to compare differences in tumor incidence, followed by the log-rank test. Tumor incidence was also analysed just for post-DMBA weeks 13 and 22, and in this analysis nulliparous control rats were compared to parous control rats and either parous rats exposed to leptin or E2 were included to the analysis. Final tumor incidence was determined using Chi-square test. All tests were performed using the SPSS SigmaStat software, and differences were considered significant if the p-value was less than 0.05. All probabilities were two-tailed.
Results
Effects on weight gain and pregnancy hormone levels
Neither E2 nor leptin affected weight gain during pregnancy (Table 1). Birth weights of the pups also were similar, as were the numbers of pups born per litter (Table 1). The concentrations of circulating E2 and leptin, measured in serum samples collected on day 19 of pregnancy, are shown in Figure 1. Leptin levels were significantly higher in the leptin exposed dams when compared to either the E2 or control dams (p<0.001). Circulating E2 levels were significantly higher in the E2-treated group, when compared to the control or leptin-treated dams (p=0.004).
Table 1. Effects of exposure to leptin or estradiol on rat dams’ pregnancy weight gain.
Rats were administered DMBA at 50 days of age and mated 2 weeks later. Pregnant dams were exposed to leptin, estradiol or vehicle control between days 8 of and 19 of gestation. Body weight values (in grams) collected at base-line and on the last week of gestation are expressed as the mean ± SEM. There were no significant differences in pregnancy weight gain among the groups.
| Treatment | Baseline (g) (mean ± SEM) |
Third week of gestation (g) (mean ± SEM) |
Net weight gain (g) (mean ± SEM) |
|---|---|---|---|
| Control | 223.98 ± 2.15 | 267.92 ± 3.04 | 43.20 ± 4.16 |
| Leptin | 224.39 ± 2.52 | 266.44 ± 2.82 | 42.59 ± 1.53 |
| Estradiol | 224.00 ± 1.94 | 264.76 ± 2.13 | 40.90 ± 1.83 |
Figure 1.
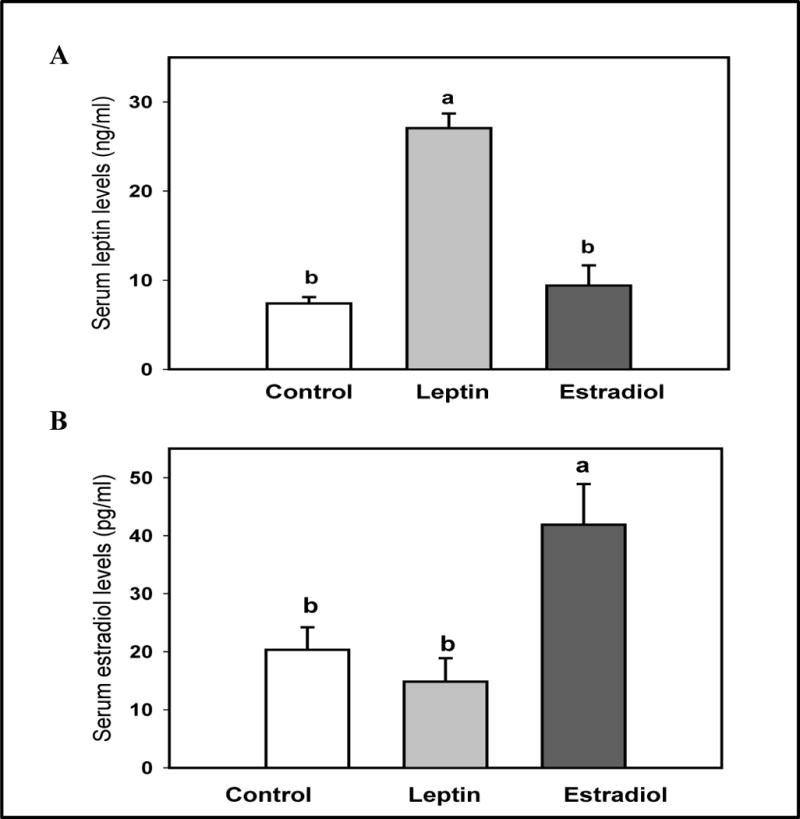
Serum (A) leptin and (B) estradiol levels on day 19 of pregnancy in rat dams exposed to 15 μg of leptin or 10 mg of estradiol in between days 8 and 19 of gestation. All values are expressed as the mean ± SEM of 5–7 rats/group. Means with a different letter are significantly different from each other: p<0.05.
Effects on mammary tumorigenesis
Because pregnancy has a transient and long-term effect on breast cancer risk, we considered tumors which developed between weeks 1 and 12 after DMBA as early appearing tumors, and those developing on week 13 or after as long-term. Twelve weeks post DMBA treatment coincided with completion of mammary gland involution in parous rats, as the rats became pregnant two weeks after DMBA, gave birth five weeks after DMBA and started undergoing involution 8 weeks after DMBA. It then takes 4 weeks for the rat mammary gland to return to a pre-pregnancy stage (13, 35); i.e., this occurred on week 12 in our study.
Effect of E2 and leptin exposures in nulliparous rats
We first determined whether a two-week exposure of nulliparous rats to E2 or leptin alters mammary tumorigenesis. Table 2 indicates that the mean mammary tumor latency in nulliparous control rats is about 13 weeks. Tumor latency did not differ among the nulliparous control, E2 or leptin exposed rats. In the vehicle-treated nulliparous rats, 41% of the tumors become palpable during weeks 1 and 12, and 59% during weeks 13–22. The majority of the tumors in the leptin group (79%) were detected before week 13, while in the E2 group 23% of the tumors were detected early and 77% were detected after week 12 (p<0.004). Final mammary tumor incidence and multiplicity were similar in the three groups of nulliparous rats exposed to vehicle, leptin or E2. These results are shown in Table 2 and Figure 2A.
Table 2. Effects of a 3-week exposure to leptin or estradiol on later mammary carcinogenesis in nulliparous and parous rats.
Nulliparous rats were administered DMBA at 50 days of age and 3 weeks later treated with vehicle, leptin or estradiol for 2 weeks. Parous rats were also administered DMBA at 50 days of age, and mated 2 weeks later. Pregnant dams were exposed to leptin or estradiol between days 8 of and 19 of gestation. Values for tumor latency and multiplicity are expressed as the mean ± SEM. Total tumor incidence values are expressed as the number of rats with mammary tumors per group, and between weeks 0–12 or 13–22 after DMBA as number and percentage of all tumors per group. Values marked with a different letter are significantly different from each other: p<0.05.
| Treatment | Tumor incidence (%) Weeks after treatment |
Tumor multiplicity (mean±SEM) |
Tumor latency (weeks) (mean±SEM) |
||
|---|---|---|---|---|---|
| 0–22 | 0–12 | 13–22 | |||
| Nulliparous rats | |||||
| Control | 17/29 59% |
7/17 41% |
10/17a 59% |
1.88 ± 0.40 | 13.29 ± 1.09 |
| Leptin | 14/28 50% |
11/14 79% |
3/14b 21% |
1.43 ± 0.14 | 11.14 ± 1.22 |
| Estradiol | 22/41 54% |
5/22 23% |
17/22c 77% |
1.73 ± 0.24 | 14.9 ± 0.84 |
| Parous rats | |||||
| Control | 14/43b 32% |
13/14 93% |
1/14a 7% |
1.93 ± 0.29 | 7.07 ± 0.76a |
| Leptin | 26/40a 65% |
14/26 54% |
12/26b 46% |
1.33 ± 0.12 | 12.27 ± 1.11b |
| Estradiol | 19/42a,b 45% |
9/19 47% |
10/19b 53% |
1.37 ± 0.18 | 12.31 ± 1.38b |
Figure 2.
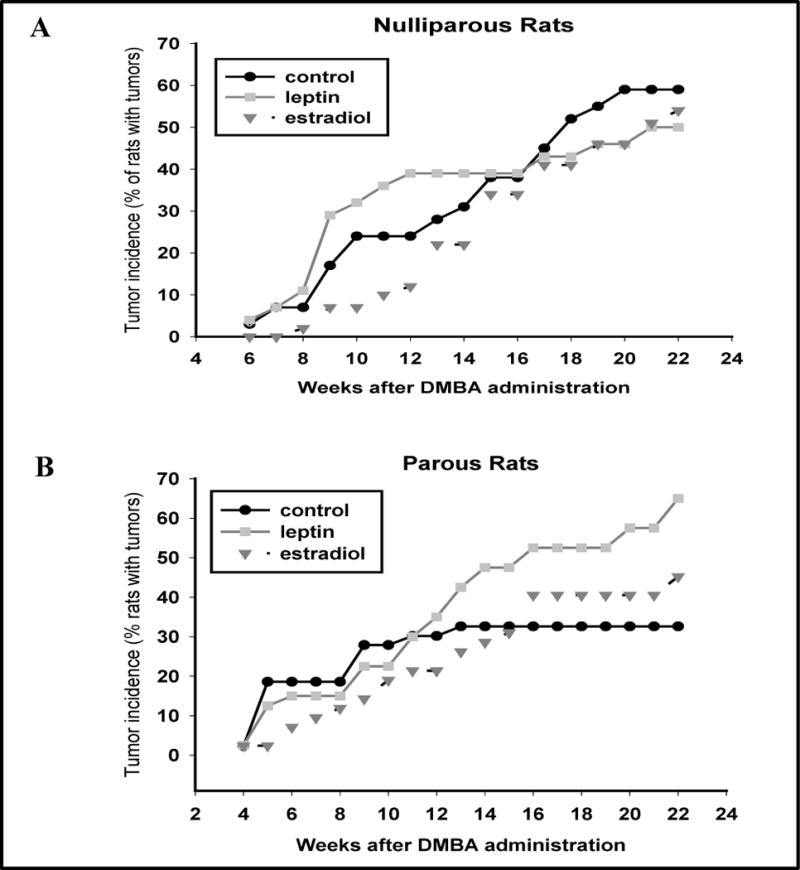
Effects of a daily exposure to leptin or estradiol, starting 3 weeks after DMBA administration and continuing for two weeks on mammary tumorigenesis (A) in nulliparous rats, or (B) in parous rats (received hormonal treatments between gestation days 8 and 19) on mammary tumorigenesis. Tumor incidence values are expressed as percentage of animals with mammary tumors in the control, leptin or E2 groups. No statistical significance was seen among the nulliparous rats, but parous rats exposed to leptin during pregnancy exhibited significantly higher mammary tumor incidence than vehicle treated parous controls (p<0.039).
Effect of parity
Next, we compared mammary tumorigenesis in the vehicle (control) treated nulliparous and parous rats. Latency of mammary tumor appearance was shorter in the parous than nulliparous rats (p<0.005). In the parous control rats, 93% of the tumors appeared during weeks 1 and 12, compared to 41% in the nulliparous group (p<0.001). The final tumor incidence during weeks 1 and 22 (p<0.001) and during weeks 13 and 22 (p<0.001) in the nulliparous controls was higher than in the parous rats, but tumor multiplicity was similar (Table 2). Thus, similar to women, we found that after a transient increase in mammary cancer risk, pregnancy provided protection against breast cancer in rats.
Effect of E2 and leptin exposures during pregnancy
In the parous control group, all but one (7%) of 14 tumors became palpable within 12 weeks of DMBA exposure, while 12 (46%) of the 26 tumors in the leptin group and 10 (53%) of the 19 tumors in the E2 group appeared after week 12 of pregnancy (p<0.018) (Table 2). This is similar than what was seen in nulliparous control rats of which 59% developed mammary tumors after week 12. Thus, although the mean tumor latency period was longer in both the leptin (p<0.001) and E2 treated parous rats (p<0.002) than in the vehicle treated parous rats, it did not differ between the parous hormone treated rats and nulliparous control rats; i.e., the treatments did not delay tumor development.
To determine whether an exposure to leptin or E2 during pregnancy affected mammary tumorigenesis, differences were assessed between week 13 and 22. Both the leptin (p<0.001) and E2 groups (p<0.0037) exhibited significantly higher mammary tumor incidence than the parous control rats (Figure 2), but neither group differed from nulliparous control rats. At the end of the monitoring period, final tumor incidence was higher in the parous rats exposed during pregnancy to either leptin (65%) or E2 (45%), when compared to the controls (33%) (Table 2), but this difference reached statistical significance in the leptin group (p<0.039). However, tumor incidence Tumor multiplicity among the groups was not statistically significant (Table 2).
Effects on mammary cell proliferation and apoptosis
Cell proliferation and apoptosis were determined in mammary glands obtained from rats sacrificed 22 weeks after exposure to DMBA. Figure 3 shows that the proliferation index, determined by PCNA staining, was significantly higher in the mammary glands of E2 treated parous rats compared to those of vehicle treated parous control rats (p<0.001). The number of apoptotic cells present in the mammary glands of rats in the two treatment groups and controls were determined using the ISOL assay. There were no significant differences among these two treatment groups, when compared to the controls (p=0.17) (Figure 4).
Figure 3.
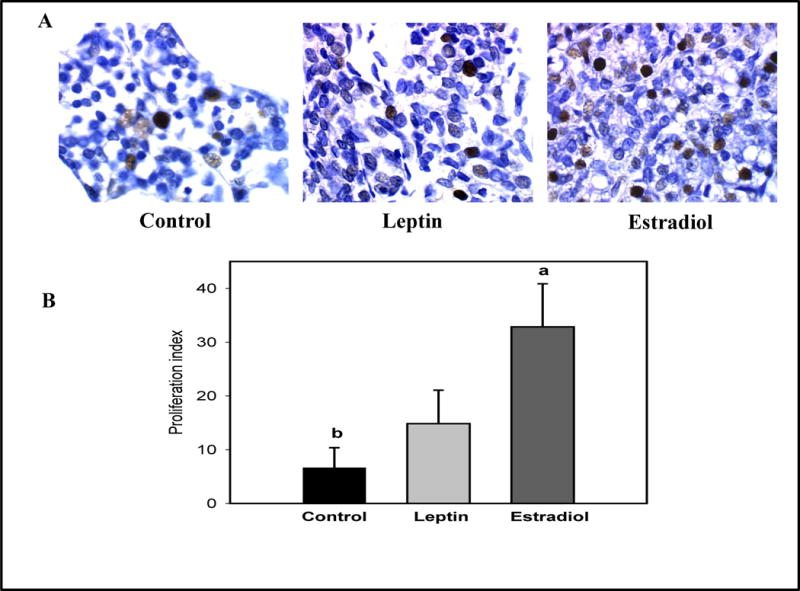
Effects of an exposure to leptin or estradiol during pregnancy on mammary gland cell proliferation, determined 17 weeks post pregnancy. (A) PCNA staining (dark nuclei) in representative mammary gland sections (400× magnification), and (B) proliferation index (percentage of PCNA positive cells/1000 cells). All values are expressed as the mean ± SEM, n = 6 rats/group. Means with a different letter are significantly different from each other: p<0.05.
Figure 4.
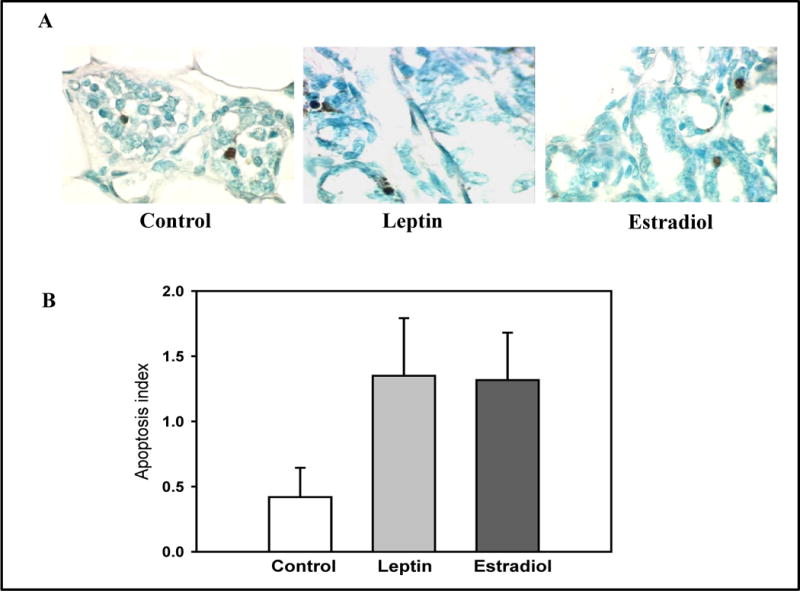
Effects of an exposure to leptin or estradiol during pregnancy on mammary gland apoptosis, determined 17 weeks post pregnancy. (A) ISOL staining in representative mammary gland sections (400× magnification), and (B) apoptosis index (percentage of ISOL positive cells/1000 cells). All values are expressed as the mean ± SEM of 6 rats/group.
Gene microarray analysis
To explore the long-term effects on gene expression in the mammary glands of rats exposed to E2 or leptin during pregnancy, microarray experiments were performed using RNA extracted from mammary glands collected 22 weeks after DMBA exposure. In the comparison between the control and leptin groups, 352 genes were found to be differentially expressed (criteria for differential expression was 2-fold difference and p<0.05). The comparison between the control and E2 groups revealed 252 differentially expressed genes. We then compared the E2 and leptin groups, and found only 11 genes to be differentially expressed between these two groups. For this reason, these two groups were combined into one high risk group and compared to controls. In this analysis, we identified 143 genes associated with changes in tumorigenesis between the control and high risk groups. Of those, 62 genes were down-regulated (Table 3) and 80 genes up-regulated (Table 4) in the high risk group compared to controls.
Table 3.
Genes downregulated in parous rats treated with E2 or leptin during pregnancy, compared to vehicle exposed parous rats
| Gene Name | Symbol | Accession # | Function | Category | Fold |
|---|---|---|---|---|---|
| Cytochrome c oxidase, subunit Va | Cox5a | rc_AI104513_at | Electron transport | Metabolism | 0.41 |
| Glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase | Gne | rc_AI145931_at | Amino sugar biosynthesis | Metabolism | 0.49 |
| Parathymosin | Ptms | M33025_s_at | Regulation of glycolysis | Metabolism | 0.34 |
| Acidic nuclear phosphoprotein 32 family, member A | Anp32a | rc_AI070967_at | Nucleocytoplasmic transport | Transporter/differentiation | 0.42 |
| Apolipoprotein E | Apoe | X04979_at | Lipid transport | Transporter | 0.42 |
| Aquaporin 5 | Aqp5 | U16245_g_at | Carbon dioxide and water transport | Transporter | 0.42 |
| Calcium channel, voltage-dependent, L type, alpha 1D subunit | Cacna1d | D38101_s_at | Calcium ion transport | Transporter | 0.47 |
| Epsin 2 | Epn2 | AF096269_at | Endocytosis/Notch signal transduction | Transporter/ signal transduction/ embryonic development | 0.4 |
| Ferritin light polypeptide | Ftl1 | rc_AI231807_g_at | Iron transport | Transporter | 0.44 |
| Lectin, galactoside-binding, soluble, 9 | Lgals9 | U72741_at | Ion transport/NFκB signaling | Transporter/Signal Transduction | 0.41 |
| Low density lipoprotein receptor-related protein 3 | Lrp3 | AB009463_g_at | Lipoprotein receptor/endocytosis | Transporter | 0.43 |
| PDZK1 interacting protein 1 | Pdzk1ip1 | rc_AA892264_at | Glucose transport | Transporter | 0.37 |
| Rab38, member RAS oncogene family | Rab38 | rc_AI136175_at | Vesicle mediated transport | Transporter | 0.37 |
| Secretogranin V (7B2 protein) | Scg5 | M63901_g_at | Regulation of hormone secretion | Transporter | 0.34 |
| Sodium channel, nonvoltage-gated 1 gamma | Scnn1g | X77933_at | Sodium ion transport | Transporter | 0.47 |
| Integrin beta 4 | Itgb4 | U60096_at | Integrin signaling | ECM/cell motility | 0.49 |
| Syndecan 4 | Sdc4 | S61868_g_at | Focal adhesion assembly | ECM | 0.5 |
| Transforming growth factor, beta 3 | Tgfb3 | U03491_g_at | Growth Factor | ECM/EMT | 0.34 |
| Tropomyosin 3, gamma | Tpm3 | L24776_at | Actin binding | Structural | 0.11 |
| Troponin T type 2 cardiac | Tnnt2 | M80829_at | Actin binding | Muscle contraction | 0.35 |
| Alpha-2-macroglobulin | A2m | X13983mRNA_at | Inflammatory response | Immune | 0.33 |
| Granzyme F | Gzmf | U57063_at | Protease | Immune | 0.45 |
| Kininogen 1 | Kng1 | K02814_g_at | Inflammatory response | immune | 0.47 |
| Lipopolysaccharide binding protein | Lbp | L32132_at | Inflammatory response | Immune | 0.43 |
| Lipocalin 2 | Lcn2 | rc_AA946503_at | Protease and iron binding/ transporter | Immune/Apoptosis | 0.47 |
| Myxovirus (influenza virus) resistance 2 | Mx2 | X52713_at | Antiviral | Immune | 0.36 |
| Protein tyrosine phosphatase, non-receptor type 1 | Ptpn1 | M33962_at | Endocytosis/Insulin receptor signaling | UPR/Signal Transduction | 0.5 |
| Heat shock protein 1A | Hspa1a | Z27118cds_s_at | Stress-inducible chaperone | UPR/Antiproliferative | 0.35 |
| B-cell CLL/lymphoma 2 | Bcl2 | L14680_g_at | Intrinsic apoptotic pathway | Antiapoptosis | 0.5 |
| Cystatin C | Cst3 | rc_AI231292_g_at | Cystein protease inhibitor | Apoptotsis | 0.49 |
| Guanine nucleotide binding protein (G protein), beta polypeptide 1 | Gnb1 | U88324_at | GTPase | Apoptosis/Proliferation/Signal transduction | 0.33 |
| G protein-coupled receptor kinase 1 | Grk1 | U63971_at | G protein coupled receptor signaling | Apoptosis/Signal transduction | 0.36 |
| Nitric oxide synthase 3, endothelial cell | Nos3 | U02534_at | NO synthesis | Apoptosis/Angiogenesis | 0.42 |
| SMAD family member 3 | Smad3 | U66479_at | Transcription factor/ TGFβ signaling | Apoptosis/ Signal transduction | 0.48 |
| Adrenergic receptor, alpha 2b | Adra2b | M32061_at | G-protein coupled receptor | Signal transduction/ Angiogenesis | 0.43 |
| ArfGAP with dual PH domains 1 | Adap1 | U51013_at | Inositol phosphate-mediated signaling | Signal transduction | 0.48 |
| Cbp/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 1 | Cited1 | AF104399_g_at | Transcription factor/ Estrogen receptor- and TGFβ signaling | Signal Transduction | 0.48 |
| GNAS complex locus | Gnas | L10326_at | G-protein alpha subunit | Signal transduction/ differentiation | 0.48 |
| G protein-coupled estrogen receptor 1 | Gper | U92802_at | Estrogen receptor activity | Signal Transduction | 0.43 |
| Guanylate cyclase 1, soluble, beta 2 | Gucy1b2 | M57507_at | Guanylate cyclase | Signal transduction | 0.31 |
| High mobility group box 1 | Hmgb1 | rc_AI029805_at | Transcription Factor/cytokine | Signal transduction/immune | 0.38 |
| Mitogen activated protein kinase kinase 1 | Map2k1 | rc_AI178835_at | MAPK signaling | Signal Transduction/ Differentiation | 0.45 |
| Prepronociceptin | Pnoc | S79730_s_at | Opiod-like orphan receptor ligand | Signal transduction | 0.43 |
| Ret proto-oncogene | Ret | AF042830_at | Proto-oncogene/ receptor tyrosine kinase | Signal transduction/ embryonic development | 0.45 |
| Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, epsilon polypeptide | Ywhae | rc_AA965154_at | Calcium/ calmodulin- dependent signal transduction | Signal transduction | 0.48 |
| Brain abundant, membrane attached signal protein 1 | Basp1 | D14441_at | Transcriptional corepressor | Differentiation | 0.46 |
| Ceruloplasmin (ferroxidase) | Cp | Y12178_at | Copper transport | Differentiation | 0.44 |
| Casein alpha s1 | Csn1s1 | J00710_at | Milk protein | Differentiation | 0.46 |
| D4, zinc and double PHD fingers family 1 | Dpf1 | X66022mRNA#3_i_at | Transcription factor | Differentiation | 0.41 |
| Jun-B proto-oncogene | Junb | X54686cds_at | Transcription factor/proto-oncogene | Differentiation/response to hormone stimulus | 0.34 |
| Keratin 19 | Krtt19 | X81449cds_at | Keratin | Differentiation/response to estrogen stimulus | 0.36 |
| Lactalbumin alpha | Lalba | X00461_at | Lactose biosynthesis | Differentiation | 0.49 |
| Growth hormone releasing hormone | Ghrh | U41183_at | Growth hormone receptor binding | Growth factor | 0.31 |
| H3 histone, family 3B | H3f3b | rc_AA875069_at | Nucleosome assembly | Reponse to hormone stimulus/gene expression | 0.25 |
| Prolactin family 8, subfamily a, member 4 | Prl8a4 | AB009889_f_at | Hormone activity | Hormone | 0.32 |
| FEV (ETS oncogene family) | FEV | U91679_at | Transcription factor | Gene expression | 0.48 |
| Forkhead box O1A | Foxo1a | rc_AA893671_at | Transcription factor | Gene expression/proliferation/Antiapoptosis | 0.46 |
| Proline-rich nuclear receptor coactivator-1 | Pnrc1 | rc_AI235492_at | Nuclear receptor coactivator | Gene expression | 0.45 |
| Ribosomal protein L10 | Rpl10 | rc_AA945611_at | Translation | Gene expression | 0.48 |
| Ribosomal protein L30 | Rpl30 | D84480_s_at | Translation | Gene expression | 0.47 |
| Ribosomal protein S14 | Rps14 | rc_AA945806_at | Translation | Gene expression/ differentiation | 0.49 |
| Calpain 8 | Capn8 | D14478_s_at | Proteolysis | Protein regulation | 0.39 |
Table 4.
Genes upregulated in parous rats treated with E2 or leptin during pregnancy, compared with vehicle treated parous rats
| Gene name | Symbol | Accession # | Function | Category | Fold |
|---|---|---|---|---|---|
| Acyl-CoA dehydrogenase, short/branched chain | Acadsb | U64451_at | Fatty acid metabolism | Metabolism | 3.3 |
| Aminolevulinate, delta-, synthase 1 | Alas1 | J03190_g_at | Heme biosynthesis/ Amino acid metabolism | Metabolism | 2 |
| Antizyme inhibitor 1 | Azin1 | D89983_at | Polyamine biosynthesis | Metabolism | 2.2 |
| Biotinidase | Btd | rc_AI236721_r_at | Nitrogen compound metabolism | Metabolism | 2.9 |
| Fumarate hydratase | Fh | rc_AI171734_s_at | Tricarboxylic acid cycle | Metabolism | 2.4 |
| Glutamate cysteine ligase, modifier subunit | Gclm | rc_AI233261_i_at | Glutathione metabolism | Metabolism | 2.2 |
| Glutathione S-transferase, mu 5 | Gstm5 | U86635_g_at | Glutathione metabolism | Metabolism/response to estrogen stimulus | 2.2 |
| Hypoxanthine phosphoribosyltransferase 1 | Hprt1 | M63983_s_at | Purine metabolism | Metabolism | 2.2 |
| Methionine adenosyltransferase II, alpha (Mat2a) | Mat2a | J05571_s_at | Methionine metabolism | Metabolism | 2.1 |
| Nicotinamide nucleotide transhydrogenase | Nnt | rc_AA891872_at | Proton transport | Metabolism | 2 |
| Ornithine aminotransferase | Oat | rc_AA893325_at | Amino acid metabolism | Metabolism | 2 |
| Phosphoribosyl pyrophosphate synthetase 1 | Prps1 | X16554_at | Purine biosynethesis | Metabolism | 2.1 |
| Protein-L-isoaspartate (D-aspartate) O-methyltransferase 1 | Pcmt1 | M26686_g_at | Amino acid metabolism | Metabolism | 2.3 |
| Pyruvate dehydrogenase E1 alpha 1 | Pdha1 | Z12158cds_at | Glycolysis | Metabolism | 2.1 |
| Blocked early in transport 1 homolog | Bet1 | U42755_at | Vesicular membrane trafficking | Transporter | 2.3 |
| Exocyst complex component | Exoc5 | U79417_at | Exocytosis | Transporter | 2.6 |
| Nucleoporin 155 | Nup155 | Z21780_at | Nucleocytoplasmic transport | Transporter | 2.1 |
| Solute carrier family 11 member 2 | Slc11a2 | AF008439_at | Iron transport | Transporter | 2.5 |
| Solute carrier family 16, member 1 (monocarboxylic acid transporter 1) | Slc16a1 | rc_AI145680_s_at | Organic anion transport | Transporter | 2.1 |
| Synaptosomal-associated protein 23 | Snap23 | rc_AA892759_at | Exocytosis/protein transport | Transporter | 2 |
| Syntaxin 12 | Stx12 | AF035632_s_at | Vesicle mediated transport | Transporter | 2.2 |
| Trans-golgi network protein 1 | Tgoln1 | X53565_at | Endosome transport | Transporter | 3.1 |
| Transmembrane emp24 domain trafficking protein 2 | Tmed2 | X92097_at | Protein transport | Transporter | 2.2 |
| Uncoupling protein 3 | Ucp3 | AF035943_at | Hydrogen ion transmembrane transporter/Oxidative phoshorylation uncoupling | Transporter/Response to hormone stimulus | 2.6 |
| Annexin A4 | Anxa4 | rc_AI171167_at | Exocytosis | ECM | 2.1 |
| CD36 molecule (thrombospondin receptor) | Cd36 | AB005743_g_at | Thrombospondin receptor | ECM | 4.5 |
| Collagen, type 1, alpha 1 | Col1a1 | Z78279_at | ECM structural protein | ECM/EMT | 2.5 |
| Discs, large homolog 1 | Dlgh1 | U14950_at | Cell adhesion | ECM | 2.1 |
| Fat tumor suppressor homolog 1 (Drosophila) | Fat1 | L41684mRNA_at | Cell-cell adhesion | ECM | 2.3 |
| Prolyl 4-hydroxylase aplpha polypeptide 1 | P4ha1 | X78949_at | Collagen fibril organization | ECM | 2 |
| caldesmon 1 | Cald1 | rc_AI180288_s_at | Actin binding | Structural | 2 |
| LIM motif-containing protein kinase 1 | Limk1 | D31873_g_at | Kinase involved in actin dynamics | Structural | 2.1 |
| Cathepsin C | Ctsc | D90404_g_at | Aminopeptidase | Immune | 2.2 |
| Proteasome (prosome, macropain) subunit, alpha type 2 | Psma2 | E03358cds_at | Antiviral response | Immune | 2.3 |
| Protein phosphatase, Mg2+/Mn2+ dependent 1B | Ppm1b | S90449_at | Serine/Threonine phosphatase/Antiviral | Immune | 3.1 |
| Calnexin | Canx | L18889_at | Protein folding | UPR | 2 |
| Eukaryotic translation initiation factor 2-alpha kinase 2 | Eif2ak2 | rc_AI013987_s_at | Interferon-inducible RNA-dependent protein kinase/antiviral response | UPR/immune | 2.6 |
| Nuclear factor, erythroid derived 2, like 2 | Nfe2l2 | rc_AI177161_g_at | Transcription factor | UPR/antioxidant response | 2.1 |
| Stress-associated endoplasmic reticulum protein 1 | Serp1 | AF100470_g_at | Protein glycosylation | UPR | 2.4 |
| Cytochrome c, somatic | Cycs | rc_AI008815_s_at | Electron transport chain | Apoptosis/metabolism | 2.9 |
| Dynamin 1-like | Dnm1l | AF019043_at | Membrane fission | Apoptosis | 2 |
| Mitogen-activated protein kinase 9 | Mapk9 | rc_AI231354_at | Cysteine-type endopeptidase/MAPK signaling | Apoptosis/Signal Transduction | 2.5 |
| Glutamate cysteine ligase, modifier subunit | Gclm | rc_AI233261_i_at | Glutathione metabolism | Antiapoptosis | 2.2 |
| Serpine1 mRNA binding protein 1 | Serbp1 | rc_AA893338_at | mRNA 3′-UTR binding | Antiapoptotic | 2.2 |
| Heme oxygenase (decycling) 1 | Hmox1 | J02722cds_at | Heme catabolism | Angiogenesis/Antiapoptosis | 2.1 |
| Vascular endothelial growth factor A human | Vegfa | L20913_s_at | Growth factor | Angiogenesis | 3.2 |
| v-Crk sarcoma virus CT10 oncogene homolog (avian) | Crk | D44481_at | Oncoprotein/adaptor protein/actin cytoskeleton organization | Signal transduction/structural | 2.4 |
| Guanylate cyclase 2G | Gucy2g | U33847_at | cGMP biosynthesis | Signal transduction | 3.3 |
| Multiple inositol- polyphosphate phosphatase 1 | Minpp1 | rc_AI111401_s_at | Phosphatase | Signal transduction | 2 |
| Neuroblastoma RAS viral (v-ras) oncogene homolog | Nras | rc_AA943331_s_at | GTPase/actin cytoskeleton organization | Signal transduction/structural | 2.3 |
| O-linked N-acetylglucosamine (GlcNAc) transferase (UDP-N-acetylglucosamine:polypeptide-N-acetylglucosaminyl transferase) | Ogt | U76557_at | O-GlcNAcylation of proteins | Signal transduction/Insulin receptor signaling | 2 |
| Protein kinase, cAMP dependent regulatory, type II beta | Prkar2b | M12492mRNA#1_g_at | PKA signaling/fatty acid metabolism | Signal transduction/ metabolism | 2.2 |
| RAB28, member RAS oncogene family | Rab28 | X78606_at | GTPase | Signal transduction | 2.1 |
| Arginine-glutamic acid dipeptide repeats | Rere | U44091_at | Chromatin remodeling/transcription factor | Signal transduction | 2.2 |
| calpastatin | Cast | Y13591_s_at | Endopeptidase inhibitor | Cell cycle | 4.3 |
| Eukaryotic translation initiation factor 4E | Eif4 | X83399_at | Translation initiation | Cell cycle | 2.2 |
| Cyclin G1 | Ccng1 | X70871_at | Cyclin | Cell growth/mitosis | 2.9 |
| Dynein, cytoplasmic 1 light intermediate chain 2 | Dync1li2 | AB008521_s_at | Centrosome localization/retrograde organelle transport | Mitosis/transport | 2.3 |
| Microtubule-associated protein, RP/EB family, member 1 | Mapre1 | U75920_at | Microtubule binding | Mitosis | 2.4 |
| Growth hormone receptor | Ghr | Z83757mRNA_g_at | Growth hormone signaling | Growth-promoting | 2.9 |
| Hydroxysteroid (17-beta) dehydrogenase 12 | Hsd17b12 | U81186_at | Estrogen biosynethesis/extracellular matrix organization | Growth-promoting/ECM | 2.6 |
| Pleiotrophin | Ptn | rc_AI102795_at | Growth factor | Growth promoting | 2.3 |
| Peroxisomal biogenesis factor 2 | Pex2 | E03344cds_s_at | Peroxisome biogenesis | Antiproliferative | 2.1 |
| Heterogeneous nuclear ribonucleoprotein K | Hnrpk | D17711cds_s_at | mRNA processing | Gene expression | 2.1 |
| Iron responsive element binding protein 2 | Ireb2 | U20181_at | Transcriptional regulation | Gene expression /Iron homeostasis | 2.3 |
| Pleiomorphic adenoma gene-like 1 | Plagl1 | rc_AA900750_s_at | DNA binding/ transcriptional regulation | Gene expression | 2.5 |
| Ring finger protein 4 | Rnf4 | AF022081_at | Nuclear receptor coregulator | Gene expression | 2.5 |
| Splicing factor proline/glutamine-rich | Sfpq | AF036335_g_at | mRNA splicing | Gene expression | 2.5 |
| Transformer 2 beta homolog (Drosophila) | Tra2b | rc_AA851749_s_at | mRNA splicing | Gene expression/response to ROS | 2.3 |
| Aspartyl-tRNA synthetase | Dars | rc_AI009682_s_at | Protein biosynethesis | Protein regulation | 3.1 |
| Phosducin-like | Pdcl | L15354_s_at | Protein folding | Protein regulation | 2.6 |
| Praja ring finger 2 | Pja2 | rc_AA894089_s_at | Regulation of protein kinase A signaling/protein ubiquitination | Protein regulation | 2.3 |
| Prostaglandin F2 receptor negative regulator | Ptgfrn | rc_AI145502_s_at | Prostaglandin inhibitor/negative regulation of translation | Protein regulation | 2 |
| Tripeptidyl peptidase II | Tpp2 | rc_AI071507_s_at | Proteolysis | Protein regulation | 2.3 |
| Ubiquitin-conjugating enzyme E2D 2 | Ube2d2 | U13176_at | Protein degradation | Protein regulation | 2 |
| Ubiquitin-conjugating enzyme E2G 1 | Ubc7 | AF099093_at | Protein degradation | Protein regulation | 2.1 |
| Delta-like 1, Drosophila | Dll1 | U78889_at | Notch receptor ligand | Development/Differentiation/cell signaling | 2.9 |
| Cytochrome c oxidase subunit Vb | Cox5b | D10952_i_at | Cytochrome-c oxidase activity | Response to hormone stimulus | 2.2 |
| Oxidation resistance 1 | Oxr1 | rc_H33461_at | Cell wall macromolecule catabolic process | Response to oxidative stress | 2.6 |
| Protein phospatase 3, regulatory subunit B, alpha isoform,type 1 | Ppp3r1 | D14568_at | Protein phosphatase | EMT | 2.7 |
Confirmation of changes in gene expression by qRT PCR
Several of the genes that were differentially expressed in the mammary glands of parous rats exposed to either leptin or E2 during pregnancy, compared to controls, are involved in cell growth, survival and angiogenesis. These genes included Mapk9 (mitogen activated protein kinase 9), Nras (neuroblastoma ras oncogene), Ptn (pleiotrophin), Vegfa (vascular endothelial growth factor) and Eif4e (eukaryotic initiation translation factor 4e), which were up-regulated in the mammary gland of rats exposed to leptin or E2 during pregnancy when compared to vehicle treated controls (Table 4). We also found that the expression of genes inducing mammary epithelial differentiation, such as α-lactalbumin and α-casein, were down-regulated in the leptin or E2 exposed dams (Table 3).
Differential expression of these genes was confirmed by real-time PCR. As illustrated in Figure 5, transcripts for Vegfa and Ptn were more abundant in the rats treated with either leptin or E2 during pregnancy than in the controls (p<0.001 and p<0.001, respectively). Vegfa levels were 3.8 and 6.8-fold higher in mammary glands of leptin and E2 treated dams than in the controls, respectively (Figure 5A). Ptn mRNA levels were 3.3-fold higher in leptin-treated and 21-fold higher in E2-treated dams than in the controls (Figure 5B).
Figure 5.
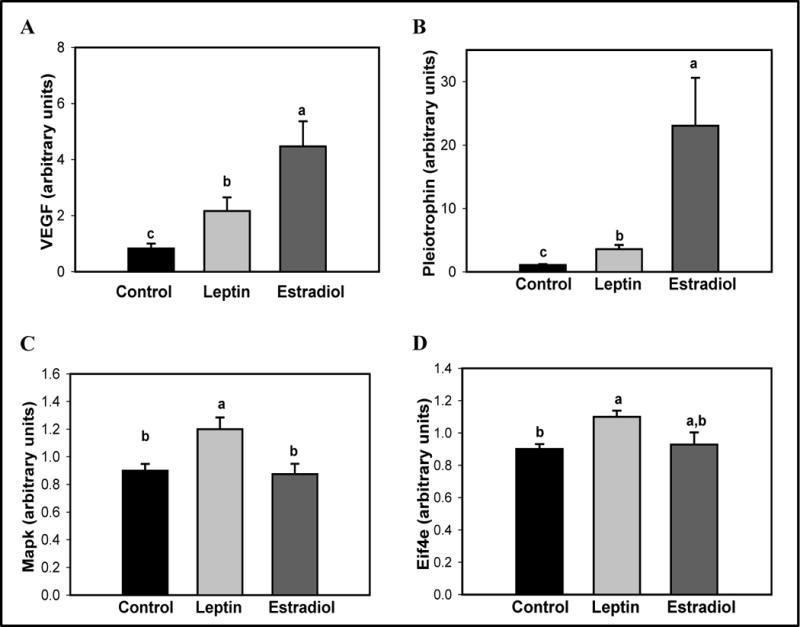
Effects of an exposure to leptin or estradiol during pregnancy on mammary gland mRNA levels of (A) Vegfa, (B) Ptn, (C) Mapk 9, and (D) Eif4e, determined 17 weeks after pregnancy. All values are expressed as the mean ± SEM of 6–8 rats/group. Means with a different letter are significantly different from each other: p<0.05.
RT-PCR data indicated that the levels of Mapk9 mRNA were 1.3-fold higher in the leptin-treated parous rats than in the vehicle- or E2-treated dams (Figure 5C) (p=0.008). Transcription levels of Eif4e were 1.2-fold higher in mammary glands of leptin treated animals compared to the controls (p=0.003) (Figure 5D). Differential expression of Nras in the microarray was not confirmed by real-time PCR.
Comparison to data obtained in previous studies assessing effect of parity on gene expression
Several earlier studies have outlined a gene expression signature characterizing the effect of parity on the mammary gland. We investigated whether there were any similarities between these signatures and changes in gene expression induced by an exposure to excess leptin or E2 during pregnancy. For that purpose, we used the tables of differentially expressed genes between parous and nulliparous rat and mouse strains generated in studies by D’Cruz et al. (13) and Blakely et al. (14), and humans by Asztalos et al. (36).
Several common genes in the parous rats exposed to E2 or leptin versus vehicle, and parous versus nulliparous animals and women were identified. The genes identified in this comparison are shown in Table 5. Importantly, genes that were up-regulated (or down-regulated) in parous rats, compared to nulliparous rats, were also up-regulated (or down-regulated) in vehicle-treated parous rats, compared to parous rats treated with E2 or leptin during pregnancy, suggesting that these hormonal exposures prevented parous-induced signaling changes in the mammary glands. For example, TGFβ3 has been reported to be up-regulated in parous animals and humans, compared to nulliparous controls, and we found that is was also up-regulated in parous control rats, compared to parous rats treated with leptin or E2 during pregnancy. The down-regulated genes are those that induce differentiation (Casein alpha s1, Csn1, Ceruloplasmin, Cp, and Lactalbumin alpha, Lalba) or regulate immune functions (Lipocalin 2, Lcn2, and Lipopolysaccharide binding protein, Lbp), while the up-regulated genes are those that promote growth (Growth hormone receptor, Ghr, and Ptn) and angiogenesis (VegfA) and induce epithelial to mesenchymal transition (Collagen type 1 alpha 1, Col1a1).
Table 5.
| Genes up-regulated in parous human breast and/or rodent mammary gland, but down-regulated in parous mammary gland of rats treated with E2 or leptin during pregnancy | ||||
|---|---|---|---|---|
| Gene name | Symbol | Reference | Category | Fold-change (p<0.05) |
| Aquaporin 5 | Aqp5 | (14) | Transporter | 0.42 |
| Casein alpha s1 | Csn1 | (13) | Differentiation | 0.46 |
| Ceruloplasmin | Cp | (14) | Differentiation | 0.44 |
| Lactalbumin | Lalba | (13) | Differentiation | 0.49 |
| Lipocalin 2 | Lcn2 | (14) | Immune | 0.47 |
| Lipopolysaccharide binding protein | Lbp | (14, 36) | Immune | 0.43 |
| Transforming growth factor, beta 3 | Tgfb3 | (13, 14, 36) | Growth inhibition | 0.34 |
| Genes down-regulated in parous human breast and/or rodent mammary gland, but up-regulated in parous mammary gland of rats treated with E2 or leptin during pregnancy | ||||
|---|---|---|---|---|
| Gene name | Symbol | Reference | Category | Fold-change (p<0.05) |
| Collagen, type 1, alpha 1 | Col1a1 | (14, 36) | ECM, Cancer progression | 2.5 |
| Growth hormone receptor | Ghr | (14) | Growth factor | 2.9 |
| Pleiotrophin | Ptn | (13) | Growth promoter, Angiogenesis inducer | 2.3 |
| Solute carrier family 11 member 2 | Slc11a2 | (14) | Transporter | 2.5 |
| Vascular endothelial growth factor A | VegfA | (36) | Angiogenesis | 3.2 |
Only one gene, Cited 1, was found to be altered into a similar manner both in the parous animals (compared to nulliparous animals) and in the leptin or E2 exposed parous rats (compared to control parous rats) in our study. Cited1 is a transcriptional co-regulator of ER-α and affects estrogen sensitivity in a gene-specific manner (37). Therefore, pregnancy suppresses ER-α signaling; with increasing suppression the higher the hormone levels were during pregnancy. However, we did not observe any changes in the expression of ER-α between the parous rats which received E2 or leptin during pregnancy and parous control rats. Instead, G protein-coupled estrogen receptor 1 (Gper) that localizes to the endoplasmic reticulum and binds estrogen, was down-regulated in the parous E2 and leptin treated rats. This protein is involved in the rapid nongenomic signaling events observed with estrogen.
Discussion
Results obtained in our study indicate that parous control rats had a lower mammary tumor incidence than nulliparous rats which is consistent with the protective effect of pregnancy against breast cancer in women who have their first child before age 20 (5) and previous reports in rats (27, 38). Importantly, the majority of mammary tumors in parous rats in our study appeared before mammary gland involution had been completed. These findings are in accordance with the transient increase in breast cancer risk caused by pregnancy in women (1–4). An exposure to E2 or leptin during pregnancy increased mammary cancer risk in parous rats. Specifically, E2 or leptin treated parous rats continued to develop mammary tumors also after the initial transient increase in risk. Thus, the pattern of mammary tumor development in the rats treated with E2 or leptin during pregnancy mimicked that of nulliparous rats, suggesting that the hormonal exposures prevented the protective effects of parity on mammary cancer risk.
The increase in mammary cancer risk in rats exposed to E2 or leptin during pregnancy is consistent with findings reported in humans. Women who took the synthetic estrogen DES during pregnancy are at an increased risk of developing breast cancer (18, 19). Further, women who exhibit the highest pregnancy estrogen levels, either in the first trimester of gestation (16) or third (17), are at elevated breast cancer risk later in life. We are not aware of any studies that have investigated whether leptin levels during pregnancy affect later breast cancer risk among mothers, but indirect parameters of high leptin levels, such as obesity or weight gain (23–25) indicate that parous women who had the highest leptin levels during pregnancy also are at an increased risk of developing breast cancer. Excessive weight gain during pregnancy is common: close to 50% of pregnant women gain more than recommended by the IOM (26, 39). Since these women are at an increased risk of developing breast cancer after menopause (26), the results obtained in our animal model suggest that high leptin levels during pregnancy are responsible, at least partly, for this finding.
The mechanisms responsible for the association between elevated E2 or leptin levels during pregnancy and increased breast cancer risk remain to be elucidated. We performed microarray analysis to identify differentially expressed genes in the mammary glands between the parous control rats and parous rats exposed to E2 or leptin during pregnancy. Intriguingly, only 11 (0.05%) of >7,000 genes were differentially expressed between the rats that were exposed to E2 or leptin during pregnancy, although both groups exhibited a number of differentially expressed genes compared to controls. The similarity of gene expression in the two hormone-treated groups may reflect the close association between leptin and estrogen signaling in the mammary gland (28–31, 33). We therefore focused on the 142 differentially expressed genes, shown in Table 4, between the mammary glands of rats exposed to vehicle or E2/leptin during pregnancy.
The differentially expressed genes included Eif4e, Mapk9, Nras, Ptn and Vegfa. All these genes have been linked to breast cancer. Deregulation of protein synthesis is a hallmark of many cancers, and overexpression of eukaryotic translation factor Eif4e contributes to the deregulation. It is overexpressed in breast cancers and high expression is linked to an elevated risk of recurrence (40). When overexpressed, Eif4e may enable the translation of a select pool of mRNAs encoding for proteins involved in malignant growth, such as those for cyclin D1, c-MYC, VEGF and matrix metalloprotease-9 (MMP-9) (41). Mapk9 regulates cell proliferation and apoptosis (42) and inhibition of its activity reduces cell proliferation in breast cancer cells (43). Ptn is overexpressed in at least 60% of human breast cancers (43), and this overexpression is linked to high risk of metastasis (44). Vegfa is often up-regulated in breast tumors, especially in those expressing HER-2/neu (45) or mutant p53 (46). Further, both leptin and estrogens activate Vegfa (47, 48). Leptin itself can induce angiogenesis in vitro and in vivo (49), and a neutralizing anti-leptin receptor monoclonal antibody suppresses leukemia cell growth by inhibiting angiogenesis in rats (50). Thus, we were able to confirm up-regulation of Eif4e, Mapk9, Ptn and Vegfa in the mammary glands of parous rats exposed to leptin or E2 during pregnancy, compared to parous control rats, and these changes may be associated with their increased mammary tumorigenesis. Increase in Nras expression in the microarray analysis was not confirmed by qRT PCR.
In addition to these genes, several others were differentially expressed between control and E2/leptin exposed parous rats. We were particularly interested in those genes that have been suggested to explain the protective effect(s) of pregnancy in rodents (13, 14) and humans (36). Thirteen of them were identified as differentially expressed between the parous control and E2/leptin exposed rats. Importantly, genes that have been reported to be up-regulated in the parous women/rodents, compared with nulliparous women/rodents, were up-regulated in the parous control rats in our study, compared to parous rats treated with E2 or leptin during pregnancy. Thus, gene expression patterns in the E2/leptin treated parous rats resembled those in the nulliparous rats. The same applied to down-regulated genes: those that are found to be down-regulated in parous versus nulliparous women/rodents were down-regulated in parous control rats, compared to parous rats treated with E2 or leptin during pregnancy. Most of the down-regulated genes (that are up-regulated by parity) in the mammary glands of parous rats treated with E2 or leptin during pregnancy were those that induce differentiation (Csn1, Cp, and Lalba), inhibit growth (Tgfβ3) or regulate immune functions (Lcn2, and Lbp). Among the up-regulated genes in the parous E2/leptin rats (and down-regulated in parous women and rodents) were VegfA, and Ghr and Ptn that promote growth, and Col1a1 that induces cancer progression by stimulating epithelial mesenchymal transition. Our findings suggest that high levels of E2 and leptin during pregnancy may prevent parity-induced reduction in breast cancer risk by preventing protective signaling changes in their mammary gland.
The parity-induced protective signaling patterns are likely to induce or reflect functional changes that result in reduced breast cancer risk. During pregnancy, the mammary gland undergoes substantial morphological changes, but after the gland has involuted, it returns to a stage resembling that seen in nulliparous animals (51, 52) or humans (12, 53). Findings in mice suggest that pregnancy promotes functional differentiation at a cellular level, and causes a reduction in the proportion of mammary epithelial stem/progenitor cells and an increase in differentiated luminal and myoepithelial cells (54, 55). Since breast cancer is thought to be initiated in epithelial stem/progenitor cells or differentiated cells that acquire stem cell like properties (56), reduction in stem/progenitor cell population may explain why early pregnancy reduces later breast cancer risk. In our study, parous rats exposed to E2 or leptin during pregnancy exhibited a sustained increase in cell proliferation, compared with parous control rats. Proliferating cells represent a progenitor cell population (57), and thus it is possible that a high hormonal environment during pregnancy prevents a pregnancy-induced reduction in stem cells. Although we did not determine whether there were less differentiated cells in the mammary glands of rats treated with E2 or leptin during pregnancy than in the controls, microarray analysis indicated that several genes that induce differentiation were down-regulated, and those increasing cell proliferation were up-regulated (Table 5). In addition to the ones already discussed above, these included down-regulation of Keratin 19 that is a marker of differentiated luminal cells (58).
Conclusion
In our study, parous rats treated with E2 or leptin during pregnancy exhibited higher mammary cancer risk than parous control rats, consistent with the findings in humans showing that women exposed to DES (19), having the highest pregnancy E2 levels (16, 17), or gaining more weight during pregnancy than recommended by the IOM (26), are at an increased risk of developing breast cancer. Parous control rats exhibited a transient increase in mammary cancer risk that lasted until their mammary gland had completed involution. After this transient period, the risk of developing mammary cancer was very low. However, in the parous rats treated with E2 or leptin during pregnancy the risk of developing mammary tumors remained elevated past the transient increase. Thus, the pattern of mammary tumor appearance in the parous E2 and leptin exposed rats was similar to that seen in nulliparous rats, suggesting that parity does not protect against breast cancer if the levels of E2 or leptin during pregnancy are excessive. This conclusion is supported by comparing the pregnancy-induced protective mRNA signature obtained in earlier microarray analyses in rodents and humans (13, 14, 36) to the signature in parous rats treated with E2 or leptin during pregnancy. Gene expression in the mammary glands of E2 or leptin treated parous rats was similar to that of nulliparous individuals. Thus, an exposure to excess E2 or leptin during pregnancy prevents the protective effects of pregnancy on the mammary gland and increases subsequent breast cancer risk. These findings suggest that pregnant women should avoid being exposed to the highest levels of E2 and leptin during pregnancy, caused for example by gaining excessive amounts of weight during pregnancy, because it may not only put them in a risk of for example developing gestational diabetes and hypertension (59), but also increase later breast cancer risk.
Acknowledgments
Grant support: This study was supported by National Cancer Institute (1P30-CA51008; R01 89950; 1R01CA164384; U54 CA000970 and U54 CA149147) for L Hilakivi-Clarke.
List of abbreviations
- DES
Diethylstilbestrol
- DMBA
7,12-dimethylbenz(a)anthracene
- E2
Estradiol
- EIF4E
Eukaryotic initiation translation factor 4e
- ER
Estrogen receptor
- GD
Gestation day
- GHR
Growth hormone receptor
- IGF-1
Insulin like growth factor 1
- MAPK9
Mitogen activated protein kinase 9
- NRAS
Neuroblastoma ras oncogene
- PCNA
Proliferating cell nuclear antigen
- PTN
Pleiotrophin
- qRT-PCR
quantitative real-time reverse transcription-PCR
- TGFβ3
Transforming growth factor a
- VEGFa
Vascular endothelial growth factor a
Footnotes
Conflict of Interest: LH-C has served as an expert witness in a case involving DES daughters
Competing Interests
Authors declare that they have no competing interests.
Authors’ contributions
SDA carried-out animal studies, tissue collection, hormone measurements, mammary gland morphology analysis, cell proliferation and apoptosis assays. MW carried-out microarray analyses and together with LJ interpreted the gene expression data. KB prepared Table 3 and edited the manuscript. LHC was responsible for the study design, interpretation of data, and together with SDA, performed data analyses and prepared the manuscript. All authors read and approved the final manuscript.
Reference List
- 1.Bruzzi P, Negri E, La Vecchia C. Short term increase in risk of breast cancer after full term pregnancy. BMJ. 1988;297:1096–8. doi: 10.1136/bmj.297.6656.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams EMI, Jones L, Vessey MP, McPherson K. Short term increase in risk of breast cancer associated with full pregnancy. BMJ. 1990;300:578–9. doi: 10.1136/bmj.300.6724.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albrektsen G, Heuch I, Hansen S, Kvale G. Breast cancer risk by age at birth, time since birth and time intervals between births: exploring interaction effects. Br J Cancer. 2005;92:167–75. doi: 10.1038/sj.bjc.6602302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Q, Wuu J, Lambe M, Hsieh SF, Ekbom A, Hsieh CC. Transient increase in breast cancer risk after giving birth: postpartum period with the highest risk (Sweden) Cancer Causes Control. 2002;13:299–305. doi: 10.1023/a:1015287208222. [DOI] [PubMed] [Google Scholar]
- 5.Bernstein L. Epidemiology of endocrine-related risk factors for breast cancer. J Mammary Gland Biol Neoplasia. 2002;7:3–15. doi: 10.1023/a:1015714305420. [DOI] [PubMed] [Google Scholar]
- 6.MacMahon B, Cole P, Lin TM, Lowe CR, Mirra AP, Ravnihar B, et al. Age at first birth and breast cancer. Bull World Health Organ. 1970;43:209–21. [PMC free article] [PubMed] [Google Scholar]
- 7.Lyons TR, Schedin PJ, Borges VF. Pregnancy and breast cancer: when they collide. J Mammary Gland Biol Neoplasia. 2009;14:87–98. doi: 10.1007/s10911-009-9119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma H, Bernstein L, Pike MC, Ursin G. Reproductive factors and breast cancer risk according to joint estrogen and progesterone receptor status: a meta-analysis of epidemiological studies. Breast Cancer Res. 2006;8:R43. doi: 10.1186/bcr1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lord SJ, Bernstein L, Johnson KA, Malone KE, McDonald JA, Marchbanks PA, et al. Breast cancer risk and hormone receptor status in older women by parity, age of first birth, and breastfeeding: a case-control study. Cancer Epidemiol Biomarkers Prev. 2008;17:1723–30. doi: 10.1158/1055-9965.EPI-07-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ursin G, Bernstein L, Lord SJ, Karim R, Deapen D, Press MF, et al. Reproductive factors and subtypes of breast cancer defined by hormone receptor and histology. Br J Cancer. 2005;93:364–71. doi: 10.1038/sj.bjc.6602712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Britt K, Ashworth A, Smalley M. Pregnancy and the risk of breast cancer. Endocr Relat Cancer. 2007;14:907–33. doi: 10.1677/ERC-07-0137. [DOI] [PubMed] [Google Scholar]
- 12.Russo IH, Russo J. Pregnancy-induced changes in breast cancer risk. J Mammary Gland Biol Neoplasia. 2011;16:221–33. doi: 10.1007/s10911-011-9228-y. [DOI] [PubMed] [Google Scholar]
- 13.D’Cruz CM, Moody SE, Master SR, Hartman JL, Keiper EA, Imielinski MB, et al. Persistent parity-induced changes in growth factors, TGF-beta3, and differentiation in the rodent mammary gland. Mol Endocrinol. 2002;16:2034–51. doi: 10.1210/me.2002-0073. [DOI] [PubMed] [Google Scholar]
- 14.Blakely CM, Stoddard AJ, Belka GK, Dugan KD, Notarfrancesco KL, Moody SE, et al. Hormone-induced protection against mammary tumorigenesis is conserved in multiple rat strains and identifies a core gene expression signature induced by pregnancy. Cancer Res. 2006;66:6421–31. doi: 10.1158/0008-5472.CAN-05-4235. [DOI] [PubMed] [Google Scholar]
- 15.Belitskaya-Levy I, Zeleniuch-Jacquotte A, Russo J, Russo IH, Bordas P, Ahman J, et al. Characterization of a genomic signature of pregnancy identified in the breast. Cancer Prev Res (Phila) 2011;4:1457–64. doi: 10.1158/1940-6207.CAPR-11-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukanova A, Surcel HM, Lundin E, Kaasila M, Lakso HA, Schock H, et al. Circulating estrogens and progesterone during primiparous pregnancies and risk of maternal breast cancer. Int J Cancer. 2012;130:910–20. doi: 10.1002/ijc.26070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peck JD, Hulka BS, Poole C, Savitz DA, Baird D, Richardson BE. Steroid hormone levels during pregnancy and incidence of maternal breast cancer. Cancer Epidemiol Biomarkers Prev. 2002;11:361–8. [PubMed] [Google Scholar]
- 18.Greenberg ER, Barnes AB, Resseguie L, Barrett JA, Burnside S, Lanza LL, et al. Breast cancer in mothers given diethylstilbestrol in pregnancy. N Engl J Med. 1984;311:1393–8. doi: 10.1056/NEJM198411293112201. [DOI] [PubMed] [Google Scholar]
- 19.Colton T, Greenberg R, Noller K, Resseguie L, Bennekom Cv, Heeren T, et al. Breast cancer in mothers prescribed diethylstilbestrol in pregnancy. JAMA. 1993;269:2096–100. [PubMed] [Google Scholar]
- 20.Bukowski R, Chlebowski RT, Thune I, Furberg AS, Hankins GD, Malone FD, et al. Birth weight, breast cancer and the potential mediating hormonal environment. PLoS ONE. 2012;7:e40199. doi: 10.1371/journal.pone.0040199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al Atawi FS, Addar MH, Warsy AS, Babay ZA. Leptin concentration during different trimesters of pregnancy and its relation to other pregnancy hormones. Saudi Med J. 2004;25:1617–22. [PubMed] [Google Scholar]
- 22.Stock SM, Sande EM, Bremme KA. Leptin levels vary significantly during the menstrual cycle, pregnancy, and in vitro fertilization treatment: possible relation to estradiol. Fertil Steril. 1999;72:657–62. doi: 10.1016/s0015-0282(99)00321-0. [DOI] [PubMed] [Google Scholar]
- 23.Hardie L, Trayhurn P, Abramovich D, Fowler P. Circulating leptin in women: a longitudinal study in the menstrual cycle and during pregnancy. Clin Endocrinol (Oxf) 1997;47:101–6. doi: 10.1046/j.1365-2265.1997.2441017.x. [DOI] [PubMed] [Google Scholar]
- 24.Schubring C, Englaro P, Siebler T, Blum WF, Demirakca T, Kratzsch J, et al. Longitudinal analysis of maternal serum leptin levels during pregnancy, at birth and up to six weeks after birth: relation to body mass index, skinfolds, sex steroids and umbilical cord blood leptin levels. Horm Res. 1998;50:276–83. doi: 10.1159/000023290. [DOI] [PubMed] [Google Scholar]
- 25.Festa A, Shnawa N, Krugluger W, Hopmeier P, Schernthaner G, Haffner SM. Relative hypoleptinaemia in women with mild gestational diabetes mellitus. Diabet Med. 1999;16:656–62. doi: 10.1046/j.1464-5491.1999.00122.x. [DOI] [PubMed] [Google Scholar]
- 26.Kinnunen TI, Pasanen M, Aittasalo M, Fogelholm M, Hilakivi-Clarke L, Weiderpass E, et al. Preventing excessive weight gain during pregnancy – a controlled trial in primary health care. Eur J Clin Nutr. 2007;61:884–91. doi: 10.1038/sj.ejcn.1602602. [DOI] [PubMed] [Google Scholar]
- 27.de Assis S, Wang M, Goel S, Foxworth A, Helferich WG, Hilakivi-Clarke L. Excessive weight gain during pregnancy increases carcinogen-induced mammary tumorigenesis in Sprague-Dawley and lean and obese Zucker rats. J Nutr. 2005;136:998–1004. doi: 10.1093/jn/136.4.998. [DOI] [PubMed] [Google Scholar]
- 28.Catalano S, Marsico S, Giordano C, Mauro L, Rizza P, Panno ML, et al. Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J Biol Chem. 2003;278:28668–76. doi: 10.1074/jbc.M301695200. [DOI] [PubMed] [Google Scholar]
- 29.Catalano S, Mauro L, Marsico S, Giordano C, Rizza P, Rago V, et al. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J Biol Chem. 2004;279:19908–15. doi: 10.1074/jbc.M313191200. [DOI] [PubMed] [Google Scholar]
- 30.Garofalo C, Sisci D, Surmacz E. Leptin interferes with the effects of the antiestrogen ICI 182,780 in MCF-7 breast cancer cells. Clin Cancer Res. 2004;10:6466–75. doi: 10.1158/1078-0432.CCR-04-0203. [DOI] [PubMed] [Google Scholar]
- 31.Alonso A, Fernandez R, Moreno M, Ordonez P, Diaz F, Gonzalez C. Leptin and its receptor are controlled by 17beta-estradiol in peripheral tissues of ovariectomized rats. Exp Biol Med (Maywood) 2007;232:542–9. [PubMed] [Google Scholar]
- 32.Hu X, Juneja SC, Maihle NJ, Cleary MP. Leptin–a growth factor in normal and malignant breast cells and for normal mammary gland development. J Natl Cancer Inst. 2002;94:1704–11. doi: 10.1093/jnci/94.22.1704. [DOI] [PubMed] [Google Scholar]
- 33.Dieudonne MN, Machinal-Quelin F, Serazin-Leroy V, Leneveu MC, Pecquery R, Giudicelli Y. Leptin mediates a proliferative response in human MCF7 breast cancer cells. Biochem Biophys Res Commun. 2002;293:622–8. doi: 10.1016/S0006-291X(02)00205-X. [DOI] [PubMed] [Google Scholar]
- 34.Ando S, Catalano S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat Rev Endocrinol. 2012;8:263–75. doi: 10.1038/nrendo.2011.184. [DOI] [PubMed] [Google Scholar]
- 35.Uehara N, Unami A, Kiyozuka Y, Shikata N, Oishi Y, Tsubura A. Parous mammary glands exhibit distinct alterations in gene expression and proliferation responsiveness to carcinogenic stimuli in Lewis rats. Oncol Rep. 2006;15:903–11. [PubMed] [Google Scholar]
- 36.Asztalos S, Gann PH, Hayes MK, Nonn L, Beam CA, Dai Y, et al. Gene expression patterns in the human breast after pregnancy. Cancer Prev Res (Phila) 2010;3:301–11. doi: 10.1158/1940-6207.CAPR-09-0069. [DOI] [PubMed] [Google Scholar]
- 37.Yahata T, Shao W, Endoh H, Hur J, Coser KR, Sun H, et al. Selective coactivation of estrogen-dependent transcription by CITED1 CBP/p300-binding protein. Genes Dev. 2001;15:2598–612. doi: 10.1101/gad.906301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hilakivi-Clarke L, Onojafe I, Raygada M, Cho E, Clarke R, Lippman M. Breast cancer risk in rats fed a diet high in n-6 polyunsaturated fatty acids during pregnancy. J Natl Cancer Inst. 1996;88:1821–7. doi: 10.1093/jnci/88.24.1821. [DOI] [PubMed] [Google Scholar]
- 39.Olson CM. Achieving a healthy weight gain during pregnancy. Annu Rev Nutr. 2008;28:411–23. doi: 10.1146/annurev.nutr.28.061807.155322. [DOI] [PubMed] [Google Scholar]
- 40.McClusky DR, Chu Q, Yu H, Debenedetti A, Johnson LW, Meschonat C, et al. A prospective trial on initiation factor 4E (eIF4E) overexpression and cancer recurrence in node-positive breast cancer. Ann Surg. 2005;242:584–90. doi: 10.1097/01.sla.0000184224.55949.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De BA, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–99. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 42.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–49. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 43.Mingo-Sion AM, Marietta PM, Koller E, Wolf DM, Van den Berg CL. Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene. 2004;23:596–604. doi: 10.1038/sj.onc.1207147. [DOI] [PubMed] [Google Scholar]
- 44.Fang W, Hartmann N, Chow DT, Riegel AT, Wellstein A. Pleiotrophin stimulates fibroblasts and endothelial and epithelial cells and is expressed in human cancer. J Biol Chem. 1992;267:25889–97. [PubMed] [Google Scholar]
- 45.Konecny GE, Meng YG, Untch M, Wang HJ, Bauerfeind I, Epstein M, et al. Association between HER-2/neu and vascular endothelial growth factor expression predicts clinical outcome in primary breast cancer patients. Clin Cancer Res. 2004;10:1706–16. doi: 10.1158/1078-0432.ccr-0951-3. [DOI] [PubMed] [Google Scholar]
- 46.Linderholm BK, Lindahl T, Holmberg L, Klaar S, Lennerstrand J, Henriksson R, et al. The expression of vascular endothelial growth factor correlates with mutant p53 and poor prognosis in human breast cancer. Cancer Res. 2001;61:2256–60. [PubMed] [Google Scholar]
- 47.Misztal-Dethloff B, Stepien H, Komorowski J. Effect of leptin on proliferative activity and vascular endothelial growth factor (VEGF) secretion from cultured endothelial cells HECa10 in vitro. Endocr Regul. 2004;38:161–6. [PubMed] [Google Scholar]
- 48.Nakamura J, Savinov A, Lu Q, Brodie A. Estrogen regulates vascular endothelial growth/permeability factor expression in 7,12-dimethylbenz(a)anthracene-induced rat mammary tumors. Endocrinology. 1996;137:5589–96. doi: 10.1210/endo.137.12.8940388. [DOI] [PubMed] [Google Scholar]
- 49.Sierra-Honigmann MR, Nath AK, Murakami C, Garcia-Cardena G, Papapetropoulos A, Sessa WC, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281:1683–6. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- 50.Iversen PO, Drevon CA, Reseland JE. Prevention of leptin binding to its receptor suppresses rat leukemic cell growth by inhibiting angiogenesis. Blood. 2002;100:4123–8. doi: 10.1182/blood-2001-11-0134. [DOI] [PubMed] [Google Scholar]
- 51.Rajkumar L, Guzman RC, Yang J, Thordarson G, Talamantes F, Nandi S. Short-term exposure to pregnancy levels of estrogen prevents mammary carcinogenesis. Proc Natl Acad Sci U S A. 2001;98:11755–9. doi: 10.1073/pnas.201393798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sivaraman L, Stephens LC, Markaverich BM, Clark JA, Krnacik S, Conneely OM, et al. Hormone-induced refractoriness to mammary carcinogenesis in Wistar-Furth rats. Carcinogenesis. 1998;19:1573–81. doi: 10.1093/carcin/19.9.1573. [DOI] [PubMed] [Google Scholar]
- 53.Russo J, Rivera R, Russo IH. Influence of age and parity on the development of the human breast. Breast Cancer Res Treat. 1992;23:211–8. doi: 10.1007/BF01833517. [DOI] [PubMed] [Google Scholar]
- 54.Siwko SK, Dong J, Lewis MT, Liu H, Hilsenbeck SG, Li Y. Evidence that an early pregnancy causes a persistent decrease in the number of functional mammary epithelial stem cells–implications for pregnancy-induced protection against breast cancer. Stem Cells. 2008;26:3205–9. doi: 10.1634/stemcells.2008-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tiede BJ, Owens LA, Li F, DeCoste C, Kang Y. A novel mouse model for non-invasive single marker tracking of mammary stem cells in vivo reveals stem cell dynamics throughout pregnancy. PLoS ONE. 2009;4:e8035. doi: 10.1371/journal.pone.0008035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 57.Bussard KM, Boulanger CA, Kittrell FS, Behbod F, Medina D, Smith GH. Immortalized, premalignant epithelial cell populations contain long-lived, label-retaining cells that asymmetrically divide and retain their template DNA. Breast Cancer Res. 2010;12:R86. doi: 10.1186/bcr2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petersen OW, Polyak K. Stem cells in the human breast. Cold Spring Harb Perspect Biol. 2010;2:a003160. doi: 10.1101/cshperspect.a003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Institute of Medicine (US) and National Research Council (US) Committee to Reexamine IOM Pregnancy Weight Guidelines. Weight Gain During Pregnancy: Reexamining the Guidelines. Washington DC: National Academies Press (US); 2009. [Google Scholar]