

SUMMARY
Deregulation of synaptic plasticity may contribute to the pathogenesis of developmental cognitive disorders. In particular, exaggerated mGluR-dependent LTD is featured in fragile X syndrome, but the mechanisms that regulate mGluR-LTD remain incompletely understood. We report that conditional knockout of Cdh1, the key regulatory subunit of the ubiquitin ligase Cdh1-anaphase promoting complex (Cdh1-APC), profoundly impairs mGluR-LTD in the hippocampus. Mechanistically, we find that Cdh1-APC operates in the cytoplasm to drive mGluR-LTD. We also identify the fragile X syndrome protein FMRP as a substrate of Cdh1-APC. Endogenous Cdh1-APC forms a complex with endogenous FMRP, and knockout of Cdh1 impairs mGluR-induced ubiquitination and degradation of FMRP in the hippocampus. Knockout of FMRP suppresses, and expression of an FMRP mutant protein that fails to interact with Cdh1 phenocopies, the Cdh1 knockout phenotype of impaired mGluR-LTD. These findings define Cdh1-APC and FMRP as components of a novel ubiquitin-signaling pathway that regulates mGluR-LTD in the brain.
INTRODUCTION
Synaptic plasticity is thought to play a fundamental role in the adaptive responses of the nervous system to experience. Two forms of synaptic plasticity, long-term potentiation (LTP) and long-term depression (LTD), have been characterized at several synapses in the mammalian brain and may represent physiological correlates of learning and memory (Bliss and Lomo, 1973; Ito and Kano, 1982; Mulkey and Malenka, 1992). Synaptic plasticity is also affected in distinct neurological diseases including developmental disorders of cognition. In particular, alterations of LTD have been implicated in the pathogenesis of fragile X syndrome, the most common known monogenic cause of intellectual disability and autism spectrum disorder (Bassell and Warren, 2008; Bear et al., 2004). Loss of the fragile X syndrome protein FMRP triggers exaggeration of a form of LTD that is induced by the activation of metabotropic glutamate receptors (mGluR-LTD) (Bhakar et al., 2012; Huber et al., 2002). On the other hand, activation of mGluRs in neurons triggers degradation of FMRP (Hou et al., 2006; Nalavadi et al., 2012), suggesting that FMRP and mGluR signaling have a mutually opposing relationship. However, the E3 ubiquitin ligase that targets FMRP to proteasome-dependent degradation in neurons has remained unknown.
Among E3 ubiquitin ligases, the anaphase-promoting complex (APC) has emerged as a critical and pleiotropic regulator of neuronal morphogenesis and synaptic connectivity in the nervous system (Yamada et al., 2013a; Yang et al., 2010). Cdh1 and the Cdh1-related protein Cdc20 represent the key regulatory and coactivating subunits of the APC. Cdh1-APC and Cdc20-APC control the morphogenesis of axons and dendrites, respectively, in the rodent cerebellar cortex. Whereas Cdh1-APC acts in the nucleus to limit axon growth (Konishi et al., 2004; Stegmuller et al., 2006), Cdc20-APC acts at the centrosome to drive the elaboration of dendrite arbors (Kim et al., 2009). These findings suggest that spatial control of the APC plays a critical role in determining its pleiotropic functions (Yamada et al., 2013a). Cdh1-APC also controls synaptic plasticity including EphA4-dependent homeostatic plasticity in forebrain neurons and late phase long-term potentiation (LTP) in the hippocampus (Fu et al., 2011; Li et al., 2008; Pick et al., 2013). However, the role of Cdh1-APC in long-term depression (LTD) has remained unexplored.
In this study, we report a novel signaling link between the major ubiquitin ligase Cdh1-APC and the fragile X syndrome protein FMRP that governs mGluR-LTD in the mammalian brain. Conditional knockout of Cdh1 in the forebrain profoundly impairs the induction of mGluR-LTD, but not NMDAR-LTD, at the CA1 synapse in the hippocampus. Structure-function analyses of Cdh1 in the background of Cdh1 knockout mice in vivo reveal that Cdh1-APC operates in the cytoplasm rather than the nucleus to drive mGluR-LTD. Importantly, we identify FMRP as a novel substrate of Cdh1-APC in the regulation of mGluR-LTD. Endogenous Cdh1-APC interacts with endogenous FMRP in the hippocampus, and mutation of a conserved Cdh1 recognition motif, the D-box, within FMRP disrupts the interaction of FMRP with Cdh1. Knockout of Cdh1 impairs mGluR-induced ubiquitination and degradation of FMRP in the hippocampus. In epistatic analyses in mice, knockout of FMRP suppresses the conditional Cdh1 knockout-induced phenotype of impaired mGluR-LTD. Expression of an FMRP protein in which the Cdh1 recognition motif is mutated impairs mGluR-LTD in the hippocampus, phenocopying the conditional Cdh1 knockout phenotype. These findings define cytoplasmic Cdh1-APC and FMRP as components of a novel ubiquitin-signaling pathway that regulates mGluR-dependent synaptic plasticity, with potential implications for our understanding of fragile X and related syndromes.
RESULTS
Cdh1-APC plays an essential role in mGluR-LTD in the hippocampus
To determine the function of the ubiquitin ligase Cdh1-APC in the forebrain, we generated conditional Cdh1 knockout mice by crossing mice harboring a floxed allele of the Cdh1 gene, (Cdh1loxP/loxP) (Garcia-Higuera et al., 2008) with mice carrying the recombinase Cre expressed under the control of the forebrain-specific driver Emx. In Emx-Cre mice, Cre is expressed in neocortical and hippocampal excitatory neurons but not GABAergic interneurons (Gorski et al., 2002). In immunoblotting analyses, we confirmed that disruption of the Cdh1 gene in Emx-Cre;Cdh1loxP/loxP mice (also referred to as Cdh1 cKO in figures) led to the loss of Cdh1 protein in the hippocampus (Figure 1A). Notably, Cdh1 downregulation in conditional knockout mice became marked after the second postnatal week and continued into adulthood (Figure 1A). Although Cdh1 knockout at early embryonic stages leads to impaired neurogenesis (Delgado-Esteban et al., 2013), forebrain-specific conditional knockout of Cdh1 using the Emx-Cre driver had little effect on the proliferation of neural precursor cells in the ventricular zone (Figures S1A and S1B). Conditional knockout of Cdh1 also had little or no effect on neuronal migration in the forebrain (Figure S1C). Consistent with these results, conditional knockout of Cdh1 in the Emx-Cre;Cdh1loxP/loxP mice did not appear to alter the structure of the forebrain including the hippocampus (Figure 1B).
Figure 1. Hippocampal mGluR-LTD is impaired in conditional Cdh1 knockout mice.
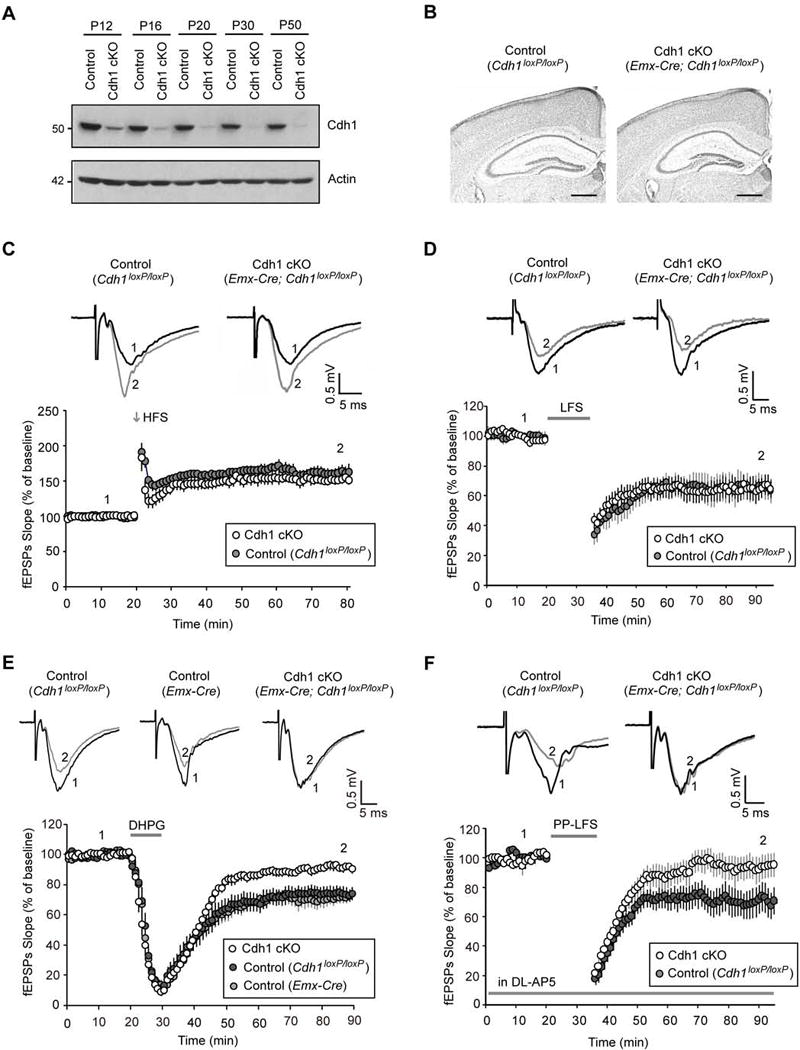
(A) Lysates of hippocampus from control or conditional Cdh1 knockout mice were immunoblotted with the Cdh1 and Actin antibodies, the latter to serve as loading control.
(B) Coronal brain sections from P22 control or conditional Cdh1 knockout mice were subjected to Nissl staining. Scale bars represent 500 μm.
(C) One train of high frequency stimulation (100 pulses at 100 Hz, 1 s) induced hippocampal early-LTP in both control and conditional Cdh1 knockout mice (P=0.94; control: 160.4 ± 9.3% of baseline, n=8 slices from 4 animals; Cdh1 cKO: 161.3 ± 9.6% of baseline, n=8 slices from 4 animals). In this and all subsequent electrophysiology figures, evoked synaptic responses over the course of the experiment were normalized to the average of baseline responses. The percent LTP or LTD was quantified from the average of the responses 50–60 minutes after induction. Representative recording traces from control or conditional Cdh1 knockout mice were taken at times indicated by the numbers 1 and 2. Trace 1 represents baseline responses and trace 2 represents responses one hour after LTP or LTD induction.
(D) Low frequency stimulation (900 pulses at 1 Hz, 15 min) induced hippocampal NMDAR-LTD in both control and conditional Cdh1 knockout mice (P=0.98; control: 65.4 ± 8.0% of baseline, n=8 slices from 5 animals; Cdh1 cKO: 65.6 ± 6.9% of baseline, n=10 slices from 6 animals).
(E) DHPG (50 μM, 10 min) induced significantly reduced hippocampal mGluR-LTD in conditional Cdh1 knockout mice as compared to the Cdh1loxP/loxP control mice (P<0.01) or Emx-Cre control mice (P<0.001) (Cdh1 cKO: 92.0 ± 2.4% of baseline, n= 13 slices from 7 animals; Cdh1loxP/loxP_ control: 74.5 ± 4.5% of baseline, n=8 slices from 5 animals; Emx-Cre_control: 71.6 ± 3.5% of baseline, n=10 slices from 5 animals).
(F) Paired-pulse low frequency stimulation (900 pairs at 1 Hz with 50 ms paired-pulse interval, 15 min), in the presence of the NMDAR antagonist DL-AP5 (100 μM), induced significantly reduced hippocampal mGluR-LTD in conditional Cdh1 knockout mice as compared to control mice (P<0.01; Cdh1 cKO: 99.5 ± 10.0% of baseline, n=9 slices from 5 animals; Control: 65.2 ± 9.2% of baseline, n=10 slices from 6 animals).
Supplemental Figure 1 and Supplemental Figure 2 are related to Figure 1. See also Figure S1 and S2.
To assess the role of Cdh1-APC in synaptic plasticity, we characterized LTP and LTD in the CA1 region of the hippocampus in control (Cdh1loxP/loxP) and conditional Cdh1 knockout (Emx-Cre;Cdh1loxP/loxP) littermate mice. By applying one train of high frequency stimulation (HFS, 100 pulses at 100Hz) or low frequency stimulation (LFS, 900 pulses at 1 Hz) to Schaffer collaterals, we induced LTP or LTD, respectively, at CA1 synapses in the hippocampus in control Cdh1loxP/loxP mice (Figures 1C and 1D). The induction of these forms of synaptic plasticity is thought to depend on activation of NMDA receptors (Bliss and Collingridge, 1993). Conditional knockout of Cdh1 had little or no effect on HFS-induced early-LTP or LFS-induced LTD in the hippocampus (Figures 1C and 1D), suggesting that Cdh1-APC is dispensable for these forms of NMDAR-dependent synaptic plasticity.
We next asked whether Cdh1-APC might play a role in non-NMDAR-dependent synaptic plasticity. Besides LFS-induced NMDAR-dependent LTD, another form of LTD is triggered upon activation of group 1 metabotropic glutamate receptors (mGluR-LTD) (Huber et al., 2001). Notably, mGluR-LTD is thought to contribute to learning flexibility and is deregulated in certain types of neurodevelopmental disorders of cognition (Auerbach et al., 2011; Bateup et al., 2011; D’Antoni et al., 2014). The selective group 1 mGluR agonist 3,5-dihydroxyphenylglycine (DHPG) effectively induces mGluR-LTD (Huber et al., 2001). In control experiments, the mGluR1 and mGluR5 antagonists LY367385 and MPEP blocked the ability of DHPG to induce phosphorylation of the protein kinase ERK1/2 in the hippocampus (Figures S2A and S2B), confirming DHPG is a group 1 mGluR agonist. In other control experiments, the group 1 mGluR antagonists LY367385 and MPEP blocked DHPG-induced mGluR-LTD in the hippocampus (Figure S2C), consistent with published results (Volk et al., 2006) and validating that DHPG triggers mGluR-LTD via mGluR stimulation.
We characterized DHPG-induced mGluR-LTD in control and conditional Cdh1 knockout mice. Strikingly, knockout of Cdh1 significantly impaired DHPG-induced mGluR-LTD at hippocampal CA1 synapses (Figure 1E). Exposure of hippocampal slices from control mice to DHPG at 50μM for 10min induced robust mGluR-LTD at CA1 synapses (Figure 1E). However, in conditional Cdh1 knockout mice, the level of DHPG-induced mGluR-LTD was substantially attenuated (Figure 1E). In control experiments, expression of the recombinase Cre alone in Emx-Cre mice failed to impair hippocampal mGluR-LTD (Figure 1E). These results suggest that Cdh1 is required for mGluR-LTD in the hippocampus.
In addition to mGluR activation with DHPG, electrical stimulation at Schaffer collaterals by a paired-pulse low frequency stimulation paradigm (PP-LFS, 900 pairs at 1 Hz with 50 ms paired-pulse interval) in the presence of the NMDAR antagonist AP5 also triggers mGluR-LTD in the hippocampus (Auerbach et al., 2011). PP-LFS triggered robust mGluR-LTD in the control mice (Figure 1F). By contrast, PP-LFS-induced mGluR-LTD was profoundly reduced in conditional Cdh1 knockout mice (Figure 1F). Together, based on independent pharmacological and electrical modes of inducing synaptic plasticity, we conclude that Cdh1 plays an essential role in mGluR-dependent LTD in the hippocampus.
Cdh1 stimulates the catalytic activity of the APC and is not known to regulate biological processes in the absence of the APC (Chang and Barford, 2014; Peters, 2006). To further assess whether the catalytic activity of Cdh1-APC drives synaptic plasticity, we measured the effect of expression of the protein Emi1, which inhibits the catalytic activity of the APC (Frye et al., 2013), on mGluR-LTD. We performed in utero electroporation of E15 mouse embryos followed by whole-cell patch clamp electrophysiological analyses of transfected neurons in the hippocampus from three-week old mouse pups. Together with the Emi1 expression plasmid or control vector, we included an expression plasmid encoding mCherry to identify the transfected neurons in the hippocampus. These analyses revealed that expression of Emi1 inhibited the ability of DHPG to induce mGluR-LTD in CA1 hippocampal neurons (Figure 2A). In control experiments, expression of Emi1 had little or no effect on the amplitude and frequency of spontaneous excitatory postsynaptic currents (sEPSCs) in CA1 hippocampal neurons (Figures S4A–C). Together, these data support the conclusion that Cdh1 acts in concert with the APC to drive mGluR-LTD in the hippocampus.
Figure 2. Cdh1-APC drives mGluR-LTD.
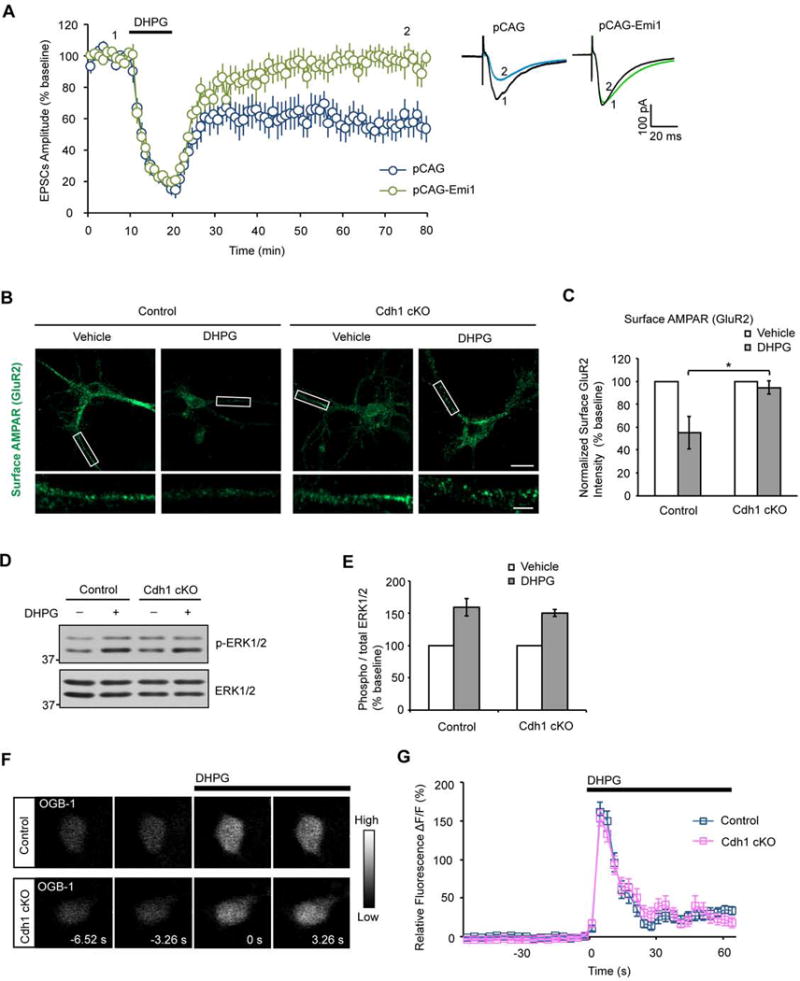
(A) E15 mouse embryos were electroporated with the Emi1 expression plasmid or its control vector together with an mCherry expression plasmid followed by whole-cell patch clamp analyses in transfected mCherry-positive CA1 hippocampal neurons in three-week old mice. DHPG-induced mGluR-LTD was significantly reduced in CA1 neurons upon expression of Emi1 compared to control vector-transfected neurons (P<0.01; pCAG-Emi1: 96.2±8.2% of baseline, n=14 from 6 animals; pCAG vector: 57.9±8.7% of baseline, n=11 from 6 animals).
(B) Immunofluorescence of surface AMPAR subunit, GluR2, in primary hippocampal neurons 14 days after plating from P0 control or conditional Cdh1 knockout mice. Neurons were treated with or without DHPG (50 μM, 10 min). 20 min after DHPG washout, neurons were fixed and subjected to immunofluorescence analyses under nonpermeabilizing condition with the antibody recognizing the ectodomain of GluR2. DHPG induced internalization of surface GluR2 in control but not conditional Cdh1 knockout hippocampal neurons. Scale bars represent 20 μm in the top panel and 5 μm in the bottom panel.
(C) Quantification of surface GluR2 after DHPG treatment reveals significantly reduced surface GluR2 in control but not in conditional Cdh1 knockout hippocampal neurons (P< 0.05; Control: 55.2±14.2% of baseline, n=54 neurons from 4 animals; Cdh1 cKO: 94.6±5.7% of baseline; n=48 neurons from 4 animals).
(D) Lysates of acute hippocampal slices from control or conditional Cdh1 knockout mice were treated with or without DHPG (50 μM for 10 min) and immunobloted with the ERK1/2 and phospho-ERK1/2 (Thr202/Tyr204) antibodies.
(E) DHPG induction of phospho-ERK1/2 immunoreactivity is indistinguishable between control and conditional Cdh1 knockout mice (P=0.56; Control: 159.5±13.4% of baseline, n=3 animals; Cdh1 cKO: 150.4±5.3% of baseline, n=3 animals).
(F) Calcium imaging on primary hippocampal neurons 14 days after plating from P0 control or conditional Cdh1 knockout mice. Neurons were loaded with Oregon Green 488 BAPTA-1AM (OGB-1) to measure the intracellular calcium. DHPG (50 μM) led to increased levels of intracellular calcium in both control and conditional Cdh1 knockout hippocampal neurons.
(G) Quantification of the relative fluorescence ΔF/F from calcium imaging analyses as in (F). DHPG-induced increase in intracellular calcium was indistinguishable between control and conditional Cdh1 knockout hippocampal neurons (P=0.64; Control: 161.67±12.8% of baseline, n=36 neurons from 3 animals; Cdh1 cKO: 153.8±10.5% of baseline, n=33 neurons from 3 animals).
Supplemental Figure 3 is related to Figure 2. See also Figure S3.
Because endocytosis of AMPARs is thought to play a critical role in the expression of mGluR-LTD (Davidkova and Carroll, 2007; Zhang et al., 2008), we asked whether DHPG-induced AMPARs internalization is altered in Cdh1 knockout hippocampal neurons. In immunofluorescence analyses, DHPG induced the downregulation of cell surface expression of the AMPAR subunit GluR2 in control but not Cdh1 knockout hippocampal neurons (Figures 2B and 2C). These results suggest DHPG-induced AMPAR internalization is attenuated in Cdh1 knockout hippocampal neurons, consistent with the phenotype of impaired mGluR-LTD in the conditional Cdh1 knockout mice.
We next performed several series of control experiments to determine whether the impairment of mGluR-LTD in conditional Cdh1 knockout mice might be secondary to abnormalities in mGluR signaling, synaptic morphogenesis, or basal synaptic transmission and intrinsic neuronal excitability. Conditional knockout of Cdh1 had little or no effect on the ability of DHPG to induce the phosphorylation of ERK1/2 in the hippocampus (Figures 2D and 2E). In other analyses in which we measured the levels of intracellular calcium using Oregon Green 488 BAPATA-1, representing another readout of mGluR signaling in neurons (Flint et al., 1999), conditional knockout of Cdh1 had little or no effect on the ability of DHPG to increase the intracellular levels of calcium in hippocampal neurons (Figures 2F and 2G). Together, these data show that Cdh1 knockout does not affect general mGluR function.
In morphological assays, the density, length and width of dendritic spines in hippocampal CA1 neurons from P18 animals were not altered in conditional Cdh1 knockout mice as compared to control mice (Figures S1D–F). In analyses of basal synaptic transmission, no significant differences were detected in the amplitude and frequency of sEPSCs in conditional Cdh1 knockout and control mice (Figures S3A and S3B). Likewise, there was little or no difference in synaptic strength in response to distinct electrical stimulation intensities at the population level (Figures S3C and S3D) or at the single synapse level (Figures S3E and S3F). In measures of intrinsic excitability of hippocampal CA1 neurons, Cdh1 knockout had little or no effect on the action potential firing rate and amplitude (Figures S3G–I). Collectively, our findings suggest that Cdh1-APC specifically regulates mGluR-LTD independently of alterations of basal synaptic neurotransmission, synapse morphology, and general mGluR signaling.
Cdh1-APC operates in the cytoplasm to regulate mGluR-LTD
Having identified a critical function for the ubiquitin ligase Cdh1-APC in synaptic plasticity, we next determined the mechanism underlying the novel function of Cdh1-APC in the brain. We first characterized the subcellular site of action of Cdh1-APC in mGluR-LTD. In granule neurons of the cerebellar cortex, Cdh1-APC resides predominantly but not exclusively in the nucleus, where Cdh1-APC promotes the ubiquitination and consequent degradation of transcriptional regulators and thereby controls axon growth (Konishi et al., 2004; Lasorella et al., 2006; Stegmuller et al., 2006). Although Cdh1-APC has been implicated in other processes in the nervous system beyond the control of axon growth (Fu et al., 2011; Pick et al., 2013; Yang et al., 2010), the subcellular site of Cdh1-APC action in these functions has largely remained to be defined. These observations raised the question of whether Cdh1-APC operates in the nucleus or cytoplasm to regulate mGluR-LTD.
In fractionated lysates of the hippocampus, Cdh1 and the core subunit of APC, Cdc27, were present in both the nuclear and cytoplasmic fractions (Figure 3A). mGluR activation by DHPG had little or no effect on the subcellular localization of Cdh1 and Cdc27 in hippocampal neurons (Figure 3A). To determine whether the nuclear or cytoplasmic pool of Cdh1-APC regulates synaptic plasticity, we performed structure-function analyses of Cdh1 in the background of conditional Cdh1 knockout mice. We employed an in utero electroporation approach in conditional Cdh1 knockout mice to express Cdh1 targeted to the cytoplasm using a nuclear export signal (GFP-NES-Cdh1) or Cdh1 targeted to the nucleus using a nuclear localization signal (GFP-NLS-Cdh1). We performed in utero electroporations at E15 and whole-cell patch clamp eletrophysiological analyses from transfected neurons in hippocampal slices from three-week old mice (illustrated in Figure 3B). First, we confirmed that GFP-NES-Cdh1 and GFP-NLS-Cdh1 were expressed in the cytoplasmic and nuclear compartments, respectively, in hippocampal CA1 neurons from three-week old mice (Figure 3C). Notably, although GFP-NLS-Cdh1 displayed modest expression in the soma in addition to robust expression in the nucleus, GFP-NES-Cdh1 was restricted to the cytoplasm and excluded from the nucleus in CA1 neurons (Figure 3C). GFP-NES-Cdh1 and GFP-NLS-Cdh1 were expressed at comparable levels in CA1 neurons (Figure 3C) and in heterologous cells (Stegmuller et al., 2006).
Figure 3. Cdh1-APC operates in the cytoplasm rather than the nucleus to regulate mGluR-LTD.
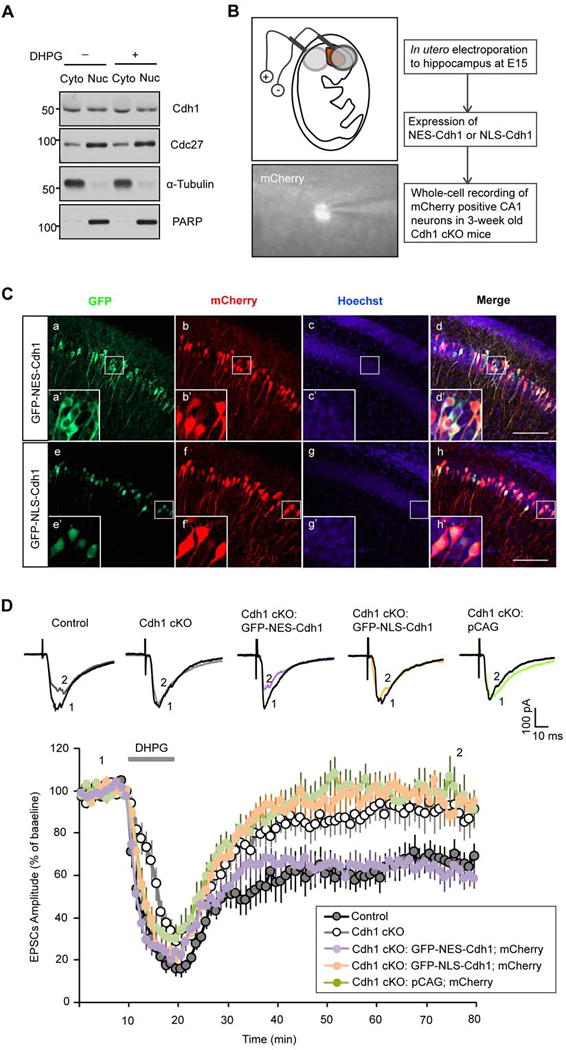
(A) Cytosolic (Cyto) and nuclear (Nuc) fractionations were prepared from hippocampus of P15 wild type mice and immunoblotted with the Cdh1, Cdc27, α-tubulin and PARP antibodies. PARP and α-tubulin served as the nuclear and cytoplasmic marker, respectively. Equal volume of cytosolic and nuclear fractionation was loaded in western blot.
(B) Schematic diagram of in utero electroporation to hippocampus at E15 embryos and whole-cell patch clamp recording of mCherry-positive CA1 neurons in three-week old conditional Cdh1 knockout mice.
(C) E15 mouse embryos electroporated with a plasmid expressing GFP-NES-Cdh1 (a–d) or GFP-NLS-Cdh1 (e–h) together with a mCherry expressing plasmid were allowed to develop until P20. Brain sections were subjected to immunofluorescence analyses with the GFP and DsRed antibodies and the DNA dye bisbenzimide (Hoechst 33258). GFP-NES-Cdh1 and GFP-NLS-Cdh1 appeared to be predominantly in the cytoplasm and nucleus, respectively. Areas inside the white box (a–h) are enlarged in a’–h’. Scale bars represent 100 μm. Notably, although GFP-NLS-Cdh1 displayed modest expression in the soma in addition to robust expression in the nucleus, GFP-NES-Cdh1 was restricted to the cytoplasm and excluded from the nucleus in CA1 neurons.
(D) The Cdh1 knockout-induced mGluR-LTD deficit was effectively reversed by expression of cytoplasmic Cdh1 (GFP-NES-Cdh1) (P<0.01), but not by nuclear Cdh1 (GFP-NLS-Cdh1) (P>0.05), compared with expression of pCAG empty vector in the Cdh1 knockout mice. mGluR-LTD in CA1 hippocampal neurons expressing GFP-NES-Cdh1 in the background of Cdh1 knockout is comparable in magnitude to neurons from control mice. (Control: 65.0 ± 6.8% of baseline, n=12 from 7 animals; Cdh1 CKO: 91. 6 ± 7.0% of baseline, n=12 from 6 animals; GFP-NES-Cdh1-expressing neurons in Cdh1 knockout background: 64. 9 ± 4.5% of baseline, n=11 from 7 animals; GFP-NLS-Cdh1-expressing neurons in Cdh1 knockout background: 96.3 ± 4.4% of baseline, n=12 from 6 animals; Neurons transfected with pCAG empty vector in Cdh1 knockout background: 99.8 ± 6.1% of baseline, n=12 from 5 animals).
Supplemental Figure 4 is related to Figure 3. See also Figure S4.
In whole-cell patch-clamp recording analyses, conditional knockout of Cdh1 strongly impaired DHPG-induced mGluR-LTD (Figure 3D). Importantly, in analyses of mCherry-positive CA1 neurons in Cdh1 knockout mice, expression of cytoplasmic Cdh1 (GFP-NES-Cdh1) effectively reversed the Cdh1 knockout-induced mGluR-LTD deficit (Figure 3D). By contrast, expression of nuclear Cdh1 (GFP-NLS-Cdh1) failed to reverse the mGluR-LTD deficit in Cdh1 knockout mice (Figure 3D). In control analyses, electroporation with the pCAG expression vector had little or no effect on the impairment of mGluR-LTD (Figure 3D). In other control experiments, expression of GFP-NLS-Cdh1 and GFP-NES-Cdh1 had little or no effect on the amplitude and frequency of sEPSCs in CA1 hippocampal neurons (Figures S4D–F). Together, our data reveal that Cdh1-APC operates in the cytoplasm rather than the nucleus to promote hippocampal mGluR-LTD.
Cdh1-APC triggers the ubiquitination and degradation of FMRP
The importance of the cytoplasmic localization of Cdh1-APC in the regulation of synaptic plasticity suggests that relevant targets of Cdh1-APC reside in the cytoplasm. Because Cdh1-APC promotes the ubiquitination and consequent degradation of protein substrates, we reasoned that a physiologically relevant substrate of Cdh1-APC in the regulation of synaptic plasticity should suppress mGluR-LTD. Growing evidence suggests that the fragile X syndrome protein FMRP suppresses mGluR-LTD in the hippocampus (Bear et al., 2004; Huber et al., 2002). In addition, FMRP undergoes degradation in neurons upon mGluR activation (Hou et al., 2006; Nalavadi et al., 2012), but the E3 ubiquitin ligase that triggers FMRP ubiquitination and consequent degradation has remained unknown. We asked whether Cdh1-APC might regulate mGluR-LTD via the ubiquitination and degradation of FMRP.
We first determined whether Cdh1 interacts with FMRP. In reciprocal coimmunoprecipitation analyses, Cdh1 formed a complex with FMRP (Figures 4A and 4B). Whereas Cdh1 interacted strongly with FMRP (Figures 4A and 4B), the Cdh1-related APC coactivator Cdc20 failed to interact with FMRP (Figure 4C), suggesting that FMRP specifically interacts with Cdh1-APC. Importantly, in endogenous coimmunoprecipitation analyses in which the core APC subunit, Cdc27, was immunoprecipitated, endogenous Cdc27 formed a complex with endogenous FMRP and endogenous Cdh1 in the hippocampus (Figure 4E). In reciprocal endogenous coimmunoprecipitation analyses in which FMRP was immunoprecipitated, endogenous FMRP formed a complex with endogenous Cdh1 in the hippocampus (Figure 4F). These data indicate that Cdh1-APC interacts with FMRP in the hippocampus.
Figure 4. Cdh1-APC interacts with FMRP.
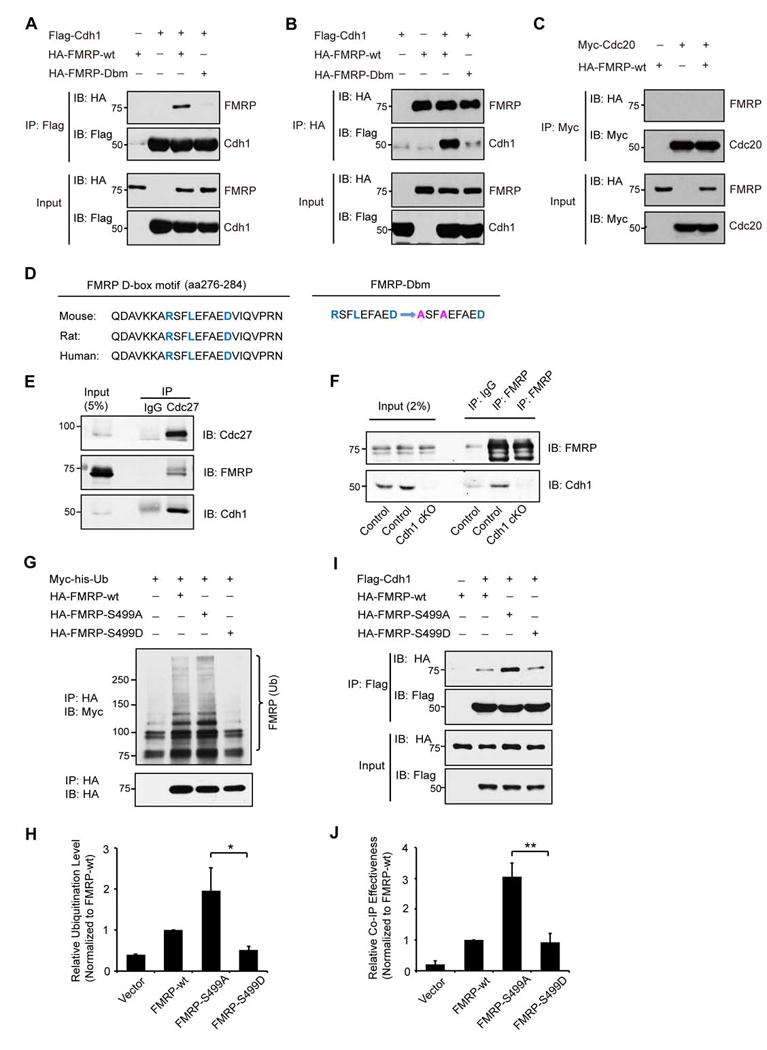
(A) Lysates of 293T cells expressing Flag-Cdh1, HA-FMRP-wt and HA-FMRP-Dbm were immunoprecipitated with the Flag agarose beads followed by immunoblotting with the HA and Flag antibodies. Input was also immunoblotted with the HA and Flag antibodies. Wild type FMRP, but not D-box mutant of FMRP, formed a complex with Cdh1.
(B) Lysates of 293T cells expressing Flag-Cdh1, HA-FMRP-wt and HA-FMRP-Dbm were immunoprecipitated with the HA agarose beads followed by immunoblotting with the HA and Flag antibodies. Input was also immunoblotted with the HA and Flag antibodies. Cdh1 formed a complex with wild type FMRP but not D-box mutant of FMRP.
(C) Lysates of 293T cells expressing Myc-Cdc20 and HA-FMRP-wt were immunoprecipitated with the Myc antibody followed by immunoblotting with the HA and Myc antibodies. Cdc20 failed to form a complex with FMRP.
(D) Conserved sequences of FMRP D-box motif from mouse, rat and human are listed. The amino acid sequence change of D-box mutation (Dbm) in FMRP is illustrated.
(E) Lysates of hippocampus from wild type mice were immunoprecipitated with the Cdc27 antibody or IgG control followed by immunoblotting with the Cdh1, FMRP and Cdc27 antibodies. Endogenous Cdc27 formed a complex with endogenous Cdh1 and endogenous FMRP.
(F) Lysates of hippocampus from control or conditional Cdh1 knockout mice were immunoprecipitated with the FMRP antibody or IgG control followed by immunoblotting with the Cdh1 and FMRP antibodies. Endogenous FMRP formed a complex with endogenous Cdh1 in control but not conditional Cdh1 knockout mice.
(G) Lysates of N2A cells transfected with HA-FMRP-wt, HA-FMRP-S499A or HA-FMRP-S499D together with Myc-his-Ub and pretreated with MG132 were immunopreicipitated with the HA agarose beads followed by immunoblotting with the Myc and HA antibodies. FMRP S499A was ubiquitinated at a higher level than FMRP S499D.
(H) Quantification of analyses as in (G). The FMRP S499A mutant is significantly more ubiquitinated than the FMRP S499D mutant protein (P<0.05).
(I) Lysates from N2A cells transfected with HA-FMRP-wt, HA-FMRP-S499A or HA-FMRP-S499D, together with Flag-Cdh1, were immunopreicipitated with the Flag agarose beads followed by immunoblotting with the HA and Flag antibodies. FMRP S499A interacted with Cdh1 more effectively than FMRP S499D.
(J) Quantification of analyses in (I). The FMRP S499A mutant associates with Cdh1 significantly more effectively than the FMRP S499D mutant protein (P<0.01).
Interrogation of FMRP by sequence gazing revealed a conserved D-box motif (Figure 4D), which represents a Cdh1 recognition motif (Peters, 2006). Mutation of the D-box in FMRP (Dbm), whereby the sequence RSFLEFAED was changed to ASFAEFAED (Figure 4D), disrupted the ability of FMRP to interact with Cdh1 in coimmunoprecipitation analyses (Figures 4A and 4B). These results suggest that FMRP specifically interacts with Cdh1 via the FMRP D-box motif.
Notably, mGluR-induced ubiquitination and consequent degradation of FMRP is facilitated by the dephosphorylation of FMRP at Ser499 (Nalavadi et al., 2012). We asked whether FMRP dephosphorylation might regulate the interaction of FMRP with Cdh1-APC. The inhibitory effect of the Ser499 phosporylation and conversely the facilitating effect of Ser499 dephosphorylation on the ubiquitination and degradation of FMRP are mimicked with the Ser499D and Ser499A mutations, respectively (Ceman et al., 2003; Nalavadi et al., 2012). We confirmed that FMRP Ser499A was ubiquitinated at a higher level than FMRP Ser499D in cells (Figures 4G and 4H), consistent with published results (Nalavadi et al., 2012). In coimmunoprecipitation analyses, FMRP Ser499A interacted with Cdh1 more effectively than FMRP Ser499D or wild type FMRP (Figures 4I and 4J). These results suggest that mGluR-induced dephosphorylation of FMRP at Ser499 promotes its interaction with Cdh1-APC.
The interaction of FMRP with Cdh1-APC at their endogenous levels in the hippocampus, and the roles of the Cdh1 recognition motif and Ser499 dephosphorylation in the interaction of FMRP with Cdh1 suggested that FMRP might represent a substrate of the ubiquitin ligase Cdh1-APC. We asked whether ubiquitination of endogenous FMRP is dependent on endogenous Cdh1-APC in neurons. In immunoprecipitation analyses of FMRP followed by immunoblotting with an antibody that recognizes ubiquitin, FMRP was ubiquitinated in DHPG-treated hippocampal slices from control mice (Figure 5A). Strikingly, the level of ubiquitinated FMRP was substantially reduced in DHPG-treated conditional Cdh1 knockout hippocampal slices (Figure 5A). Likewise, the level of ubiquitinated FMRP was substantially reduced in microdissected CA1 region of the hippocampus in conditional Cdh1 knockout mice compared to control mice (Figure 5B). The specificity of the FMRP antibody for endogenous immunoprecipitation was verified in control experiments using FMRP knockout mice (Figures S5A and S5B). Together, these findings suggest that endogenous FMRP is ubiquitinated by endogenous Cdh1-APC in the hippocampus.
Figure 5. Cdh1-APC triggers the ubiquitination and degradation of FMRP in the hippocampus.
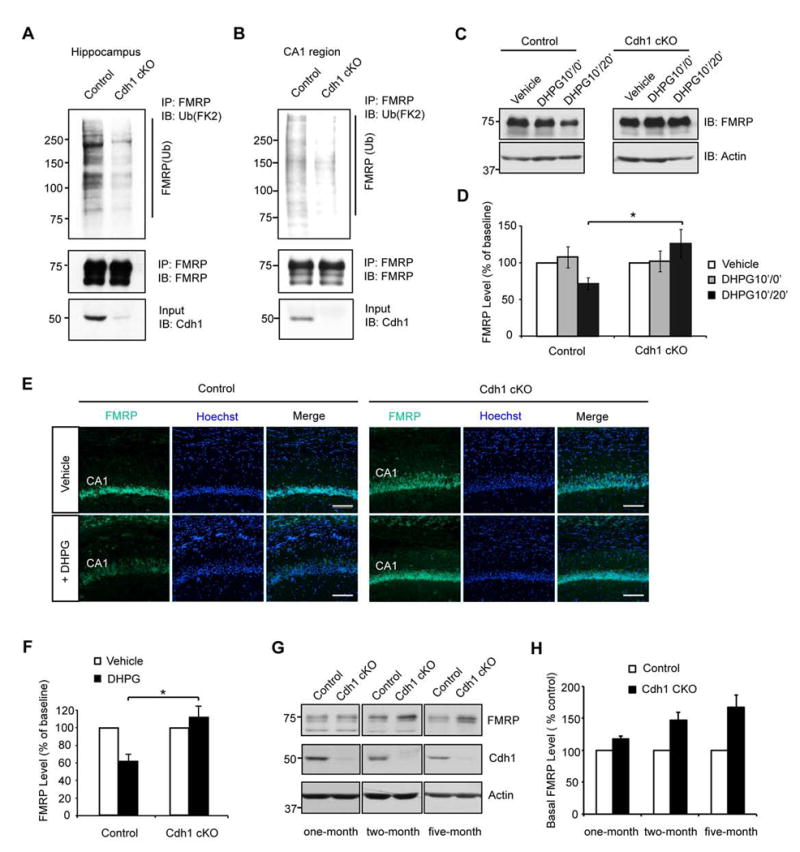
(A) Lysates of acute hippocampal slices from control or conditional Cdh1 knockout mice pretreated with MG132 (20 μM for 1 h) and then treated with DHPG (50 μM for 10 min) were immunoprecipitated with the FMRP antibody followed by immunoblotting with the ubiquitin (FK2), FMRP and Cdh1 antibodies. Endogenous level of ubiquitinated FMRP was substantially reduced in hippocampal slices from conditional Cdh1 knockout mice as compared to control mice.
(B) Lysates of microdissected CA1 region from control or conditional Cdh1 knockout mice were analyzed as in (A).
(C) Lysates of acute hippocampal slices from control or conditional Cdh1 knockout mice, pretreated with anisomycin (25 μM for 1 h), incubated with or without DHPG (50 μM for 10 min), and collected immediately after or 20 min after DHPG washout, were immunoblotted with FMRP and Actin antibodies. DHPG10′/0′ represents 0 min after DHPG (10 min) treatment; DHPG10′/20′ represents 20 min after DHPG (10 min) treatment. DHPG induced the downregulation of endogenous FMRP from control but not conditional Cdh1 knockout mice.
(D) Quantification of DHPG-induced downregulation of FMRP protein levels as in (C). FMRP was normalized to Actin level in each sample. Endogenous. FMRP was significantly reduced 20 min after DHPG treatment in control mice but not in conditional Cdh1 knockout mice (P<0.05; Control: 71.7 ± 8.1% of baseline, n=3 animals; Cdh1 cKO: 126.5 ± 19.2% of baseline, n=3 animals).
(E) Immunofluorescence analyses of acute hippocampal slices from control or conditional Cdh1 knockout mice, pretreated with anisomycin (25 μM for 1 h), incubated with or without DHPG (50 μM for 10 min) and collected 20 min after drug washout. Sections cut from acute slices were subjected to immunofluorescence analyses with the FMRP antibody (Ab17722) and the Hoechst 33258. DHPG triggered the downregulation of endogenous FMRP in the CA1 region from control but not conditional Cdh1 knockout mice. Scale bars represent 100 μm.
(F) Quantification of DHPG-induced downregulation of FMRP fluorescent intensities at CA1 region as in (E). Endogenous FMRP was significantly decreased in response to DHPG treatment in control mice but not in conditional Cdh1 knockout mice (P<0.05; Control: 61.7 ± 8.4 % of baseline, n=3 animals; Cdh1 cKO: 112.2 ± 13.0% of baseline, n=3 animals).
(G) Lysates of hippocampus from control or conditional Cdh1 knockout littermates were immunoblotted with the FMRP, Cdh1 and Actin antibodies. The basal levels of FMRP were increased in the hippocampus upon conditional Cdh1 knockout at two and five months of age.
(H) Quantification of basal levels of FMRP as in (G). FMRP level was normalized to Actin level in each sample and then normalized to the level of control littermate (one-month old Cdh1 cKO: 117.3±4.9% of control littermates, n=3 pairs of animals; two-month old Cdh1 cKO: 146.9±12.4% of control littermates, n=3 pairs of animals; five-month old Cdh1 cKO: 166.9±19.9% of control littermates, n=4 pairs of animals).
Supplemental Figure 5 is related to Figure 5. See also Figure S5.
We next asked if Cdh1-APC triggers the degradation of endogenous FMRP in the hippocampus. Because FMRP undergoes both synthesis and degradation in response to mGluR activation (Hou et al., 2006; Nalavadi et al., 2012; Weiler et al., 1997), to facilitate analysis of FMRP degradation, acute brain slices were pretreated with the protein synthesis inhibitor anisomycin (Nalavadi et al., 2012). In immunoblotting assays, exposure of hippocampal slices from three-week old control mice to DHPG induced the downregulation of FMRP (Figures 5C and 5D). By contrast, DHPG failed to induce the downregulation of FMRP in hippocampal slices from conditional Cdh1 knockout mice (Figures 5C and 5D). In immunofluorescence analyses, DHPG triggered the downregulation of endogenous FMRP in the CA1 region of the hippocampus from control but not conditional Cdh1 knockout mice (Figures 5E and 5F). In control experiments, the specificity of the FMRP antibody for immunohistochemistry was confirmed using FMRP knockout mice (Figures S5C). Together, these data suggest that Cdh1-APC triggers the ubiquitination and consequent degradation of endogenous FMRP in the hippocampus. Conditional knockout of Cdh1 did not alter the basal levels of FMRP in the hippocampus from one-month old mice (Figures 5G and 5H), suggesting that Cdh1-APC regulates FMRP degradation in the hippocampus of these mice specifically upon mGluR stimulation. Notably, conditional knockout of Cdh1 moderately increased the basal levels of FMRP in the hippocampus from two-month and five-month old mice (Figures 5G and 5H), suggesting that Cdh1-APC may regulate basal levels of FMRP in an age-dependent manner. Together, our data strongly support the conclusion that FMRP represents a novel substrate of Cdh1-APC in the hippocampus.
FMRP operates downstream of Cdh1-APC in the regulation of mGluR-dependent LTD
The identification of FMRP as a novel substrate of the ubiquitin ligase Cdh1-APC led us to the fundamental question of whether the degradation of FMRP mediates the ability of Cdh1-APC to drive mGluR-LTD. To address this question, we generated Cdh1 and FMRP double knockout mice. We compared the ability of DHPG to induce mGluR-LTD in control (Cdh1loxP/loxP), conditional Cdh1 knockout (Emx-Cre;Cdh1loxP/loxP), FMRP knockout (FMR1−/y;Cdh1loxP/loxP), and Cdh1 and FMRP double knockout (FMR1−/y;Emx-Cre;Cdh1loxP/loxP) mice, respectively. In FMRP knockout mice, mGluR-dependent LTD was exaggerated (Figures 6A and 6B), consistent with published results (Hou et al., 2006; Huber et al., 2002). In conditional Cdh1 knockout mice, mGluR-LTD was substantially impaired (Figures 6A and 6B). Remarkably, in Cdh1 and FMRP double knockout mice, mGluR-LTD was exaggerated and indistinguishable from the phenotype in FMRP knockout mice (Figures 6A and 6B). Thus, knockout of FMRP suppressed the Cdh1 knockout-induced impaired mGluR-LTD phenotype. In control experiments, knockout of FMRP or double knockout of Cdh1 and FMRP had little or no effect on the ability of DHPG to induce ERK1/2 phosphorylation in the hippocampus as compared to control mice (Figures S6A and S6B), suggesting that general mGluR function is not impaired in these mice. These findings support the conclusion that FMRP operates downstream of Cdh1-APC in mGluR-LTD.
Figure 6. FMRP operates downstream of Cdh1-APC in the regulation of mGluR-dependent LTD.
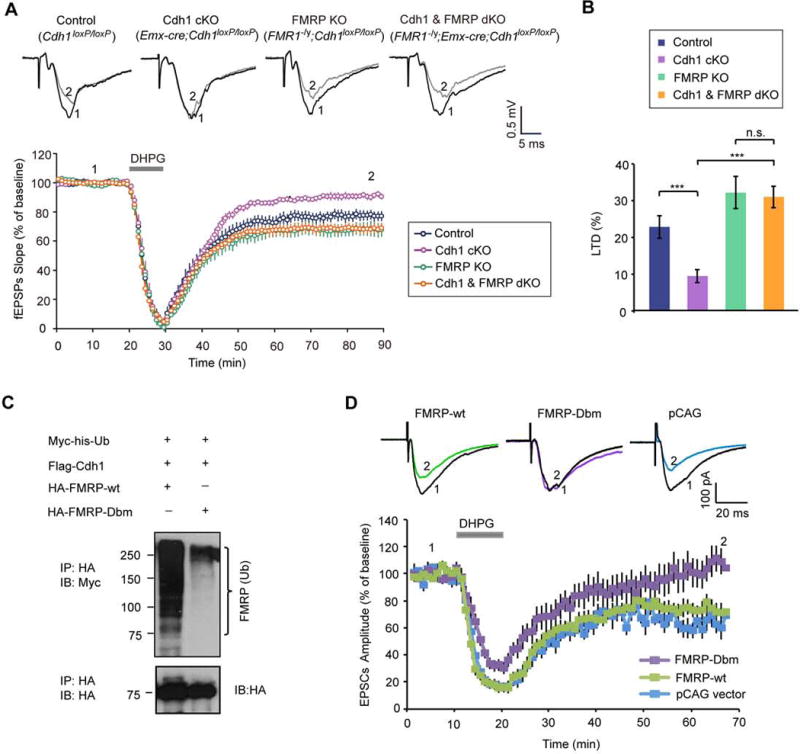
(A) Measurement of DHPG-induced hippocampal mGluR-LTD in control (Cdh1loxP/loxP), conditional Cdh1 knockout (Emx-Cre;Cdh1loxP/loxP), FMRP knockout (FMR1−/y;Cdh1loxP/loxP), Cdh1 and FMRP double knockout (FMR1−/y;Emx-Cre;Cdh1loxP/loxP) mice, respectively. DHPG-induced mGluR-LTD in Cdh1 and FMRP double knockout mice was significantly increased as compared to Cdh1 knockout mice (P<0.001), but indistinguishable in magnitude as compared to FMRP knockout mice. (Control: 76.0 ± 3.4% of baseline, n=12 slices from 6 animals; Cdh1 cKO: 90.9 ± 1.6% of baseline, n=13 slices from 7 animals; FMRP KO: 68.0 ± 4.2% of baseline, n=12 slices from 7 animals; Cdh1 and FMRP dKO: 68.5 ± 2.9% of baseline, n=17 slices from 8 animals).
(B) LTD level shown in (A) was quantified as percent reduction from baseline responses (average of the last ten minutes of recording). mGluR-LTD in Cdh1 and FMRP double knockout mice was significantly increased as compared to Cdh1 knockout mice (P<0.001), and indistinguishable in magnitude as compared to FMRP knockout mice (Cdh1 and FMRP dKO: 31.5 ± 2.9% of LTD, n=17 slices from 8 animals; Cdh1 cKO: 9.1 ± 1.6% of LTD, n=13 slices from 7 animals; FMRP KO: 32.0 ± 4.2% of LTD, n=12 slices from 7 animals; Control: 24.0 ± 3.4% of LTD, n=12 slices from 6 animals).
(C) Lysates of 293T cells expressing wild type FMRP (HA-FMRP-wt) or the D-box mutant FMRP (HA-FMRP-Dbm) together with Myc-his-Ubiquitin and Flag-Cdh1 were immunoprecipitated with the HA agarose beads followed by immunoblotting with the Myc and HA antibodies. The D-box mutation inhibits the ubiquitination of FMRP.
(D) E15 mouse embryos were electroporated with wild type FMRP (HA-FMRP-wt), D-box mutant FMRP (HA-FMRP-Dbm) or control vector (pCAG) together with an mCherry expression plasmid followed by whole-cell patch clamp analyses in transfected mCherry-positive CA1 hippocampal neurons of three-week old mice. DHPG-induced mGluR-LTD was significantly reduced in CA1 neurons upon expression of the FMRP D-box mutant protein when compared to wild type FMRP (P<0.05) or the control vector (P<0.05) (FMRP-Dbm: 102.3±11.4% of baseline, n=11 from 7 animals; FMRP-wt: 72.6±6.5% of baseline, n=10 from 6 animals; pCAG vector: 62.9±6.0% of baseline, n=8 from 4 animals).
Supplemental Figure 6 is related to Figure 6. See also Figure S6.
We next performed structure-function analyses of FMRP to determine whether the relationship of FMPR as a substrate of Cdh1-APC is critical to the ability of FMRP to regulate mGluR-LTD. We tested whether expression of an FMRP protein in which the Cdh1 recognition motif, the D-box, is mutated impairs mGluR-LTD in the hippocampus. First, we confirmed that the D-box mutation inhibited the ubiquitination of FMRP in cells (Figure 6C). We next employed in utero electroporation to transfect the hippocampus of E15 mouse embryos with the FMRP D-box mutant or wild type FMRP expression plasmid or their control vector followed by whole-cell patch clamp eletrophysiological analyses of transfected CA1 neurons in the hippocampus. Expression of the FMRP D-box mutant protein inhibited DHPG-induced mGluR-LTD in CA1 hippocampal neurons, when compared to expression of wild type FMRP or the control vector (Figure 6D). Thus, expression of the FMRP D-box mutant protein in the hippocampus phenocopies the conditional Cdh1 knockout-induced phenotype of impaired mGluR-LTD. In control RNA-immunoprecipitation (RIP) followed by RT-PCR or RT-quantitative real-time PCR analyses, the D-box mutation had little or no effect on the ability of FMRP to associate with the mRNA targets MAP1b, Arc, and PSD95 (Figure S6C and S6D), suggesting that the D-box mutation of FMRP does not impair FMRP’s association with mRNA targets. Together, our data suggest that Cdh1-APC-induced ubiquitination and degradation of FMRP plays a critical role in hippocampal mGluR-LTD. Collectively, we have identified a novel Cdh1-APC/FMRP ubiquitin signaling pathway that regulates mGluR-dependent synaptic plasticity.
DISCUSSION
In this study, we have discovered a novel signaling link between the major ubiquitin ligase Cdh1-APC and the fragile X syndrome protein FMRP that regulates synaptic plasticity in the mammalian brain. Conditional knockout of Cdh1 in the forebrain profoundly impairs the induction of LTD in CA1 hippocampal neurons upon mGluR activation. Structure-function analyses of Cdh1 in the background of conditional Cdh1 knockout mice reveal that cytoplasmic Cdh1-APC drives mGluR- LTD in the hippocampus. We have also uncovered FMRP as a novel substrate of Cdh1-APC. Endogenous FMRP interacts with endogenous Cdh1-APC in the hippocampus, and a Cdh1 recognition motif, the D-box, within FMRP is required for the interaction of FMRP with Cdh1. Knockout of Cdh1 impairs the ubiquitination and degradation of FMRP in mGluR-stimulated hippocampal neurons. Finally, FMRP knockout suppresses the conditional Cdh1 knockout-induced impaired mGluR-LTD phenotype in the hippocampus, and expression of an FMRP protein in which the D-box is mutated phenocopies the Cdh1 knockout phenotype of impaired mGluR-LTD. Our findings define a novel Cdh1-APC/FMRP ubiquitin signaling pathway that governs mGluR-dependent LTD, with important implications for the study of synaptic plasticity and potentially for our understanding of fragile X syndrome.
The identification of a function for the major ubiquitin ligase Cdh1-APC in mGluR-dependent LTD sheds light on the mechanisms of protein degradation in synaptic plasticity. Although the ubiquitin-proteasome system is thought to play a critical role in adaptive responses of neurons (Hegde, 2010), Cdh1-APC represents to our knowledge the first ubiquitin ligase that regulates mGluR-LTD in the brain. As with other forms of synaptic plasticity, mGluR-LTD is thought to represent a correlate of learning and memory. In particular, mGluR-LTD is thought to play a role in learning flexibility (Gladding et al., 2009). In addition, growing evidence suggests that mGluR-LTD is altered in intellectual disability and autism spectrum disorders (Auerbach et al., 2011; Bateup et al., 2011; Bhakar et al., 2012). Thus, by regulating mGluR-LTD, Cdh1-APC may contribute to adaptive functions of the nervous system and its deregulation may contribute to neurodevelopmental disorders of the brain.
Cdh1-APC has been reported to play a role in hippocampal late-phase LTP. Constitutive Cdh1 heterozygous knockout mice and conditional Cdh1 knockout mice using the Nse-cre driver impaired late-phase LTP in the hippocampus (Li et al., 2008; Pick et al., 2013). However, no impairment of early-phase LTP in the hippocampus was observed in these Cdh1 knockout mice (Li et al., 2008; Pick et al., 2013). Consistent with the latter results, conditional knockout of Cdh1 in forebrain excitatory neurons has little or no effect on early-phase LTP in the hippocampus. Further, our results suggest that Cdh1-APC selectively drives mGluR-dependent LTD but not NMDAR-dependent LTD, suggesting that Cdh1-APC is crucial to mGluR-dependent synaptic plasticity in the brain.
The identification of the fragile X syndrome protein FMRP as a novel substrate of the ubiquitin ligase Cdh1-APC bears significant implications for our understanding of the biology of both of these widely studied proteins. Because FMRP represses translation in neurons, in future studies, it will be interesting to determine whether and how the Cdh1-APC/FMRP signaling link regulates protein translation in neurons. It will be also interesting in future studies to determine whether Cdh1-APC acts via additional substrates to regulate synaptic plasticity. The finding that FMRP abundance is intimately controlled by Cdh1-APC suggests that FMRP may contribute to other biological processes beyond synaptic plasticity that are orchestrated by Cdh1-APC.
The finding that Cdh1-APC controls the abundance of FMRP in neurons also bears potential implications for our understanding of fragile X syndrome. In fragile X syndrome patients, an expansion of the trinucleotide CGG repeats (>200) in the 5′ untranslated region induces the hypermethylation and transcriptional silencing of the Fmr1 gene. However, in a substantial fraction of fragile X syndrome patients, transcriptional silencing is incomplete (Tassone et al., 2000). Likewise, carriers of the FMR1 premutation (55–200) have reduced but not absent FMRP and display neuropsychological symptoms including autism spectrum disorders (Farzin et al., 2006). It will be interesting to determine whether Cdh1-APC regulation of FMRP degradation may provide clues for better understanding of variants of fragile X syndrome that express reduced but not absent FMRP.
EXPERIMENTAL PROCEDURES
Materials
Antibodies, reagents and plasmids are described in the Supplemental Experimental Procedures.
Animals
All animal procedures in this study were conducted under the institutional guidelines and approved by Institutional Animal Care and Use Committee at Harvard Medical School and Washington University School of Medicine. Emx-Cre;Cdh1loxP/loxP mice were mated with Cdh1loxP/loxP mice to generate the conditional Cdh1 knockout mice (Emx-Cre;Cdh1loxP/loxP) and control mice (Cdh1loxP/loxP). Female Fmr1−/+;Cdh1loxP/loxP were bred with male Emx-Cre;Cdh1loxP/loxP to generate FMRP and Cdh1 double knockout mice (FMR1−/y;Emx-Cre;Cdh1loxP/loxP), FMRP knockout mice (FMR1−/y; Cdh1loxP/loxP), conditional Cdh1 knockout mice (Emx-Cre;Cdh1loxP/loxP), and control mice (Cdh1loxP/loxP).
Electrophysiology
We used P21–P27 age- and gender- matched littermate mice for measurement of HFS-LTP, DHPG-LTD and PP-LFS-LTD. We used P17–P21 mice for measurement of LFS-LTD, basal synaptic transmission and intrinsic neuronal excitability. Acute hippocampal slices (350 μm) were prepared by vibratome (Leica VT1000S) in cold high-Mg2+ and low-Ca2+ slicing solution containing (in mM) NaCl 124, KCl 2.5, CaCl2 0.5, MgCl2 8, NaHCO3 26, and D-Glucose 17, and then allowed to recover for 1 hour at 35° in ACSF containing (in mM) NaCl 124, KCl 5, NaH2PO4 1.25, NaHCO3 26, MgCl2 1, CaCl2 2, and D–Glucose 17, saturated in 95% O2/5% CO2. For recording, slices were perfused with O2-saturated ACSF at a rate of 2 ml/min, and maintained at 30°. Whole-cell patch-clamp recordings were performed as described (Huang et al., 2010). Recording electrodes (3–4 MΩ) were filled with internal solution containing (in mM) K-Gluconate 125, KCl 15, HEPES 10, ATP-Mg 2, GTP-Na3 0.3, Na2-Phosphocreatine 10, and EGTA 0.2. Series resistance in whole-cell patch-clamp recording was <30 MΩ and remained stable during the time course of experiments. Electrophysiological signals were acquired by Axon-700B MultiClamp Amplifier, digitized at 10 kHz by Digidata 1440A D–A Converter, and Bessel filtered at 2 kHz.
Electrical stimulations were delivered from a stimulus isolator (WPI A360) through a concentric bipolar electrode (FHC, CBAEC75) placed on the Schaffer collaterals. Electrical stimulations intensity was determined by the induction of around 50–60% of the maximum response. LTP was induced by one train of high frequency stimulation (HFS, 100 pulses at 100 Hz for 1 s). NMDAR-LTD was induced by single-pulse low-frequency stimulation (LFS, 900 pulses at 1 Hz for 15 min). mGluR-LTD was induced by bath application of 50 μM DHPG for 10 min, or by paired-pulse low-frequency stimulation (PP-LFS, 900 pairs at 1 Hz for 15 min with 50 ms paired-pulse interval in the presence of NMD AR antagonist DL-AP5 (100 μM)).
In utero electroporation
In utero electroporations were performed as described (Zhang et al., 2013). For details, see Supplemental Experimental Procedures.
Subcellular fractionation
Nuclear and cytoplasmic fractionation experiments from P15 mouse hippocampus are described in the Supplemental Experimental Procedures.
Immunohistochemistry
Immunofluorescence staining was performed as described (Yamada et al., 2013b). For details, see Supplemental Experimental Procedures.
Calcium Imaging
Calcium imaging analyses using Oregon Green 488 BAPATA-1 are described in the Supplemental Experimental Procedures.
Immunoprecipitation analyses
Tissues or cells were lysed in a lysis buffer containing 150 mM NaCl, 20 mM Tris-HCl pH 7.5, 1 mM ethylenediaminetetraacetic acid (EDTA), 1% nonyl phenoxypolyethoxylethanol 40 (NP40), protease inhibitor cocktail (Sigma Aldrich) and 1mM DTT. For immunoprecipitating Flag- or HA-tagged proteins, lysates were incubated with anti-Flag M2 or anti-HA (HA-7) agarose beads (Sigma Aldrich) for 3 hours at 4° and washed 5 times with lysis buffer. For endogenous co-IP experiments, acute hippocampal slices were fast crosslinked with 1.2% formaldehyde for 7 minutes and quenched with 1.25M glycine/PBS before being lysed. For immunoprecipitating endogenous proteins, lysates were incubated with the appropriate primary antibody overnight at 4°. The antibody-protein complexes were purified with Protein G Sepharose 4 Fast Flow beads (GE Healthcare) 2 hours at 4° and washed 5 times with lysis buffer. The immunopreciptated protein complexes were then analyzed by SDS-PAGE and transferred to a nitrocellulose membrane for immunoblotting analyses with the appropriate primary antibodies and HRP-conjugated secondary antibodies (Jackson ImmunoResearch).
FMRP ubiquitination and degradation
For FMRP ubiquitination assays, acute hippocampal slices were prepared and pretreated with MG132 (20μM) for 1 hour, and then treated with DHPG (50μM) for 10min. Twenty minutes after DHPG washout, the entire hippocampal region or specific CA1 region from the acute slices were microdissected and lysed in buffer containing 1% SDS, 1% Np40, 0.5% Na-deoxycholate,150mM NaCl, 50mM Tris-HCl pH 7.4, 2mM EDTA, 40mM N-ethylmaleimide, 1mM DTT and proteinase inhibitor cocktail (1:100 Sigma). Lysates were then diluted to reduce the SDS to 0.1%. Immuoprecipitation of endogenous FMRP was performed with the anti-FMRP (Ab17722) antibody (2.5μg/each), followed by immunoblotting with anti-ubiquitin FK2 antibody (1:1000).
For analyses of endogenous FMRP levels in response to DHPG treatment, acute hippocampal slices were pretreated with the protein synthesis inhibitor anisomycin (25μM) for 1 hour, and then treated with DHPG (50uM) for 10min. The entire hippocampal region was microdissected immediately and 20min after DHPG washout, tissues were lysed in RIPA buffer containing 0.1% SDS, 1% Np40, 0.5% Na-deoxycholate, 150mM NaCl, 50mM Tris-HCl pH 7.4, 2mM EDTA, 1mM DTT and proteinase inhibitor cocktail (1:100 Sigma), followed by immunoblotting with anti-FMRP (1C3) antibody (1:2000).
Data analysis and statistics
Student’s t-test was used to compare the means of two independent samples in analyses that contained two samples. Analysis of variance (ANOVA) followed by the Bonferroni post-hoc test was used for pairwise comparisons within multiple samples. The Kolmogorov-Smirnov test was used to compare the cumulative distribution of two samples. Data were presented as mean ± SEM in all the figure legends. Statistical difference was displayed as ***(P<0.005), **(P<0.01) and *(P<0.05), whereas n.s. indicates no significant difference. Electrophysiological data analyses were carried out with IGOR Pro or Clampfit 10.1 software.
Supplementary Material
HIGHLIGHTS.
Hippocampal mGluR-LTD is markedly impaired in conditional Cdh1 knockout mice.
Cdh1-APC operates in the cytoplasm rather than the nucleus to drive mGluR-LTD.
Fragile X syndrome protein FMRP is a substrate of the ubiquitin ligase Cdh1-APC.
FMRP operates downstream of Cdh1-APC in the regulation of mGluR-LTD.
Acknowledgments
We thank the members of the Bonni laboratory for helpful advice and critical reading of the manuscript and Ivy Jong for guidance in setting up calcium imaging analyses. Supported by NIH grant NS051255 (A.B.) and a Harvard Medical School-Portugal Program Award (A.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes six Supplemental Figures and Supplemental Experimental Procedures.
References
- Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, Sabatini BL. Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. The Journal of neuroscience. 2011;31:8862–8869. doi: 10.1523/JNEUROSCI.1617-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends in neurosciences. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Dolen G, Bear MF. The pathophysiology of fragile X (and what it teaches us about synapses) Annual review of neuroscience. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. The Journal of physiology. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceman S, O’Donnell WT, Reed M, Patton S, Pohl J, Warren ST. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Human molecular genetics. 2003;12:3295–3305. doi: 10.1093/hmg/ddg350. [DOI] [PubMed] [Google Scholar]
- Chang L, Barford D. Insights into the anaphase-promoting complex: a molecular machine that regulates mitosis. Current opinion in structural biology. 2014;29C:1–9. doi: 10.1016/j.sbi.2014.08.003. [DOI] [PubMed] [Google Scholar]
- D’Antoni S, Spatuzza M, Bonaccorso CM, Musumeci SA, Ciranna L, Nicoletti F, Huber KM, Catania MV. Dysregulation of group-I metabotropic glutamate (mGlu) receptor mediated signalling in disorders associated with Intellectual Disability and Autism. Neuroscience and biobehavioral reviews. 2014 doi: 10.1016/j.neubiorev.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidkova G, Carroll RC. Characterization of the role of microtubule-associated protein 1B in metabotropic glutamate receptor-mediated endocytosis of AMPA receptors in hippocampus. The Journal of neuroscience. 2007;27:13273–13278. doi: 10.1523/JNEUROSCI.3334-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Esteban M, Garcia-Higuera I, Maestre C, Moreno S, Almeida A. APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nature communications. 2013;4:2879. doi: 10.1038/ncomms3879. [DOI] [PubMed] [Google Scholar]
- Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, Gane L, Tassone F, Hagerman P, Hagerman R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. Journal of developmental and behavioral pediatrics: JDBP. 2006;27:S137–144. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- Flint AC, Dammerman RS, Kriegstein AR. Endogenous activation of metabotropic glutamate receptors in neocortical development causes neuronal calcium oscillations. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:12144–12149. doi: 10.1073/pnas.96.21.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye JJ, Brown NG, Petzold G, Watson ER, Grace CR, Nourse A, Jarvis MA, Kriwacki RW, Peters JM, Stark H, et al. Electron microscopy structure of human APC/C(CDH1)-EMI1 reveals multimodal mechanism of E3 ligase shutdown. Nature structural & molecular biology. 2013;20:827–835. doi: 10.1038/nsmb.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu AK, Hung KW, Fu WY, Shen C, Chen Y, Xia J, Lai KO, Ip NY. APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nature neuroscience. 2011;14:181–189. doi: 10.1038/nn.2715. [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I, Manchado E, Dubus P, Canamero M, Mendez J, Moreno S, Malumbres M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nature cell biology. 2008;10:802–811. doi: 10.1038/ncb1742. [DOI] [PubMed] [Google Scholar]
- Gladding CM, Fitzjohn SM, Molnar E. Metabotropic glutamate receptor-mediated long-term depression: molecular mechanisms. Pharmacological reviews. 2009;61:395–412. doi: 10.1124/pr.109.001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. The Journal of neuroscience. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN. The ubiquitin-proteasome pathway and synaptic plasticity. Learning & memory. 2010;17:314–327. doi: 10.1101/lm.1504010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huang J, Zhang W, Qiao W, Hu A, Wang Z. Functional connectivity and selective odor responses of excitatory local interneurons in Drosophila antennal lobe. Neuron. 2010;67:1021–1033. doi: 10.1016/j.neuron.2010.08.025. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis–dependent long-term depression in hippocampal area CA1. Journal of neurophysiology. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- Ito M, Kano M. Long-lasting depression of parallel fiber-Purkinje cell transmission induced by conjunctive stimulation of parallel fibers and climbing fibers in the cerebellar cortex. Neuroscience letters. 1982;33:253–258. doi: 10.1016/0304-3940(82)90380-9. [DOI] [PubMed] [Google Scholar]
- Kim AH, Puram SV, Bilimoria PM, Ikeuchi Y, Keough S, Wong M, Rowitch D, Bonni A. A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell. 2009;136:322–336. doi: 10.1016/j.cell.2008.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Stegmuller J, Matsuda T, Bonni S, Bonni A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 2004;303:1026–1030. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Stegmuller J, Guardavaccaro D, Liu G, Carro MS, Rothschild G, de la Torre-Ubieta L, Pagano M, Bonni A, Iavarone A. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature. 2006;442:471–474. doi: 10.1038/nature04895. [DOI] [PubMed] [Google Scholar]
- Li M, Shin YH, Hou L, Huang X, Wei Z, Klann E, Zhang P. The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nature cell biology. 2008;10:1083–1089. doi: 10.1038/ncb1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Nalavadi VC, Muddashetty RS, Gross C, Bassell GJ. Dephosphorylation-induced ubiquitination and degradation of FMRP in dendrites: a role in immediate early mGluR-stimulated translation. The Journal of neuroscience. 2012;32:2582–2587. doi: 10.1523/JNEUROSCI.5057-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nature reviews Molecular cell biology. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Pick JE, Wang L, Mayfield JE, Klann E. Neuronal expression of the ubiquitin E3 ligase APC/C-Cdh1 during development is required for long-term potentiation, behavioral flexibility, and extinction. Neurobiology of learning and memory. 2013;100:25–31. doi: 10.1016/j.nlm.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmuller J, Konishi Y, Huynh MA, Yuan Z, Dibacco S, Bonni A. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron. 2006;50:389–400. doi: 10.1016/j.neuron.2006.03.034. [DOI] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Loesch DZ, Lachiewicz A, Taylor AK, Hagerman PJ. Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. American journal of medical genetics. 2000;94:232–236. doi: 10.1002/1096-8628(20000918)94:3<232::aid-ajmg9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Daly CA, Huber KM. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. Journal of neurophysiology. 2006;95:2427–2438. doi: 10.1152/jn.00383.2005. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Bonni A. Spatial organization of ubiquitin ligase pathways orchestrates neuronal connectivity. Trends in neurosciences. 2013a;36:218–226. doi: 10.1016/j.tins.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Huang J, Coppola G, Geschwind DH, Bonni A. Sumoylated MEF2A coordinately eliminates orphan presynaptic sites and promotes maturation of presynaptic boutons. The Journal of neuroscience. 2013b;33:4726–4740. doi: 10.1523/JNEUROSCI.4191-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kim AH, Bonni A. The dynamic ubiquitin ligase duo: Cdh1-APC and Cdc20-APC regulate neuronal morphogenesis and connectivity. Current opinion in neurobiology. 2010;20:92–99. doi: 10.1016/j.conb.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Mejia LA, Huang J, Valnegri P, Bennett EJ, Anckar J, Jahani-Asl A, Gallardo G, Ikeuchi Y, Yamada T, et al. The X-linked intellectual disability protein PHF6 associates with the PAF1 complex and regulates neuronal migration in the mammalian brain. Neuron. 2013;78:986–993. doi: 10.1016/j.neuron.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Venkitaramani DV, Gladding CM, Zhang Y, Kurup P, Molnar E, Collingridge GL, Lombroso PJ. The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. The Journal of neuroscience. 2008;28:10561–10566. doi: 10.1523/JNEUROSCI.2666-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.