

Abstract
An enantioselective N-heterocyclic carbene (NHC)-catalyzed β-protonation through the orchestration of three distinct organocatalysts has been developed. This cooperative catalyst system enhances both yield and selectivity, compared to only the NHC-catalyzed process. This new method allows for the efficient conversion of a large scope of aryl-oxobutenoates to highly enantioenriched succinate derivatives and demonstrates the benefits of combining different activation modes in organocatalysis.
Advances in sustainable and selective reaction development for applications in bioactive molecule construction, chemical synthesis, and material science rely on translating innovative concepts into new catalytic asymmetric approaches.1 The orchestration of independent catalysts to promote unique transformations is a powerful strategy for reaction discovery.2 Within the field of N-heterocyclic carbene (NHC) catalysis,3 cooperative catalysis has been demonstrated as a viable strategy for improving yield, selectivity, and expanding substrate scope.2d,4 Of particular interest is the ability of NHCs to generate homoenolates, or carbonyl β-anions.5 These unique nucleophiles have been utilized in various C═X π systems to form C–C/C–N bonds, allowing access to a wide array of heterocycles and bioactive compounds.4b,6 Yet, C–H bond formation through the β-protonation of homoenolates has only been achieved with low enantioselectivity.7 The proton is effectively the simplest functional group in chemistry, and its manipulation is the basis of many modern catalytic processes.1a,8 The development of catalytic asymmetric α-protonations of enolates through malonates, silyl enol ethers, ketenes, and α,β-unsaturated carbonyls has led to novel strategies for the synthesis of compounds of interest possessing tertiary carbon stereocenters (Figure 1).1a,2b,9 By contrast, to the best of our knowledge a highly enantioselective β-protonation has not been achieved. In addition to general considerations of enantioselective α-protonation—racemization under protonation conditions and selectively generating pure E- or Z-enolates—β-protonation encounters the additional challenges of generating the necessary homoenolate under catalytic conditions and imparting enantioinduction through interactions with remote functional groups of the chiral catalyst.9b–d Hence, a selective β-protonation would be an enabling and distinctive addition to the broad class of asymmetric protonation transformations.
Figure 1.

Asymmetric protonation.
We decided to pursue cooperative NHC/H-bond donor (HBD)3e,10 activation for an enantioselective β-protonation of α,β-unsaturated aldehydes. If successful, this process would offer an organocatalytic alternative to chiral auxiliary and metal-mediated transformations,11 as well as direct access to easily differentiated succinate products. An unexplored route to chiral succinic esters is asymmetric β-protonation of 2-substituted-oxobutenoates. Compared to standard alkyl/aryl substituted homoenolates, the ester/aryl substitution of oxobutenoates carries two important functions: to provide an opportunity for cooperative carbonyl activation and to promote rapid NHC-homoenolate equivalent formation.4b We proposed that HBD cocatalyst coordination would impart greater steric bulk near the β-position and therefore enhance enantioselectivity. This new approach would provide a conceptually distinct and complementary Umpolung strategy12 to known reductions of β,β-disubstituted carbonyl substrates11a,13 and access chiral succinic esters in the process.
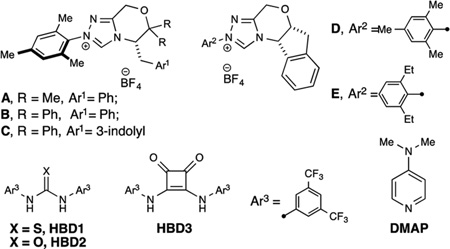
Our investigation commenced by examining the enantioselective β-protonation of β-ethyl ester cinnamaldehyde derivative 1. Combining 1 with ethanol in the presence of Hünig’s base and triazolium salt A produced saturated bis-ester 2 in 80% isolated yield and 66:34 er (Table 1, entry 1). We then directed our efforts toward optimizing the reaction through cooperative catalysis. Addition of thiourea HBD1, which has demonstrated high reactivity among achiral H-bond donors,10a,14 produced a significant increase in enantioselectivity but also resulted in diminished yield (entry 2). Conducting the reaction at a lower temperature provided an increase in enantioselectivity, though the yield was further suppressed (entry 3). We hypothesized that side reactions were occurring following the β-protonation event (but prior to catalyst regeneration) thereby furnishing unproductive byproducts. To test this hypothesis we introduced an acyl transfer agent to increase the rate of catalyst regeneration.15 Gratifyingly, addition of a DMAP cocatalyst incorporated higher enantioselectivity and good yield (entry 4) compared to the initial single catalyst system. After a survey of triazolium precatalysts showed no improvement in either yield or selectivity (entries 5–8), the HBD was evaluated. The urea analog (HBD2) provided a significant increase in yield, with minimal impact on enantioselectivity (entry 9). Further inquiries into the core structure of the carbonyl-activation cocatalyst led to the investigation of squaramides, which Rawal has demonstrated to be excellent H-bond donor catalysts.16 Incorporation of HBD3 provided high yields with a slight erosion of enantioselectivity (entry 10). While the omission of DMAP provided the highest levels of enantioselectivity (entry 11), addition of only 5 mol % of DMAP provided moderately improved yields and, more importantly, a significant decrease in reaction time (entry 12, 24 h vs 36 h for 100% conversion).
Table 1.
Optimization of Reaction Conditions
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Azolium | Temp. (°C) | Additive (mol %) | yield (%)a | erb | time (h) |
| 1 | A | 23 | none | 80 | 66:34 | 12 |
| 2 | A | 23 | HBD1 (30) | 44 | 88:12 | 12 |
| 3 | A | 0 | HBD1 (30) | 41 | 93:7 | 12 |
| 4 | A | 0 | HBD1 (30), DMAP (10) | 71 | 92:8 | 10 |
| 5 | B | 0 | HBD1 (30), DMAP (10) | 40 | 84:16 | - |
| 6 | C | 0 | HBD1 (30), DMAP (10) | 17 | 66:34 | - |
| 7 | D | 0 | HBD1 (30), DMAP (10) | 42 | 73:27 | - |
| 8 | E | 0 | HBD1 (30), DMAP (10) | 40 | 74:26 | - |
| 9 | A | 0 | HBD1 (30), DMAP (10) | 84 | 92:8 | 20 |
| 10 | A | 0 | HBD1 (30), DMAP (10) | 94 | 91:9 | 24 |
| 11 | A | 0 | HBD1 (30) | 81 | 96:4 | 36 |
| 12 | A | 0 | HBD1 (30), DMAP (5) | 85 | 94:6 | 24 |
 | ||||||
Isolated yields.
Determined by HPLC (chiral stationary phase).
With an optimized system that balances selectivity and reactivity, the scope for the asymmetric β-protonation was explored (Table 2).17 Both electron-rich and -poor substituents about the aryl ring were well tolerated (6–12). Ortho-substitution on the aryl group led to lower enantioselectivity (10). Changing the steric size of the ester functionality had a positive impact on enantioselectivity (15–17), with tert-butyl ester (17) giving the highest level of enantioselectivity. Finally, the catalytic system was tolerant of different nucleophiles necessary for catalyst turnover (19–22).18,19
Table 2.
Substrate Scopea
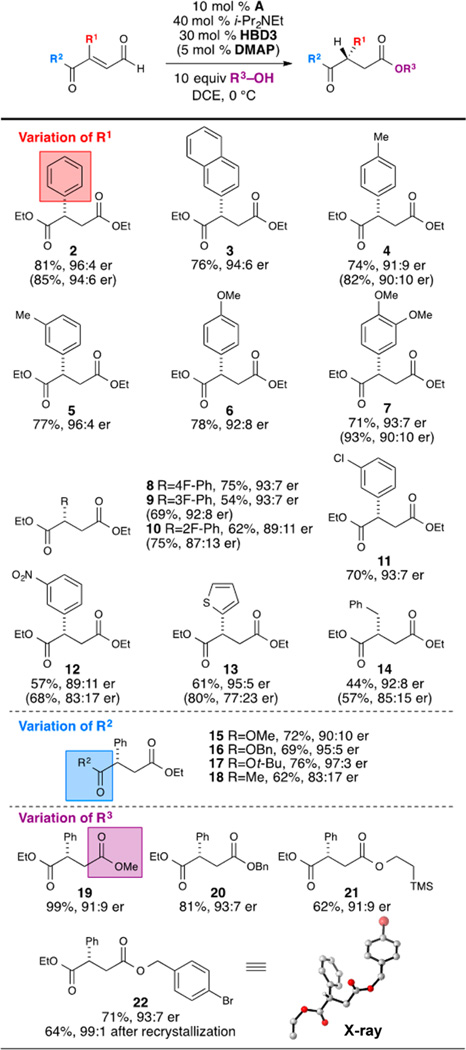 |
Yields are of isolated product after column chromatography; er was determined by chiral HPLC analysis. Yield and er values given in parentheses represent substrates run with 5 mol % DMAP cocatalyst.
A practical advantage of this catalytic strategy is the ease of chemoselectively elaborating these succinic esters. Succinic acids are valuable chiral building blocks for the synthesis of bioactive natural products, peptidomimics, and β-amino acids,20 and succinate derivatives have various therapeutic applications.21 The ability to independently synthesize the ester groups allows for complete regioselectivity in the synthesis of butanoic acids, γ-butyrolactones, and β-amino esters (Scheme 1). Starting with benzyl ester 16 or 20, hydrogenolysis furnishes the α- or β-aryl-butanoic acid (23 and 24, respectively) quantitatively. Selective borane reduction of the acid followed by Lewis acid catalyzed lactonization gave 2- and 3-aryl-γ-butyrolactone derivatives (25 and 26). Alternatively, a Curtius rearrangement in tert-butanol affords Boc-protected β2- and β3-amino esters (27 and 28). From simple starting materials, our NHC/HBD cooperative catalysis system allows easy synthetic differentiation to access privileged regioisomers rapidly, efficiently, and selectively.
Scheme 1.

Succinate Differentiationa
aReagents: (a) BH3·THF; (b) BF3·OEt2; (c) DPPA, t-BuOH.
In key control experiments to probe the complementary roles of the base and proton source, we observed low deuterium incorporation at the β-position (Scheme 2, eq 1). This suggests a strong kinetic isotope effect as a result of proton exchange between the azolium salt, base, and Breslow intermediate.22 Additionally, while the base presumably acts as a proton shuttle to facilitate the overall transformation, the chirality of the base does not affect the stereoselectivity of the protonation step (eqs 2 and 3). Finally, this catalytic system is amenable to larger scale reactions (5 mmol), with a negligible difference in yield or enantioselectivity. Furthermore, on a larger scale, the HBD cocatalyst can be efficiently recovered by precipitation from the unpurified reaction mixture by dilution with CH2Cl2 (eq 4).
Scheme 2.

Control Experimentsa
aReagents: (cond A) 10 mol % A, 30 mol % HBD3, 40 mol % i-Pr2Net, 72 h; (cond B) 10 mol % A, 30 mol % HBD3, 10 equiv EtOH; (cond C) 10 mol % A, 30 mol % HBD3, 40 mol % i-Pr2NEt, 5 mol % DMAP.
To gain insight into the roles of each catalyst, we combined our DFT calculations and experimental evidence to propose a model for facial selectivity of the NHC homoenolate (Figure 2). This observed stereochemical assignment was corroborated by DFT modeling of ground state structures, which proposed homoenolate intermediate NHC_HE2 as more energetically favorable. The benzyl group of the NHC framework blocks the re-face thereby allowing for an si-face protonation. NMR spectroscopy provided evidence that the HBD is involved in the protonation event: the intermolecular interaction of substrate and HBD was observed through 1D NOESY where the methylene protons of 1 exhibited an NOE with the ortho-aryl-hydrogens of HBD1 (Figure 3).23 Combining the DFT calculations, observed NOE, knowledge of NHC catalysis, and the known activation modes of HBDs, intermediate I (Figure 3) emerges as the working stereochemical model for activation and enantioinduction. The coordination of the HBD presumably increases the steric interaction proximal to the β-position of the homoenolate and allows for more selective protonation. This hypothesis is further supported by our substrate scope, which demonstrated increased enantioselectivity as the β-ester increased in steric size (Table 2, 15–17) and decreased enantioselectivity when there were competing sites for hydrogen bonding (12).24
Figure 2.

DFT calculations of homoenolate, computed with Schrödinger interface using Jaguar DFT with B3LYP/6-31G**.
Figure 3.

Stereoinduction model.
Given the data above, we propose the following reaction pathway (Scheme 3): initial deprotonation of A gives the active catalyst species, the free carbene (NHC). Following addition of the NHC to 1, a formal [1,2] proton shift gives extended Breslow intermediate I. HBD3 coordinates to the ester, providing additional steric interactions near the β-position, and enhances facial selectivity. β-protonation and subsequent tautomerization affords acyl azolium II. Catalyst turnover can be enhanced by acyl transfer catalyst DMAP, which forms pyridinium III and regenerates the NHC catalyst. Finally, acylation of the alcohol regenerates DMAP and furnishes chiral succinate 2.
Scheme 3.

Proposed Reaction Pathway
This novel cooperative process is a new, metal-free route to succinic esters and the strategy of deploying multiple catalysts in unison expands the concepts and utility of organocatalysis. Ultimately, this catalytic system delivers the first highly enantioselective, high yielding β-protonation of β,β-disubstituted enals, due in part to unique contributions of all three catalysts: the NHC, HBD, and acyl transfer species. This system leverages distinct reactivity modes modeled from different organocatalysis strategies (nucleophilic catalysis + hydrogen bond donor activation) in a synergistic manner to efficiently promote a challenging bond-forming reaction. The efficient and operational simplicity of utilizing distinct, compatible catalysts versus complex, elaborated single structures with multiple activation sites could lend itself to many catalytic systems in the future.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by the NIH NIGMS (GM073072).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures, spectral data, and crystallographic data. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b02887.
The authors declare no competing financial interest.
REFERENCES
- 1.(a) Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. New York: Springer; 1999. [Google Scholar]; (b) Shang G, Li W, Zhang X, Ojima I. Catalytic Asymmetric Synthesis. New York: John Wiley & Sons; 2010. [Google Scholar]
- 2.(a) Lee JM, Na Y, Han H, Chang S. Chem. Soc. Rev. 2004;33:302. doi: 10.1039/b309033g. [DOI] [PubMed] [Google Scholar]; (b) Berkessel A, Groger H. Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis. Oxford: Blackwell Science Publishers; 2005. [Google Scholar]; (c) Allen AE, MacMillan DWC. Chem. Sci. 2012;3:633. doi: 10.1039/C2SC00907B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cohen DT, Scheidt KA. Chem. Sci. 2012;3:53. doi: 10.1039/C1SC00621E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Phillips EM, Chan A, Scheidt KA. Aldrichimica Acta. 2009;42:55. [PMC free article] [PubMed] [Google Scholar]; (b) Campbell CD, Ling KB, Smith AD. N-Heterocyclic Carbenes in Organocatalysis. Vol. 32. Dordrecht: Springer; 2011. [Google Scholar]; (c) Grossmann A, Enders D. Angew. Chem. Int. Ed. 2012;51:314. doi: 10.1002/anie.201105415. [DOI] [PubMed] [Google Scholar]; (d) Izquierdo J, Hutson GE, Cohen DT, Scheidt KA. Angew. Chem. Int. Ed. 2012;51:11686. doi: 10.1002/anie.201203704. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hopkinson MN, Richter C, Schedler M, Glorius F. Nature. 2014;510:485. doi: 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]
- 4.(a) Raup DEA, Cardinal-David B, Holte D, Scheidt KA. Nat. Chem. 2010;2:766. doi: 10.1038/nchem.727. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao X, DiRocco DA, Rovis T. J. Am. Chem. Soc. 2011;133:12466. doi: 10.1021/ja205714g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dugal-Tessier J, O’Bryan EA, Schroeder TBH, Cohen DT, Scheidt KA. Angew. Chem. Int. Ed. 2012;51:4963. doi: 10.1002/anie.201201643. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mo J, Chen X, Chi YR. J. Am. Chem. Soc. 2012;134:8810. doi: 10.1021/ja303618z. [DOI] [PubMed] [Google Scholar]
- 5.For a recent review, see: Nair V, Menon RS, Biju AT, Sinu CR, Paul RR, Jose A, Sreekumar V. Chem. Soc. Rev. 2011;40:5336. doi: 10.1039/c1cs15139h.
- 6.For selected examples, see: Nair V, Varghese V, Babu BP, Sinu CR, Suresh E. Org. Biomol. Chem. 2010;8:761. doi: 10.1039/b922981g. White NA, DiRocco DA, Rovis T. J. Am. Chem. Soc. 2013;135:8504. doi: 10.1021/ja403847e. McCusker EOB, Scheidt KA. Angew. Chem. Int. Ed. 2013;52:13616. doi: 10.1002/anie.201307292. Guo C, Sahoo B, Daniliuc CG, Glorius F. J. Am. Chem. Soc. 2014;136:17402. doi: 10.1021/ja510737n.
- 7.(a) Maki BE, Chan A, Scheidt KA. Synthesis. 2008:1306. doi: 10.1055/s-2008-1072516. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maki BE, Patterson EV, Cramer CJ, Scheidt KA. Org. Lett. 2009;11:3942. doi: 10.1021/ol901545m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu J-Q, Shi Z, editors. C–H Activation. Vol. 292. Berlin: Springer Berlin Heidelberg; 2010. [Google Scholar]
- 9.(a) Ishihara K, Nakamura S, Kaneeda M, Yamamoto H. J. Am. Chem. Soc. 1996;118:12854. [Google Scholar]; (b) Mohr JT, Hong AY, Stoltz BM. Nat. Chem. 2009;1:359. doi: 10.1038/nchem.297. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Poisson T, Kobayashi S. In: Stereoselective Synthesis of Drugs and Natural Products. Andrushko V, Andrushko N, editors. Hoboken, NJ: John Wiley & Sons, Inc.; 2013. [Google Scholar]; (d) Oudeyer S, Brière J-F, Levacher V. Eur. J. Org. Chem. 2014;2014:6103. [Google Scholar]
- 10.For selected reviews, see: Doyle AG, Jacobsen EN. Chem. Rev. 2007;107:5713. doi: 10.1021/cr068373r. Auvil TJ, Schafer AG, Mattson AE. Eur. J. Org. Chem. 2014;2014:2633. For examples of combining NHCs with HBDs, see: Brand JP, Siles JIO, Waser J. Synlett. 2010;2010:881. Jin Z, Xu J, Yang S, Song B-A, Chi YR. Angew. Chem. Int. Ed. 2013;52:12354. doi: 10.1002/anie.201305023. Nawaz F, Zaghouani M, Bonne D, Chuzel O, Rodriguez J, Coquerel Y. Eur. J. Org. Chem. 2013;2013:8253. Youn SW, Song HS, Park JH. Org. Lett. 2014;16:1028. doi: 10.1021/ol5000617.
- 11.For selected examples of asymmetric hydrogenation, see: Etayo P, Vidal-Ferran A. Chem. Soc. Rev. 2013;42:728. doi: 10.1039/c2cs35410a. Bernasconi M, Müller M-A, Pfaltz A. Angew. Chem. Int. Ed. 2014;53:5385. doi: 10.1002/anie.201402034. For a selected example of a chiral auxiliary, see: Evans DA, Wu LD, Wiener JJM, Johnson JS, Ripin DHB, Tedrow JS. J. Org. Chem. 1999;64:6411.
- 12.(a) Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; (b) Shen B, Makley DM, Johnston JN. Nature. 2010;465:1027. doi: 10.1038/nature09125. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bugaut X, Glorius F. Chem. Soc. Rev. 2012;41:3511. doi: 10.1039/c2cs15333e. [DOI] [PubMed] [Google Scholar]
- 13.(a) Yang JW, Hechavarria Fonseca MT, Vignola N, List B. Angew. Chem. Int. Ed. 2005;44:108. doi: 10.1002/anie.200462432. [DOI] [PubMed] [Google Scholar]; (b) Ouellet SG, Walji AM, Macmillan DWC. Acc. Chem. Res. 2007;40:1327. doi: 10.1021/ar7001864. [DOI] [PubMed] [Google Scholar]
- 14.(a) Schreiner PR, Wittkopp A. Org. Lett. 2002;4:217. doi: 10.1021/ol017117s. [DOI] [PubMed] [Google Scholar]; (b) Connon SJ. Synlett. 2009;2009:354. [Google Scholar]; (c) Schreiner PR, Lippert KM, Hof K, Gerbig D, Ley D, Hausmann H, Guenther S. Eur. J. Org. Chem. 2012;2012:5919. [Google Scholar]
- 15.(a) Xu S, Held I, Kempf B, Mayr H, Steglich W, Zipse H. Chem. – Eur. J. 2005;11:4751. doi: 10.1002/chem.200500398. [DOI] [PubMed] [Google Scholar]; (b) Müller CE, Schreiner PR. Angew. Chem. Int. Ed. 2011;50:6012. doi: 10.1002/anie.201006128. [DOI] [PubMed] [Google Scholar]; (c) Klauber EG, De CK, Shah TK, Seidel D. J. Am. Chem. Soc. 2010;132:13624. doi: 10.1021/ja105337h. [DOI] [PubMed] [Google Scholar]; (d) Birrell JA, Desrosiers J-N, Jacobsen EN. J. Am. Chem. Soc. 2011;133:13872. doi: 10.1021/ja205602j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malerich JP, Hagihara K, Rawal VH. J. Am. Chem. Soc. 2008;130:14416. doi: 10.1021/ja805693p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.All substrates were evaluated with the NHC-HBD cooperative catalyst conditions. If the isolated yields were deemed lower than a practical level of ~60%, then 5 mol % DMAP was employed (yield and er given in parentheses).
- 18.The absolute configuration of 22 was established by X-ray diffraction. The remainder products were assigned by analogy.
- 19.The use of the Z-alkene isomer of 1 undergoes an intramolecular cyclization to provide a substituted furanone (not shown; see Supporting Information for details).
- 20.Enantioselective Synthesis of β-Amino Acids. 2nd ed. Oxford: Blackwell Science Publishers; 2005. [Google Scholar]
- 21.Arason KM, Bergmeier SC. Org. Prep. Proced. Int. 2002;34:337. [Google Scholar]
- 22.A full analysis of this result is currently underway.
- 23.The propensity of ureas and squaramides to form insoluble aggregates (vs more soluble thiourea variants) prevented the observation of the analogous interactions under similar, relevant conditions. Despite those limitations, all of the HBDs most likely share similar function in our catalyst system, as evidenced by their advantageous contribution to enantioselectivity (vide supra, Table 1).
- 24.(a) Amendola V, Fabbrizzi L, Mosca L. Chem. Soc. Rev. 2010;39:3889. doi: 10.1039/b822552b. [DOI] [PubMed] [Google Scholar]; (b) Chauhan P, Mahajan S, Kaya U, Hack D, Enders D. Adv. Synth. Catal. 2015;357:253. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.