

Abstract
Organic N-containing compounds, including amines, are essential components of many biologically and pharmaceutically important molecules. One strategy for introducing nitrogen into substrates with multiple reactive bonds is to insert a monovalent N fragment (nitrene or nitrenoid) into a C–H bond or add it directly to a C=C bond. However, it has been challenging to develop well-defined catalysts capable of promoting predictable and chemoselective aminations solely through reagent control. Herein, we report remarkable chemoselective aminations that employ a single metal (Ag) and a single ligand (phenanthroline) to promote either aziridination or C–H insertion by manipulating the coordination geometry of the active catalysts.
Amines are present in a multitude of pharmaceuticals and natural products with useful biological activities. As a result, the development of synthetic methodologies for the chemo-, regio-, and stereoselective introduction of C–N bonds has been vigorously pursued.1a–e One attractive approach is the direct insertion of a nitrene or nitrenoid species into a C–H or C=C bond of an unsaturated substrate, and many catalysts based on Rh, Ru, Fe, Co, Cu, Mn, Au, and Ag have been exploited in this context.2a–m However, chemoselective C–N bond formation in substrates bearing both reactive C–H and C=C bonds is a particularly challenging task, as these compounds (Figure 1) often give rise to multiple products or exhibit substratecontrolled selectivity.3a–g
Figure 1.

“Static” vs “dynamic” chemoselective amination.
One strategy employed to overcome the problem of substrate control in metal-catalyzed amination is to change the identity of the transition metal. For example, Ru- and Fe-based catalysts favor C–H amination over the aziridination pathway that is preferred using Rh(II) carboxylates.4a,b A second tactic is to utilize different supporting ligands with a single metal, but this has been only marginally successful for chemoselective amination.3a–d Finally, the nature of the nitrene precursor can influence the reaction outcome.5a–c We refer to strategies employing a single, well-defined complex to control a specific amination event as ‘static’ approaches to catalysis (Figure 1, top).
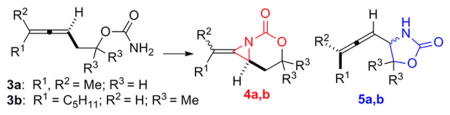
Our previous studies on the chemoselective aziridination of homoallenic carbamates to bicyclic methylene aziridines (Table 1) showed that Ag complexes supported by bidentate N ligands provided superior chemoselectivity for aziridination compared to Rh2(esp)2, irrespective of the substrate identity (compare entry 1 vs 2–7 and 9 vs 10–15).6,7a–f However, a tridentate ligand reversed this selectivity (entries 8 and 16). This result stimulated our curiosity, and a further perusal of the literature showed that Ag has the unique ability to change coordination geometry in response to changes in the Ag counteranion, the ligand identity, or the metal/ligand ratio.8 If these changes in the coordination geometries of the Ag catalysts were indeed responsible for inducing divergent chemoselectivity, a ‘dynamic approach’ to catalytic amination could be envisioned (Figure 1, bottom). In this scenario, treatment of a single Ag salt with a single ligand would yield a mixture of several potential catalytic species. Simple perturbation of the equilibrium of this mixture could give different catalytic species capable of promoting divergent amination using reagent control.
Table 1.
Chemoselective Aziridination and C–H Amination of Homoallenic Carbamates Catalyzed by Silver Catalysts
| ||||||||
|---|---|---|---|---|---|---|---|---|
| entrya | catalystb,c |
|
4a | 5a | entrye |
|
4b | 5b |
| 1 | Rh2(esp)2 |
|
35% | 17% | 9 |
|
5% | 80% |
| 2 | AgOTf/phen |
|
79% | --- | 10 |
|
80% | 14% |
| 3 | AgOTf/bipy |
|
60% | --- | 11 |
|
68% | 11% |
| 4 | AgOTf/bathophen |
|
57% | --- | 12 |
|
84% | 12% |
| 5 | AgOTf/p-MeObipy |
|
72% | --- | 13 |
|
73% | 11% |
| 6 | AgOTf/dafone |
|
32% | --- | 14 |
|
59% | 22% |
| 7 | AgOTf/p-Ph-bipy |
|
66% | --- | 15 |
|
62% | 20% |
| 8 | AgOTf/terpy |
|
27% | 35% | 16 |
|
9% | 61% |
Substrate 3a.
5 mol % Rh2(esp)2, 2 equiv PhlO.
Ag: 20 mol % AgOTf, 25 mol % ligand, 2 equiv PhIO, 4 Å MS, CH2Cl2.
A: aziridination. I: insertion.
Substrate 3b.
In order to test the potential for developing a dynamic catalyst system, the metal:ligand stoichiometry of a AgOTf:phen catalyst system was varied to determine the effect on chemoselectivity.8a–g Phenanthroline was chosen for its high yield in the amination and its relatively low cost. To our delight, a clear impact on the amination of 3b was observed (Table 2). AgOTf:phen ratios close to 1:1 (entries 1–4) promoted aziridination to 4b as the major reaction pathway, while increasing the amount of phen gave C–H insertion to 5b as the dominant mode of reactivity (entries 5, 6). The dramatic reversal in the reaction outcome suggests that an equilibrium between Ag(phen)OTf and Ag(phen)2OTf exists and that each complex favors a different mode of reactivity.
Table 2.
Effect of AgOTf:phen Stoichiometry on the Aziridination/Insertion Ratio
|
| ||||
|
|
||||
| entrya | equiv AgOTf/phen |
4b:5b (
|
4b (5b)b |
|
|
| ||||
| 1 | 0.2/0.1 | 5:1 | 60% (12%) | |
| 2 | 0.2/0.2 | 5.8:1 | 75% (13%) | |
| 3 | 0.2/0.25 | 6.2:1 | 80% (13%) | |
| 4 | 0.2/0.3 | 5.8:1 | 70% (12%) | |
| 5 | 0.2/0.4 | 1:4 | 18% (72%) | |
| 6 | 0.2/0.6 | 1:38 | 2% (76%) | |
|
|
||||
Reactions were carried out at 0.125 M 3b in CH2Cl2, 2 equiv PhIO, AgOTf/phen, rt.
NMR yields with mesitylene as the internal standard.
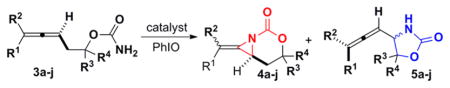
The scope of the ‘dynamic’ amination was explored using homoallenic carbamates (Table 3). In all cases, a 1:1.25 ratio of AgOTf:phen favored aziridination,6 while a 1:3 ratio of AgOTf:phen yielded mainly C–H insertion. Trisubstituted allenes (entries 1, 6–7) exhibited good selectivity under both conditions, while less substituted allenes (entries 2–5, 8) usually gave better selectivity in C–H insertion. Interestingly, the addition of 10 mol % of 2,6-bis(1,1-dimethylethyl)-4-methylphenol (BHT) appeared to improve the conversion of the C–H insertion (entries 4, 8–10).7f
Table 3.
Tunable Amination of Homoallenic Carbamates
| |||||
|---|---|---|---|---|---|
| entry | allene | AgOTf:phena,b |
|
yield 4 | 5 |
| 1 |
|
1:1.25 |
|
79% | <4% |
| 1:3 |
|
0% | 81% | ||
| Rh2(esp)2 |
|
35% | 17% | ||
| 2 |
 3b
3b
|
1:1.25 |
|
79% | 13% |
| 1:3 |
|
1% | 76% | ||
| 3 |
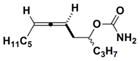 3c
3c
|
1:1.25 |
|
80% | 9% |
| 1:3 |
|
0% | 76% | ||
| Rh2(esp)2 |
|
34% | 34% | ||
| 4 |
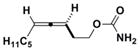 3d
3d
|
1:1.25 |
|
72% | 18% |
| 1:3 |
|
5% | 65% | ||
| 1:3e |
|
<1% | 71% | ||
| 5 |
|
1:1.25c |
|
67% | 18% |
| 1:3 |
|
0% | 83% | ||
| 6 |
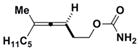 3f
3f
|
1:1.25 |
|
87% | 7% |
| 1:3 |
|
0% | 88% | ||
| Rh2(esp)2 |
|
34% | 44% | ||
| 7 |
|
1:1.25 |
|
70% | 4% |
| 1:3 |
|
0% | 78% | ||
| 8 |
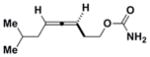 3h
3h
|
1:1.25 |
|
57% | 12% |
| 1:3 |
|
4% | 74% | ||
| 1:3e |
|
<1% | 68% | ||
| 9 |
|
1:1.25 |
|
61% | 0% |
| 1:3 |
|
3% | 74% | ||
| 1:3e |
|
<1% | 71% | ||
| 10 |
|
1:1.25 |
|
58% | 0% |
| 1:3 |
|
0% | 36% | ||
| 1:3e |
|
2% | 62% | ||
Aziridination: 20 mol % AgOTf, 25 mol % phen, 2 equiv PhlO, 4 Å MS, CH2Cl2.
C–H insertion: 10 mol % AgOTf, 30 mol % phen, 3.5 equiv PhIO, 4 Å MS, CH2Cl2.
2,2′-bipyridine ligand.
I = insertion. A = aziridination,
10 mol % BHT added
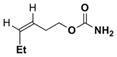
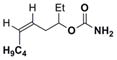
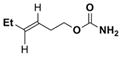
Simple changes in the AgOTf:phen stoichiometry also provided good chemoselectivity in the amination of homoallylic carbamates (Table 4). The cis-disubstituted 6a showed increased selectivity for aziridination in switching from Rh2(OAc)4 to 1:1.25 AgOTf:phen (entry 1), while changing the AgOTf:phen ratio to 1:3 promoted exclusive insertion. This trend held for both the cis-disubstituted 6b (entry 2) containing substitution in the tether and the trans-disubstituted 6c (entry 3). The stereochemistry of the olefin was transferred to the resulting aziridines and allylic amines with no detectable isomerization. The 1,1′-disubstituted 6d gave better selectivity and yield for aziridination compared to Rh2(OAc)4, although the C–H insertion was moderate. Substrate 6e gave poor results using Ag(phen)OTf, but good selectivity for insertion.
Table 4.
Selective Amination of Homoallylic Carbamates
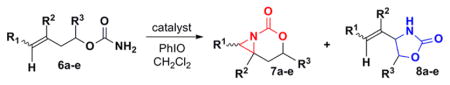
| ||||||
|---|---|---|---|---|---|---|
| entry | substrate | catalysta,b,c |
|
7 | 8 | dr (cis:trans) |
| 1 |
 6a |
Rh2(OAc)4 |
|
58% 7a | 18% 8a | (100:0) |
| 1:1.25 AgOTf:phen |
|
67% 7a | 4% 8a | --- | ||
| 1:3 AgOTf:phen |
|
0% 7a | 93% 8a | --- | ||
| 2 |
 6b |
Rh2(esp)2 |
|
45% 7b | 25% 8bd | nd |
| 1:1.25 AgOTf:phen |
|
89% 7b | 9% 8b | 3.2:1 | ||
| 1:3 AgOTf:phen |
|
4% 7a | 87% 8b | 3:1 | ||
| 3 |
 6c |
Rh2(OAc)4 |
|
68% 7c | 14% 8c | (0:100) |
| 1:1.25 AgOTf:phen |
|
88% 7c | 3% 8c | --- | ||
| 1:3 AgOTf:phen |
|
11% 7c | 73% 8c | --- | ||
| 4 |
6d |
Rh2(OAc)4 |
|
35% 7d | 5% 8d | --- |
| 1:1.25 AgOTf:phen |
|
85% 7d | <1% 8d | --- | ||
| 1:3 AgOTf:phen |
|
23% 7d | 66% 8d | --- | ||
| 5 |
6e |
Rh2(esp)2 |
|
25% 7e | 21% 8ed | nd |
| 1:1.25 AgOTf:phen |
|
54% 7e | 39% 8ed | nd | ||
| 1:3 AgOTf:phen |
|
0% 7e | 68% 8e | 2.4:1 | ||
Rh cat.: 3 mol %, 2 equiv PhIO, 4 Å MS, CH2Cl2.
Aziridination: 20 mol % AgOTf, 25 mol % phen, 2 equiv PhlO, 4 Å MS, CH2Cl2.
C–H insertion: 10 mol % AgOTf, 30 mol % phen, 3.5 equiv PhlO, 4 Å MS, CH2Cl2.
NMR yields with mesitylene as the internal standard.
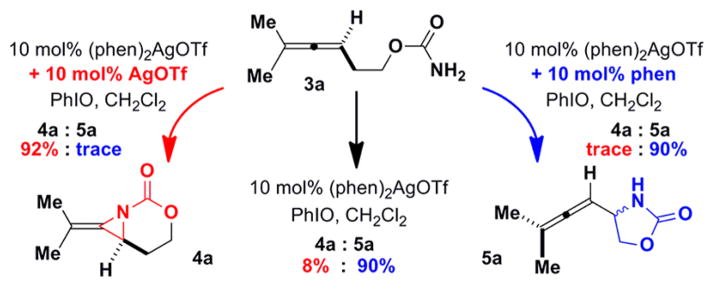
Attempts to isolate the proposed 1:1 and 1:2 AgOTf:phen complexes in the solid state (Table 2) resulted in the recovery of only Ag(phen)2OTf.9 Nonetheless, Ag(phen)2OTf was capable of dissociating and reassembling into two distinct catalytic species capable of divergent amination (Scheme 1). Reaction of 3a with the preformed Ag(phen)2OTf gave a 90:8 mixture of products in favor of the C–H insertion, consistent with the results described in Table 2. Addition of 10 mol % AgOTf to the initial Ag(phen)2OTf complex completely reversed the chemoselectivity to provide 4a in 92% yield, while an extra 10 mol % of phen shut down the competing aziridination pathway, giving 5a in 90% yield.
Scheme 1.
Solution-State Behavior of a Preformed Ag(phen)2OTf Catalyst for Chemoselective Amination
Attempts to corroborate the proposed solution state geometries for the two Ag catalysts illustrated in Table 2 were carried out using NMR titration experiments with 4,4′-di-tert-butyl-2,2′-bipyridine (tBu-bipy, substituted for phen to improve solubility) and AgOTf. Unfortunately, rapid dynamic exchange, even at temperatures as low as −85 °C, prevented direct observation of the individual species present in solution (see Supporting Information (SI) for details). However, the averaged 1H, 13C chemical shifts indicated that the major species in the mixture changed as the ratio of ligand:AgOTf was increased. A combination of pulse gradient spin echo (PGSE) and MALDI MS experiments showed that a monomeric Ag(L)OTf complex was the major species in solution when a 1:1 AgOTf:ligand ratio was used, while a monomeric Ag(L)2OTf complex predominated when a 1:2 AgOTf:ligand ratio was used (details in SI). The additional equivalent of ligand serves to perturb the equilibrium of the Ag(L)OTf:Ag(L)2OTf mixture to favor the latter.
With information about the nature of the two catalytic species in hand, we wanted to understand the factors responsible for our unexpected and tunable chemoselectivity. The exact mechanisms of metal-catalyzed aminations have been notoriously difficult to unravel and often involve multiple reaction pathways.2,7f Yet, we felt even preliminary insights into the mechanism could help extend our dynamic catalysis beyond the scope here.
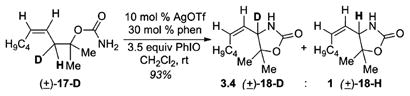
Experiments to determine whether Ag-catalyzed amination proceeds through a stepwise or concerted mechanism were carried out using the stereochemical probes (±)-9 and (±)-12 (Scheme 2). Only (±)-10 and (±)-14 were observed and no isomerization was detected, suggesting a concerted event. A substrate 15 containing a radical trap yielded only 16 and no ring-opened product, arguing against long-lived radical intermediates. A kinetic isotope effect (KIE) experiment yielded a 3.4 ± 0.1 mixture of isotopomers (±)-18-D and (±)-18-H (eq 1). KIEs in
Scheme 2.

Stereochemical Probes for Radical Intermediates
 |
(1) |
the range of 1–3 are believed to correspond to a concerted pathway, while KIEs in the range of 6–12 usually signal a stepwise process involving potential radical intermediates.3c,11a–c This suggests that C–H insertion favors a singlet nitrene pathway over hydrogen atom abstraction.
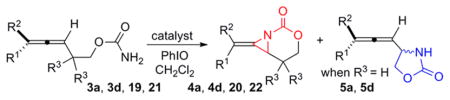
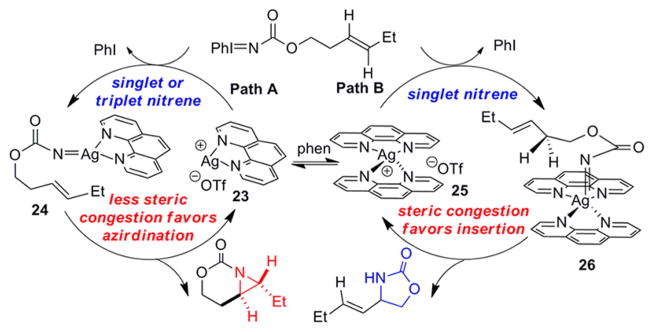
To shed light on the differences between Ag-catalyzed pathways promoting aziridination vs insertion, initial rates were measured for four homoallenic carbamates 3a, 3d, 19, and 21 (Table 5). As expected, the initial rate of aziridination was faster than C–H insertion for both tri- and disubstituted allenes (compare entries 4 and 6, as well as 11 and 13). When sites for potential C–H insertion were blocked in substrates 19 and 21, the Ag(phen)OTf catalyst still gave aziridination (entries 1 and 8), but the Ag(phen)2OTf complex gave either no reaction (entry 2) or significantly decreased reactivity (entry 9). This suggests that the steric congestion around the Ag center plays an important role in dictating the chemoselectivity, with a more hindered Ag center promoting insertion over the aziridination which is favored by a less sterically congested Ag center. This is likely due to the difficulty of overcoming steric clashing with the ligands when the substrate attempts to adopt the appropriate orientation of the olefin for reaction with the nitrene (see 26 in Figure 2, vide infra). When two ligands are coordinated to the Ag center, insertion into the C–H bond of 26 presents a more favorable pathway, in contrast to the aziridination that occurs through the proposed 1:1 Ag:L complex 24.
Table 5.
Relative Rates of Aziridination and C–H Insertion
| ||||
|---|---|---|---|---|
| entry | R1-R3 | catalyst (AgOTf, phen) | product(s) | init rate (mmol/min*mL)a |
| 1 | Me, Me, Me 19 |
20 mol%, 25 mol% | 20 | 1.8×10−3 (98% yield) |
| 2 | 10 mol%, 30 mol% | 20 | no reaction | |
| 3 | 20 mol%, 25 mol%, 20 mol% BHT | 20 | 1.4×10−3 (48% yield) | |
|
| ||||
| 4 | Me, Me, H 3a |
20 mol%, 25 mol% | 4a | 1.3×10−3 |
| 5 | 20 mol%, 25 mol%, 20 mol% BHT | 5a | 9.6×10−4 | |
| 6 | 20 mol%, 60 mol% | 5a | 2.8×10−4 | |
| 7 | 20 mol%, 60 mol%, 20 mol% BHT | 5a | 2.1×10−4 | |
|
| ||||
| 8 | C5H11, H, Me 21 |
20 mol%, 25 mol% | 22 | 9.85×10−4 (88% yield) |
| 9 | 20 mol%, 60 mol% | 22 | 36% yieldb | |
| 10 | 20 mol%, 25 mol%, 20 mol% BHT | 22 | 6.15×10−4 | |
|
| ||||
| 11 | C5H11, H, H 3d |
20 mol%, 25 mol% | 4d | 5.73×10−4 |
| 12 | 20 mol%, 25 mol%, 20 mol% BHTc |
4d (26%) 5d (34%) |
3.31×10−4 2.31×10−4 |
|
| 13 | 20 mol%, 60 mol% | 5d | 1.58×10−4 | |
| 14 | 20 mol%, 60 mol%, 20 mol% BHT | 5d | 2.94×10−4 | |
The rate of product formation was monitored by 1H NMR using mesitylene as the internal standards. The indicated initial rates are the average of the three runs, and the standard deviations are included in the SI.
Yield after 21 h, 73% conversion.
The ratio of 4d:5d was 1:1.
Figure 2.
Possible mechanisms for Ag-catalyzed divergent, chemoselective amination.
BHT was initially employed to ascertain the impact of a radical inhibitor on the amination (Table 3), where the presence of this additive appeared to improve the conversion of disubstituted homoallenic carbamates to allenic amines. Closer examination of the role of BHT (Table 5) showed that the initial rates in the aziridination of 19 and 21 were decreased in the presence of the radical inhibitor (compare entries 1 and 3, and entries 8 and 10). However, the effect of BHT on the rate of C–H insertion was variable (compare entries 6–7 and 13–14) and did result in an increase in the rate of insertion when a disubstituted allene was employed (entries 13 vs 14). Interestingly, the addition of BHT to 3d in the presence of 1:1.25 AgOTf:phen (entry 12) gave a 1:1 ratio of aziridine 4d to allenic amine 5d. This suggests BHT may also play a role in altering the Ag(L)OTf:Ag(L)2OTf equilibrium by shifting it toward Ag(L)2OTf, but further study will be needed to completely understand its impact on the reaction.
The retention of stereochemistry at a chiral center, the low KIE value, the lack of isomerization in the reactions of homoallylic carbamates (Table 4), and the absence of ring opening in the cyclopropane 15 all support a concerted pathway involving a singlet nitrene for the C–H insertion (Figure 2, Path B).3b,c,7e,f However, the aziridination Path A could involve either singlet or triplet nitrene intermediates, or perhaps both. The differences in energies between these two states can be very small; Pérez and co-workers have recently reported that Ag-catalyzed olefin aziridination may involve both paths.7f,12a–c The decrease in the rates of aziridination in the presence of BHT implies there may a triplet nitrene involved, but the lack of isomerization in the aziridination of homallylic carbamates (Table 4) argues against this and further mechanistic studies will need to be carried out to clarify this issue. Irrespective of the exact reaction pathways, the best explanation for Ag-catalyzed chemoselectivity resides in the dramatic steric differences in the coordination geometries adopted by Ag(phen)OTf and Ag(phen)2OTf, respectively.
In conclusion, we have developed a simple Ag-based catalyst system that represents the only method to date capable of employing the same metal and the same ligand to accomplish either aziridination or C–H insertion in good yields. The ability for Ag to readily adopt multiple coordination geometries provides a new approach to identify catalysts that can promote other types of chemoselective aminations, including choosing between two different C–H bonds. In addition, the ease with which this methodology can be implemented and hopefully extended to other chemoselective C–heteroatom and C–C bond formations opens a potential gateway in reaction discovery. Computational and further mechanistic studies are currently underway to unveil the electronic and steric nature of the reactive species in these Ag-catalyzed chemoselective aminations.
Supplementary Material
Acknowledgments
This research was supported by start-up funds provided by the UW—Madison. Dr. Charles Fry of the UW—Madison is thanked for help with NMR spectroscopy, and Professors John Berry, Hans Reich, and Chuck Casey of UW—Madison are also thanked for helpful comments and discussion.
Footnotes
Notes
The authors declare no competing financial interest.
Experimental procedures and characterization for new compounds are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Lawrence SA. Amines: Synthesis, Properties and Applications. Cambridge University Press; New York, NY: 2004. [Google Scholar]; (b) Nugent TC, El-Shazly M. Adv Synth Catal. 2010;352:753. [Google Scholar]; (c) Smith MB. Compendium of Organic Synthetic Methods. Vol. 12 Wiley; New York, NY: 2009. [Google Scholar]; (d) Emerson WS. Organic Reactions. Wiley; New York, NY: 2004. The Preparation of Amines by Reductive Alkylation. [Google Scholar]; (e) Afagh NA, Yudin AK. Angew Chem Int Ed. 2010;49:262. doi: 10.1002/anie.200901317. [DOI] [PubMed] [Google Scholar]
- 2.(a) Zalatan DN, Du Bois J. Top Curr Chem. 2010;292:347. doi: 10.1007/128_2009_19. [DOI] [PubMed] [Google Scholar]; (b) Roizen JL, Harvey ME, Du Bois J. Acc Chem Res. 2012;45:911. doi: 10.1021/ar200318q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Müller P, Fruit C. Chem Rev. 2003;103:2905. doi: 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]; (d) Li ZG, He C. Eur J Org Chem. 2006;19:4313. [Google Scholar]; (e) Halfen JA. Curr Org Chem. 2005;9:657. [Google Scholar]; (f) Dequirez G, Pons V, Dauban P. Angew Chem Int Ed. 2012;51:7384. doi: 10.1002/anie.201201945. [DOI] [PubMed] [Google Scholar]; (g) Davies HML, Morton D. Chem Soc Rev. 2011;40:1857. doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]; (h) Davies HML, Du Bois J, Yu JQ. Chem Soc Rev. 2011;40:1855. doi: 10.1039/c1cs90010b. [DOI] [PubMed] [Google Scholar]; (i) Collet F, Dodd RH, Dauban P. Chem Commun. 2009;34:5061. doi: 10.1039/b905820f. [DOI] [PubMed] [Google Scholar]; (j) Collet F, Lescot C, Dauban P. Chem Soc Rev. 2011;40:1926. doi: 10.1039/c0cs00095g. [DOI] [PubMed] [Google Scholar]; (k) Collet F, Lescot C, Liang CG, Dauban P. Dalton Trans. 2010;39:10401. doi: 10.1039/c0dt00283f. [DOI] [PubMed] [Google Scholar]; (l) Lescot C, Darses B, Collet F, Retailleau P, Dauban P. J Org Chem. 2012;77:7232. doi: 10.1021/jo301563j. [DOI] [PubMed] [Google Scholar]; (m) Gómez-Emeterio BP, Urbano J, Díaz-Requejo MM, Pérez PJ. Organometallics. 2008;27:4126. [Google Scholar]
- 3.(a) Hayes CJ, Beavis PW, Humphries LA. Chem Commun. 2006:4501. doi: 10.1039/b611662k. [DOI] [PubMed] [Google Scholar]; (b) Fiori KW, Du Bois J. J Am Chem Soc. 2007;129:562. doi: 10.1021/ja0650450. [DOI] [PubMed] [Google Scholar]; (c) Fiori KW, Espino CG, Brodsky BH, Du Bois J. Tetrahedron. 2009;65:3042. [Google Scholar]; (d) Zalatan DN, Du Bois J. J Am Chem Soc. 2008;130:9220. doi: 10.1021/ja8031955. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kornecki KP, Berry JF. Eur J Inorg Chem. 2012;3:562. [Google Scholar]; (f) Srivastava RS, Tarver NR, Nicholas KM. J Am Chem Soc. 2007;129:15250. doi: 10.1021/ja0751072. [DOI] [PubMed] [Google Scholar]; (g) Barman DN, Nicholas KM. Eur J Org Chem. 2011;5:908. [Google Scholar]
- 4.(a) Harvey ME, Musaev DG, Du Bois J. J Am Chem Soc. 2011;133:17207. doi: 10.1021/ja203576p. [DOI] [PubMed] [Google Scholar]; (b) Paradine SM, White MC. J Am Chem Soc. 2012;134:2036. doi: 10.1021/ja211600g. [DOI] [PubMed] [Google Scholar]
- 5.(a) Adams CS, Boralsky LA, Guzei IA, Schomaker JM. J Am Chem Soc. 2012;134:10807. doi: 10.1021/ja304859w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boralsky LA, Marston D, Grigg RD, Hershberger JC, Schomaker JM. Org Lett. 2011;13:1924. doi: 10.1021/ol2002418. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Grigg RD, Schomaker JM, Timokhin V. Tetrahedron. 2011;67:4318. [Google Scholar]
- 6.Rigoli JW, Weatherly CD, Vo VT, Neale S, Meis AR, Schomaker JM. Org Lett. 2013;15:290. doi: 10.1021/ol303167n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Cui Y, He C. J Am Chem Soc. 2003;125:16202. doi: 10.1021/ja038668b. [DOI] [PubMed] [Google Scholar]; (b) Cui Y, He C. Angew Chem Int Ed. 2004;43:4210. doi: 10.1002/anie.200454243. [DOI] [PubMed] [Google Scholar]; (c) Li Z, Capretto DA, Rahaman RH, He C. Angew Chem Int Ed. 2007;46:5184. doi: 10.1002/anie.200700760. [DOI] [PubMed] [Google Scholar]; (d) Harmata M, editor. Silver in Organic Chemistry. Wiley; Hoboken, NJ: 2010. [Google Scholar]; (e) Llaveria J, Beltran A, Diaz-Requejo MM, Matheu MI, Castillon S, Perez PJ. Angew Chem Int Ed. 2010;49:7092. doi: 10.1002/anie.201003167. [DOI] [PubMed] [Google Scholar]; (f) Maestre L, Sameera WMC, Diaz-Requejo MM, Maseras F, Perez PJ. J Am Chem Soc. 2013;135:1338. doi: 10.1021/ja307229e. [DOI] [PubMed] [Google Scholar]
- 8.(a) Hung-Low F, Renz A, Klausmeyer KK. Polyhedron. 2009;28:407. [Google Scholar]; (b) Hung-Low F, Renz A, Klausmeyer KK. J Chem Cryst. 2011;41:1174. [Google Scholar]; (c) Du JL, Hu TL, Zhang SM, Zeng YF, Bu XH. CrystEngComm. 2008;10:1866. [Google Scholar]; (d) Zhang H, Chen L, Song H, Zi G. Inorg Chim Acta. 2011;366:320. [Google Scholar]; (e) Hung-Low F, Renz A, Klausmeyer KK. J Chem Cryst. 2009;39:438. [Google Scholar]; (f) Levason W, Spicer MD. Coord Chem Rev. 1987;76:45. [Google Scholar]; (g) Leschke M, Rheinwald G, Lang H. Z Anorg Allg Chem. 2002;628:2470. [Google Scholar]
- 9.Price WS. Concepts Magn Reson. 1997;9:299. [Google Scholar]
- 10.(a) Casey CP, Johnson JB, Singer SW, Cui Q. J Am Chem Soc. 2005;127:3100. doi: 10.1021/ja043460r. [DOI] [PubMed] [Google Scholar]; (b) Valentini M, Pregosin PS, Rüegger H. Organometallics. 2000;19:2551. [Google Scholar]
- 11.(a) Au SM, Huang JS, Yu WY, Fung WH, Che CM. J Am Chem Soc. 1999;121:9120. [Google Scholar]; (b) Au SM, Huang JS, Che CM, Yu WY. J Org Chem. 2000;65:7858. doi: 10.1021/jo000881s. [DOI] [PubMed] [Google Scholar]; (c) Leung SK, Tsui WM, Huang JS, Che CM, Liang JL, Zhu N. J Am Chem Soc. 2005;127:16629. doi: 10.1021/ja0542789. [DOI] [PubMed] [Google Scholar]
- 12.(a) Harvey JN, Aschi M, Schwartz H, Koch W. Theor Chem Acc. 1998;99:95. [Google Scholar]; (b) Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Chem Rev. 2005;105:2279. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]; (c) Meunier P, de Visser SP, Shaik S. Chem Rev. 2004;104:3947. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.