

Abstract
Progression from colorectal adenoma to carcinoma is strongly associated with an accumulation of genomic alterations, including gain of chromosome 13. This gain affects the whole q arm and is present in 40%–60% of all colorectal cancers (CRCs). Several genes located at this amplicon are known to be overexpressed in carcinomas due to copy number dosage. A subset of these genes, including the mir-17~92 cluster, are functionally involved in CRC development. The present study set out to explore whether apart from mir-17~92, other miRNAs located at the 13q amplicon show a copy number dependent dosage effect that may contribute to 13q-driven colorectal adenoma-to-carcinoma progression. Integration of publically available miRNA expression, target mRNA expression and DNA copy number data from 125 CRCs yielded three miRNAs, miR-15a, -17, and -20a, of which high expression levels were significantly correlated with a 13q gain and which influenced target mRNA expression. These results could be confirmed by qRT-PCR in a series of 100 colon adenomas and carcinomas.Functional analysis of both mature miRNAs encoded by mir-15a, i.e. miR-15a-5p and miR-15a-3p, showed that silencing of miR-15a-3p significantly inhibited viability of CRC cells. Integration of miR-15a expression levels with mRNA expression data of predicted target genes identified mitochondrial uncoupling protein 2 (UCP2) and COP9 signalosome subunit 2 (COPS2) as candidates with significantly decreased expression in CRCs with 13q gain. Upon silencing of miR-15a-3p, mRNA expression of both genes increased in CRC cells, supporting miR-15a-3p mediated regulation of UPC2 and COPS2 expression. In conclusion, significant overexpression of miR-15a-3p due to gain of 13q is functionally relevant in CRC, with UCP2 and COPS2 as candidate target genes. Taken together our findings suggest that miR-15a-3p may contribute to adenoma-to-carcinoma progression.
Introduction
The development of colorectal cancer (CRC) is marked by the accumulation of several recurrent chromosomal alterations, including gains of 8q, 13q, and 20q and losses of 8p, 15q, 17p and 18q [1–3], which can lead to altered expression of oncogenes and tumour suppressor genes [4,5]. In fact, for a number of genes identified to be differentially expressed in a copy number dependent manner, functional relevance has been demonstrated. For instance, DIS3, LNX2 and CDK8 at 13q were recently described to play a role in CRC development due to 13q gain-dependent overexpression [6–8]. The same holds true for AURKA and TPX2 located at 20q, which were found to promote 20q amplicon-driven colorectal adenoma to carcinoma progression [9].
In addition to protein-encoding genes, DNA copy number changes may also affect expression of microRNAs (miRNAs) [10,11]. MiRNAs are a family of small non-coding RNA molecules that play an important role in the regulation of many cellular processes by targeting the 3’ UTR of mRNA molecules, thereby leading to gene silencing [12]. Dysregulation of miRNA expression has been shown to play an important role in several human diseases, including cancer [13].
In CRC, altered expression of a number of miRNAs, due to chromosomal alterations or other mechanisms like epigenetic modifications, has been described. A well-documented example is the altered expression of the oncogenic miRNA cluster, mir-17~92 [14]. We previously showed that increased mir-17~92 expression was linked to copy number gain of 13q and increased c-MYC expression during colorectal adenoma to carcinoma progression [15]. Moreover, upregulation of mir-17~92 by c-MYC showed pro-angiogenic activity in colonocytes [16].
Chromosome 13 is gained in 40–60% of CRCs and is strongly associated with colorectal adenoma to carcinoma progression [1,2,5]. This gain mostly encompasses the entire q-arm of chromosome 13 [6,17], raising the question whether next to coding genes and the mir-17~92 cluster, expression of other miRNAs located at this region is also affected by copy number dosage and may contribute to colorectal adenoma to carcinoma progression.
The present study aimed to identify additional candidate oncomiRs located at 13q in CRC. To this end, as an unbiased approach, we analysed all miRNAs mapping at 13q. Following validation of DNA copy number-dependent overexpression of 13q miRNAs by integrative analysis of DNA copy number and expression data-sets, the functional role of selected miRNAs was investigated in CRC cell lines using loss-of-function assays. Candidate target genes were identified by integration of miRNA expression levels with mRNA expression levels.
Materials and Methods
TCGA data
Data of 125 CRC samples from The Cancer Genome Atlas (TCGA) on DNA-copy number, mRNA and miRNA expression were downloaded from the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/tcgaDataType.jsp). Additional information of all the cases used in this study (sample ID, platform, data level and file name) can be found in S1 Table. In all cases level 2 data (i.e. normalised signals per probe or probe set) were obtained, except for the SNP data, for which only level 3 was available (i.e. segmented DNA copy number data). The latter were re-formatted to represent the copy number measurements on 30,000 equally spaced locations on the genome. More detailed information on the data types downloaded can be found at the TCGA Data Portal.
Integration of expression and copy number data
To investigate 13q miRNAs of which expression was regulated by DNA copy number changes, associations between DNA copy number and miRNA expression levels were studied. To this end we used an integrated approach, based on the use of covariate sets rather than single covariates, to determine subtle consistent associations with copy number alterations of a chromosomal region. The covariate set was defined by considering each miRNA probe individually and all copy number measurements within a 2Mb-window around the genomic start position of the miRNA. The analysis pipeline and definition of the covariate set is graphically presented in Fig 1 (association 1). The method used is implemented in the BioConductor package SIM [18]. This procedure finds miRNAs of which expression is likely to be regulated by DNA copy number changes within this 2MB-window, by assigning one p-value to each miRNA. Multiple testing correction was done using the false-discovery rate (FDR) step-down procedure of Benjamini & Hoghberg [19]. This analysis yielded a list of FDR values for miRNAs, of which expression is likely to be regulated by copy number changes in these samples. MiRNAs with FDR<0.05 were considered significant.
Fig 1. Schematic overview of in silico analysis pipeline used in this study.
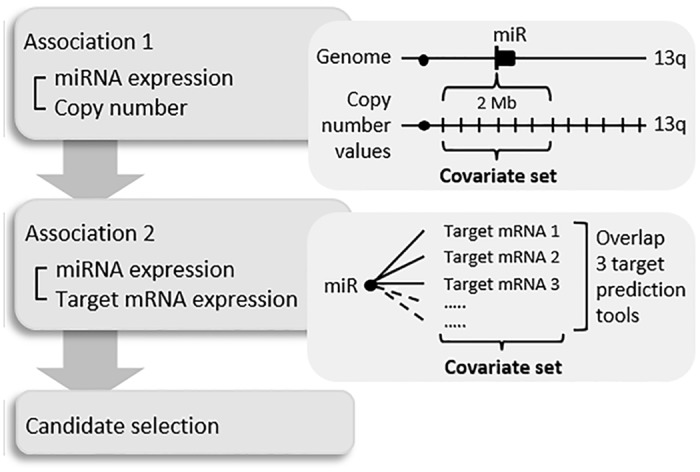
First level (association 1) shows a graphical representation of integration analysis of miRNA expression with copy number (CN) data. CN values for miRNAs on 13q were determined by combining values within a 2 Mb window surrounding the start of the miRNA. These values defined the covariate set used to determine association with miRNA expression. Second level (association 2) gives a graphical representation of the association of miRNA expression with predicted target mRNA expression. For each miRNA on 13q target mRNAs were determined by combining three or more target prediction tools. Expression levels of these target mRNAs defined the covariate set used to determine association with miRNA expression.
Target mRNA and miRNA expression integration
To identify which miRNAs are more likely to be functional, we studied their association with expression of predicted target mRNAs in the 125 CRC TCGA samples. To this end we used the approach developed by van Iterson et al [20]. In short, for each miRNA, results of 4 target-prediction tools, namely TargetScan, PITA, microCosm and Mirtarbase [21–24], were combined to yield a list of possible mRNA targets. For further (i.e. second) analysis, candidate mRNA targets that were predicted by three or more prediction tools were selected. This list of target mRNAs defined the covariate set for this second analysis (Fig 1, association 2). Subsequently, a global test was used [25] to test for associations between miRNA and mRNA expression, yielding one p-value per miRNA. Here the same statistical test was used as for the miRNA-copy number analysis, but now using the mRNA target list as covariate set. These p-values were corrected for multiple testing using the Benjamini & Hochberg FDR [19]. FDR values <0.05 were considered significant. For each significant miRNA, we subsequently prioritised the mRNA targets using the p-value derived from an additional global test applied to the miRNA and each individual mRNA target. We selected for further analyses mRNA targets that had an individual p-value of <0.01.
In order to analyse expression patterns of several selected target mRNAs, previously generated expression data were used of a panel of colorectal adenomas (n = 37) and carcinomas (n = 31), available at GEO under accession number GSE8067 [5]. Expression levels in both groups were compared using the Mann-Whitney U-test, considering p-values <0.05 as significant.
Tumour samples
A series of 100 snap-frozen colorectal tumour samples (48 adenomas and 52 carcinomas) prospectively collected at the VU University Medical Center in Amsterdam was used in this study. RNA from these samples was isolated using TRIzol (Invitrogen, Life technologies, Bleiswijk, The Netherlands), following the supplier’s instructions. For a subset of these samples (12 adenomas and 15 carcinomas) whole genome DNA copy number status was known (5). For the remaining samples copy number status of chromosome 13q was determined by multiplex ligation-dependent probe amplification (MLPA) as described previously [26].
Quantitative reverse transcription-PCR
MiRNA expression levels in colon adenomas and carcinomas of miR-15a-5p, miR-15a-3p, miR-17 and miR-20a were analysed using TaqMan MicroRNA Assays (assays: 000389, 002419, 002308, 000580 respectively) according to the manufacturer’s instructions (Applied Biosystems, Life Technologies). Relative miRNA expression levels were determined using the 2(-ΔΔCt)-method [27]. Data were normalised using miR-16 as a reference gene (Applied Biosystems, Life technologies).
In cell lines miRNA expression levels of miR-15a-3p, miR-15a-5p and miR-17 were analysed using the miRCURY LNA Universal RT microRNA PCR (Exiqon, Vedbaek, Denmark) following the manufacturer’s instructions. PCRs were run on an ABI 7500 Fast Real-time PCR system (Life technologies). Relative miRNA expression levels were determined using the 2(-ΔΔCt)-method [27]. Data were normalised using the small nucleolar RNA transcript RNU43 as a reference gene (Exiqon).
Messenger RNA (mRNA) expression levels of predicted targets (i.e. UCP2 and COPS2) were analysed following cDNA synthesis using a mix of oligo(dT) and random hexamers (iScript cDNA Synthesis Kit; Bio-Rad, Veenendaal, The Netherlands). PCR reactions were performed using SYBR Green PCR mastermix (Life Technologies) with 0.5 μM of each primer (UCP2: 5’-CACCGTGAGACCTTACAAAGCC-3’, 3’-TGCTACGTCCCAGGAGATGG-5’; COPS2: 5’-CGCCAGTTACATCAGTCGTGC-3’, 3’-AGTGGATGAGGGATGGCAGAC-5’) and 20 ng of cDNA. Amplification reactions were performed for 50 cycles with an annealing temperature of 60°C, using a ViiA7 PCR system (Life Technologies). Data were normalised using β-2-microglobulin as a reference gene [28] and relative mRNA expression levels were determined using the 2(-ΔΔCt)-method [27]. Expression levels of miRNAs and target genes were compared using Student’s t-test, considering p-values <0.05 as significant.
Cell culture and transfections
The CRC cell line SW480 was kindly provided Dr. G.J. Peters, Department of Oncology, VU University Medical Center. SW480 was cultured in Dulbecco’s modified Eagle’s medium (Lonza, Wijchen, The Netherlands) supplemented with 10% fetal bovine serum (PAA, Cölbe, Germany), 2 mM L-glutamine (Life Technologies), 100 IU/ml sodium-penicillin and 100 μg/ml streptomycin. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2.
SW480 was transiently transfected with 40 nM of mirCURY LNA microRNA power inhibitor directed against miR-15a-3p, miR-15a-5p and miR-17 (Assays: 426841–00, 426842–00, 426848–00; Exiqon). Negative control A was used as non-targeting control (Assays: 199020–04 and 199020–00; Exiqon). Cells were seeded in 6-wells cell-culture plates and transfections were performed after 18 to 24 hours. Lipofectamine 2000 Reagent (Life technologies) was used as transfection reagent following manufacturer’s instructions.
Cell viability
Cell viability was measured using the 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyl tetrazolium bromide assay (MTT; MP Biomedicals, Eindhoven, The Netherlands). Twenty-four hours post-transfection with the antagomirs, cells were seeded in 96-wells culture plates for the MTT assay. Simultaneously, RNA was extracted from a subset of the cells to determine the miRNA expression levels. MTT measurements were performed at t = 0 and subsequently after 3 and 6 days. The relative viability was determined by subtracting the measurement at day 0 from subsequent time points. All measurements were performed in triplicate (technical replicates) and at least 2 independent experiments were performed (biological replicates). Relative viability of cells was compared using Student’s t-test, with p-values <0.05 considered significant.
Results
MiR-15a shows copy number-associated differential expression in CRC
In the present study, publically available TCGA data from 125 CRC samples were used to analyse both miRNA expression levels and DNA copy number levels of the loci involved on chromosome 13 [17]. According to miRBase release 21 (www.mirbase.org), there are 40 miRNA located at chromosome 13 [29]. For 14 out of these 40 miRNAs on 13q, expression data were available, and 6 of these were significantly overexpressed in CRCs due to copy number gain of 13q, i.e. miR-15a, -17, -19a, -20a, 19b-1 and -92a-1 (Table 1; FDR<0.05). These miRNAs all belong to two well-known clusters of miRNAs on chromosome 13, i.e. mir-15a/16 and mir-17~92.
Table 1. Association of miRNA expression with 13q copy number status and target mRNA expression.
| miRNA | Chr | Start (bp position) | Copy number (FDR) | Target mRNA exp (FDR) |
|---|---|---|---|---|
| hsa-mir-20a | 13q | 90801327 | 0.000 | 0.000 |
| hsa-mir-92a-1 | 13q | 90801618 | 0.000 | n.d. |
| hsa-mir-17 | 13q | 90800874 | 0.000 | 0.001 |
| hsa-mir-15a | 13q | 49521268 | 0.000 | 0.010 |
| hsa-mir-19a | 13q | 90801194 | 0.001 | 0.052 |
| hsa-mir-19b-1 | 13q | 90801463 | 0.002 | n.d. |
| hsa-mir-16-1 | 13q | 49521123 | 0.160 | n.d. |
| hsa-mir-18a | 13q | 90801011 | 0.255 | 0.017 |
| hsa-mir-320d-1 | 13q | 40199964 | 0.360 | n.d. |
| hsa-mir-622 | 13q | 89681504 | 0.428 | 0.633 |
| hsa-mir-623 | 13q | 98806372 | 0.779 | 0.050 |
| hsa-mir-1297 | 13q | 53784118 | 0.807 | 0.984 |
| hsa-mir-1267 | 13q | 106981565 | 0.964 | 0.989 |
| hsa-mir-621 | 13q | 40282935 | 0.969 | 0.906 |
MiRNAs are ordered by significance of FDR of miRNA expression versus copy number. Chr, Chromosome; bp, base pair; exp, expression; n.d., no data; Significance considered at FDR<0.05.
Next to miRNA expression data, mRNA expression data were also available for the same panel of 125 CRCs. Using these data we examined whether overexpression of these miRNAs on 13q resulted in changes in the expression of predicted target genes [20]. This analysis showed that, for miR-15a, -17 and -20a, overexpression due to copy number also significantly influenced predicted target mRNA expression (Table 1; FDR<0.05).
Next we analysed by qRT-PCR the expression levels of the top candidate miRNAs found in the in silico analyses in a series of 100 colon adenoma and carcinoma samples. Given the fact that mir-15a encodes for two mature miRNA strands, miR-15a-5p and miR-15a-3p, both were studied using strand-specific qRT-PCR. In line with the in silico data, expression levels of miR-15a-3p, miR-17 and miR-20a, were significantly higher in carcinomas compared to adenomas (Fig 2A). However, expression of miR-15a-5p was not significantly increased. Comparison of tumours with 13q gain with tumours without 13q gain showed that expression levels of all four miRNAs were significantly higher in tumours with gain (Fig 2B), corroborating the results from the in silico analysis.
Fig 2. Analysis of miRNA expression in colorectal adenomas and carcinomas.

A) Box plots comparing relative expression determined by qRT-PCR in adenomas (n = 48) versus carcinomas (n = 52) of four top candidate miRNAs, miR-15a-3p, miR-15a-5p, miR-17 and miR-20a. B) Box plots comparing miRNA expression between tumours (adenomas and carcinomas) with (n = 24) or without (n = 73) 13q gain. P-values, determined by Mann-Whitney U-test, ≤ 0.05 are considered significant (* p ≤ 0.05; ** p < 0.01).
MiR-15a-3p inhibits viability of CRC cells
Based on these analyses, miR-15a, -17 and -20a were identified as candidate oncogenic miRNAs involved in CRC. Of these, both miR-17 and -20a have recently been described as oncogenes in CRC and prostate cancer [30–32], confirming the validity of the approach. MiR-15a on the other hand has been described to function as a tumour suppressor in chronic lymphocytic leukaemia (CLL) [33,34] and, more recently, in CRC [35]. To further support our current observations pointing to an oncogenic role of miR-15a in CRC, we set out to determine the functional relevance of miR-15a overexpression in CRC. Both strands of miR-15a, miR-15a-5p and miR-15a-3p, were studied using strand-specific miRNA inhibitors (antagomirs). An antagomir directed against miR-17, silencing of which was shown to decrease viability of CRC cells, was included as a positive control [30]. A non-targeting antagomir was included as a negative control.
The antagomirs were introduced into the SW480 CRC cell line, which has a gain of 13q encompassing the miR-15a locus. Efficiency of silencing varied per experiment between 75%-97%, 60–80% and 94%-96% for miR-15a-5p, miR-15a-3p and miR-17, respectively (Fig 3A). Silencing of miR-15a-3p and miR-17 led to a significant decrease in cell viability (Fig 3B). Silencing of miR-15a-5p also decreased cell viability significantly, although results were less consistent throughout replicate experiments (data not shown).
Fig 3. Functional analysis of the effect of miR-15a-5p, -3p and miR-17 silencing in CRC cell line SW480.
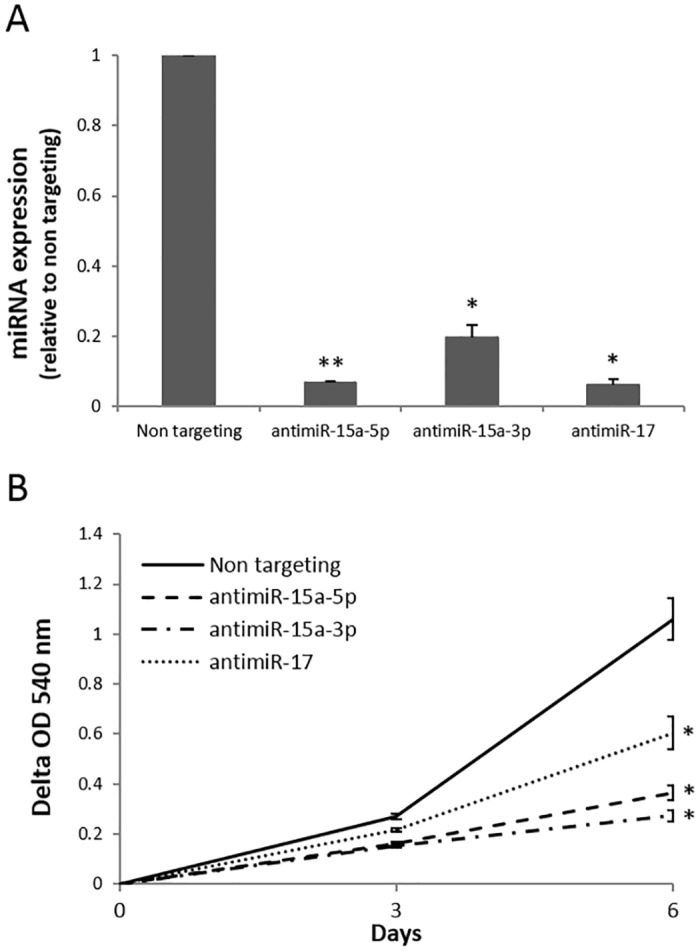
A) Representative example of expression levels of miRNAs upon silencing using antagomirs compared to a non-targeting control (**p<0.01; *p<0.05). B) Cell viability determined by MTT assay upon miRNA silencing in SW480 compared to non-targeting control (* p<0.05).
UCP2 and COPS2 as potential targets of miR-15a-3p in CRC
By integrating miRNA expression profiles with mRNA expression profiles of predicted targets in 125 CRCs, we identified candidate target genes of miR-15a [20]. The top five are shown in Table 2 (p <0.01). Of these five candidate target genes, four genes (TYRO3, UCP2, COPS2, RASGEF1B) were negatively associated with miR-15a expression, whereas (RNF125) was positively associated.
Table 2. Significantly affected predicted target genes of miR-15a.
| Gene symbol | Chr | p-value | Association |
|---|---|---|---|
| TYRO3 | 15q | 0.001 | - |
| UCP2 | 11q | 0.001 | - |
| RNF125 | 18q | 0.001 | + |
| RASGEF1B | 4q | 0.008 | - |
| COPS2 | 15q | 0.010 | - |
Genes are ordered by significance (NCBI build 37). Significance at p-value <0.01. Positive and negative association denotes association of mRNA target expression versus expression of miR-15a. Chr, chromosome.
Since copy number gain of chromosome 13 has been associated with progression from colorectal adenoma-to-carcinoma, and current data show that this gain is associated with overexpression of miR-15a, we next evaluated whether the expression levels of the top five predicted target genes would show a decrease in carcinomas compared to adenomas, in conjunction with 13q gain. To this end, previously obtained array-CGH and mRNA expression microarray data of a panel of 37 colon adenomas and 31 carcinomas were used [5]. In this set, expression of UCP2 was significantly decreased in carcinomas compared to adenomas (Fig 4A). Both UCP2 and COPS2 were significantly decreased in adenomas and carcinomas with a 13q gain compared to those without (Fig 4B). The comparison of adenomas without 13q gain to carcinomas with 13q gain, showed for both UCP2 and COPS2 significantly decreased expression in carcinomas with 13q gain (Fig 4C). In the case of TYRO3, a significant increase in expression was observed in both carcinomas (compared to adenomas), and in samples with 13q gain (compared to tumours without 13q gain). RNF125 and RASGEF1B expression levels were not related to progression or 13q copy number gain.
Fig 4. Analysis of target gene expression in colorectal adenomas and carcinomas.

A) Box plots comparing relative mRNA expression (determined by expression microarray) in adenomas (n = 37) versus carcinomas (n = 31) of 5 target genes, TYRO3, UCP2, RNF125, RASGEF1B and COPS2. B) Box plots comparing mRNA expression between tumours (adenomas and carcinomas) with (n = 18) or without (n = 44) 13q gain. C) Box plots comparing mRNA expression in adenomas without 13q gain (n = 30) to carcinomas with 13q gain (n = 14). P-values, determined by Mann-Whitney U-test, ≤ 0.05 are considered significant (* p ≤ 0.05; ** p < 0.01; ns, not significant).
To further establish a role of miR-15a overexpression in UCP2 and COPS2 downregulation in CRC, mRNA expression levels of both genes were determined in SW480 cells, in which either miR15a-3p, miR15a-5p or control miR-17 were silenced. Compared to the non-targeting control, mRNA expression levels of UCP2 and, to a lesser extent, COPS2 were increased upon silencing of miR-15a-3p (Fig 5). Hardly any effect was seen when using antagomirs against miR-15a-5p or miR-17 (Fig 5). These results indicate that miR-15a-3p indeed targets both UCP2 and COPS2 in SW480 CRC cells.
Fig 5. Analysis of mRNA expression levels of target genes in colorectal cancer cell line SW480.
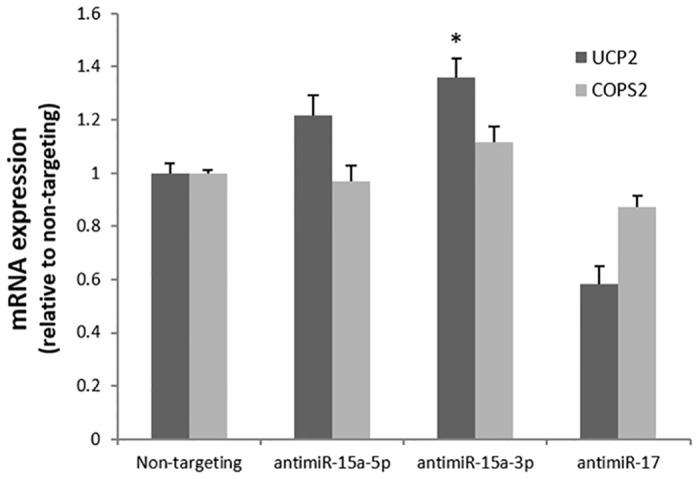
Relative expression of UCP2 and COPS2 in SW480 cells silenced for miR-15a-5p, -3p and miR-17 compared to a non-targeting control (* p<0.05).
Discussion
Here we investigated miRNAs that are overexpressed in CRC due to copy number gain of chromosome 13q. Analysis of publicly available TCGA miRNA expression and DNA copy number data of 125 CRC samples [17] revealed that miR-15a, -17 and -20a were significantly upregulated in CRC in association with copy number gain of 13q, and that miR-15a, -17 and -20a significantly influenced expression of target genes. For both miR-20a and -17, an oncogenic role in CRC has been established [30,36]. Interestingly, for miR-15a actually a tumour suppressor role has been claimed [35]. The present results however indicate that miR-15a, in particular miR-15a-3p, functions as an oncogenic miRNA in CRC. Silencing of miR-15a-3p significantly decreased viability of CRC cells.
MiR-15a is part of the mir-15a/16-1 cluster located at chromosome 13q14, which is encoded by its host gene deleted in leukaemia 2 (DLEU2) [34,37]. To date, members of the mir-15a/16-1 cluster have been described as tumour suppressor miRNAs in several types of cancer [34,35,38–40]. In chronic lymphocytic leukaemia, inactivation of these miRNAs occurs through a chromosomal loss at the locus of the mir-15a/16-1 cluster [34]. In multiple myeloma, expression of mir-15a/16-1 was decreased whereas expression of its target gene VEGF-A was increased. Moreover, in these tumours expression of mir-15a/16-1 was shown to inhibit tumour formation by targeting pro-angiogenic factor VEGF-A [39]. Similarly, in prostate cancer the expression of the miRNA cluster was downregulated, which was associated with upregulation of target genes BCL2, CCND1 and WNT3A. Knockdown of miR-15a and miR-16 in an in vitro model enhanced cancer progression by affecting cell survival, proliferation and invasion [38].
In CRC, miR-16 of this cluster had already been described to have a function as tumour suppressor [41,42]. More recently however, the entire mir-15a/16-1 cluster was investigated and found to target AP4 in CRC, downregulation of which resulted in induction of mesenchymal-epithelial transition (MET), inhibition of migration and invasion, and induction of cell cycle arrest [35]. Additionally, expression of DLEU2, the gene encoding the mir-15a/16-1 cluster, was repressed in CRC samples compared to normal colon tissue and inversely correlated to AP4 expression. Altogether, those data suggest that miR-15a expression is decreased in CRC compared to normal tissue, which seems contradictory to the findings in the current study. In fact, the TCGA data presented here, also show that in CRC samples the expression of miR-15a actually increases in association with a DNA copy number gain of chromosome 13q. This gain is a common event in CRC, and in particular, in the progression from colorectal adenoma to carcinoma [2,6,17].
With respect to the role of the mir-15a/16-1 cluster in CRC, it is important to realize that the present study specifically focused on the association with a 13q gain involved in adenoma to carcinoma progression, whereas the above mentioned study of Shi et al. focused on CRC metastasis, representing a later event in the carcinogenic process [35]. More importantly, we specifically identified miR-15a-3p as the driver strand and did not find evidence for the whole cluster to be involved. Recent research also showed that miR-15a-3p plays a tumour suppressor role in several cancer cell lines by targeting BCL-X L. However, no significant effect was seen in CRC cell lines [40]. It is known that both the 5p and 3p strands of a miRNA can be expressed in cells and regulate target gene expression [43,44]. The expression of either strand can also be tissue-specific [45]. We therefore speculate that expression of different mature miRNAs from the mir-15a/16-1 cluster may have different roles in CRC, perhaps depending on the stage of CRC.
Evaluation of miR-15a-3p expression in a series of 48 adenomas and 52 carcinomas, for which 13q copy number status was known, showed that miR-15a-3p was significantly higher expressed in carcinomas compared to adenomas in a DNA copy number-dependent manner. These results corroborate an oncogenic role for increased miR-15a-3p expression on the 13q amplicon.
By combining the results of target prediction algorithms with mRNA expression profile data of CRCs, UCP2 and COPS2 were identified as candidate target genes of miR-15a. In support of this observation, upregulation of UCP2 and, to a lesser extent, of COPS2 was found in CRC cells when miR-15a-3p was silenced, compared to control transfectants. The absence of a similar effect in miR-15a-5p silenced CRC cells is consistent with our functional data showing that both strands seem to act independently. The present data indicate that UCP2, and potentially also COPS2, may function as tumour suppressors in CRC. Interestingly, overexpression of UCP2 in human pancreatic cancer and glioblastoma cell lines was recently described to repress malignancy by controlling mitochondrial function and redirecting energy production away from glycolysis [46]. Also COPS2 was described as a putative tumour suppressor gene in a panel of different cancer types, where loss-of-function showed partial bypass of cellular senescence [47]. Altogether, one could hypothesize that tumour cells would benefit from the inhibition of these genes through miRNA-mediated modulation.
According to the most recent release of the miRNA database (miRBase release 21), there are 40 miRNAs located at chromosome 13q. However, for this study we only had expression data of 14 13q-mapping miRNAs available, as expression levels of the remaining miRNAs were undetectable or not measured. Therefore, it cannot be excluded that additional miRNAs located at 13q may also be involved in CRC.
In summary, miR-15a-3p is gained and overexpressed in CRC, and this overexpression affects cell viability, supporting an oncogenic role of this miRNA in CRC. As a consequence, miR-15a-3p may play a role in the 13q gain-driven colorectal adenoma-to-carcinoma progression.
Supporting Information
Additional information on the data of 125 cases used from the TCGA. For all cases the sample ID, type of data (platform type), platform used, data level and the corresponding file names are listed. More information on the data used can be found at the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/tcgaDataType.jsp).
(DOC)
Data Availability
Expression microarray data from colorectal cancer cases are available at Gene Expression Omnibus (GEO) data repository, under accession number GSE8067. All other relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was funded by the VUmc Cancer Center Amsterdam (CCA) and performed within the framework of CTMM, the Center for Translational Molecular Medicine. DeCoDe project (grant 03O-101).
References
- 1. Meijer GA, Hermsen MAJA, Baak JPA, van Diest PJ, Meuwissen SGM, Belien JAM, et al. Progression from colorectal adenoma to carcinoma is associated with non-random chromosomal gains as detected by comparative genomic hybridisation. J Clin Pathol. 1998;51: 901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hermsen M, Postma C, Baak J, Weiss M, Rapallo A, Sciutto A, et al. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology. 2002;123: 1109–1119. [DOI] [PubMed] [Google Scholar]
- 3. Nakao K, Mehta KR, Fridlyand J, Moore DH, Jain AN, Lafuente A, et al. High-resolution analysis of DNA copy number alterations in colorectal cancer by array-based comparative genomic hybridization. Carcinogenesis. 2004;25: 1345–1357. [DOI] [PubMed] [Google Scholar]
- 4. Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3: 695–700. [DOI] [PubMed] [Google Scholar]
- 5. Carvalho B, Postma C, Mongera S, Hopmans E, Diskin S, van de Wiel M, et al. Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression. Gut. 2009;58: 79–89. 10.1136/gut.2007.143065 [DOI] [PubMed] [Google Scholar]
- 6. De Groen FLM, Krijgsman O, Tijssen M, Vriend LEM, Ylstra B, Hooijberg E, et al. Gene-dosage dependent overexpression at the 13q amplicon identifies DIS3 as candidate oncogene in colorectal cancer progression. Genes Chromosomes Cancer. 2014;53: 339–48. 10.1002/gcc.22144 [DOI] [PubMed] [Google Scholar]
- 7. Camps J, Pitt JJ, Emons G, Hummon AB, Case CM, Grade M, et al. Genetic amplification of the NOTCH modulator LNX2 upregulates the WNT/β-catenin pathway in colorectal cancer. Cancer Res. 2013;73: 2003–2013. 10.1158/0008-5472.CAN-12-3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455: 547–551. 10.1038/nature07179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sillars-Hardebol AH, Carvalho B, Tijssen M, Beliën JA, de Wit M, Delis-van Diemen PM, et al. TPX2 and AURKA promote 20q amplicon-driven colorectal adenoma to carcinoma progression. Gut. 2012;61: 1568–1575. [DOI] [PubMed] [Google Scholar]
- 10. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101: 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wilting SM, Snijders PJF, Verlaat W, Jaspers a, van de Wiel M a, van Wieringen WN, et al. Altered microRNA expression associated with chromosomal changes contributes to cervical carcinogenesis. Oncogene. 2013;32: 106–16. 10.1038/onc.2012.20 [DOI] [PubMed] [Google Scholar]
- 12. Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004;116: 281–297. [DOI] [PubMed] [Google Scholar]
- 13. Jansson MD, Lund AH. MicroRNA and cancer. Mol Oncol. Elsevier B.V; 2012;6: 590–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013;20: 1603–14. 10.1038/cdd.2013.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diosdado B, van de Wiel M, Terhaar Sive Droste J, Mongera S, Postma C, Meijerink W, et al. MiR-17-92 cluster is associated with 13q gain and c-myc expression during colorectal adenoma to adenocarcinoma progression. Br J Cancer. 2009;101: 707–14. 10.1038/sj.bjc.6605037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38: 1060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487: 330–337. 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Menezes RX, Boetzer M, Sieswerda M, van Ommen G-JB, Boer JM. Integrated analysis of DNA copy number and gene expression microarray data using gene sets. BMC Bioinformatics. 2009;10: 203 10.1186/1471-2105-10-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benjamini Y and Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Statist. Soc. B. 1995;57(1): 289–300. [Google Scholar]
- 20. Van Iterson M, Bervoets S, de Meijer EJ, Buermans HP, ‘t Hoen PAC, Menezes RX, et al. Integrated analysis of microRNA and mRNA expression: adding biological significance to microRNA target predictions. Nucleic Acids Res. 2013;41: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39: 1278–84. [DOI] [PubMed] [Google Scholar]
- 22. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36: 154–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19: 92–105. 10.1101/gr.082701.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hsu S-D, Lin F-M, Wu W-Y, Liang C, Huang W-C, Chan W-L, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39: 163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goeman JJ, van de Geer SA, de Kort F, van Houwelingen HC. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20: 93–9. [DOI] [PubMed] [Google Scholar]
- 26. Schouten JP, Mcelgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25: 402–408. [DOI] [PubMed] [Google Scholar]
- 28. Dydensborg AB, Herring E, Auclair J, Tremblay E, Beaulieu J-F. Normalizing genes for quantitative RT-PCR in differentiating human intestinal epithelial cells and adenocarcinomas of the colon. Am J Physiol-Gastr L. 2006;290: 1067–1074. [DOI] [PubMed] [Google Scholar]
- 29. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42: 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luo H, Zou J, Dong Z, Zeng Q, Wu D, Liu L. Up-regulated miR-17 promotes cell proliferation, tumour growth and cell cycle progression by targeting the RND3 tumour suppressor gene in colorectal carcinoma. Biochem J. 2012;442: 311–21. 10.1042/BJ20111517 [DOI] [PubMed] [Google Scholar]
- 31. Xu Z, Zhao L, Zhu L-Y, He M, Zheng L, Wu Y. MicroRNA-17, 20a regulates the proangiogenic function of tumor-associated macrophages via targeting hypoxia-inducible factor 2α. Cho WCS, editor. PLoS One. Public Library of Science; 2013;8: e77890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qiang X-F, Zhang Z-W, Liu Q, Sun N, Pan L-L, Shen J, et al. miR-20a promotes Prostate cancer invasion and migration through targeting ABL2. J Cell Biochem. 2014;115: 1269–1276. 10.1002/jcb.24778 [DOI] [PubMed] [Google Scholar]
- 33. Cimmino A, Calin GA, Fabbri M, Iorio M V, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102: 13944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99: 15524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shi L, Jackstadt R, Siemens H, Li H, Kirchner T, Hermeking H. p53-induced miR-15a/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2014;74: 532–42. 10.1158/0008-5472.CAN-13-2203 [DOI] [PubMed] [Google Scholar]
- 36. Chai H, Liu M, Tian R, Li X, Tang H. miR-20a targets BNIP2 and contributes chemotherapeutic resistance in colorectal adenocarcinoma SW480 and SW620 cell lines. Acta Biochim Biophys Sin (Shanghai). 2011;43: 217–25. [DOI] [PubMed] [Google Scholar]
- 37. Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17: 28–40. 10.1016/j.ccr.2009.11.019 [DOI] [PubMed] [Google Scholar]
- 38. Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Memeo L, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14: 1271–7. 10.1038/nm.1880 [DOI] [PubMed] [Google Scholar]
- 39. Sun C-Y, She X-M, Qin Y, Chu Z-B, Chen L, Ai L-S, et al. miR-15a and miR-16 affect the angiogenesis of multiple myeloma by targeting VEGF. Carcinogenesis. 2013;34: 426–35. 10.1093/carcin/bgs333 [DOI] [PubMed] [Google Scholar]
- 40. Druz A, Chen Y-C, Guha R, Betenbaugh M, Martin SE, Shiloach J. Large-scale screening identifies a novel microRNA, miR-15a-3p, which induces apoptosis in human cancer cell lines. RNA Biol. 2013;10: 287–300. 10.4161/rna.23339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Young LE, Moore AE, Sokol L, Meisner-Kober N, Dixon DA. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol Cancer Res. 2012;10: 167–80. 10.1158/1541-7786.MCR-11-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma Q, Wang X, Li Z, Li B, Ma F, Peng L, et al. microRNA-16 represses colorectal cancer cell growth in vitro by regulating the p53/survivin signaling pathway. Oncol Rep. 2013;29: 1652–1658. 10.3892/or.2013.2262 [DOI] [PubMed] [Google Scholar]
- 43. Yang X, Du WW, Li H, Liu F, Khorshidi A, Rutnam ZJ, et al. Both mature miR-17-5p and passenger strand miR-17-3p target TIMP3 and induce prostate tumor growth and invasion. Nucleic Acids Res. 2013;41: 9688–704. 10.1093/nar/gkt680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guo L, Lu Z. The fate of miRNA* strand through evolutionary analysis: implication for degradation as merely carrier strand or potential regulatory molecule? PLoS One. Public Library of Science; 2010;5: e11387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ro S, Park C, Young D, Sanders KM, Yan W. Tissue-dependent paired expression of miRNAs. Nucleic Acids Res. 2007;35: 5944–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Esteves P, Pecqueur C, Ransy C, Esnous C, Lenoir V, Bouillaud F, et al. Mitochondrial retrograde signaling mediated by UCP2 inhibits cancer cell proliferation and tumorigenesis. Cancer Res. 2014;74: 3971–3982. 10.1158/0008-5472.CAN-13-3383 [DOI] [PubMed] [Google Scholar]
- 47. Leal JFM, Fominaya J, Cascón A, Guijarro M V, Blanco-Aparicio C, Lleonart M, et al. Cellular senescence bypass screen identifies new putative tumor suppressor genes. Oncogene. 2008;27: 1961–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional information on the data of 125 cases used from the TCGA. For all cases the sample ID, type of data (platform type), platform used, data level and the corresponding file names are listed. More information on the data used can be found at the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/tcgaDataType.jsp).
(DOC)
Data Availability Statement
Expression microarray data from colorectal cancer cases are available at Gene Expression Omnibus (GEO) data repository, under accession number GSE8067. All other relevant data are within the paper and its Supporting Information files.