

Abstract
Helicobacter pylori infection causes chronic active gastritis, ulcer disease, and gastric cancer. Current eradication regimens use a proton pump inhibitor (PPI) and two antibiotics. Triple therapy now has a success rate less than 80%, below the cutoff for efficacious eradication. Antibiotic resistance, inconsistent acid control by PPIs, and poor patient compliance contribute to the failure rate. H. pylori is a neutralophile that has developed special acid acclimation mechanisms to colonize its acidic gastric niche. Identifying the components of these mechanisms will provide novel bactericidal drug targets. Alternatively, better 24-hour acid control would increase the efficacy of antibiotics, leading to dual therapy with improved PPIs and amoxicillin. Studies of acid acclimation by H. pylori have identified several potential eradication targets including UreI, α-carbonic anhydrase, and a two-component system. Continuing improvement of PPIs has led to the development of at least three candidate drugs with improved 24-hour acid control.
Introduction
It has been nearly 25 years since the association of Helicobacter pylori infection with gastritis and peptic ulcer disease was discovered. During this time, much effort has been spent investigating the role of the organism in gastric and extragastric diseases, development of eradication therapies, and basic biology. In addition to its role in gastric inflammation and ulcer disease, H. pylori is classified as a class 1 carcinogen for its role in gastric cancer, one of the mostly deadly cancers. At least two forms of gastric cancer are associated with H. pylori infection: adenocarcinoma and, less commonly, mucosa-associated lymphoid tissue (MALT) lymphoma [1]. Eradication of H. pylori in patients with duodenal or gastric ulcer cures the ulcers and reduces the risk of cancer [2,3].
Controversy exists whether the organism should be eradicated in asymptomatic individuals (the “test-andtreat” approach). It is claimed that the organism is not a pathogen but a commensal [4]. Evidence for this idea is the putative worsening of GERD after eradication, but most data suggest neither an increased incidence of GERD after eradication nor increased acid secretion or reflux [5]. In this article, we regard the risk of gastric adenocarcinoma as sufficient justification for a test-and-treat strategy, and focus on current and future eradication therapies.
Current H. pylori eradication therapy requires a proton pump inhibitor (PPI) and at least two antibiotics (triple therapy). This approach requires treatment for 7 to 14 days and has an inadequate success rate (< 80%) according to the Maastricht III consensus report [6]. The development of antimicrobial resistance by H. pylori is a major factor in unsuccessful eradication with triple therapy. Treatment with clarithromycin or metronidazole may also contribute to development of antibiotic resistance of other important bacterial pathogens [7,8]. In H. pylori, primary resistance to amoxicillin has not been described. In a recent meta-analysis, primary resistance to clarithromycin decreased the eradication rate by 50%, whereas primary resistance to metronidazole decreased the rate of eradication by 37% [9].
A meta-analysis of eradication failure categorized subjects according to their rate of metabolism of the PPI used in the therapy. Mutations in CYP219, a situation more common in Japan than in the West, provided information that improved acid suppression results from increased plasma residence time of omeprazole and consequently highly efficient eradication of H. pylori with a PPI and only amoxicillin twice daily [10].
Given the decreasing efficacy of triple therapy and its contribution to the global problem of increasing antibiotic resistance, novel therapy is needed for H. pylori eradication. Compliance is another issue: patients find twice-daily consumption of three tablets or capsules burdensome. Another confounding factor is that when a symptomatic patient is treated, the PPI relieves the symptoms, thus discontinuation of the medication may occur. This article therefore focuses on new potential eradication strategies.
Gastric Biology of H. pylori
To identify a better regimen for H. pylori eradication, an understanding of the gastric biology of the organism is needed. H. pylori is a neutralophile that colonizes the acidic gastric environment, and it has developed mechanisms to combat high acidity. Therefore, identifying the components of the acid resistance mechanisms and how these components are regulated may provide bactericidal drug targets by exploiting its acidic gastric niche.
Gastric Environment of H. pylori
The gastric lumen in humans with normal acid secretion has a median pH of 1.4 with pH elevations after dinner, breakfast. and lunch because of the buffering action of food [11]. The luminal pH of the stomach is not disputed; however, considerable controversy exists as to the gastric surface pH and the pH within the mucus layer, the sites of H. pylori infection [12]. Historically, it was proposed that a pH gradient exists through the gastric mucus, with the luminal face being acidic and the gastric surface being neutral because of restricted proton back diffusion by the mucus or secretion of HCO3- [13].
A considerable body of literature exists using either glass-tipped or open-tip microelectrodes, suggesting that a mucus “barrier” was present and hindered proton back diffusion [12,14]. More recently, however, use of fluorescent pH probes and confocal microscopy showed no difference between surface and luminal pH when the luminal pH was set at 3.0 [15]. Using pH microelectrodes in the mouse model, no gastric barrier was detected with H. pylori infection [16••]. Likewise, in the gerbil model, transcriptome analysis of infecting organisms showed that the gene profile reflected an acidity less than pH 4.5 [17••]. Hence, for colonization, H. pylori must be able to grow under acidic conditions encountered at the site of infection.
Acid Resistance of Neutralophiles
For an organism to pass through the stomach and access the small intestine or colon, it must survive both low and high acidity if ingested with or without food. For an organism to remain viable, it must maintain an inward electrochemical gradient of protons, the proton-motive force (PMF)—the algebraic sum of the inward pH gradient and the inner membrane electrical potential difference [18,19]. In Escherichia coli, the PMF is ~ -170 mV, moving protons inward to enable ATP synthesis via the F1F0 ATPase and allowing proton-coupled uptake or export of solutes [18]. Loss of an adequate inner membrane potential interferes with folding of membrane proteins and solute homeostasis. Therefore, in a neutralophile under acidic conditions cell growth is abolished, but the organism can still survive provided excessive acidification of the cytoplasm is prevented.
General Acid Responses of Neutralophiles
Various mechanisms have evolved to allow gastric transit by pathogenic bacteria. E. coli can survive extreme acid stress because of expression of 12 genes located at the acid fitness island [20]. One group of genes consists of glutamate, arginine, or lysine decarboxylases (gadA/B, adiA, cadA) along with the product antiporters (γ-amino butyric acid, agmatine, and cadaverine (GadC, AdiC, CadC). Their activity results in the consumption of a proton with each coupled cycle.
Acid Acclimation by H. pylori
H. pylori is a gram-negative neutralophile and shares some of the cytoplasmic pH regulatory systems with other neutralophiles. However, the acid acclimation pathways developed by this organism are unique, enabling H. pylori to maintain its periplasmic pH in acidic medium at levels where both the cytoplasmic pH and membrane potential are compatible with growth [21]. The several systems used to maintain periplasmic pH form the basis for acid acclimation and growth in the gastric environment.
The Urease System of H. pylori
H. pylori expresses a neutral pH optimum urease at higher levels than any other known bacteria, accounting for as much as 8% of the total protein [22]. Urease activity generates NH3 and CO2, providing acid neutralizing and buffering capacity. Urease-negative mutants are unable to colonize animal models, showing that this urease is essential for gastric habitation [23]. The urease gene cluster consists of seven genes: ureA and ureB (the structural subunits of the enzyme), followed by the urease accessory genes ureI, ureE, ureF, ureG, and ureH [24]. UreA and UreB form a hexameric heterodimer requiring Ni2+ insertion for activity. Nickel insertion is mediated by a pair of complexes—UreE/UreG and UreF/UreH [25]. Regulation of urease occurs at several levels: biosynthesis of the structural genes, insertion of Ni2+ into the apoenzyme, and regulation of urea access to urease.
UreA and UreB are present largely as apoenzyme; therefore, regulation of Ni2+ concentration inside the organism plays a vital role in acid survival. NixA is a specific Ni2+ transporter in the inner membrane of H. pylori. Deletion of nixA reduces, but does not prevent, infection of the mouse stomach, but many other genes may also be implicated in uptake and storage of this essential cation [26]. HypA and HypB are involved in the insertion of nickel into hydrogenase. Deletion of these nickel assembly genes not only affected hydrogenase activity but sharply reduced urease activity by 200-fold. This finding is attributed to a role for these genes in nickel incorporation into urease [27].
Role of Urease in Acid Acclimation
At neutral pH, urease activity of intact organisms is low, in contrast to that of total cell lysate. However, as the ambient pH is reduced from pH 6.5 to 5.5 and down to pH 2.5 the urease activity of the intact organism increases 10-20 fold. Thus acidic pH activates a urea transporter in the inner membrane, increasing urea access to cytoplasmic urease. Stable urease activity down to pH 2.5 also shows that even at this medium pH, cytoplasmic pH does not fall; otherwise urease activity would be inhibited [28]. Deletion of ureI, an integral membrane protein, prevented the pH-dependent increase of urease activity [28]. Hence, UreI is an acid-activated urea transporter providing urea to the cytoplasmic urease. Expression of ureI is essential for colonization of mouse and gerbils [29,30].
The ureI gene appears to be uniquely expressed by gastric Helicobacter spp [28]. Topographic analysis showed that this protein is a six-transmembrane segment polytopic membrane protein, and fractionation studies localized it to the inner membrane of the organism [31]. Expression of UreI in Xenopus oocytes determined its urea transport properties [32]. UreI displayed characteristics of a pH gated urea channel (nonsaturable, voltage independent), having low open probability at neutral pH and high open probability at pH 5.0 and below with half maximal opening at pH 5.9.
Consequences of UreI Activation
H. pylori is suitable for the study of its inner membrane potential and cytoplasmic pH using fluorescent dyes. At a medium pH of 7.4, the membrane potential was found to be ~ -180 mV, and as the pH of the medium was decreased, the membrane potential fell as expected from the increase of the inward ΔpH, reaching 0 mV at a medium pH of 3.5. The addition of urea under acidic medium pH conditions between pH 3.0 and 6.0 elevated inner membrane potential to a relatively constant value of ~ -101 mV, sufficient for maintenance of growth.
H. pylori cytoplasmic pH, measured with 2’,7’-bis-(2-carboxyethyl)-5-(and -6)-carboxyfluorescein, acetoxymethyl ester (BCECF), was determined over a range of medium pH from 7.4 to 4.5. In the absence of urea, at neutral medium pH, cytoplasmic pH was estimated to be about 8.0 and fell to 5.3 at a medium pH of 4.5. At medium pH 4.5, the cytoplasmic pH was restored to pH 6.5 with urea addition. Hence, at neutral pH, either in the absence or presence of urea, the PMF is ~ -40 mV (ΔpH) – 180 mV (ΔΨ) = ~ -220 mV. At pH 4.5 in the absence of urea, the PMF is ~ -48 mV – 45 mV = ~ -93 mV, but with the addition of urea at this pH the PMF rises back to -120 mV – 101 mV = 221 mV [21]. Hence, urease activity permits H. pylori to maintain a normal PMF at acidic pH. The key distinction between H. pylori and presumably other gastric Helicobacter spp and other acidresistant neutralophiles is the ability to maintain periplasmic pH at a level that allows a PMF to be maintained that allows cell growth to continue in the gastric environment.
Although these data clarified the role of UreI in the acid acclimation processes of H. pylori, it would be extremely unlikely that this protein along with urease would be the only means of combating the stress of gastric habitation. Hence, acid-induced gene regulation was studied using microarray analysis at pH 7.4, 6.2, 5.5 and 4.5 in the absence or presence of urea [21]. About 187 genes were up regulated by exposure to acidic pH and 100 were down regulated. Other laboratories have used microarray data, some in agreement with the data from this laboratory and others not, but thus far, the data presented here are unique in investigating the effect of urea on gene expression in different pH media [33-35].
Changes in gene expression that occur at a medium pH of 6.2 in the absence but not in the presence of urea are likely responses to periplasmic pH because the addition of urea reverses the gene regulation, indicating that normal periplasmic pH and membrane potential have been restored. Under these conditions, several genes, which might be considered as pH homeostatic genes, are able to produce NH3 such as urease, asparaginase, aspartate ammonia lyase, and the periplasmic α-carbonic anhydrase (HP1186) able to produce HCO3- as a periplasmic buffer. Also, three hydrogenase expression/forming genes, hypA, hypB, and hypC, were up regulated; they have a role in nickel uptake and sequestration, essential for adequate urease enzyme activity [36].
Because urease produces 2NH3 + CO2 from urea, α-carbonic anhydrase was investigated in detail. When the periplasmic carbonic anhydrase gene was deleted, many of the properties of the wild type were lost, similar to ureI deletion. For example, the addition of urea no longer restored membrane potential in acidic media. There was also complete loss of periplasmic buffering in the carbonic anhydrase mutant. The addition of acetazolamide to the wild-type organism, after regeneration of the membrane potential by urea, inhibited membrane potential, restoring it to baseline [37]. Further, acid survival was impaired, either with deletion of the enzyme or by addition of acetazolamide. There was a 3-log decrease in survival in the knockout mutants or in the presence of acetazolamide at pH 2.0 in the presence of urea. Recent in vivo data demonstrate a loss of infectivity in mice in the absence of carbonic anhydrase activity [38•]. These data, along with the gene analysis, show that the acid acclimation response of H. pylori is significantly more complicated than simply the activation of urease by acid activation of UreI.
Although the NH3 produced by urease activity is able to neutralize entering protons, the pKa of the NH4+/NH3 couple is 9.2 and would not effectively buffer the periplasm to a relatively neutral pH. Therefore, NH3 alone would not account for the finding that the periplasmic pH is relatively constant at 6.1 in the presence of urea, down to at least pHout 3.0. On the other hand, pH 6.1 is the effective pKa of HCO3-. Hence, both NH3 and CO2 production from intrabacterial urease activity enable acid acclimation by H. pylori, the NH3 to neutralize entering protons and the HCO3- to buffer the periplasm. At pH 4.0 and above, deletion of carbonic anhydrase had little effect on survival, hence the pH of the habitat of H. pylori in the mouse stomach must be less than 4.0.
Two-component Systems
Prokaryotes use two-component systems (TCS) to relay environmental stimuli to the cytoplasm, resulting in cellular responses similar to the G protein–coupled receptors (GPCR) in eukaryotes. TCS are minimally composed of a histidine kinase sensor protein, usually located in the inner membrane, and a response regulator located in the cytoplasm. Responding to an environmental stimulus, the sensor component autophosphorylates, followed by phospho-transfer from the kinase sensor to an aspartyl side chain of the response regulator. Although specificity of response to specific input signals is typical, evidence is mounting for cross-talk between a sensor kinase and noncognate response regulators. It may well be that this cross-talk is designed for optimal response to an environmental change, such as pH. Further, differences in affinity exist between sets of genes between the phosphorylated and nonphosphorylated forms of the regulator, allowing a graded response to a stimulus [39,40]. Thus, TCS, if vital for infection or colonization, make novel targets for eradication therapies.
H. pylori expresses four histidine kinases and six response regulators, including those with known response regulators, HP0165/HP0166 (ArsRS, an acid-responsive TCS), HP0244/HP0703 (FlgRS) involved in intermediate flagellar development, and HP1364/HP1365 [41,42]. Knockout mutants lacking the sensor kinase component of the TCS did not impair urease activity at neutral pH and only the HP0244 knockout mutation affected motility. However, none of the sensor kinase knockouts were able to infect the mouse stomach, a finding expected for the HP0244 deletion mutant because motility is required for infection [43]. Deletion of the response regulator HP1365 impaired growth in vitro at pH 5.5 [44].
ArsRS
Transcriptomal analysis of H. pylori, HP0165 deletion mutants at pH 5.0, found most genes were repressed [39]. 109 genes were affected by the deletion at this pH, including nine acid acclimation genes, seven envelope genes, and genes shown to be up regulated in vitro at acidic pH and in the gerbil stomach [17••, 42].
The acid acclimation gene cluster is only one set of genes used by H. pylori enabling gastric habitation, and the wide range of genes that change expression in acid or in the stomach attests to the broad response of this gastric denizen just to acidification. The general model resulting from the studies discussed above is shown in Figure 1, encompassing the results on urease, UreI, and carbonic anhydrase and regulation by the TCS ArsRS. Apparently, HP0165 mutants are not able to infect a mouse model.
Figure 1.
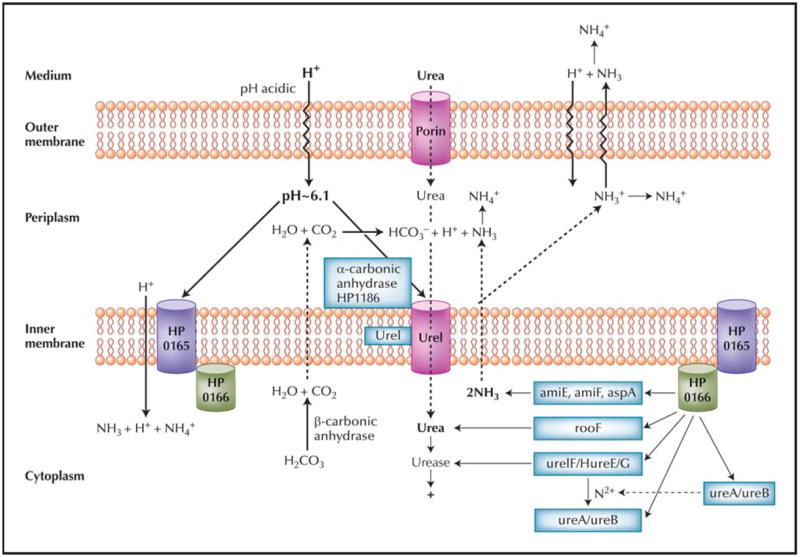
A model of HP0165/HP0166 regulation of periplasmic pH of Helicobacter pylori. Under acidic conditions, urea enters the periplasm through the outer membrane via porins. Urea entry into the cytoplasm is accelerated by the H+-gated urea channel, UreI, increasing urease activity. Periplasmic acidity also activates the histidine kinase sensor, HP0165, by phosphate transfer to the response element, HP0166, thus increasing transcription of the “acid acclimation” genes, with the exception of hypB and aspA. Arginase produces cytoplasmic urea and the amidases and aspartase, cytoplasmic NH3. Urease activity is increased by nickel insertion into the UreA/UreB apoenzyme by UreE, F, G, and H in concert with HypA and HypB, hydrogenase accessory proteins. Cytoplasmic urease generates 2NH3 + H2CO3- which is converted to CO2 + H2O by cytoplasmic β-carbonic anhydrase. The membrane-permeant NH3 and CO2 diffuse into the periplasm. The CO2 is converted to H+ + HCO3- by the membrane-bound periplasmic α-carbonic anhydrase that buffers the periplasm to pH about 6.1. The NH3 absorbs the proton released by carbonic anhydrase and the other NH3 is able to absorb entering protons or can alkalinize the medium. The cooperative upregulation of these genes and the role of the HP0165/HP0166 two-component system illustrate not only that the organism is exposed to acidic pH, but also that several genes of this regulon are involved in maintaining gastric infection.
HP0244
More recent studies examined the role of a cytoplasmic histidine kinase HP0244, already known to regulate expression of intermediate flagellar genes via the HP0703 regulon. Surprisingly, HP0244 deletion mutants failed to survive a pH of 2.0 even in the presence of 10-mM urea. Cytoplasmic pH and membrane potential were signifi cantly impaired, accounting for the loss of survival. Therefore, H. pylori senses both periplasmic and cytoplasmic pH via sensor kinases to regulate gene transcription in response to an acid challenge.
Blue native gel electrophoresis showed that UreA and UreB, soluble cytoplasmic proteins, were associated with UreI, a membrane protein [25]. Further investigation using immunoelectron microscopy showed that this association was increased at an acidic medium pH of 5.5 [45]. Hence, urea is immediately available to the urease under acidic conditions. It is also possible that UreI is able to transport NH3 more rapidly than free diffusion across the lipid bilayer, allowing even more rapid periplasmic neutralization of entering protons
Direct measurement of NH3 and CO2 permeability of H. pylori wild type and ureI deletion mutants did indeed show that this urea channel, similar to the aquaporin water channel, was able to transport NH3, NH4+, and CO2. Hence, the complex detected by electrophoresis and electron microscopy probably has relevant physiologic function.
Improved Acid Suppression
The drawback to current PPIs is that extending acid suppression into the night is very difficult because new pumps are being synthesized and the drug disappears within 2 hours of the evening meal. Improved acid suppression rests on the concept that amoxicillin requires the organism to be in growth phase for its action. At relatively high acidity, many organisms are in stationary phase and inhibition of acid secretion with PPI administration increases the fraction of the bacteria in growth phase. However, with the return of nocturnal acid secretion, the acidity is sufficient to keep many in stationary phase. The benefit of amoxicillin is that thus far there have been no reports of resistance to this antibiotic. Because the effectiveness of current triple therapy has fallen below 75%, improved acid inhibition in combination with amoxicillin should result in greater than 90% eradication. Currently, three candidate PPIs and a reversible inhibitor of the gastric acid pump show promise in extending acid suppression into the night.
Kapidex
Kapidex is a delayed-release form of the R enantiomer of lansoprazole with a double coating to allow biphasic release [46]. If administered before breakfast and again at bedtime, perhaps acid suppression would be sufficient to inhibit nocturnal breakthrough.
Tenatoprazole
Tenatoprazole is a long half-life PPI; early data show that administration at night of the R enantiomer improves nighttime acid suppression [47]. Administration of this drug twice daily may allow eradication with amoxicillin in dual therapy.
AGN904
A novel form of omeprazole, AGN904 is designed to prolong absorption. Phase 1 clinical trial data have shown greatly improved acid suppression over 24 hours compared with esomeprazole, 40 mg, particularly at night. It is predicted that twice-daily administration of this drug would maintain a nearly neutral intragastric pH for 24 hours, preventing highly acidic nighttime pH excursions. Hence, dual therapy is likely to succeed.
Reversible inhibitors of the gastric H,K ATPase
A long-acting reversible inhibitor of the gastric acid pump is in development. This compound is expected to give better acid inhibition at night than current PPIs. Administration twice daily in combination with amoxicillin might provide dual therapy.
Novel Eradication Targets
Carbonic anhydrase
As discussed earlier, the need for α-carbonic anhydrase activity for acid survival of H. pylori is required. High levels of acidity anticipated at the gastric surface are a requirement for loss of H. pylori survival in the presence of the carbonic anhydrase inhibitor, acetazolamide. The gastric pH of the gerbil is about pH 1.0, similar to that of the human stomach. Recently, using the gerbil model of H. pylori infection, acetazolamide treatment for just 3 days resulted in a 66% eradication rate and significantly reduced H. pylori gastric load by 95% in the remaining animals. However, this protocol was not optimized and it is anticipated that an increase in the duration of treatment would result in eradication rates approaching 100%. The presence of acetazolamide would be most effective at high levels of acidity, which occurs at night. Hence, controlledrelease acetazolamide at bedtime would optimize the acid-dependent bactericidal effect of the drug.
In 1976, treatment of peptic ulcers using acetazolamide showed healing rates of 91% to 97% with a relapse rate of only 6% over 2 years [48]. These results were attributed to inhibition of acid secretion. It is clear that these results are not the result of inhibition of acid secretion alone because the relapse rates for the PPIs and H2-receptor antagonists for the treatment of peptic ulcer disease are about 60% after 1 year, and both are much more potent antisecretory agents than acetazolamide [8]. Because work on acetazolamide was performed before the isolation and culture of H. pylori in 1982 by Marshall and Warren [49], the contribution of eradication of this gastric pathogen to these data was not recognized.
In light of the novel findings that carbonic anhydrase found in the periplasm of H. pylori is required for acid acclimation in vitro, and that the α-carbonic anhydrase inhibitor acetazolamide eradicated H. pylori in the gerbil model, and the apparently profound effect of acetazolamide on the healing of ulcer disease and subsequent low rate of relapse, acetazolamide treatment may result in eradication of H. pylori in humans. Furthermore, this medication will be much more affordable than current therapy and the simplicity of treatment would allow worldwide eradication of this type 1 carcinogen. Eradication is important not only for the prevention and cure of peptic ulcer disease but also for the prevention of gastric adenocarcinoma, the third leading cause of cancer deaths in the United States. Furthermore, because acetazolamide is an enzyme inhibitor, resistance would not be expected.
UreI
UreI, a proton-gated urea channel, is unique to H. pylori among human pathogens and its activity is essential for acid acclimation and gastric colonization [29,30,50]. An in vitro high-throughput screen is available for inhibitors of this channel and, given the carcinogenic consequences of infection, development of a compound able to inhibit urea uptake by the organism would appear worthwhile.
HP0165
HP0165, a two-component histidine kinase sensor, has its sensor domain exposed to the periplasm. It is unique to gastric Helicobacter spp. Development of an inhibitor preventing the pH response of this sensor would impair gastric survival.
Conclusions
The analysis of acid acclimation by H. pylori suggests several targets for eradication. The first—improved acid suppression for eradication with a PPI and only amoxicillin— requires availability of a longer-acting PPI whose acid suppression extends into the night. The second—inhibition of carbonic anhydrase activity—also requires enzyme inhibition at night. The third—inhibition of novel targets such as UreI or HP0165—requires the development of new chemical entities.
Footnotes
Disclosure
No potential conflicts of interest relevant to this article were reported.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as:
-
•
Of importance
-
••
Of major importance
- 1.Blaser MJ. Helicobacter pylori and gastric diseases. BMJ. 1998;316:1507–1510. doi: 10.1136/bmj.316.7143.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akre K, Signorello LB, Engstrand L, et al. Risk for gastric cancer after antibiotic prophylaxis in patients undergoing hip replacement. Cancer Res. 2000;60:6376–6380. [PubMed] [Google Scholar]
- 3.Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 4.Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136:1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loffeld RJ, Werdmuller BF, Kuster JG, et al. Colonization with cagA-positive Helicobacter pylori strains inversely associated with reflux esophagitis and Barrett’s esophagus. Digestion. 2000;62:95–99. doi: 10.1159/000007801. [DOI] [PubMed] [Google Scholar]
- 6.Malfertheiner P, Megraud F, O’Morain C, et al. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut. 2007;56:772–781. doi: 10.1136/gut.2006.101634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Megraud F. Resistance of Helicobacter pylori to antibiotics. Aliment Pharmacol Ther. 1997;11(Suppl 1):43–53. doi: 10.1046/j.1365-2036.11.s1.11.x. [DOI] [PubMed] [Google Scholar]
- 8.Penston JG, McColl KE. Eradication of Helicobacter pylori: an objective assessment of current therapies. Br J Clin Pharmacol. 1997;43:223–243. doi: 10.1046/j.1365-2125.1997.00551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dore MP, Leandro G, Realdi G, et al. Effect of pretreatment antibiotic resistance to metronidazole and clarithromycin on outcome of Helicobacter pylori therapy: a meta-analytical approach. Dig Dis Sci. 2000;45:68–76. doi: 10.1023/a:1005457226341. [DOI] [PubMed] [Google Scholar]
- 10.Furuta T, Shirai N, Takashima M, et al. Effects of genotypic differences in CYP2C19 status on cure rates for Helicobacter pylori infection by dual therapy with rabeprazole plus amoxicillin. Pharmacogenetics. 2001;11:341–348. doi: 10.1097/00008571-200106000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Teyssen S, Chari ST, Scheid J, Singer MV. Effect of repeated boluses of intravenous omeprazole and primed infusions of ranitidine on 24-hour intragastric pH in healthy human subjects. Dig Dis Sci. 1995;40:247–255. doi: 10.1007/BF02065405. [DOI] [PubMed] [Google Scholar]
- 12.Schreiber S, Konradt M, Groll C, et al. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc Natl Acad Sci U S A. 2004;101:5024–5029. doi: 10.1073/pnas.0308386101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Code CF. Defense mechanisms of the gastric mucosa. Scand J Gastroenterol Suppl. 1981;67:201–204. [PubMed] [Google Scholar]
- 14.Schade C, Flemstrom G, Holm L. Hydrogen ion concentration in the mucus layer on top of acid-stimulated and -inhibited rat gastric mucosa. Gastroenterology. 1994;107:180–188. doi: 10.1016/0016-5085(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 15.Baumgartner HK, Montrose MH. Regulated alkali secretion acts in tandem with unstirred layers to regulate mouse gastric surface pH. Gastroenterology. 2004;126:774–783. doi: 10.1053/j.gastro.2003.11.059. [DOI] [PubMed] [Google Scholar]
- 16••.Henriksnas J, Phillipson M, Storm M, et al. Impaired mucusbicarbonate barrier in Helicobacter pylori-infected mice. Am J Physiol Gastrointest Liver Physiol. 2006;291:G396–G403. doi: 10.1152/ajpgi.00017.2006. This article showed for the first time that H. pylori infection disrupted the putative pH gradient through the gastric mucus using pH-sensitive glass microeletrodes. This important finding shows that H. pylori colonize an acidic gastric mucosa. [DOI] [PubMed] [Google Scholar]
- 17••.Scott DR, Marcus EA, Wen Y, et al. Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proc Natl Acad Sci U S A. 2007;104:7235–7240. doi: 10.1073/pnas.0702300104. This study used in vivo transcriptomal analysis of H. pylori and found that changes in gene expression mirrored that of acidic in vitro transcriptomal analysis, showing that the pH at the site of H. pylori infection is less than 4.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchell P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol Rev Camb Philos Soc. 1966;41:445–502. doi: 10.1111/j.1469-185x.1966.tb01501.x. [DOI] [PubMed] [Google Scholar]
- 19.Padan E, Zilberstein D, Schuldiner S. pH homeostasis in bacteria. Biochim Biophys Acta. 1981;650:151–166. doi: 10.1016/0304-4157(81)90004-6. [DOI] [PubMed] [Google Scholar]
- 20.Foster JW. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol. 2004;2:898–907. doi: 10.1038/nrmicro1021. [DOI] [PubMed] [Google Scholar]
- 21.Wen Y, Marcus EA, Matrubutham U, et al. Acid-adaptive genes of Helicobacter pylori. Infect Immun. 2003;71:5921–5939. doi: 10.1128/IAI.71.10.5921-5939.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mobley HL, Island MD, Hausinger RP. Molecular biology of microbial ureases. Microbiol Rev. 1995;59:451–480. doi: 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eaton KA, Brooks CL, Morgan DR, Krakowka S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect Immun. 1991;59:2470–2475. doi: 10.1128/iai.59.7.2470-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cussac V, Ferrero RL, Labigne A. Expression of Helicobacter pylori urease genes in Escherichia coli grown under nitrogenlimiting conditions. J Bacteriol. 1992;174:2466–2473. doi: 10.1128/jb.174.8.2466-2473.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voland P, Weeks DL, Marcus EA, et al. Interactions among the seven Helicobacter pylori proteins encoded by the urease gene cluster. Am J Physiol Gastrointest Liver Physiol. 2003;284:G96–G106. doi: 10.1152/ajpgi.00160.2002. [DOI] [PubMed] [Google Scholar]
- 26.Nolan KJ, McGee DJ, Mitchell HM, et al. In vivo behavior of a Helicobacter pylori SS1 nixA mutant with reduced urease activity. Infect Immun. 2002;70:685–691. doi: 10.1128/iai.70.2.685-691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olson JW, Mehta NS, Maier RJ. Requirement of nickel metabolism proteins HypA and HypB for full activity of both hydrogenase and urease in Helicobacter pylori. Mol Microbiol. 2001;39:176–182. doi: 10.1046/j.1365-2958.2001.02244.x. [DOI] [PubMed] [Google Scholar]
- 28.Scott DR, Marcus EA, Weeks DL, et al. Expression of the Helicobacter pylori ureI gene is required for acidic pH activation of cytoplasmic urease. Infect Immun. 2000;68:470–477. doi: 10.1128/iai.68.2.470-477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mollenhauer-Rektorschek M, Hanauer G, Sachs G, Melchers K. Expression of UreI is required for intragastric transit and colonization of gerbil gastric mucosa by Helicobacter pylori. Res Microbiol. 2002;153:659–666. doi: 10.1016/s0923-2508(02)01380-3. [DOI] [PubMed] [Google Scholar]
- 30.Skouloubris S, Thiberge JM, Labigne A, De Reuse H. The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect Immun. 1998;66:4517–4521. doi: 10.1128/iai.66.9.4517-4521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weeks DL, Gushansky G, Scott DR, Sachs G. Mechanism of proton gating of a urea channel. J Biol Chem. 2004;279:9944–9950. doi: 10.1074/jbc.M312680200. [DOI] [PubMed] [Google Scholar]
- 32.You G, Smith CP, Kanai Y, et al. Cloning and characterization of the vasopressin-regulated urea transporter. Nature. 1993;365:844–847. doi: 10.1038/365844a0. [DOI] [PubMed] [Google Scholar]
- 33.Ang S, Lee CZ, Peck K, et al. Acid-induced gene expression in Helicobacter pylori: study in genomic scale by microarray. Infect Immun. 2001;69:1679–1686. doi: 10.1128/IAI.69.3.1679-1686.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bury-Mone S, Thiberge JM, Contreras M, et al. Responsiveness to acidity via metal ion regulators mediates virulence in the gastric pathogen Helicobacter pylori. Mol Microbiol. 2004;53:623–638. doi: 10.1111/j.1365-2958.2004.04137.x. [DOI] [PubMed] [Google Scholar]
- 35.Merrell DS, Goodrich ML, Otto G, et al. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect Immun. 2003;71:3529–3539. doi: 10.1128/IAI.71.6.3529-3539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehta N, Olson JW, Maier RJ. Characterization of Helicobacter pylori nickel metabolism accessory proteins needed for maturation of both urease and hydrogenase. J Bacteriol. 2003;185:726–734. doi: 10.1128/JB.185.3.726-734.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marcus EA, Moshfegh AP, Sachs G, Scott DR. The periplasmic alpha-carbonic anhydrase activity of Helicobacter pylori is essential for acid acclimation. J Bacteriol. 2005;187:729–738. doi: 10.1128/JB.187.2.729-738.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38•.Bury-Mone S, Mendz GL, Ball GE, et al. Roles of alpha and beta carbonic anhydrases of Helicobacter pylori in the urease-dependent response to acidity and in colonization of the murine gastric mucosa. Infect Immun. 2008;76:497–509. doi: 10.1128/IAI.00993-07. This study shows that in the absence of α-carbonic anhydrase, H. pylori are unable to colonize the mouse. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beier D, Frank R. Molecular characterization of two-component systems of Helicobacter pylori. J Bacteriol. 2000;182:2068–2076. doi: 10.1128/jb.182.8.2068-2076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wen Y, Feng J, Scott DR, et al. The HP0165-HP0166 two-component system (ArsRS) regulates acid-induced expression of HP1186 α-carbonic anhydrase in Helicobacter pylori by activating the pH-dependent promoter. J Bacteriol. 2007;189:2426–2434. doi: 10.1128/JB.01492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niehus E, Gressmann H, Ye F, et al. Genome-wide analysis of transcriptional hierarchy and feedback regulation in the flagellar system of Helicobacter pylori. Mol Microbiol. 2004;52:947–961. doi: 10.1111/j.1365-2958.2004.04006.x. [DOI] [PubMed] [Google Scholar]
- 42.Pflock M, Finsterer N, Joseph B, et al. Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J Bacteriol. 2006;188:3449–3462. doi: 10.1128/JB.188.10.3449-3462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ottemann KM, Lowenthal AC. Helicobacter pylori uses motility for initial colonization and to attain robust infection. Infect Immun. 2002;70:1984–1990. doi: 10.1128/IAI.70.4.1984-1990.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pflock M, Muller S, Beier D. The CrdRS (HP1365-HP1364) two-component system is not involved in pH-responsive gene regulation in the Helicobacter pylori strains 26695 and G27. J Bacteriol. 2007;54:320–324. doi: 10.1007/s00284-006-0520-9. [DOI] [PubMed] [Google Scholar]
- 45.Hong W, Sano K, Morimatsu S, et al. Medium pH-dependent redistribution of the urease of Helicobacter pylori. J Med Microbiol. 2003;52:211–216. doi: 10.1099/jmm.0.05072-0. [DOI] [PubMed] [Google Scholar]
- 46.Metz DC, Vakily M, Dixit T, Mulford D. Review article: dual delayed release formulation of dexlansoprazole MR, a novel approach to overcome the limitations of conventional single release proton pump inhibitor therapy. Aliment Pharmacol Ther. 2009;29:928–937. doi: 10.1111/j.1365-2036.2009.03984.x. [DOI] [PubMed] [Google Scholar]
- 47.Galmiche JP, Bruley Des Varannes S, Ducrotte P, et al. Tenatoprazole, a novel proton pump inhibitor with a prolonged plasma half-life: effects on intragastric pH and comparison with esomeprazole in healthy volunteers. Aliment Pharmacol Ther. 2004;19:655–662. doi: 10.1111/j.1365-2036.2004.01893.x. [DOI] [PubMed] [Google Scholar]
- 48.Puscas I, Paun R, Ursea N, et al. Carbonic anhydrase inhibitors in the treatment of gastric and duodenal ulcers [in French, author’s transl] Arch Fr Mal App Dig. 1976;65:577–583. [PubMed] [Google Scholar]
- 49.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 50.Scott DR, Marcus EA, Weeks DL, Sachs G. Mechanisms of acid resistance due to the urease system of Helicobacter pylori. Gastroenterology. 2002;123:187–195. doi: 10.1053/gast.2002.34218. [DOI] [PubMed] [Google Scholar]