

Abstract
The endoplasmic reticulum (ER) has emerged as a critical regulator of cell fate. IRE1 is a transmembrane protein with kinase and RNase activities that is localized to the ER and that promotes resistance to ER stress. Here we showed a mechanism by which IRE1 conferred protection against ER stress-mediated cell death. IRE1 signaling prevented ER membrane permeabilization mediated by Bax and Bak and cell death under ER stress conditions. Suppression of IRE1 signaling led to the accumulation of the BH3 domain-containing protein Bnip3, which in turn triggered the oligomerization of Bax and Bak in the ER membrane and ER membrane permeabilization. As a result, cells deficient in IRE1 were susceptible to leakage of ER contents in response to ER stress, which was associated with the accumulation of calcium in mitochondria, oxidative stress in the cytosol, and cell death. Our results reveal a role for IRE1 in preventing an initial step of cell death emanating from the ER and provide a potential target for treating diseases characterized by ER stress, including diabetes and Wolfram syndrome.
Introduction
The endoplasmic reticulum (ER) is a membranous network in cells that is involved in multiple functions including production of secretory proteins, calcium storage, and regulation of cellular redox state (1). Homeostatic alterations in the ER play roles in the pathogenesis of chronic human disorders, such as type 1 and type 2 diabetes, myocardial infarction, stroke, and neurodegeneration, as well as inherited disorders including Wolfram syndrome, which is characterized by β cell death and neurodegeneration (2, 3). Under ER stress conditions, cell fate is controlled by the three major regulators of the unfolded protein response (UPR): inositol requiring enzyme 1α (IRE1α), protein kinase R-like ER kinase (PERK), and activating transcription factor 6α (ATF6α) (4, 5). The opposing effects of IRE1α and PERK determine whether ER stressed cells live or die. IRE1α activation confers protection against cell death through the regulated IRE1-dependent decay (RIDD) of death receptor 5 (DR5), whereas prolonged activation of PERK induces cell death mediated by CCAAT/enhancer-binding protein homologous protein (CHOP) and DR5 under pathological ER stress (6, 7). Bax- and Bak-dependent ER membrane permeabilization plays a role in ER stress-mediated cell death (8), which prompted us to study the relationship between the UPR and ER membrane permeabilization.
Results
IRE1 signaling suppresses ER membrane permeabilization
ER luminal proteins distribute to the cytosol by Bax- and Bak-dependent ER membrane permeabilization under ER stress conditions (8). To confirm this finding, we monitored the redistribution of ER luminal proteins in wild-type and Bax/Bak double knockout (DKO) MEFs treated with tunicamycin and thapsigargin. As expected, the redistribution of GRP78 and GRP94 to the cytosol was attenuated in DKO MEFs (Figure 1A). However, we observed some leakage of ER contents in DKO MEFs treated with thapsigargin, suggesting that there could be a minor pathway mediating the leakage of ER contents independently of Bax and Bak. We also found that ectopic expression of Bak caused the redistribution of GRP78 and GRP94 to the cytosol in DKO MEFs treated with tunicamycin (Figure 1B). Electron microscopic imaging revealed dilated ER under ER stress conditions; however, pores in ER membranes were not obvious (Figure S1A).
Figure 1. ER stress induces ER membrane permeabilization.

(A) Immunoblot analysis of GRP94 and GRP78 (ER luminal), VAPB (ER membrane), GAPDH (cytosolic) in cytosolic and membrane fractions of wild-type (WT) and DKO MEFs treated with tunicamycin (TM) or thapsigargin (TG) or untreated (Untx). (B) Left: Immunoblot analysis of GRP94 and GRP78 (ER luminal), VAPB (ER membrane) and GAPDH (cytosolic) in cytosolic and membrane fractions of wild-type (WT), DKO MEFs and DKO MEFs rescued with WT-Bak (DKO+Bak) treated with TM or untreated. Right: Quantification of cytosolic GRP78 in wild-type (WT), DKO MEFs and DKO MEFs rescued with Bak (DKO+Bak) treated with TM or untreated. (C) Immunoblot analysis of GRP94, GRP78, Calreticulin (CRT) and protein disulfide isomerase (PDI) (ER lumen), IRE1α and VAPB (ER membrane) and GAPDH in cytosolic and membrane fractions of wild-type MEFs treated with or without TM. (D) (Upper) Immunoblot analysis of GAPDH and HA-tagged A1AT-NHK mutant expressed in NSC34 cells cultured with or without kifunensine (Kif.) in the presence of cycloheximide (CHX). (Lower) Quantitation of A1AT-NHK in immunoblots. (E) Immunoblot analysis of GRP94, GRP78, Calreticulin (CRT), VAPB and GAPDH in cytosol or membrane fraction of wild-type MEFs treated with TM with or without kifunenesine (kif). A white arrowhead: non-specific signal. (F) Immunoblot analysis of ERDj5 and GAPDH in INS1 832/13 cells transfected with siRNA scramble control (si-Cont) or siRNA against ERDj5 (si-ERDj5). (G) Immunoblot analysis of A1AT-NHK and GAPDH in INS1 832/13 cells transfected with siRNA scramble control (si-Cont) or siRNA against ERDj5 (si-ERDj5), treated with CHX or untreated (Untx). (H) Quantitation of A1AT-NHK shown in (G). Statistical significance was calculated by Student t test. (I) Immunoblot analysis of GRP78, calreticulin (CRT) and GAPDH in cytosol of INS1 832/13 cells transfected with siRNA scramble control (si-Cont) or siRNA against ERDj5 (si-ERDj5), treated with TM or untreated. (J) Quantification of cytosolic GRP78 and CRT shown in (I). (K) Immunoblot analysis of A1AT-NHK, IRE1α and GAPDH in whole cell lysates (WCL), cytosol or membrane fractions of NSC34 cells overexpressing A1AT-NHK mutant. An arrow shows glycosylated A1AT-NHK and an arrowhead shows A1AT-NHK de-glycosylated by Endoglycosydase H (Endo-H). (L) Immunoprecipitation (IP) of cytosolic fractions and membrane fractions of wild-type MEFs treated with TM or untreated with anti-GRP78 antibody followed by immunoblot analysis with anti-Ubiquitin (Ub) and anti-GRP78 antibodies. Left panels show input for IP. N=at least 3 biological replicates for (A) to (L). Representative blots are shown. Unless otherwise stated, statistical significance was calculated by one-way analysis of variance (ANOVA) followed by Tukey’s test. *: p<0.05; **:p<0.01; n.s.: not significant. Error bars show SD.
We examined the leakage of various ER luminal and membrane proteins. All the examined ER luminal proteins, including GRP94, GRP78, Calreticulin and protein disulfide isomerase, leaked from the ER, whereas ER membrane proteins, including IRE1α and VAPB, did not leak to the cytosol (Figure 1C). We therefore speculated that there was no selectivity for the leakage of ER content. To exclude the possibility that ER luminal contents were retrotranslocated from the ER lumen through ER-associated degradation (ERAD) pathway, we tested whether a small molecule inhibitor for ERAD or knockdown of an ERAD component affected the leakage of ER contents to the cytosol. As reported previously, Kifunensine inhibited the degradation of the ERAD substrate A1AT-NHK (alpha 1 antitrypsin Null Hong Kong) mutant, an ERAD substrate (9) (Figure 1D), but did not affect the leakage of ER contents (Figure 1E). We next examined the effect of RNAi-mediated knockdown of ERDj5 also known as DNAJC10 (DNAJ (Heat Shock Protein 40) homolog, subfamily C, member 10)), which is required for ERAD (10). Although knockdown of ERDj5 inhibited the degradation of A1AT-NHK (Figure 1G and 1H), it did not affect the leakage of ER contents (Figure 1I and 1J). To exclude the possibility that the leakage of ER luminal proteins was due to the abnormal translation of ER luminal proteins in cytosol, we overexpressed the A1AT-NHK mutant, which is misfolded in the ER lumen and causes ER stress (9). A1AT-NHK is also glycosylated in the ER (11), which occurs only in the ER; thus, if the A1AT-NHK in the cytosol is glycosylated, the protein is translated in the ER and then leaked from the ER. We detected glycosylated A1AT-NHK in the cytosol, indicating that this protein was not translated in the cytosol (Figure 1K). Finally, we determined that cytosolic GRP78 was not polyubiquitinated (Figure 1L), indicating that it was not a substrate for ERAD. These results indicate that the Bax- and Bak-dependent ER membrane permeabilization, but not the ERAD pathway, causes the redistribution of ER luminal proteins to the cytosol.
Dysregulation of ER homeostasis is linked to the activation of the UPR, raising the possibility that the ER membrane permeabilization might be modified by the regulators of the UPR, including IRE1α, PERK, and ATF6α (1). To test this notion, we analyzed ER membrane permeabilization in wild-type, Ire1α, Perk, and Atf6α knockout MEFs treated with thapsigargin for 6 h, which induces mild leakage of ER contents in wild-type MEFs (Figure 2A and Figure S1B). Cell fractionation analysis revealed that Ire1α knockout MEFs were more susceptible than wild-type, Perk knockout, or Atf6α knockout MEFs to thapsigargin-induced leakage of ER luminal proteins (Figure 2A). Ectopic expression of IRE1α in Ire1α knockout MEFs decreased the leakage of the ER contents to the cytosol (Figure 2B). As further confirmation of these observations, we found that thapsigargin exposure enhanced leakage of ER content in rodent INS-1 832/13 β cells with transient knock down of endogenous Ire1α by RNAi (Figure 2C). Conversely, overexpression of wild-type IRE1α in INS-1 832/13 cells significantly suppressed ER content leakage in response to thapsigargin (Figure 2D and 2E), suggesting that IRE1α protects against ER membrane permeabilization.
Figure 2. IRE1 signaling suppresses ER membrane permeabilization.

(A) Left: Immunoblot analysis of GRP94, GRP78, calreticulin (CRT), VAPB, and GAPDH in cytosolic and membrane fractions of wild-type (WT), Atf6α knockout (Atf6−/−), Ire1α knockout (Ire1α−/−), and Perk knockout MEFs (Perk−/−) treated with TG or untreated (Untx). Right: Quantification of cytosolic GRP78 in WT, Atf6−/−, Ire1α−/−, and Perk−/− MEFs treated with TG or untreated (Untx). (B) Immunoblot analysis of GRP94 and GRP78 (ER luminal), VAPB and IRE1α (ER membrane) and GAPDH (cytosolic) in cytosolic and membrane fractions of WT, Ire1α−/− and Ire1α−/− rescued with WT-IRE1α (Ire1α−/−res.), treated with TG or untreated (Untx). (C) Left: Immunoblot analysis of GRP94, GRP78 and Calreticulin (CRT) (ER luminal), VAPB and IRE1α (ER membrane) and GAPDH (cytosolic) in cytosolic and membrane fractions of INS1 832/13 cells transiently transfected with scrambled control siRNA (si-Cont) or siRNA against IRE1α (si-IRE1α). Right: Quantification of cytosolic GRP78 and Calreticulin (CRT). (D) Immunoblot analysis of GRP94, GRP78, Calreticulun (CRT), VAPB, GAPDH and IRE1α in the cytosolic or membrane fractions of INS1 832/13 cells transduced with LacZ or wild-type IRE1α, treated with TG or untreated (Untx). (E) Quantitation of cytosolic GRP78 and Calreticulin (CRT). (F) Immunoblot analysis of the pro-apoptotic proteins Bax and Bak, the anti-apoptotic proteins Bcl2 and Bcl-xL, the pro-apoptotic BH3 only proteins PUMA, BimEL, Bad, Bid, and Bnip3, in Ire1α+/+ and Ire1α−/− MEFs treated with TM, TG or untreated (Untx). N=at least 3 biological replicates for (A) to (F). Representative blots are shown. Statistical significance was calculated by one-way ANOVA followed by Tukey’s test. *: p<0.05; **: p<0.01. Error bars show S.D.
Transcriptional regulation and/or post-translational modification of BH-3 only proteins, including BAD, Bim, Puma, Bid, and Bnip3 (Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3), results in the activation of Bax and Bak (12) (13). We hypothesized that this group of proteins might play a role in the ER membrane permeabilization mediated by Bax and Bak and that such a mechanism might be enhanced in Ire1α knockout MEFs as compared to control MEFs. We found that the abundance of Bnip3 was increased in Ire1α knockout MEFs treated with thapsigargin and tunicamycin (Figure 2F) Bnip3 is a BH3 only pro-apoptotic protein localized to the mitochondria and ER through its C-terminal transmembrane domain, and induces apoptotic cell death by interfering with the anti-apoptotic proteins Bcl-2 and Bcl-xL (14) after ischemia/reperfusion and under oxidative stress conditions (15–18). Immunoblot analysis revealed multiple bands corresponding to monomers, dimers and oligomers of Bnip3 (Figure S2A), as reported previously (19, 20). Treatment of the cell lysates with lambda protein phosphatase to dephosphorylate proteins resulted in a single band corresponding to the monomeric form of Bnip3, suggesting that Bnip3 was a highly phosphorylated protein (Figure S2A and S2B). Tunicamycin induced accumulation of highly phosphorylated Bnip3 (Figure S2C).
Bnip3 induces ER membrane permeabilization
We next sought to verify that ectopic expression of Bnip3 could induce the ER membrane permeabilization. Ectopic expression of Bnip3 in wild-type and Ire1α knockout MEFs induced the redistribution of GRP94 to the cytosol under normal conditions, which was enhanced by thapsigargin treatment (Figure 3A). Fractionation analysis further confirmed that ectopic expression of Bnip3 induced the redistribution of GRP94 to the cytosol in Ire1α knockout MEFs in a dose-dependent manner (Figure 3B). Furthermore, ectopic expression of Bnip3 was associated with calcium depletion of the ER (Figure S3A and S3B). Because Bnip3 is localized to the ER and mitochondria (21), we wanted to determine whether Bnip3 was involved in the activation of the ER membrane permeabilization mediated by Bax and Bak. The interaction of Bnip3 with Bcl-2 induces the activation of Bax and Bak and apoptosis (14, 22). As expected from previous studies, the interaction between Bnip3 and Bcl-2 was enhanced by tunicamycin treatment in wild-type and Ire1α knockout MEFs (Figure 3C). Bnip3 formed a heterodimer with Bcl-2 even in untreated Ire1α knockout MEFs, suggesting that this mechanism could underlie the susceptibility of these cells to ER membrane permeabilization (Figure 3C). To further confirm that Bnip3 forms a heteromer with Bcl-2 on the ER membrane, we first performed subcellular fractionation to obtain pure ER fractions without mitochondrial contamination (Figure 3D). Multimeric and presumably highly phosphorylated forms of Bnip3 were present in the ER fraction (Figure S4A). Immunoprecipitation analysis of the purified ER fraction also showed that phosphorylated Bnip3 interacted with Bcl-2, an interaction that was enhanced by tunicamycin treatment (Figure S4B). In addition, ectopic expression of Bnip3 induced the oligomerization of ER-localized Bak, but not of GRP94, in Ire1α knockout MEFs (Figure 3E and Figure S5A), and oligomerization was further enhanced by thapsigargin treatment (Figure 3E). To further confirm that ER-localized Bak was activated by Bnip3, we performed immunoprecipitations with an antibody that recognizes the active conformation of Bak (23) using ER fractions from Ire1α knockout MEFs. The amount of ER-localized active Bak was modestly increased by ectopic expression of Bnip3 and enhanced by thapsigargin treatment (Figure 3F). Ectopic expression of Bnip3 did not increase the redistribution of GRP78 and calreticulin to the cytosol in DKO MEFs, whereas it increased the distribution of GRP78 and calreticulin to the cytosol in wild-type MEFs or DKO MEFs also expressing wild-type Bak, indicating that the ability of Bnip3 to promote ER membrane permeabilization was Bax- and Bak-dependent (Figure 3G). Finally, we found that short-hairpin RNA (shRNA)-mediated knock down of Bnip3 reduced the redistribution of GRP94 and GRP78 into the cytosol in Ire1α knockout MEFs treated with thapsigargin, suggesting that Bnip3 knockdown prevented ER membrane permeabilization (Figure 3H). Collectively, these results indicate that Bnip3 promotes the ER membrane permeabilization mediated by Bax and Bak.
Figure 3. Bnip3 induces ER membrane permeabilization.

(A) Left: Immunoblot analysis of GRP94, VAPB, and GAPDH in cytosolic and membrane fractions of Ire1α+/+ and Ire1α knockout (Ire1α−/−) MEFs transduced with adenovirus expressing LacZ or Bnip3 and then treated with TG or untreated (Untx). Right: Quantitation of cytosolic GRP94. (B) Immunoblot analysis of GRP94, VAPB, and GAPDH in cytosolic and membrane fractions of Ire1α−/− MEFs transduced with adenovirus expressing LacZ or Bnip3 at indicated multiplicity of infection (MOI). (C) Co-immunoprecipitation analysis of endogenous Bnip3 and Bcl-2 in Ire1α+/+ and Ire1α−/− MEFs treated with TM or untreated (Untx). Arrows indicate monomer, black arrowheads indicate dimer, and white arrowheads indicate oligomer. (D) Examples of mitochondria and ER fractions of Ire1α−/− MEFs treated with TG or untreated (Untx). Tomm 20 and PDI were used as mitochondrial and ER markers. (E) Gel filtration analysis of endogenous Bak in the ER fractions of Ire1α−/− MEFs transduced with adenovirus expressing LacZ or Bnip3 treated with or without TG. (F) Immunoprecipitation of activated Bak using anti-Bak-N-terminus antibody from the ER fraction of Ire1α−/− MEFs transduced with adenovirus expressing LacZ or Bnip3 treated with TG or untreated (Untx). (G) Immunoblot analysis of GRP78, calreticulin (CRT), VAPB and GAPDH in cytosolic and membrane fractions of wild-type (WT), DKO MEFs (DKO) and DKO MEFs rescued with Bak (DKO + Bak) transduced with adenovirus expressing LacZ or Bnip3 and then treated with TM. (H) Left: Immunoblot analysis of GRP94, Calreticulun (CRT) and GAPDH in cytosolic fractions of Ire1α−/− MEFs stably expressing shRNA directed against Luciferase (shLuc) or Bnip3 treated with TG or untreated (Untx). Right: Quantitation of cytosolic GRP94 and CRT. N=at least 3 biological replicates for (A) to (H). Representative blots are shown. N=3 biological replicates. Statistical significance was calculated by one-way ANOVA followed by Tukey’s test. *: p<0.05. Error bars show S.D.
Impaired IRE1 signaling leads to accumulation of Bnip3
In response to ER stress, IRE1α activates multiple signals through its endonuclease and kinase domains. The endonuclease domain of IRE1α promotes splicing of the mRNA encoding X-box binding protein 1 (XBP1) and regulates IRE1α-dependent decay of mRNAs including that encoding DR5 (7). The kinase domain of IRE1α promotes the activation of c-jun N-terminal kinase (JNK) by recruiting tumor necrosis factor α receptor-associated factor-2 (TRAF2) (24), which prompted us to test the relationship between the IRE1α-TRAF2-JNK cascade and ER membrane permeabilization. To determine the enzymatic activity of IRE1α that contributes to the suppression of ER membrane permeabilization, we monitored the leakage of ER luminal proteins into the cytosol in INS1 832/13 cells treated with tunicamycin and STF-083010, an IRE1α endonuclease specific inhibitor or SP600125, a JNK inhibitor (25). SP600125, but not STF-083010, increased the leakage of ER luminal contents to the cytosol, suggesting that the kinase domain of IRE1α played a role in preventing the ER membrane permeabilization through JNK activation (Figure 4A and S6A). To further confirm this observation, we used an endonuclease-dead K907A-mutant of IRE1α and a kinase-dead K599A mutant that can still interact with TRAF2 (26–28). Wild-type and K907A-mutant IRE1α, but not K599A-mutant IRE1α, suppressed ER membrane permeabilization in thapsigargin-treated Ire1α knockout MEFs, indicating that the kinase activity of IRE1α was essential to inhibit ER membrane permeabilization (Figure 4B). We also monitored ER membrane permeabilization in wild-type MEFs (Xbp1+/+) and XBP1 knockout MEFs (Xbp1−/−) treated with thapsigargin (Figure S7A). Consistent with the observations above, ablation of Xbp1 did not affect ER membrane permeabilization. Next, we monitored the leakage of ER luminal proteins to the cytosol in Traf2 knockout MEFs, which were more susceptible to tunicamycin-induced leakage of ER luminal proteins as compared to control cells (Figure 4C). Finally, we noted that the phosphorylation of JNK induced by ER stress preceded ER membrane permeabilization (Figure S8A). These results indicate that the IRE1-TRAF2-JNK pathway prevents the ER membrane permeabilization.
Figure 4. Impaired IRE1 signaling leads to accumulation of Bnip3.

(A) Left: Immunoblot analysis of GRP94, GRP78, VAPB, phosphorylated JNK (p-JNK), JNK and GAPDH in cytosolic and membrane fractions of INS1 832/13 cells untreated (Untx) and/or treated with TM, STF-083010 (STF) or SP600125 (SP) as indicated. Right: Quantitation of GRP78 in the cytosolic fractions of INS1 832/13 cells untreated (Untx) and/or treated with TM, STF-083010 (STF) or SP600125 (SP) as indicated. (B) Left: Immunoblot analysis of GRP94, Calreticulin (CRT), GAPDH and IRE1α in cytosolic and membrane (mem) fractions of Ire1α−/− MEFs transduced with adenovirus encoding LacZ, wild-type (WT) IRE1α, K599A-IRE1α or K907A-IRE1α, treated with TG or untreated (Untx). Right: Quantification of cytosolic CRT. (C) Immunoblot analysis of GRP94, GRP78, Calreticulin (CRT), VAPB and GAPDH in cytosolic and membrane fractions of MEFs containing floxed TRAF2 alleles, transduced with adenovirus encoding LacZ or Cre-recombinase (Cre), treated with TM or untreated (Untx). Statistical significance was determined by one-way ANOVA followed by Tukey’s test. (D) Quantitative PCR of Bnip3 of Ire1α+/+ and Ire1α−/− MEFs treated with TM or untreated (Untx). Gene expression was normalized to β-actin mRNA. (E) (Left) Immuno-gold labeling of Bnip3 in human embryo kidney 293 cells. mito: mitochondria. (Right) Immuno-gold double labeling of Bnip3 and LC3 in human embryo kidney 293 cells. Bnip3 was labeled with 18 nm gold particles and LC3 was labeled with 12 nm gold particles. Imaging was performed in two independent experiments. (F) Ire1α+/+ and Ire1α−/− MEFs transduced with adenovirus expressing GFP-LC3 treated with TM or untreated (Untx). The scale bars show 20 μm. Imaging was performed in three independent experiments. (G) Immunoblot analysis of LC3 and GAPDH of Ire1α+/+ and Ire1α−/− MEFs, treated with TM or untreated (Untx). (H) Cycloheximide (CHX) chase of endogenous Bnip3 in wild-type, Ire1α knockout (Ire1α−/−) and Atg5 knockout (Atg5−/−) MEFs treated with TM and CHX. (I) Mean intensities of Bnip3 in wild-type, Ire1α−/− and Atg5−/− MEFs treated with TM and CHX were plotted on a semi log graph. Statistical significance was calculated by one-way ANOVA followed by Dunnett’s test. (J) Immunoblot analysis of Bnip3 and GAPDH in wild-type MEFs in the absence or presence of Rapamycin. N=at least 3 biological replicates for (A) to (D) and (G) to (J). Representative blots and images are shown. Unless otherwise indicated, statistical significance was calculated by one-way ANOVA followed by Tukey’s test. *: p<0.05, **: p<0.01, n.s.: not significant. Error bars show S.D. The scale bar in the electron microscopic images indicates 100 nm and the scale bars in the confocal images indicate 20 μm.
We next investigated the mechanisms that mediate the increase in Bnip3 abundance in the absence of IRE1 signaling. The basal expression of Bnip3 mRNA was higher in Ire1α knockout MEFs than in wild-type MEFs, but the induction of Bnip3 mRNA by tunicamycin was higher in wild-type MEFs. As a result, the tunicamycin-induced expression of Bnip3 mRNA was comparable in both cell types (Figure 4D), raising the possibility that the accumulation of Bnip3 protein in Ire1α knockout MEFs might be due to delayed protein degradation. Bnip3 protein turnover can be regulated by autophagy (29, 30). IRE1α-mediated activation of JNK induces cytoprotective autophagy under ER stress conditions, and suppression of autophagy can render cells susceptible to death triggered by ER stress (31, 32). We therefore used LY294002, which inhibits PI3K signaling and autophagy, or MG132, an inhibitor of the ubiquitin-proteasome system, and found that both inhibitors increased the amount of Bnip3 in human embryonic kidney 293 (HEK293) cells ectopically expressing this protein (Figure S9A). The proteasomal activity of wild-type MEFs and Ire1α knockout MEFs did not significantly differ (Figure S9B), suggesting that proteasomal degradation of Bnip3 proceeds normally in Ire1α knockout MEFs. Immunogold electron microscopy revealed that Bnip3 was internalized in autophagosomes (Figure 4E), prompting us to examine the role of IRE1α-mediated autophagy in Bnip3 degradation.
Punctate LC3-GFP dots, a marker for autophagy induction, were detected in wild-type MEFs treated with tunicamycin, but not in Ire1α knockout MEFs (Figure 4F), as previously reported (31, 32). Furthermore, the conversion of LC3-I to LC3-II was increased by tunicamycin in wild-type MEFs, but was decreased in Ire1α knockout MEFs (Figure 4G), an effect that was rescued by ectopic expression of wild-type IRE1α (Figure S9C), indicating that the induction of autophagy in response to ER stress was impaired in Ire1α knockout MEFs. We next monitored the degradation of endogenous Bnip3 under ER stress conditions using cycloheximide treatment. Bnip3 protein was more stable in Ire1α knockout or Atg5 knockout MEFs (in which autophagy is impaired) than wild-type MEFs (Figure 4H and 4I). Furthermore, the steady state abundance of Bnip3 protein was decreased by rapamycin, which induces autophagy by inhibiting mTORC1 (Figure 4J).
Bax and Bak directly interact with IRE1α and regulate its activity (33). In DKO-MEFs, IRE1α activation, as well as JNK phosphorylation, was suppressed under ER stress conditions. Therefore, in DKO-MEFs, Bnip3 could accumulate due to decreased autophagy. The phosphorylation of JNK and induction of autophagy in response to tunicamycin-induced ER stress were suppressed in DKO-MEFs and as a result, the abundance of Bnip3 was increased (Figure S10A and S10B). Collectively, these results indicate that Bnip3 accumulation in Ire1α knockout MEFs was mediated by impaired autophagy induction under ER stress conditions.
IRE1 inhibits the initial step of cell death
To further study the role of IRE1 in inhibiting ER membrane permeabilization and cell death, we used Ire1α knockout MEFs stably expressing mammalian ER-targeted redox sensitive GFP (MERO-GFP). MERO-GFP changes its excitation spectrum depending on the oxidation status of two engineered cysteines. The reduction of the celluar environment leads to a change in the MERO-GFP reporter to the reduced state, which increases the MERO-GFP ratio (34). We found that tunicamycin treatment induced the leakage of MERO-GFP from the ER to the cytosol at 6 h, and that the MERO-GFP ratio was increased by more than 2 by the reducing conditions of the cytosol (Figure 5A, 5B, Figure S11A, S11B, S11C and S11D). Because the MERO-GFP ratio in cells with ER membrane permeabilization indicated by the leakage of MERO-GFP to the cytosol was increased by more than 2 whereas the MERO-GFP ratio in cells with intact ER was below 2, we defined cells with a MERO-GFP ratio greater than 2 as cells experiencing ER membrane permeabilization (Figure 5A and 5B). To study the relationship between Bax- and Bak-mediated ER membrane permeabilization and cell death, we next performed a flow cytometry analysis of the MERO-GFP ratio and the mitochondrial activity using Mitoprobe DilC1 (5) dye which is sequestered by active mitochondria. We found that cells undergoing the ER membrane permeabilization still maintained mitochondrial activity (Fig. 5C). Dual live-cell imaging with tetramethylrhodamine methyl ester (TMRM) also showed that the MERO-GFP leaked from ER and the MERO-GFP ratio was increased before mitochondrial activity decreased, indicating that ER membrane permeabilization preceded cell death (Figure 5D, 5E, 5F and S12A). To biochemically monitor the permeabilization of ER membranes and mitochondrial outer membranes under ER stress conditions, we performed subcellular fractionation of Ire1α knockout MEFs treated with tunicamycin. Consistent with the results obtained with flow cytometry, leakage of ER contents preceded the release of cytochrome c from mitochondria (Figure 5G). As further confirmation, we performed dual live-cell imaging of MERO-GFP and propidium iodide in Ire1α knockout MEFs treated with tunicamycin. We found that cells with highly reduced MERO-GFP eventually underwent cell death (Figure 5H, 5I and S12B). Furthermore, MERO-GFP that leaked to the cytosol in dying cells was eventually oxidized (Figure 5E, 5I and S12B) because the cytosol in apoptotic cells was oxidized (Figure S13A and S13B). We also verified that Ire1α knockout MEFs underwent cell death after ER membrane permeabilization (Figure 5J).
Figure 5. IRE1 inhibits the initial step of cell death.

(A) Live-cell imaging of MERO-GFP (excitation 488 nm) in Ire1α knockout (Ire1α−/−) MEFs treated with TM (upper panels). The ratio images of 488/405 are displayed in false colors (lower panels). (B) Ratio traces of Ire1α knockout (Ire1α−/−) MEFs treated with TM. N=12 cells imaged over three independent experiments. (C) Mitochondrial membrane potential (ΔΨm) as measured by Mitoprobe dye in wild-type MEFs expressing MERO-GFP treated with TG or untreated (Untx). Statistical significance was calculated by one-way ANOVA followed by Tukey’s test. (D) Dual time-lapse imaging of tetramethylrhodamine methyl ester (TMRM) and MERO-GFP (ex. 488 nm) in Ire1α−/− MEFs treated with TM for indicated times. Yellow dashed lines indicate the shape of the cell. (E) Time-lapse tracing of the MERO-GFP ratio and TMRM in Ire1α−/− MEFs treated with TM for the indicated times. (F) Traces of TMRM intensity in Ire1α−/− MEFs treated with TM during EMP. The time point when the MERO-GFP started leaking from ER was set as T=0. N=10 cells from three independent experiments. The Wilcoxon signed-rank test was performed to compare the signal intensities at 0, 20 and 40 min to those at −20 min. None of these comparisons were significantly different (p-value >0.05). (G) Left: Immunoblot analysis of GRP78, Calreticulin (CRT), cytochrome C (cyto. C) and GAPDH in cytosol fractions of Ire1α−/− MEFs treated with TM for the indicated periods. Middle and Right: Quantification of cytosolic GRP78 and cytochrome C. Statistical significance was calculated by one-way ANOVA followed by Dunnett’s test. (H) Dual live-cell imaging of MERO-GFP ratio and propidium iodide (PI) in Ire1α−/− MEFs treated with TM for the indicated times. White dashed lines indicate the shape of the cell experiencing ER membrane permeabilization (EMP). (I) The MERO-GFP ratio of each cell shown in (H) at the indicated times. (J) Fates of Ire1α−/− MEFs treated with TM. N=30 cells imaged over three independent experiments. (K) Mitochondrial calcium monitored by Rhod2 and MERO-GFP ratio in wild-type MEFs treated with TG. Statistical significance was calculated by Student t test. (L) Reactive oxygen species (ROS) and MERO-GFP ratios in wild-type MEFs treated with TG. Statistical significance was calculated by Student t test. (M) ROS generation and MERO-GFP ratios in Perk knockout (Perk−/−) MEFs treated with TG. Statistical significance was calculated by Student t test. (N) Induction of mCherry fluorescence driven by the human Chop promoter and MERO-GFP ratios in HEK293 cells treated with TG. Statistical significance was calculated by Student t test. N=at least three biological replicates. Representative blots and images are shown. *: p<0.05; **: p<0.01. Error bars show S.D. The scale bars are 20 μm.
To understand the basis of cell death mediated by the leakage of ER contents, we first analyzed the accumulation of calcium in the mitochondria. Because the ER is the largest store for intracellular calcium, ER membrane permeabilization might release calcium from the ER to the mitochondria and trigger apoptotic signals (35). The ER is also highly oxidized with hydrogen peroxide for efficient formation of disulfide bonds (36), and ER membrane permeabilization might induce cytosolic reactive oxygen species (ROS) accumulation. Flow cytometric analysis of MERO-GFP and Rhod-2 dye to measure mitochondrial calcium indicated that wild-type MEFs with ER membrane permeabilization had higher calcium concentrations in the mitochondria (Figure 5K). Overexpression of Bnip3 induced ER membrane permeabilization, leading to the leakage of calcium (Figure S3), suggesting that the source of mitochondrial calcium might be cytosolic calcium originally contained in the ER. Flow cytometric analysis of MERO-GFP and CellRox dye (which detects cytosolic ROS) revealed that ROS concentrations were increased in cell populations with higher MERO-GFP ratios (Figure 5L). Reduction of oxidative stress under ER stress conditions is mediated by the PERK-ATF4 pathway (37). In Perk−/− MEFs stably expressing MERO-GFP, ROS concentrations were increased in cells with higher MERO-GFP ratios (Figure 5M), suggesting that the increased ROS concentrations were due to ER membrane permeabilization rather than the activation of the PERK-ATF4 pathway. Furthermore, ER stress as measured by a human Chop promoter driving mCherry expression (Figure S14A and S14B) was increased in the cells undergoing ER membrane permeabilization (Figure 5N). These results suggest that IRE1-deficient cells were susceptible to alterations in cellular calcium concentrations, redox homeostasis, and ER stress. Accumulation of calcium in mitochondria causes release of cytochrome C (Figure 5G), which is an inducer of the intrinsic apoptotic cascade (39), and as a result, apoptotic cell death could be induced by ER membrane permeabilization.
We next investigated the involvement of ER membrane permeabilization in various pathophysiological conditions. Hypoxia in cerebral tissues, cardiomyocytes, and cancer cells can lead to ER stress-mediated cell death (40–42), prompting us to test if ER contents may be redistributed to the cytosol in mouse models of cerebral and myocardial infarction. Transient middle cerebral artery occlusion (TMCAO) in mice induced the distribution of ER luminal proteins to the cytosol (Figure 6A). Furthermore, ischemia-reperfusion modeling in mice caused the distribution of the ER luminal proteins to the cytosol (Fig. 6B). In addition to these disease models, we assessed ER permeabilization in WFS1 knockout mice, which are a model of Wolfram syndrome, an inherited condition characterized by diabetes and neurodegeneration and associated with ER dysfunction (43–46). Brain tissues from WFS1 knockout mice had more ER proteins in the cytosol than those from control mice (Fig. 6C). These results suggest that ER membrane permeabilization might play important roles in the pathogenesis of various diseases.
Figure 6. ER membrane permeabilization in pathophysiological conditions.
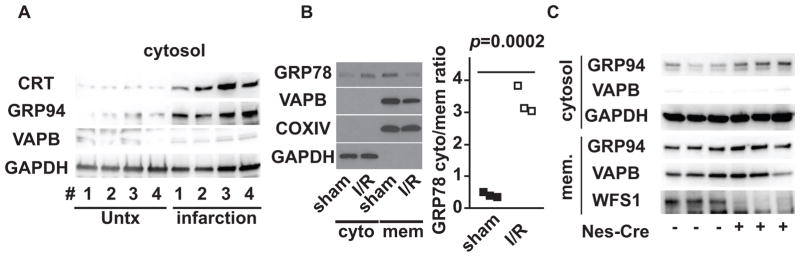
(A) Immunoblot analysis of calreticulin (CRT), GRP94, VAPB and GAPDH in cytosolic fractions of cerebrums from 4 mice with transient middle cerebral artery occlusion. # shows the pair of control cerebrum and infarcted cerebrum from same sample. (B) Left; Immunoblot analysis of GRP78, VAPB, COXIV and GAPDH in cytosolic and membrane (mem) fractions of cardiac tissue with ischemia/reperfusion (I/R) or sham operation. Right; Ratios of cytosolic GRP78 (cyto)/membrane GRP78 (mem) of three independent pairs of mice with ischemia/reperfusion (I/R) or sham operation. Statistical significance was calculated by unequal variance t-test. N=3 mice per each condition. (C) Immunoblot analysis of GRP94, VAPB and GAPDH in cytosolic and membrane (mem) fractions of cerebrums from floxed-WFS1 mice crossed with Nestin-Cre transgenic mice. Each lane represents a separate animal. N=3 mice per each condition. Representative blots are shown.
Discussion
We have demonstrated that IRE1 prevented ER membrane permeabilization mediated by the pro-apoptotic Bcl-2 family proteins Bax and Bak. IRE1-mediated cytoprotective autophagy suppressed the amount of Bnip3, its subsequent heterodimerization with Bcl-2, which in turn led to the oligomerization of Bax and Bak in the ER membrane. IRE1 played a role in suppressing Bnip3-mediated ER membrane permeabilization, which resulted in changes in cellular redox states, intracellular calcium concentrations, ER stress, and cell death (Fig. S15). Because the mitochondrial membrane potential was still maintained at the initiation of ER membrane permeabilization, our data suggest that IRE1 signaling prevented one of the earliest steps in ER stress-induced cell death. We discovered that the ER membrane permeabilization occurred in acquired pathological states associated with ER dysfunction, namely ischemia/reperfusion injury and stroke, as well as Wolfram syndrome, an inherited disease state. Thus, our results provide a potential target for ER stress-related diseases.
Materials and Methods
Animal experiments
All animal experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (A-3381-01).
Reagents
Thapsigargin, tunicamycin, CHAPS, endoplasmic reticulum isolation kit and tetramethylrhodamine methyl ester (TMRM) were purchased from Sigma (St. Louis, MO). Anti-GAPDH antibody, anti-calreticulin antibody, anti-GRP94 antibody, anti-Bim antibody, anti-Bid antibody, anti-Puma antibody, anti-Bad antibody, anti-Bax antibody, anti-Bak antibody, anti-Bcl2 antibody, anti-Bcl-xL antibody, anti-IRE1α antibody, anti-LC3A/B antibody, anti-Caspase 3 antibody and anti-cleaved caspase 3 antibody were obtained from Cell signaling technology (Danvers, MA). Anti-VAPB antibody, anti-Tomm20 antibody, anti-PDI antibody and anti-Bnip3 antibody were bought from Bethyl, Abcam, ENZO, and Genetex, respectively. Anti-GRP78 antibody and anti-WFS1 antibody were obtained from Proteintech (Chicago, IL). DMEM, Rhod-2, Fura-2, CellRox dye, Mitoprobe DilC1 (5) kit, propidium iodide solution and protein G-sepharose beads were obtained from Invitrogen (Carlsbad, CA). Anti-GFP antibody, Kifunensine and agarose-conjugated anti-Bcl2 antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Bak-NT antibody was from Millipore (Billerica, MA). Chroma spin columns were obtained from Clontech (Mountain View, CA). A small interfering RNA (siRNA) targeting rat ERDj5 (rArGrCrArArArUrArArArCrUrArGrArGrGrArUrCrGrUrUTG) and a scramble control were obtained from Origene (Rockville, MD). A siRNA targeting rat IRE1α was reported previously (47). A plasmid encoding mCherry driven by human Chop promoter was a kind gift from Dr. Quan Lu. A plasmid encoding A1AT-NHK was provided by Dr. Ron Kopito. Adenoviruses encoding wild-type, K599A and K907A- IRE1α were kind gifts from Dr. Randal Kaufman and Dr. Kezhong Zhang.
Plasmids and viral vectors
The Construction of MERO-GFP was reported previously (34). Lentivirus constructs expressing shRNA were obtained from the Genome institute of Washington University in St. Louis. The shRNA target sequences are as follows: sh-Luciferase, TCTACTGGTCTGCCTAAAGGT; mouse sh-Bnip3-1, CCATCTCTGTTACTGTCTCAT; mouse sh-Bnip3-2, AGAAGTTGAAAGTATCCTGAA. Adenovirus vectors expressing LacZ and Bnip3 were prepared as previously reported (48) and purified with Adenovirus purification Virakit (Virapur). Adenovirus vectors encoding wild-type, K599A and K907A-IRE1 were kindly provided by Dr. Randal Kaufman and Dr. Kezhong Kezhong Zhang. Titration of adenovirus was done with Adeno-X rapid titer kit following the manufacturer’s instructions (Clontech).
Cell culture and live-cell imaging
Wild-type, Ire1 knockout and Perk knockout MEFs were gifts from Dr. David Ron (University of Cambridge). Atf6 knockout MEFs were provided by Dr. Randal Kaufman (UC San Diego). Bax/Bak double knockout and Bax/Bak double knockout rescued by wild-type Bak MEFs were gifts from Dr. Navdeep Chandel (Northwestern University). Xbp1+/+ and Xbp1−/− MEFs were gifts from Dr. Laurie Glimcher (Cornell University). These MEFs were cultured in DMEM supplemented with 10% FBS DMEM and antibiotics. HEK293 cells were cultured in DMEM supplemented with 10% FBS DMEM and antibiotics. For live-cell imaging, HEK293 or Ire1α knockout MEF cells were plated on a poly-L-lysine coated glass-bottom 35 mm dish (Mattek, Ashland, MA) the day before the analysis. The cells were then cultured with 10% FBS-DMEM containing 1 or 5 μg/ml tunicamycin in association with 0.5 μg/ml of PI or 100 nM tetramethylrhodamine methyl ester (TMRM) in 5% CO2 at 37°C for 12–24 h. The images were captured every 20 min using Yokogawa spinning disk confocal on a Nikon TE-2000E2 inverted microscope with a 40x objective lens. Analyses were done on 16 bit or 32 bit images using ImageJ 1.45.
Cell fractionation
Fractionation of cytosol and membranous organelles from MEFs treated with tunicamycin (0.5 μg/ml for 16 h for MEFs and 1 μg/ml for 16 h for INS1 832/13 cells, unless indicated) or thapsigargin (0.5 μM for 16 h for MEFs and 2 μM for 4 h for INS1 832/13 cells, unless indicated) were done by ProteoExtract subcellular proteome extraction kit (Millipore) following the manufacturer’s protocol. If necessary, the cells were treated with 0.5 μM kifunenesine. After fractionation, equal volume of the lysates was subjected to SDS-PAGE followed by immunoblot analyses.
Cycloheximide chase
Wild-type MEFs, Ire1 knockout MEFs or Atg5 knockout MEFs were treated with 100 μg/ml cycloheximide in combination with 2 μg/ml tunicamycin for the indicated periods, followed by immunoblot analysis.
Co-immunoprecipitation with Bcl2
Wild-type MEFs or Ire1 knockout MEFs, untreated or treated with 5 μg/ml tunicamycin for 6 h, were lysed in cell lysis buffer (1% CHAPS, 150 mM NaCl, 10 mM Hepes pH 7.4 and protease inhibitors) followed by centrifugation at 15000 rpm to remove cellular debris. The supernatants were mixed with agarose-conjugated anti-Bcl-2 antibody and incubated at 4°C for 4 h. The beads were washed 4 times with the lysis buffer and analyzed by SDS-PAGE and immunoblot analysis. Co-immunoprecipitation with anti-Bcl-2 antibody was also performed with the ER fraction obtained from Ire1α knockout MEFs, untreated or treated with 5 μg/ml tunicamycin for 6 h using the ER isolation kit (Sigma) following the manufacturer’s protocol.
Immunoprecipitation of active Bak
Immunoprecipitation with anti-active Bak antibody (Bak-NT, Millipore) was performed with the ER fraction obtained from Ire1α knockout MEFs, untreated or treated with 1 μM thapsigargin for 6 h using the ER isolation kit (Sigma) following manufacturer’s protocol.
Immunoprecipitation of cytosolic GRP78
Immunoprecipitation with anti-GRP78 antibody (Proteintech) was performed with the cytosolic fractions from wild-type MEFs untreated or treated with 0.5 μg/ml tunicamycin for 16 h using the ProteoExtract subcellular proteome extraction kit (Millipore).
Gel filtration analysis
To detect oligomerized Bak in the ER, spin column-based gel filtration analysis was performed according to a previous report (49). Briefly, Ire1 knockout MEFs infected with adenovirus expressing LacZ or Bnip3 at moi 200, untreated or treated with 1μM thapsigargin for 6 h, were fractionated into ER or mitochondrial fraction using endoplasmic reticulum isolation kit (Sigma) following manufacture’s protocol and the ER fraction were dissolved by adding CHAPS buffer (1% CHAPS, 150 mM NaCl, 10 mM Hepes pH 7.4 and protease inhibitors) and applied to a Chroma spin column, followed by sequential gel filtration by centrifugation.
FACS analyses
For flow cytometry analyses, HEK293 cells or MEFs expressing MERO-GFP were plated onto 12-well plates, treated with each compound for indicated times, and then harvested by trypsinization. Flowcytometry analyses were performed with LSRII (BD). For measuring mitochondrial membrane potential, mitochondrial Ca2+ and cytosolic ROS, Mitoprobe DilC (5) kit (Invitrogen), Rhod2 (Invitrogen) and CellRox (Invitrogen) were used according to manufacture’s protocols. The results were analyzed by FlowJo ver.7.6.3.
Transduction of cells with Adenovirus
INS1 832/13 cells were transduced with adenovirus encoding wild-type and mutant IRE1α at MOI=10. MEFs were transuduced with adenovirus encoding Bnip3 or LacZ at MOI=200 if not mentioned.
Transient Middle Cerebral Artery Occlusion (TMCAO) procedure
Adult C57Bl/6J male mice (4 months of age) were anesthetized and left MCA occlusion was achieved using a nylon suture for 60 min under an operating microscope. Briefly, the left external carotid artery (ECA) was exposed and ligated with a 5-0 silk suture through a midline neck incision. A 14 mm 6-0 surgical monofilament nylon suture was introduced into the ECA stump, and advanced 9–10 mm from the bifurcation of the common carotid artery (CCA) into the circle of Willis to occlude the ostium of the MCA. After the desired time of MCAO, mice were re-anesthetized, and ischemia was terminated by removal of the intraluminal suture. The cerebrums were isolated 24 h after the procedure, followed by subcellular fractionation. Healthy right hemispheres were used as negative controls.
In vivo ischemia-reperfusion (I/R) model
Adult mice (8–10 weeks of age) were subjected to transient left anterior descending artery coronary ligation as described (50). Briefly, mice anesthetized were surgically prepared and ventilated. After thoracotomy, the left anterior descending artery (LAD) was ligated with a 9-0 polypropylene suture. Ischemia was confirmed by a presence of ST elevation on electrocardiogram as well as absence of blood flow visually confirmed under microscope. Thirty minutes after the occlusion, reperfusion was induced by cutting the knot of the suture. A sham operation was performed similarly without LAD ligation. One and half hours after the induction of reperfusion, the mice were euthanized, and the apical third of the heart (I/R-injured segment or sham) was subjected to subcellular fractionation.
Neuron-specific WFS1 conditional knockout mice
Mice with floxed WFS1 alleles have been previously described (51). For conditional knockout of WFS1 in neurons, WFS1-floxed mice were crossed with mice expressing Cre recombinase under the control of rat nestin promoter and enhancer (Jackson Laboratory). At the age of 8 months, the mice were sacrificed and cerebrums were isolated, followed by subcellular fractionation.
Electron microscopy of immunogold-stained Bnip3
HEK293 cells transfected with HA-Bnip3 and GFP-LC3, treated with 1 μg/ml tunicamycin for 3 h were fixed in 4% paraformaldehyde/ 0.05% glutaraldehyde (Polysciences, Warrington, PA) in 10% gelatin PIPES/0.5 mM MgCl2, pH7.2 for 1 hr at 4°C. Samples were then embedded in 10% gelatin and infiltrated overnight with 2.3M sucrose/ 20% polyvinyl pyrrolidone in PIPES/ MgCl2 at 4°C. Samples were trimmed, frozen in liquid nitrogen, and sectioned with a Leica Ultracut cryo-ultramicrotome (Leica Microsystems, Bannockburn, IL). 50 nm sections were blocked with 5% FBS/5% normal goat serum for 30 min and subsequently incubated with rabbit anti-HA antibody (Sigma) and goat anti-GFP antibody (ab5450, Abcam, Cambridge, MA) followed by secondary donkey anti-rabbit antibody conjugated to 18 nm colloidal gold and donkey anti-goat antibody conjugated with 12 nm colloidal gold (Jackson ImmunoResearch Laboratories, West Grove, PA). Sections were washed in PIPES buffer followed by a water rinse, and stained with 0.3% uranyl acetate/ 2% methyl cellulose. Samples were viewed with a JEOL 1200EX transmission electron microscpope (JEOL USA, Peabody, MA) equipped with an AMT 8 megapixel digital camera (Advanced Microscopy Techniques, Woburn, MA). All labeling experiments were conducted in parallel with controls omitting the priomary antibody. These controls were consistently negative at the concentration of colloidal gold conjugated secondary antibodies used in these studies.
Endoglycosidase H assay
The NSC34 cell lysates overexpressing A1AT-NHK was treated with 1500 units/sample of Endoglycosidase H (New England Biolab) for 1 h at 37°C following manufacturer’s protocol.
Measurement of cytosolic calcium
Cytosolic calcium concentration of wild-type MEFs transduced with adenovirus expressing LacZ or Bnip3 at moi 200 (30,000 cells/well, N=6 wells per condition) was measured with Fura-2 dye. After measurement of basal calcium, the cells were treated with 1 μM thapsigargin.
Proteasomal activity
Proteasomal activity of wild-type MEFs and Ire1α knockout (Ire1α−/−) MEFs were determined by Proteasome-Glo assay (Promega) following the manufacturer’s instructions.
Dephosphorylation of Bnip3
The proteins were dephosphorylated by Lambda protein phosphatase (New England Biolab, Ipswich, MA). Samples were reacted in the buffer containing 50 mM HEPES (pH7.5), 10 mM NaCl, 2 mM DTT, 0.01% Brij 35, 1 mM MnCl2 and 40 units/sample of lambda protein phosphatase at 30°C for 1h, followed by immunoblot analysis.
Supplementary Material
Figure S1. Leakage of ER contents to the cytosol precedes the apoptotic cascade.
Figure S2. Phosphorylation of Bnip3
Figure S3. Role of Bnip3 in the leakage of ER calcium to the cytosol.
Figure S4. Subcellular localization of Bnip3
Figure S5. Gel filtration of GRP94
Figure S6. Confirmation of attenuation of IRE1 signaling by small molecule inhibitors.
Figure S7. Leakage of ER contents to the cytosol in Xbp1+/+ cells and Xbp1−/− cells.
Figure S8. Time-course analysis of the phosphorylation of JNK and leakage of ER contents.
Figure S9. Degradation mechanism of Bnip3
Figure S10. Abundance of Bnip3, phosphorylation of JNK and LC3-II in wild-type cells and Bax/Bak DKO cells.
Figure S11. Live cell imaging of MERO-GFP-expressing IRE1 knockout cells under ER stress conditions.
Figure S12. Time-lapse ratio chase of ER membrane permeabilization in IRE1 knockout cells.
Figure S13. Time-lapse ratio imaging of HEK293 cells expressing cytosolic roGFP under ER stress conditions.
Figure S14. Fluorescent imaging of HEK293 cells expressing the mCherry-Chop reporter under ER stress conditions.
Figure S15. Scheme of ER membrane permeabilization by ER stress.
Acknowledgments
We thank Dr. Paul Furcinitti, Mrs. Cris Brown and Mrs. Mai Kanekura for technical support. We thank Dr. David Ron, Dr. Randal Kaufman, Dr. Laurie Glimcher, Dr. Kezhong Zhang, Dr. Roger Davis, Dr. Douglas Mann, Dr. Robert Brink, Dr. Herbert Virgin, Dr. Navdeep Chandel, Dr. Ron Kopito, Dr. Scott Oakes, and Dr. Feroz Papa for providing us with various reagents. We also thank Dr. Wandy Beatty for technical and scientific assistance with the electon microscopy studies.
Funding: K. Kanekura was partly supported by fellowships from the Japan Society for the Promotion of Science and the Uehara Memorial Foundation. A. Diwan is supported by grants from NIH (HL107594) and Department of Veterans Affairs (I01BX000448, I01BX001969). F. Urano is supported by grants from the National Institutes of Health (DK067493 and DK020579), Juvenile Diabetes Research Foundation (17-2013-512), American Diabetes Association (1-12-CT-61), the Team Ian, the Team Alejandro, the Ellie White Foundation for Rare Genetic Disorders, and the Jack and J.T. Snow Scientific Research Foundation.
Footnotes
Author contributions: K.K, A.D., and F.U. designed the experiments. K.K., X.M., and J.T.M. performed the experiments. L.J.Z performed statistical analyses of the data. K.K., A. D., and F.U. wrote the article.
Competing interests: The authors declare that they have no competing interests.
References and Notes
- 1.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 2.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. The Journal of cell biology. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic beta-cell death. Trends in endocrinology and metabolism: TEM. 2011;22:266–274. doi: 10.1016/j.tem.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin JH, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han D, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollien J, et al. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. The Journal of cell biology. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu M, et al. Cell death. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. 2014;345:98–101. doi: 10.1126/science.1254312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Olberding KE, White C, Li C. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell death and differentiation. 2011;18:38–47. doi: 10.1038/cdd.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hosokawa N, You Z, Tremblay LO, Nagata K, Herscovics A. Stimulation of ERAD of misfolded null Hong Kong alpha1-antitrypsin by Golgi alpha1,2-mannosidases. Biochemical and biophysical research communications. 2007;362:626–632. doi: 10.1016/j.bbrc.2007.08.057. [DOI] [PubMed] [Google Scholar]
- 10.Ushioda R, et al. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- 11.Lusch A, et al. Development and analysis of alpha 1-antitrypsin neoglycoproteins: the impact of additional N-glycosylation sites on serum half-life. Molecular pharmaceutics. 2013;10:2616–2629. doi: 10.1021/mp400043r. [DOI] [PubMed] [Google Scholar]
- 12.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 13.Boyd JM, et al. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell. 1994;79:341–351. doi: 10.1016/0092-8674(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 14.Ray R, et al. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. The Journal of biological chemistry. 2000;275:1439–1448. doi: 10.1074/jbc.275.2.1439. [DOI] [PubMed] [Google Scholar]
- 15.Althaus J, et al. Expression of the gene encoding the pro-apoptotic BNIP3 protein and stimulation of hypoxia-inducible factor-1alpha (HIF-1alpha) protein following focal cerebral ischemia in rats. Neurochemistry international. 2006;48:687–695. doi: 10.1016/j.neuint.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. American journal of physiology Heart and circulatory physiology. 2008;295:H2025–2031. doi: 10.1152/ajpheart.00552.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diwan A, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. The Journal of clinical investigation. 2007;117:2825–2833. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma X, et al. Regulation of the Transcription Factor EB-PGC1alpha Axis by Beclin-1 Controls Mitochondrial Quality and Cardiomyocyte Death under Stress. Molecular and cellular biology. 2015;35:956–976. doi: 10.1128/MCB.01091-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bocharov EV, et al. Unique dimeric structure of BNip3 transmembrane domain suggests membrane permeabilization as a cell death trigger. The Journal of biological chemistry. 2007;282:16256–16266. doi: 10.1074/jbc.M701745200. [DOI] [PubMed] [Google Scholar]
- 20.Frazier DP, et al. Acidosis regulates the stability, hydrophobicity, and activity of the BH3-only protein Bnip3. Antioxidants & redox signaling. 2006;8:1625–1634. doi: 10.1089/ars.2006.8.1625. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Li L, Liu H, Borowitz JL, Isom GE. BNIP3 mediates cell death by different pathways following localization to endoplasmic reticulum and mitochondrion. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2009;23:3405–3414. doi: 10.1096/fj.08-124354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. The Biochemical journal. 2007;405:407–415. doi: 10.1042/BJ20070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim H, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Molecular cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urano F, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 25.Papandreou I, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiu Y, et al. A crucial role for RACK1 in the regulation of glucose-stimulated IRE1alpha activation in pancreatic beta cells. Science signaling. 2010;3:ra7. doi: 10.1126/scisignal.2000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang K, et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. The EMBO journal. 2011;30:1357–1375. doi: 10.1038/emboj.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaneko M, Niinuma Y, Nomura Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biological & pharmaceutical bulletin. 2003;26:931–935. doi: 10.1248/bpb.26.931. [DOI] [PubMed] [Google Scholar]
- 29.Hanna RA, et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. The Journal of biological chemistry. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park CW, et al. BNIP3 is degraded by ULK1-dependent autophagy via MTORC1 and AMPK. Autophagy. 2013;9:345–360. doi: 10.4161/auto.23072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogata M, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Molecular and cellular biology. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding WX, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. The American journal of pathology. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hetz C, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 34.Kanekura K, Ishigaki S, Merksamer PI, Papa FR, Urano F. Establishment of a system for monitoring endoplasmic reticulum redox state in mammalian cells. Laboratory investigation; a journal of technical methods and pathology. 2013;93:1254–1258. doi: 10.1038/labinvest.2013.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Margittai E, Enyedi B, Csala M, Geiszt M, Banhegyi G. Composition of the redox environment of the endoplasmic reticulum and sources of hydrogen peroxide. Free radical biology & medicine. 2015;83:331–340. doi: 10.1016/j.freeradbiomed.2015.01.032. [DOI] [PubMed] [Google Scholar]
- 37.Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 38.Akuta N, et al. Utility of detection of telaprevir-resistant variants for prediction of efficacy of treatment of hepatitis C virus genotype 1 infection. Journal of clinical microbiology. 2014;52:193–200. doi: 10.1128/JCM.02371-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell calcium. 2003;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- 40.Roussel BD, et al. Endoplasmic reticulum dysfunction in neurological disease. Lancet neurology. 2013;12:105–118. doi: 10.1016/S1474-4422(12)70238-7. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh R, et al. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PloS one. 2010;5:e9575. doi: 10.1371/journal.pone.0009575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annual review of physiology. 2013;75:49–67. doi: 10.1146/annurev-physiol-030212-183707. [DOI] [PubMed] [Google Scholar]
- 43.Fonseca SG, et al. WFS1 Is a Novel Component of the Unfolded Protein Response and Maintains Homeostasis of the Endoplasmic Reticulum in Pancreatic {beta}-Cells. The Journal of biological chemistry. 2005;280:39609–39615. doi: 10.1074/jbc.M507426200. [DOI] [PubMed] [Google Scholar]
- 44.Fonseca SG, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. The Journal of clinical investigation. 2010;120:744–755. doi: 10.1172/JCI39678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu SM, et al. A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E5292–E5301. doi: 10.1073/pnas.1421055111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urano F. Diabetes: Targeting endoplasmic reticulum to combat juvenile diabetes. Nature reviews Endocrinology. 2014;10:129–130. doi: 10.1038/nrendo.2013.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lipson KL, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4:245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 48.Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy. 2012;8:297–309. doi: 10.4161/auto.18658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minaki H, Sasaki K, Honda H, Iwaki T. Prion protein oligomers in Creutzfeldt-Jakob disease detected by gel-filtration centrifuge columns. Neuropathology : official journal of the Japanese Society of Neuropathology. 2009;29:536–542. doi: 10.1111/j.1440-1789.2009.01007.x. [DOI] [PubMed] [Google Scholar]
- 50.Ma X, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riggs AC, et al. Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia. 2005;48:2313–2321. doi: 10.1007/s00125-005-1947-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Leakage of ER contents to the cytosol precedes the apoptotic cascade.
Figure S2. Phosphorylation of Bnip3
Figure S3. Role of Bnip3 in the leakage of ER calcium to the cytosol.
Figure S4. Subcellular localization of Bnip3
Figure S5. Gel filtration of GRP94
Figure S6. Confirmation of attenuation of IRE1 signaling by small molecule inhibitors.
Figure S7. Leakage of ER contents to the cytosol in Xbp1+/+ cells and Xbp1−/− cells.
Figure S8. Time-course analysis of the phosphorylation of JNK and leakage of ER contents.
Figure S9. Degradation mechanism of Bnip3
Figure S10. Abundance of Bnip3, phosphorylation of JNK and LC3-II in wild-type cells and Bax/Bak DKO cells.
Figure S11. Live cell imaging of MERO-GFP-expressing IRE1 knockout cells under ER stress conditions.
Figure S12. Time-lapse ratio chase of ER membrane permeabilization in IRE1 knockout cells.
Figure S13. Time-lapse ratio imaging of HEK293 cells expressing cytosolic roGFP under ER stress conditions.
Figure S14. Fluorescent imaging of HEK293 cells expressing the mCherry-Chop reporter under ER stress conditions.
Figure S15. Scheme of ER membrane permeabilization by ER stress.