

Summary
We present a newly developed method for fixing RNA–protein complexes in situ in living cells and the subsequent purification of the RNA targets. Using this approach, complex tissue such as mouse brain can be ultraviolet (UV) irradiated to covalently crosslink RNA–protein complexes. Once covalently bound, RNA–protein complexes can be purified under stringent conditions, allowing a highly specific purification scheme to be employed. After UV irradiation, the tissue is solubilized and the RNA partially digested, allowing a small fragment to remain attached to protein. RNA–protein complexes of interest are partially purified by immunoprecipitation and noncovalently associated RNA removed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). These purified RNA–protein complexes are isolated and treated with proteinase K, which removes protein but leaves intact RNA. This RNA is abundant enough, and competent for, RNA linker ligation, reverse transcriptase polymerase chain reaction (RT-PCR) amplification, and sequencing. Database matching of these short 70- to 100-nt RNA CLIP (crosslinking and immunoprecipitation of RNA–protein complexes) “tags,” which mark the native binding sites of RNA binding proteins, potentially allows the entire target repertoire of an RNA binding protein to be determined.
Keywords: CLIP, immunoprecipitation, photocrosslinking, RNA binding protein
1. Introduction
Those interested in the role RNA binding proteins play in the development and function of organisms have been limited by a paucity of techniques available to study RNA–protein interactions in vivo. Conversely, biochemists have developed potent methods for studying interactions in vitro but have not applied these to living tissues. We describe here a method in which a classical in vitro tool used by RNA biochemists—ultraviolet (UV) crosslinking—is adapted to the study of RNA–protein complexes in living tissues. We have successfully used this method, termed CLIP (crosslinking and immunoprecipitation of RNA–protein complexes) to identify a number of target RNAs of the Nova family of neuron-specific RNA binding proteins (1,2), and we are piloting its use with several other RNA binding proteins of interest in our laboratories, notably the FMRP and Hu families.
The term CLIP is largely self-explanatory, with the following addendums: We have used a living organ—specifically brain tissue—as the source for in vivo RNA–protein complexes to be CLIPed. This tissue is dissected from the animal (mouse in our experiments), rapidly dissociated on ice by trituration to allow UV light to penetrate the cells, and irradiated immediately thereafter. A series of subsequent biochemical steps is used to partially hydrolyze the RNA, allowing short (~70- to 100-nt) fragments to remain bound to the protein. We have used ribonuclease (RNase) T1, which leaves a 5′ −OH and a 2′,3′-cyclic phosphate on the RNA chain. The complex is then purified by immunoprecipitation, a step obviously critical to the protocol and dependent on an antibody good for immunoprecipitation. The RNA–protein complex is then treated with T4 polynucleotide kinase (PNK; which both adds a 5′-PO4− group and resolves the 2′,3′-cyclic phosphate to a 3′ −OH), a step that allows the introduction of radioactive tracer and prepares the RNA ends for subsequent directional linker ligation.
The complex is then further purified by boiling in sodium dodecyl sulfate (SDS)-sample buffer and running on denaturing SDS polyacrylamide gel electrophoresis (PAGE) gels. This gel purification step is critical to the success of CLIP because it partitions the RNA–protein covalent complex away from any RNA that has immunoprecipitated with the protein but was not crosslinked to the protein. While this “free” RNA pool may contain bone fide RNA targets of the protein, it may also include RNAs that have only bound the protein in vitro during the purification steps of the protocol, and thus it is critical to remove these RNAs (3). A final purification step is achieved by transferring the gel to nitrocellulose; this transfer allows purification of RNA–protein complexes from the membrane (much easier than from the gel matrix itself), and at the same time, any remaining free RNA in the gel passes through the nitrocellulose and toward the positive electrode. Radioactive bands corresponding in size to RNA–protein complexes are cut out from the nitrocellulose, and complexes are eluted and treated with proteinase K to remove the protein component. RNA is isolated, ligated to a 5′ linker harboring 5′ and 3′ −OH groups, and then ligated to a 3′ linker harboring a 5′ −OH group and a blocked 3′ end (we have used puromycin). The fully ligated RNA is then gel purified to obtain only RNAs that contain an insert size greater than 30 nt. This final pool of RNA molecules is used with a linker-specific primer and reverse transcriptase to obtain complementary DNA (cDNA), which is then PCR amplified (again with linker-specific primers), cloned, and individual transformants prepared for sequencing. Finally, the CLIP tags are of sufficient length to easily and unambiguously identify the parental RNAs by database searching.
2. Materials
2.1. Solutions
1X HBSS* buffer: 1X Hank’s balanced salt solution, Ca-Mg free (10X from Gibco, cat. no. 14186-012), 10 mM HEPES, pH 7.3.
1X PXL buffer: 1X phosphate-buffered saline (PBS; tissue culture grade; no Mg2+, no Ca2+), 0.1% SDS, 0.5% deoxycholate, 0.5% NP-40.
1X PNK+ buffer: 50 mM Tris-Cl, pH 7.4, 10 mM MgCl2, 0.5% NP-40.
1X PK buffer: 100 mM Tris-Cl, pH 7.5, 50 mM NaCl, 10 mM ethylenediaminetetraacetic acid (EDTA).
1X PK buffer/7 M urea (this buffer must be made fresh): 100 mM Tris-Cl, pH 7.5, 50 mM NaCl, 10 mM EDTA, 7 M urea.
“RNA phenol”: Pure crystalline phenol equilibrated with 0.15 M NaOAc, pH 5.2.
CHCl3 solution: 49:1 CHCl3:isoamyl alcohol.
Nucleic acid elution buffer: 1 M NaOAc, pH 5.2, 1 mM EDTA.
2.2. Enzymes, Reagents, Kits, Plasticware, Equipment
Stericups (Millipore).
Stratalinker (Stratagene).
RNasin (Promega).
RQ1 deoxyribonuclease (DNase) (Promega).
Protein A Dynabeads (Dynal).
T1 RNase, biochemistry grade, 1 U/μL (Ambion).
T4 PNK (Ambion).
Novex NuPage Bis-Tris gels (Invitrogen).
Novex LDS loading buffer.
BA-85 nitrocellulose (S&S).
RNA ligase (Fermentas).
SpinX colums (Costar).
1-cm filters (Whatman).
SuperScript III (Invitrogen).
Pfu (Stratagene).
Taq (PerkinElmer).
TOPO TA cloning kit (Invitrogen).
Glycogen (Ambion, 5 mg/mL).
2.3. Linker and Primer Sequences
RNA linkers (from Dharmacon): RL5, 5′-OH AGG GAG GAC GAU GCG G 3′-OH; RL3, 5′ -P CGA GAU GGC GGC UUC CUG C 3′-puromycin.
DNA primers (from Operon): DP5, AGG GAG GAC GAT GCG G; DP3, GCA GGA AGC CGC CAT CTC G.
3. Method
3.1. Ultraviolet Crosslinking of Tissue/Cell Lines
For our CLIP experiments with neuronal RNA binding proteins, we harvest brain and spinal cord tissue from postnatal d 8 mice (ICR strain); let the tissue sit in ice-cold HBSS* until the harvest is complete. Other rodent tissues should be harvested the same way. If your tissue source contains a lot of RNase or proteolytic activity, your best bet is to keep the tissue as cold as possible and work quickly.
When we use more than 10 brains at a time, we set up a 500-mL Stericup, remove the cellulose filter, and make a conical filter out of a sheet of 200-μm nylon mesh to replace it. The tissue can be disrupted by passing the tissue through the mesh using a flat-tip cell scraper and the remaining tissue washed through the mesh with more HBSS*. For 10 brains, the resulting cell suspension is about 100 mL. If we harvest only a small number of brains, the tissue can be triturated using a 5-mL pipet.
Next, transfer the tissue suspension to 50-mL Falcon tubes and spin at 2500 g for 5 min at 4 °C. Remove the supernatant and resuspend the tissue in approx 10X the original volume of tissue. Place the suspension in a 150-mm tissue culture dish (around 15–20 mL/plate) and irradiate the suspension for 400 mJ/cm2 in a Stratagene Stratalinker. Use a dish of ice underneath the tissue suspension as you crosslink to keep the suspension cold.
Collect the irradiated suspension in a 50-mL Falcon tube, then wash out each plate with an additional 5 mL of HBSS*, also collecting this wash. Spin down the tissue again at 2500 rpm for 5 min at 4 °C. Resuspend the tissue in 2X volume of the original tissue volume and pipet the solution into Eppendorf tubes. Spin the tubes briefly and take off the supernatant. Freeze at −80 °C until use.
For crosslinking adherent cells, grow cells in 150-mm dishes. Wash cells once with 10 mL cold 1X HBSS* and add 10 mL 1X HBSS* to the dish. Irradiate as above and collect the cells by scraping. Wash the dish with an additional 5 mL of 1X HBSS* and spin down the cell suspension as above. Collect and freeze the cells as above.
3.2. Bead Prep
We have found that protein A Dynabeads work well for CLIP. The binding capacity of the Dynabeads is not outstanding (25 μg immunoglobulin G [IgG] per 100 μL of bead solution), but this is outweighed by the ease of handling of magnetic beads during the wash steps. For our immunoprecipitations, we generally use about 100 μL of bead solution (about 50–75 μL of beads) per 300 μL of packed, crosslinked tissue. Depending on your particular protein and antibody combination for CLIP as well as the protein’s abundance in tissue/cells, these numbers could vary greatly.
Pipet 100 μL of protein A Dynabead solution. Place beads in magnetic stand to capture and wash beads with 500 μL of 1X PXL. Repeat the wash two more times. Resuspend beads in 100 μL of 1X PXL and add an appropriate amount of your anti-RNA binding protein antibody. Rock the tube for 30–45 min at room temperature to bind the antibody to the beads. Wash the beads three times with 1X PXL.
3.3. Crosslinked Lysate Workup
Lyse each 100 μL of crosslinked cells using 100 μL of ice-cold 1X PXL. Let the lysates sit on ice for 10 min. Add 10 μL RNasin and 10 μL of RQ1 DNase to each tube; incubate at 37 °C for 15 min. If you have a shaking, heating block, like an Eppendorf Thermomixer, also shake the tubes at 1000 rpm.
Add 1 μL of RNase T1 stock (1 U/μL) to the solution; incubate at 37 °C for 10 min, shaking at 1000 rpm. The actual RNase T1 dilution you use will need to be determined empirically (see Note 1).
Next, spin the lysates in a prechilled microultracentrifuge at 90K for 25 min at 4 °C. We use open-topped polycarbonate tubes in a TLA 120.2 rotor (see Note 2).
3.4. Immunoprecipitation
For immunoprecipitation, carefully remove the supernatant from the pelleted debris and add supernatant to one prepared tube of beads. Again, we use about 100 μL of beads per 300 μL of crosslinked tissue. Rock the beads/lysate for 1 h at 4 °C. Wash three times with 1 mL ice-cold 1X PXL and twice with 1 mL ice-cold 1X PNK+.
3.5. Kinase the Immunoprecipitated RNA
Resuspend beads in 80 μL of 1X PNK+ and add 5 μL of P32 γ-ATP (γ-adenosine triphosphate) (>3000 Ci/mM) and 2 μL of PNK enzyme (see Note 3). Incubate in a thermomixer at 37 °C and 1000 rpm for 20 min.
“Finish” the reaction by adding 5 μL of 1 mM ATP. Let the reaction continue another 5 min at 37 °C. Wash the beads four times with 1 mL of ice-cold 1X PNK+.
Resuspend the beads in 30 μL of 1X PNK+ and 30 μL of Novex LDS loading buffer (do not add any reducing agent). If reducing agent is used, the heavy and light chains of immunoglobulin can interfere with the migration of proteins smaller than about 75 kDa. If you are interested in performing CLIP on a very large protein, you may be best served by using reducing agents to remove intact IgG. Incubate at 70 °C for 10 min. Isolate beads in the magnetic stand and take the supernatant for loading.
3.6. SDS-PAGE Gel and Transfer
Load 2 wells/tube of a Novex NuPAGE 10% Bis-Tris gel (see Note 4). Run the gel at 150 V until the dye front is at the bottom of the gel. After the gel run, transfer gel to a piece of S&S BA-85 nitrocellulose using the Novex wet transfer apparatus (see Notes 5 and 6).
After the transfer, rinse the NC filter in 1X PBS and gently blot on Kimwipes to dry. Wrap the membrane in plastic wrap and expose to film.
3.7. Cut Out Crosslinked RNA–Protein Complex
The autoradiography of the immunoprecipitated material usually shows radio-labeled RNA–protein complexes starting at approx the molecular weight of the noncrosslinked protein and extending in a “smear” to higher molecular weights (see Note 7). In general, the RNA–protein complexes run at approximately the combined molecular weight of the protein and RNA (see Fig. 1).
Since we generally like to work with CLIP tags of about 40–80 nt (or 13,200– 26,400 kDa), we try to cut out the crosslinked material that is 10–30 kDa bigger than the migration of the protein itself. If you have titrated your RNase T1 correctly, the majority of radiolabeled signal will fall within this range. Using a scalpel blade, cut out this band and then cut the nitrocellulose into small pieces. Place these pieces into a single, clean Eppendorf tube (see Note 8).
Fig. 1.
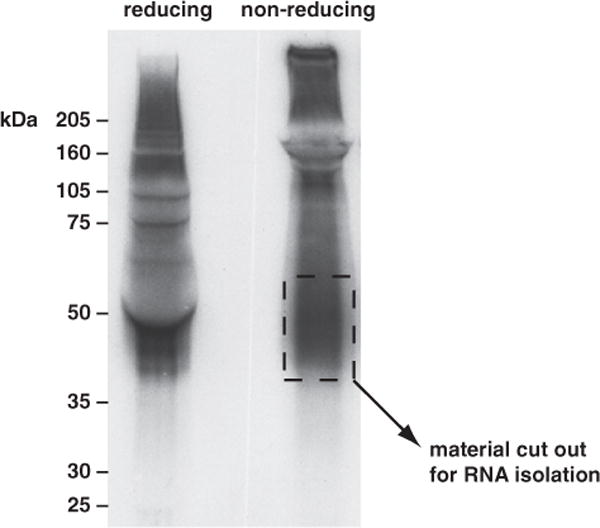
Autoradiograph of 32P-labeled RNA crosslinked to the neuronal Hu proteins. P4 mouse brain tissue was irradiated with short-wavelength ultraviolet (UV) light. The tissue was collected, and the soluble extracts were treated with ribonuclease (RNase) T1 and immunoprecipitated with antiserum specific for the neuronal Hu proteins (HuB, HuC, and HuD). The purified material was labeled with 32P and run on reducing and nonreducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The gel contents were transferred to nitrocellulose by wet transfer; the filter was blotted dry and exposed to film. The lane with reducing agent shows the interference caused by the approximately 50-kDa heavy chain “pushing” the gel contents out of its way. In the nonreducing lane, a fraction of the membrane from approximately 40 to 60 kDa was used for purification of the RNA–protein complexes. (The Hu proteins usually run at about 35 kDa.)
3.8. RNA Isolation and Purification
Make a 4-mg/mL proteinase K solution in 1X PK buffer; preincubate this stock at 37 °C for 20 min to digest any RNases. Add 200 μL of this proteinase K solution to each tube of isolated NC pieces; incubate 20 min at 37 °C with shaking (1200 rpm on Thermomixer).
Add 200 μL PK/7 M urea buffer; incubate another 20 min at 37 °C with shaking (1200 rpm on Thermomixer).
Add 400 μL RNA phenol and 130 μL of CHCl3 solution. Vortex these tubes and then incubate at 37 °C for 20 min at 1400 rpm with shaking (1200 rpm on Thermomixer). Next, spin tubes at full speed in microcentrifuge at 4 °C or room temperature for 10 min.
Put the aqueous phase from each tube in a clean tube and add 50 μL of 3 M NaOAc, pH 5.2, and 1 mL of 1:1 EtOH:isopropanol. Do not add any carrier. Precipitate overnight at −20 °C.
3.9. RNA Ligations
Spin down RNA at full speed in a cold centrifuge for 30 min. Check with a handheld Geiger counter to see if you have decent precipitation of counts; if not, you might have to add glycogen (2.5 μg) to the tube and reprecipitate (by mixing and reincubation at −20 °C). Wash the pellet with 100 μL of 75% EtOH and then dry the pellet in a SpeedVac.
Count the RNA in a scintillation counter by Cerenkov counts (we just stick in the dry, closed Eppendorf tube.) If you have 100,000 ccpm or more of RNA, you should not have any problem with the ligation step and amplification, and you may want to save about 20% to run as an unligated control for the next gel purification. If you have less, you may want to use all of it for the ligation step. If so, resuspend your RNA in 5.7 μL H2O.
For the RNA ligation, use the following (10 μL total):
1 μL 10X T4 RNA ligase buffer (Fermentas)
1 μL bovine serum albumin (BSA) (0.2 mg/mL)
1 μL ATP (10 mM)
0.3 μL T4 RNA ligase (3 U, Fermentas) (see Note 9)
1 μL RL5 RNA linker at 20 pmol/μL (see Note 10)
5.7 μL RNA resuspended in H2O
Incubate at 16 °C for 1 h, then add the following to the reaction:
1 μL RL3 RNA linker at 40 pmol/μL (see Note 11)
0.5 μL ATP (10 mM)
0.2 μL T4 RNA ligase
Incubate at 16 °C overnight, Then add the following to the reaction (100 μL total):
77 μL H2O
11 μL 10X DNase I buffer
5 μL RNasin
5 μL RQ1 DNase
Incubate at 37 °C for 20 min, Then add the following to the reaction:
300 μL H2O
300 μL RNA phenol (see Note 12)
100 μL CHCl3 solution
Vortex, spin, and take the aqueous layer. Precipitate by adding
50 μL 3 M NaOAc, pH 5.2
2 μL glycogen (20 mg/mL stock)
1 mL 1:1 EtOH:isopropanol
Precipitate overnight at −20 °C.
3.10. Size Purification of the Ligated RNA
Spin, wash, and dry RNA pellets as above. Check RNA recovery by Cerenkov counting in a scintillation counter. Pour a 20% denaturing polyacrylamide gel (1:19 bis:acrylamide, 7 M urea). Resuspend the entire ligation reaction in 5 μL H2O plus 5 μL of formamide loading buffer. Heat the resuspended RNA at 95 °C for 5 min and load the entire solution on the gel (along with preligation RNA if you have it); use radiolabeled PhiX markers for sizing the RNA.
After the gel run, place the gel on an old piece of film and wrap with plastic wrap. Place with a film at −80 °C, generally overnight for a good signal. Using the exposed film, cut out the RNA that is greater than about 65 nt (35 nt for the linkers, around 30 nt minimum for the CLIP tag itself). If you have good product past 100 nt, we generally cut out two gel slices: 65–100 nt and 100 nt and above (see Fig. 2). Place the gel slices in Eppendorf tubes; add 350 μL of nucleic acid elution buffer and crush with a 1-mL syringe plunger; incubate at 37 °C for 30 min at 1200 rpm in a Thermomixer. With a cut off P1000 tip, transfer the gel slurry to a Costar SpinX column to which you have added a 1-cm glass prefilter (Whatman). Spin the columns full speed in the microcentrifuge, take the flowthrough and place it in a clean Eppendorf tube. Add 1 mL 1:1 EtOH:isopropanol and 2 μL of glycogen (20 mg/mL stock) and precipitate overnight at −20 °C. Also, see Notes 13 and 14.
Fig. 2.
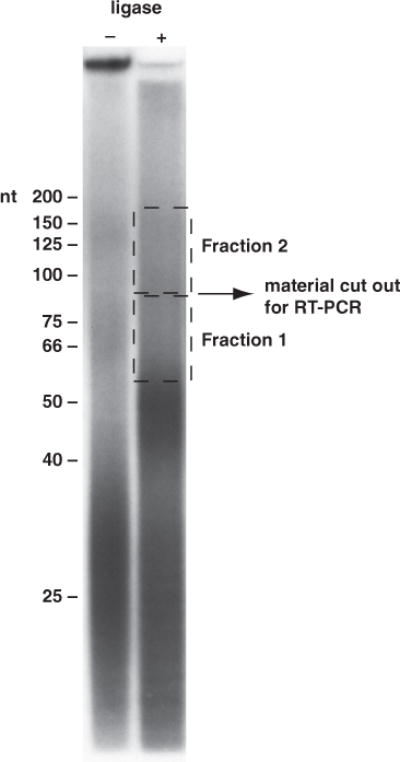
Gel purification of the CLIP (crosslinking and immunoprecipitation of RNA– protein complexes) RNA after linker ligation. RNA before and after linker ligation was run on a 7 M urea/20% polyacrylamide gel. The modal size of this RNA pool before ligation was approximately 30 nt. After ligation, the modal size of the RNA increased to about 50 nt. The postligation pool is probably composed of significant fractions of single-linker ligation (CLIP RNA of 30 nt plus one linker of 16 nt) and smaller amounts of both 5′ and 3′ linker ligations (CLIP RNA of 30 nt and linkers of 35 nt). Two fractions of RNA were isolated from this gel for reverse transcriptase polymerase chain reaction (RT-PCR). Fraction 1 is from approx 60 to approx 85 nt (with CLIP tag inserts of 25–50 nt), and fraction 2 is from approx 85 to approx 175 nt (with CLIP tag inserts of 50–140 nt).
3.11. cDNA and PCR
Spin, wash, and dry RNA as above; count the RNA again in a scintillation counter to quantitate yield. Resuspend the purified RNA in 9 μL H2O and add 2 μL of DP3 at 5 pmol/μL. Heat at 65 °C for 5 min; chill and quick spin.
Then, add
2 μL 10 mM deoxynucleotide 5′-triphosphates (dNTPs)
2 μL 0.1 M dithiothreitol (DTT)
4 μL 5X SuperScript reverse transcriptase (RT) buffer
0.5 μL RNasin
0.5 μL SuperScript III
Incubate at 55 °C for 30 min, then 90 °C for 5 min. Chill on ice, then use 3 μL of the RT reaction for the PCR step. For the PCR reaction,
4 μL 10X Pfu buffer
4 μL DP5 primer, 5 pmol/μL
4 μL DP3 primer, 5 pmol/μL
4 μL radiolabeled DP5 primer
4 μL 2.5 mM dNTPs
1 μL Pfu
16 μL water
3 μL of the RT reaction
Cycle 35 times: 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s.
Pour a 10% denaturing polyacrylamide gel and run about 10 μL of the PCR reaction on the gel; use radiolabeled markers and autoradiography gel (see Fig. 3). Cut out the major bands of about 60–100 nt (and 100 nt and up if you have it). There will probably be a major contaminant band at approx 40 nt; this is a PCR product from the linker-linker dimer (no insert), and you definitely do not want to clone any of this (see Note 15). Purify and precipitate the DNA as you did above for the RNA except let the DNA elute from the crushed gel slurry for at least 4 h. After precipitation, resuspend the purified DNA in 5–10 μL of water.
Fig. 3.
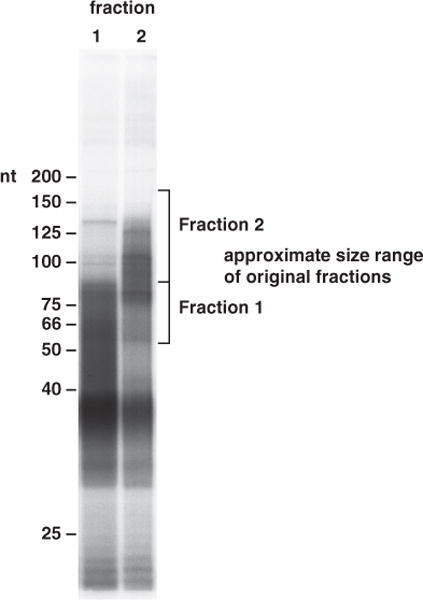
Reverse transcriptase polymerase chain reaction (RT-PCR) of purified linker-ligated CLIP (crosslinking and immunoprecipitation of RNA–protein complexes) RNA. Fractions 1 and 2 of CLIP RNA gel purified after linker ligation (Fig. 2) were used to make complementary DNA for polymerase chain reaction (PCR) amplification. The PCR products obtained from fraction 1 (lane 1) and fraction 2 (lane 2) were run on a 7 M urea/20% polyacrylamide gel. Fraction 1 PCR products primarily cover a range from 50 to 80 nt, while those from fraction 2 fall within a range of 70 to 125 nt. In general, these sizes are consistent with the sizes of RNA taken from the gel purification of the linker-ligated CLIP RNAs (both fractions of PCR products are slightly shorter than predicted). To obtain CLIP tag inserts of 30 nt or greater, only PCR products great than 65 nt should be gel purified and used for the final PCR reaction before cloning. Also note the prominent product at about 38 nt. We believe that this is a PCR product resulting from amplification of a linker-linker ligation (a very prominent side reaction of the linker ligation step). While this RNA should be removed during gel purification of the RNA, its abundance makes it very difficult to get rid of it. After gel purification of the correct PCR products, this band will disappear (thus, it is not a PCR-generated “primer-dimer”).
3.12. TOPO Cloning and Sequencing
We used three or four more rounds of PCR (one can use more if your yield is low for the first PCR). Desalt the reaction using a spin column. Then, generate the 3′ A end:
3.5 μL desalted PCR reaction
0.5 μL 10X Taq buffer
0.5 μL 10 mM deoxyadenosine 5′-triphosphate (dATP)
0.5 μL Taq polymerase (5 U)
Incubate at 72 °C for 20 min; place on ice and use immediately in the TOPO cloning reaction. For the TOPO clone,
2–4 μL of tailed PCR product
H2O to 4 μL
1 μL salt solution (from TOPO kit)
1 μL pCR4-TOPO vector
Mix gently and incubate 5 min at room temperature (store 3 μL that you do not use in first day cloning at −20 °C for potential subsequent transformation). Transform Escherichia coli as suggested by the TOPO cloning kit. Miniprep and sequence individual transformants as you would for your other sequencing reactions (see Note 16).
3.13. Database Searching
If one cannot identify the parental RNAs from which the CLIP tags are derived, the CLIP experiment is useless. Given the number of finished genomes and the number in progress, it is now unlikely that finding the origin of a CLIP tag poses any difficulty for most. We prefer to use the University of California at Santa Cruz genome entry site (genome.ucsc.edu) and the BLAT search tool for all of our CLIP tag matching. BLAT is much faster than BLAST and the “browser” view of search results is a great way to locate CLIP tags within a gene structure. For those who are doing CLIP with a genome on the University of California at Santa Cruz site, we highly recommend the BLAT search tool. For those doing BLAST searches, the National Center for Biotechnology Information (NCBI) now has a dedicated BLAST search page for those looking for short, nearly exact matches, and it is well suited for CLIP tag identification (see Notes 9 and 17).
Footnotes
The correct dilution of RNase T1 is critical; you will definitely need to do titrations of T1 to figure out which dilution will give you RNA CLIP tags of about 40–60 nt. To determine the correct T1 concentration, we generally perform the steps of the method up until the autoradiography of the SDS-PAGE filter transfer. If you run several reactions with different T1 amounts (and a T1 negative reaction), you should be able to see the effect of the amount of T1 on the migration of the RNA–protein complex on the gel (see Subheading 3.7. and Note 7).
This step clears the lysate of all high molecular weight material, like ribosomes, or very large RNPs (ribonucleoproteins).
This ensures that all RNA has a 5′ phosphate. Also, T4 PNK has a “re-solving” activity that opens up the 2′, 3′-cyclic phosphate at the 3′ end of the RNA tags to give you some fraction of free 3′ −OH (4).
The Novex gels are critical. A pour-your-own SDS-PAGE gel (Laemmli) has a pH during the run that can reach approx 9.5 and can lead to alkaline hydrolysis of the RNA. The Novex NuPAGE buffer system is close to pH 7.0.
This nitrocellulose is pure NC and is a little fragile, but it works much better for the RNA–protein extraction step.
The gel itself should be a lot less hot than the amount of radioactivity that you loaded. Most of the signal should be in the lower buffer. If there is still a lot of signal in the gel, it will likely be below a molecular weight of about 20–15 kDa. You can cut this off the gel before the transfer if your protein is greater than 30 kDa.
Use a luminescent sticker (e.g., Glogos from Stratagene) so that you can align the filter back to the autoradiograph. If a strong radioactive band is seen after 1-h exposure, you probably have enough RNA for the CLIP procedure. The protein will migrate higher than normal (and with a greater size distribution) due to the bound RNA tags (which are not of uniform length). A very sharp radioactive band is a sign of overdigestion, and usually the bound RNAs are too small to be useful for CLIP assay.
Use the film to allow you to cut out the corresponding piece of nitrocellulose using a scalpel blade. Cut this piece horizontally into as many smaller pieces as you can manage and place them all into a single Eppendorf tube. If there is signal for a molecular weight range greater than about 20 kDa, we typically divide the material into two or more sections of 20 kDa each; each section of nitrocellulose would then be cut and placed into its own individual Eppendorf tube.
Regarding contamination with fungal and bacterial sequences, we believe that some commercial RNA ligases we have tested have nontrivial amounts of nucleic acid in the enzyme preps. If there is insufficient CLIP RNA in the ligation reaction, one can end up ligating linkers to junk RNA. We have overcome this by using the Fermentas ligase (very clean in our hands) and using as much input CLIP RNA as possible. More recent work in the lab is investigating techniques to overcome this issue altogether.
It is critical to rigorously gel purify the RNA oligos for CLIP. N-1 or other non-full-length products, most notably the loss of the 3′ puromycin or the 5′ phosphate from the RL3 RNA linker, will ruin your CLIP experiment. Make sure you have performed this purification step well. We also like to purify the DNA oligos as we find the PCR reactions cleaner than when performed with “desalted”-quality oligos straight from the manufacturer.
The ligation reaction is performed sequentially (first 1-h incubation between the CLIP tag RNA and the RL5 linker, followed by the addition of the RL3 linker) to ensure that the CLIP tag RNA has sufficient time to achieve adequate ligation to the 5′ RL5 linker before the addition of the 3′ RL3 linker. Since the linkers are in vast molar excess to the CLIP RNA, leaving out this sequential step would lead to rapid “dead-end” ligation of the 5′ and 3′ linkers to each other without any CLIP tag insert. Importantly, neither linker can circularize, and the presence of a 5′ −OH on the 5′ linker and a 3′-puromycin on the 3′ linker prevents higher-order ligation products.
RNA phenol is pure phenol that has been equilibrated with 0.15 M NaOAc, pH 5.2; CHCl3 is chloroform 49:1 with isoamyl alcohol. We find that this phenol gives us more consistent results than a phenol equilibrated with buffers in the pH 4.0 range.
As the RL3 linker is blocked at its 3′ end with puromycin, it is only competent to ligate at its 5′ end. The result of the ligation will be RL5-CLIP tag-RL3 RNA and a prominent side reaction of RL5-RL3 linkers only. These will be removed by gel purification of the ligated products.
We do not believe that the presence of a single amino acid (or possibly several) at the site of crosslinking within the RNA generally interferes with reverse transcription. However, the photochemistry of short-wavelength irradiation of nucleic acid and protein is not fully understood; evidence from reverse transcription of RNAs with bromo- and iodosubstituted pyrimidines suggests that crosslinking occurs away from the basepairing faces, which are intact and competent to act as a substrate for RT (5,6).
There should be a sharp band at 36 nt and less sharp from 39 to 48 nt; these are all PCR products of the linker-linker ligation. The good bands should run in the same place as where you cut them out, 70–100 nt and 100–150 nt. It is possible that the larger fraction (the 100- to 150-nt fraction) will show products below 100 nt; this is okay.
We generally sequence around 25–50 colonies from the transformations and use this small set to check if the CLIP protocol has worked. In general, we consider a successful experiment one in which there is: (1) very little contaminating sequence in the clones (for example, if the CLIP was from mouse, we do not expect to see sequence matches to fungi, bacteria, or other organisms); (2) a high rate of matches to genomic sequence that is known to be transcribed (works only for sequenced and decently annotated genomes); (3) an absence of any sort of linker/primer contamination (generally happens when there is insufficient RNA in the ligation); and (4) the CLIP tag length is consistent with the gel purification estimates and is long enough to be used for database searching.
In our hands, BLAT or BLAST searching of CLIP tags reveals perfect or near-perfect matches to the mouse genome. Greater than 90% of our CLIP tags match the genome exactly or with one mismatch. We have speculated that the mismatches may be due to reverse transcriptase mistakes when the enzyme encounters the crosslinked nucleotide (which is still likely attached to at least a single amino acid), but we have not investigated this further.
References
- 1.Ule J, Jensen KB, Ruggiu M, et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 2.Ule J, Stefani G, Mele A, et al. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 3.Mili S, Steitz JA. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004;10:1692–1694. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cameron V, Uhlenbeck OC. 3′-Phosphatase activity in T4 polynucleotide kinase. Biochemistry. 1977;16:5120–5126. doi: 10.1021/bi00642a027. [DOI] [PubMed] [Google Scholar]
- 5.Jensen KB, Atkinson BL, Willis MC, et al. Using in vitro selection to direct the covalent attachment of human immunodeficiency virus type 1 Rev protein to high-affinity RNA ligands. Proc Natl Acad Sci U S A. 1995;92:12220–12224. doi: 10.1073/pnas.92.26.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meisenheimer KM, Koch TH. Photocross-linking of nucleic acids to associated proteins. Crit Rev Biochem Mol Biol. 1997;32:101–140. doi: 10.3109/10409239709108550. [DOI] [PubMed] [Google Scholar]