

Abstract
Significance: Epigenetic inactivation of pivotal genes involved in cell growth is a hallmark of human pathologies, in particular cancer. Histone acetylation balance obtained through opposing actions of histone deacetylases (HDACs) and histone acetyltransferases is one epigenetic mechanism controlling gene expression and is, thus, associated with disease etiology and progression. Interfering pharmacologically with HDAC activity can correct abnormalities in cell proliferation, migration, vascularization, and death. Recent Advances: Histone deacetylase inhibitors (HDACi) represent a new class of cytostatic agents that interfere with the function of HDACs and are able to increase gene expression by indirectly inducing histone acetylation. Several HDACi, alone or in combination with DNA-demethylating agents, chemopreventive, or classical chemotherapeutic drugs, are currently being used in clinical trials for solid and hematological malignancies, and are, thus, promising candidates for cancer therapy. Critical Issues: (i) Non-specific (off-target) HDACi effects due to activities unassociated with HDAC inhibition. (ii) Advantages/disadvantages of non-selective or isoform-directed HDACi. (iii) Limited number of response-predictive biomarkers. (iv) Toxicity leading to dysfunction of critical biological processes. Future Directions: Selective HDACi could achieve enhanced clinical utility by reducing or eliminating the serious side effects associated with current first-generation non-selective HDACi. Isoform-selective and pan-HDACi candidates might benefit from the identification of biomarkers, enabling better patient stratification and prediction of response to treatment. Antioxid. Redox Signal. 23, 99–126.
Shaping the Epigenome
Epigenetic mechanism(s) allow genetically identical cells to adopt different phenotypes regulating transcriptional availability of the genome through differential chromatin marking and packaging (137), creating a network of mutually reinforcing or counteracting signals (192). A key aspect of epigenetics is that chromatin marks can be preserved and/or changed according to environmental, developmental, or pathological needs. These very complex and plastic steps are achieved via the activity of initiators (such as long noncoding RNA), writers (which establish the epigenetic mark, such as histone acetyltransferases), readers (which interpret the epi-mark), and erasers (which remove the epi-mark, such as histone deacetylases, or HDACs) (41, 232). In concert, remodelers (which reposition nucleosomes) and insulators (which build boundaries between epi-domains) create, maintain, and modulate the three-dimensional structure of networking within a cell (223). It is now clear that genetic and epigenetic mechanisms influence each other, cooperating to enable the acquisition of hallmarks of human cancer (89). The frequency of epi-target mutations seen in cancers underlines the relevance of mutations in epigenetic modifiers in cancer (213) and corroborates the concept that deregulation of epigenetic control is a common characteristic of cancer (105). Conversely, these findings confirm and strengthen the crucial role of epigenetic-based drugs (so-called epidrugs) (3) in reverting the malignant phenotype. This review aims at providing a comprehensive overview of current knowledge about HDACs and histone deacetylase inhibitors (HDACi), from biochemical data to development of clinical trials. It summarizes ongoing efforts to design epi-based treatments against cancer and other human diseases. The review analyses the chemical, biochemical, and therapeutic aspects of the use of HDACi in a clinical setting (highlighting the advantages and disadvantages), guiding the reader toward a better understanding of the complex balance between acetylation/deacetylation.
HDAC Classification
HDACs catalyse the removal of acetyl groups from the amino-terminal lysine residues of histone and non-histone proteins, such as transcription factors (TFs), hormone receptors, signaling proteins, chaperones, and DNA damage response proteins (10, 47, 68, 78). Four classes of human HDACs have been defined, based on their homology to yeast HDACs (13, 108, 228), enzymatic activities, and cellular localization (Fig. 1). HDAC1, 2, 3, and 8 belong to class I and are localized in the nucleus, presenting similarity to the yeast Rpd3 (47). Class II HDACs (229) are present in both the cytoplasm and nucleus, and they shuttle between these compartments (113, 147, 234). Class II HDACs (grouped for homology to Hda1 in yeast) are divided into two subclasses: class IIa (HDAC4, 5, 7, and 9) and class IIb (HDAC6, 10). Class IV HDACs includes one member (HDAC11) (49, 71), which can be considered a “hybrid” sharing similarities to both class I and II HDACs. Sirtuins, related to Sir2 in yeast (42), have also been categorized as class III HDACs (165) (Fig. 1, lower part) and grouped as SIRT1-7 (209). These enzymes display an NAD function that is associated to a nuclear, mitochondrial, and cytoplasmic localization (31, 149). HDAC1 and 2 are highly similar with an overall identity of ∼82%. The catalytic domain on the N-terminus constitutes the main part of the proteins. Moreover, HDAC1 and 2 only act in complex with proteins required for modulating their deacetylase activity and binding to DNA (87), mediating the recruitment of HDACs to promoters (163, 190). Three protein complexes containing both HDAC1 and 2 have been characterized: Sin3, NuRD, and Co-REST (251). Both Sin3 and NuRD complexes consist of a core containing HDAC1 and 2, Rb-associated protein 48 and RbAp46 (123, 258). The core alone does not possess maximal HDAC activity, and further cofactors are needed. In addition to functioning through these complexes, HDAC1 and 2 can also bind directly to DNA-binding proteins such as Yin and Yang 1 (YY1) (249), Rb-binding protein-1 and Sp1 (51). Both activity and complex formation are regulated by phosphorylation. HDAC1 and 2 are phosphorylated (189) at a low steady-state level in resting cells, and their hyper-phosphorylation leads to a slight, but significant, increase in deacetylase activity, and, simultaneously, to disruption of the complex between HDAC1/2 and HDAC1/mSin3/YY1 (67). When hypo-phosphorylation occurs, the activity of HDAC1 and 2 decreases, although complex formation is increased (67, 173). HDAC3 (56) is closely related to HDAC8, with 34% identity, displaying the canonical domain of all class I HDACs. HDAC3 can co-precipitate with HDAC4, 5, and 7 through complex formation with SMRT and N-CoR (86). HDAC8 (69, 224, 225) consists largely of the catalytic domain with a nuclear localization signal. HDAC4, 5, and 7 represent a subgroup within class II HDACs (143). HDAC4 and 5 share an overall similarity of 70%. HDAC6 is evolutionarily most related to HDAC10. In general, however, the identity of HDAC6 with other human HDACs is low, with some resemblance to yeast HDA1, suggesting an early separation from other HDACs. HDAC6 (228) is a unique enzyme (23), in that it contains two catalytic domains in tandem, and an HDAC6-, USP3-, and Brap2-related zinc finger motif (HUB) domain at the C-terminus (264). This domain is a signal for ubiquitination, implying an involvement in degradation pathways. The catalytic domains of HDAC6 are most similar to those of HDAC9. HDAC6 and the enzyme functions as a tubulin deacetylase (98, 144, 257). Although it resides predominantly in the cytoplasm (having a nuclear export signal [NES]) to exert its function (227), HDAC6 can also be found in the nucleus along with HDAC11. HDAC9 (and its splice variants) belongs to class IIa and has a catalytic domain on the C-terminus (172, 263). HDAC10 is the most recently discovered class II HDACs (61). HDAC10 (112) is closely related (37% overall similarity) to HDAC6. HDAC10 has a catalytic domain on its N-terminus, as well as both an NES and a putative second catalytic domain on the C-terminus. HDAC10 might function as a recruiter rather than as a deacetylase (85), interacting with HDAC1, 2, 3, 4, 5, and 7, but not with HDAC6, with some contradictions present in literature. HDAC11, containing a catalytic domain at the N-terminus, is similar to HDAC3 and 8, suggesting that it might be more closely related to class I than to class II HDACs (71). In mammals, seven Sir2 homologues (SIRT1-7) have been identified, which possess primarily HDAC (SIRT1, 2, 3, and 5) or mono-ribosyltransferase activity (SIRT4 and 6) (64), targeting histone and various non-histone proteins in distinct subcellular localizations. SIRT1 has a wide range of substrates and cellular functions (171). In contrast to SIRT1, SIRT2 is predominantly cytoplasmic, deacetylating several substrates such as α-tubulin (20). For a detailed classification of SIRT3-7 (31).
FIG. 1.

HDAC and SIRT classification. Enzymes are divided into classes and sub-classes; subcellular localization and tissue distribution are reported for each member. HDAC, histone deacetylase; SIRT, sirtuin. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
HDACs and Sirtuins in Cancer: Quantitative and Functional Abnormalities
Cancer has traditionally been considered a disease of genetic defects such as gene mutations and deletions as well as chromosomal abnormalities, resulting in the loss of function of tumor-suppressor genes and/or gain of function or hyper-activation of oncogenes (121, 218). Nevertheless, the epigenetic regulation of genes and, therefore, the plasticity of the epigenome play a key role in cancer development and progression (93, 214). HDACs can act directly on chromatin, modifying histone acetylation and changing the characteristics of transcriptional factor recruitment on promoters. Consequently, HDAC deregulation frequently leads to aberrant expression of genes regulating cell proliferation, cell cycle, and apoptosis. Moreover, HDACs can promote the acetylation of structural proteins, chaperones, and TFs with a significant impact on physio-pathological pathways. Current knowledge of HDAC involvement in cancer is summarized next according to HDAC family groups.
Class I HDACs
All bonafide class I HDACs are deregulated in cancer (156). HDAC1 overexpression has been found in gastric (35, 155), breast (115), pancreatic (70, 262), hepatocellular (58, 244), lung (186), and prostate (200) malignancies, and overexpression levels correlate with prognosis and survival. High expression levels of HDAC1, 2, and 3 are associated with renal cancer (63) and Hodgkin's lymphoma (1). Interestingly, HDAC2 frame-shift mutation is the only genetic mutation identified in HDAC genes, leading to loss of HDAC2 protein and activity in human microsatellite instability (MSI)-high endometrial and MSI colon cancer cells. Unlike other class I members, HDAC8 expression appears to be cancer-type specific, as to date it has been found altered in childhood neuroblastoma (166, 167) and pancreatic ductal adenocarcinoma (48).
Class IIa HDACs
HDAC4 exerts an atypical role in cancer being overexpressed (e.g., in breast cancers) and/or down-regulated (e.g., in chondrosarcoma (204) and melanoma (252)) differently in different malignancies HDAC5 and 9 may be considered markers for medulloblastoma risk stratification (150). Their overexpression correlates with poor survival in affected patients. HDAC5 is also overexpressed in hepatocellular carcinoma (125), while HDAC7 overexpression is associated with pancreatic cancer and acute lymphoblastic leukemia (152, 169).
Class IIb HDACs
HDAC6 is implicated in breast cancer (184), diffuse large B-cell lymphomas, and peripheral T-cell lymphomas (142). Increased HDAC6 expression has been observed in more advanced—compared with early-stage cancers (239). HDAC10 expression is stronger in patients with chronic lymphocytic leukemia, and it has recently been linked to gastric cancers and reactive oxygen species (ROS) production in many cancers (130, 233).
Class III (sirtuins)
SIRT1, 4, and 7 are up-regulated in certain cancers (myeloid leukemia, prostate and non-melanoma skin cancer) (20), whereas SIRT2 is down-regulated in gliomas (94) and gastric carcinoma, as well as in melanomas (118). In melanomas, a mutation in the catalytic domain of SIRT2 (P199L) eliminates its enzymatic activity (131). Evidence suggests that SIRT2 acts as tumor suppressor and that its loss compromises the mitotic checkpoint, contributing to genomic instability and tumorigenesis. SIRT3 is, instead, up-regulated or down-regulated in different types of breast cancer (254). The quantity of the enzyme may be relevant in tumorigenesis for both loss and gain. Some studies also suggest a role for SIRT7 in breast cancer maintenance (8).
Class IV HDACs
Overexpression of HDAC11 has been found in myeloproliferative disorders as well as in Hodgkin's lymphoma (29, 198).
To date, important studies (179) have focused on aberrant HDAC recruitment to promoters through their physical association with oncogenic DNA-binding fusion proteins resulting from chromosomal translocations, or overexpression of repressive TFs that physically interact with HDACs. PML-RARα and AML1-ETO fusion proteins induce acute promyelocytic leukemia and acute myeloid leukemia (AML) by recruiting HDAC-containing repressor complexes to stably repress the expression of target genes (HDAC1 and 2) (73). HDAC6 plays a key role, by physically interacting with non-histone proteins such as HSP90, β-catenin, and epidermal growth factor (in Wnt signaling pathway) (134, 248). Altered levels of acetylation of these targets can disrupt the balance of c-Myc activation, enabling cells to escape from apoptosis. The same is true for p53 and its level of acetylation, which can induce its activity as TF. Taken together, these findings support the development of HDACi to restore the physiological pattern of gene expression and function in tumors and, possibly, other diseases.
HDAC and SIRT Modulators
HDACi represent a class of cytostatic agents that interfere with the function of HDACs and are able to modulate gene expression through indirect induction of histone acetylation (14). In particular, these compounds inhibit the proliferation of tumor cells in vivo and in vitro by inducing cell-cycle arrest, differentiation, and/or apoptosis with different kinetics and activities depending on chemical structures. Surprisingly, normal cells are often less sensitive to HDACi than are tumor cells (87). HDACi derive from natural or synthetic sources and can be classified into five main groups (16):
(i) Hydroxamates, including trichostatin A (TSA), suberoylanilide hydroxamic acid (also called Vorinostat), LAQ824, LBH589 (Panabinostat), or PXD101 (Belinostat), M344, CR2408, abexinostat hydrochloride (PCI-24781)
(ii) aliphatic acids, including sodium butyrate (NaB), valproic acid (VPA), and phenylbutyric acid
(iii) benzamides, including MS-275 (Entinostat)
(iv) tetrapeptides/depsipeptides, including Apicidin, Romidepsin, and Trapoxin B
(v) sirtuin inhibitors (SIRTi), including the pan-inhibitor nicotinamide and the specific SIRT1 and 2 inhibitors sirtinol, cambinol, and EX-527.
TSA inhibits HDAC1, 4, and 6 with IC50=6, 38, and 8.6 nM, respectively [for further information, the reader is referred to ref. (39)]. The pan-inhibitor Vorinostat inhibits (177) HDAC1 and 3 with IC50=10 and 20 nM, respectively. Vorinostat also induces marked hyperacetylation of histone H4 and inhibits growth of three prostate cancer cell lines, LNCaP, PC-3, and TSU-Pr1, at 2.5–7.0 μM concentrations. Vorinostat treatment in MCF-7 breast cancer cells inhibits cell proliferation at IC50=0.75 μM, resulting in the accumulation of cells in G1 and G2-M phase. Vorinostat treatment at 1 μM for 8 h or more is sufficient to irreversibly induce the apoptosis of human multiple myeloma (MM) cells. LAQ824 (6, 80) activates the expression of p21 cell-cycle inhibitor by activating the p21 promoter with AC50=0.3 μM. LAQ824 inhibits cell growth of H1299 and HCT116 with IC50=0.15 and 0.01 μM, respectively. The antiproliferative effect of LAQ824 is selective toward tumor cell lines while inducing only growth arrest in normal fibroblasts. Furthermore, LAQ824 induces a dose-dependent increase of p21 in A549 cells and an increase in the hypo-phosphorylated state of Rb tumor suppressor. Panobinostat (5, 174) is a broad-spectrum HDACi with IC50=5 and 20 nM in MOLT-4 and Reh cells, respectively. Panobinostat induces acetylation of histones H3K9 and H4K8 as well as p21 expression while decreasing levels of c-Myc in a dose-dependent manner. Belinostat displays (77) IC50=27 nM in HeLa extracts. In vitro Belinostat inhibits the growth of tumor cells such as A2780; HCT116 induces apoptosis through PARP cleavage and acetylation of histones H3/H4, and shows enhanced tubulin acetylation in ovarian cancer cell lines. M344 (110, 178, 235) is toxic at concentrations above 10 μM, while a maximum of only 20% of the surviving cell population are induced to differentiate. In vitro, M344 shows significant antiproliferative activities against Ishikawa and SK-OV-3 cancer cell lines with EC50=2.3 and 5.1 μM, respectively, while it displays little effect on normal human endometrial epithelial cells. In addition, M344 leads to a decreased proportion of cells in the S phase and an increased proportion in G0/G1 phases of the cell cycle, induces apoptosis, and decreases the trans-membrane potential of mitochondria. PCI-24781 (28) exhibits potent antitumor activity against a variety of cell lines with 0.15 μM<IC50>3.09 μM. PCI-24781 treatment causes dose-dependent accumulation of both acetylated histones and acetylated tubulin in HCT116 or DLD-1 cells, induces p21 expression, and leads to PARP cleavage and accumulation of γH2AX. Sodium butyrate (44) induces differentiation and prevents cell proliferation. Mechanistic studies suggest that its action is often mediated through Sp1/Sp3-associated HDAC activity, leading to transcriptional activation of the p21 gene. In addition, this inhibitor down-regulates numerous genes that are associated with cytokine signaling, specifically the interferon-γ (IFN-γ) pathway. This compound has HDAC1, 7, and 8 with IC50=140 μM. VPA has IC50≈0.4 mM and exhibits anticancer, anti-inflammatory, and neuro-protective effects. MS-275 (181, 183), class I HDACi, induces the accumulation of p21 and gelsolin in K562 cells and decreases the expression of cyclin D1 and the anti-apoptotic proteins Mcl-1 and XIAP. MS-275 inhibits the proliferation of human tumor cell lines, including A2780, Calu-3, HL-60, K562, St-4, HT-29, KB-3-1, Capan-1, 4-1St, and HCT-15 with 41.5 nM<IC50>4.71 μM. MS-275 strongly inhibits human leukemia and lymphoma cells, including U937, HL-60, K562, and Jurkat. MGCD0103 (22) inhibits at nM or low μM concentrations only a subset of the nine HDACs. MGCD0103 is active against HDAC1 and 2 in vitro and in whole cells, but it does not inhibit class II HDACs. The exocyclic amino group in MGCD0103 is necessary for inhibitory activity, as HDAC-inhibitory activity against HDAC1 and 2 is completely abolished with the desamino analogue. Apicidin (88, 219), which contains an electrophilic ketone, is a potent HDACi with IC50=0.7 nM. An in vitro activity assay demonstrates Apicidin-mediated inhibition of HDAC3/NcoRat at a much higher potency than for HDAC6 (IC50=15.8 and 665.1 nM, respectively). Romidepsin (157, 161) is an HDAC1 and 2 inhibitor with IC50=36 and 47 nM, respectively. Romidepsin treatment in HeLa cells induces histone acetylation and p21 expression with EC50=3.0 nM, G2/M arrest, cyclin D1 down-regulation, and p53-independent p21 induction, leading to inhibition of CDK and dephosphorylation of Rb (resulting in early G1 phase arrest). Although SIRTi are classified along with HDACi, they act via a nicotinamide-dependent mechanism, suggesting that they should have their own class based on their chemical functionalities. To date, a number of specific SIRT inhibitors (mainly SIRT1 and 2) have been proposed for cancer therapy. Moreover, both activators and inhibitors of sirtuins might act beneficially against different types of neurodegenerations and cancers (127). In addition to nicotinamide, some other specific inhibitors have been characterized, including splitomicin and its analogues, tenovins, AGK2, sirtinol, suramin, the indole derivative EX-257, salermide, and UVI5008. Phenol derivatives, including quercetin, piceatannol, and resveratrol, possess SIRT1-activating properties. Many other compounds have subsequently been developed such as SRT1720, SRT2183, and SRT1460. For further information on SIRTi (42, 205).
Figure 2 presents a list of well-known HDACi. Many of these compounds are in clinical trials alone or in combination with other agents for the treatment of leukemias or solid tumor malignancies. Further information with regard to compounds that are currently being used in clinical evaluation is provided in the section “HDACi and SIRTi in clinical trials against cancer.” Our understanding of the mechanistic effects of HDACi will probably change significantly, establishing a new paradigm in the field of so-called “smart drugs.” New insights will have profound implications for the design of targeted therapies in cancer and other diseases.
FIG. 2.

Structures of major HDACi belonging to classes of hydroxamic acids, benzamides, aliphatic acids, and cyclic tetrapeptides/depsipeptides. HDACi, histone deacetylase inhibitors.
Biology of HDACs in Cancer and Effects of HDACi
Many HDACs exist as components of multi-protein complexes, such as the transcriptional co-repressors mSin3, N-CoR, and SMRT (97, 215, 217, 245, 256, 259). These are then targeted to specific genomic regions by interactions with DNA-binding factors, including TFs, nuclear receptors, and other epigenetic modifiers, such as methyl-binding proteins, DNA methyltransferases, and histone methyltransferases. Along with their interactors, HDACs are able to modulate the expression of genes coding for signaling proteins and thus the function of key pathways, including apoptosis, Wnt pathway, cell-cycle progression and differentiation, regulation of the proteasome system, activation of kinases, and the unbalanced reaction between DNA damage repair and surveillance. At the chromatin level, therefore, HDACs work as transcriptional repressors in most, though not all, cases. Indeed, protein acetylation can have many different effects, as listed next (graphically represented in Fig. 3), underscoring how a defect in HDAC expression or function might affect biological processes. The readout of treatment with HDACi is a change in the expression level of about 10% of cellular proteins, leading to a global effect.
FIG. 3.

HDACs control many aspects of cancer biology. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Protein degradation
Protein stability depends on the balance between synthesis and degradation (128). Protein degradation is mediated by ubiquitination. Both acetylation and ubiquitination occur on the same lysine residues, and a direct cross-talk exists between the two (82). Thus, HDACs can decrease the half life of several substrates by exposing the lysine residue for ubiquitination (76, 82, 104, 151). Modified lysine residues mediate many interactions, and an aberrant HDAC function could, therefore, alter the assembly of multi-complexes. HDAC6 is known to interact with Hsp90, and HDACi-mediated hyperacetylation of Hsp90 induces the activation of proteasome degradation of oncogenic client proteins such as Akt, Bcr-Abl, and c-Raf (24). One of the client proteins of HSP90 is androgen receptor, which is degraded after HDACi treatment, suggesting the use of HDACi in hormone-dependent or refractory cancers. All these findings reveal that the inhibition of HDAC6 makes cells more sensitive to induced stresses, which are useful in therapeutic strategies.
Protein–protein interactions
Acetylation/deacetylation balance regulates a growing number of non-histone proteins (as p53, nuclear factor-κB [NF-κB], p65, CBP, p300, STAT3, tubulin, PC4, GATA factors, nuclear receptors, c-Myc, hypoxia-inducible factor [HIF]-1α, FOXO1, HSP-90, HMG, E2F, MyoD, Bcr–Abl, the FLT3 kinase, c-Raf kinase, and others), affecting their interactions. HDACi modulate gene regulatory activities of TFs such as E2F1, p53, STAT1, STAT2, and NF-κB via a hyperacetylated status (75). HDACi can also regulate the activity of p53 and correlated genes to induce a p53-mediated pro-apoptotic signal in cancer treatment. Thus, many genes regulated by HDACi are likewise a target of p53, suggesting the use of HDACi in tumors due to p53 (activity or expression) loss (196, 207).
Cell-cycle progression (proliferation)
Crucial stages of cell cycle are generally controlled through the transcriptional regulation of a subset of genes, which are, in turn, regulated by acetylation/deacetylation of histone and non-histone proteins (231). HDAC and sirtuin overexpression has been observed in multiple cancers (see section “HDACs and sirtuins in cancer: quantitative and functional abnormalities”). HDACi cause cell growth arrest in the G1 phase in normal and transformed cells through the activation of and promiscuous interference with many processes, and they also induce p21, which binds to and inhibits the activity of cyclin-CDK2 or -CDK1 complexes. The expression of this gene is tightly controlled by p53, which competes with HDAC1 for a binding site (SP1) on p21 promoter region (260).
Apoptosis induction
HDACs act as regulators in intrinsic and extrinsic apoptosis processes (180). The evasion of this pathway is the principal hallmark of tumor growth and progression, as confirmed by the fact that resistance to apoptosis induction has been recognized in many cancers. In the extrinsic pathway, HDACs negatively affect the transcriptional regulation of death receptors such as tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and TNF receptor superfamily member (Fas) (38, 139, 159). In the intrinsic pathway, the overexpression or aberrant function of HDACs interferes with the expression of pro-apoptotic proteins such as Bcl-family members, with the concomitant up-regulation of anti-apoptotic proteins. HDACi are capable of inducing apoptosis in tumor cells by regulating the expression of pro- and anti-apoptotic genes through the intrinsic or extrinsic pathway. In the extrinsic mechanism, HDACi enhance the sensitivity of cancer cells to TRAIL by modulating expression of the protein and its receptors (65). Relaxed chromatin, promoted by treatment with HDACi, increases the accessibility of transcriptional machinery to the TRAIL promoter region, as well as the acetylated forms of many TFs (e.g., p53), which are strongly implicated in the up-regulation of TRAIL. In the intrinsic (mitochondrial) pathway, HDACi cause the down-regulation of anti-apoptotic proteins (Bcl-2, Bcl-xl, and Mcl-1) and a change in the expression of pro-apoptotic proteins (Bax, Bak, and Bid). Oligomerization of Bax/Bak leads to mitochondrial membrane permeabilization and, consequently, the release of cytochrome c by the activation of caspases (in particular, caspase 9) and apoptosome formation (19, 191). In the intrinsic cell pathway that is induced by HDACi, an important effect is mediated by ROS production, which is increased and regulated by Bax, Hsp90, TBP-2, and Trx (220). HDACi can also induce the cell death of transformed cells by causing mitotic defects. Extremely high histone acetylation levels can disrupt centromere structure and function with the concomitant loss of heterochromatin-binding proteins. Moreover, the level of acetylation interferes with histone phosphorylation, and, consequently, the action of proteins (many of which are spindle checkpoint proteins such as BubR1, CENP-F) recognizing phospho groups is itself inhibited. Levels of Aurora A and B are also decreased after HDACi treatment, contributing to transient arrest in promethaphase after aberrant mitosis and so, the activation of apoptosis (32).
Differentiation
The deacetylated state of histones is required for the maintenance of the undifferentiated cellular state, as genes involved in lineage progression are unexpressed. A shift to an acetylated state interferes with the proper execution of the differentiating process and contributes to cancer development. Particular attention has been focused on the role of HDACs in AML. In these systems, the presence of oncofusion proteins affects the release of HDACs from promoters (e.g., thus containing RAREs), maintaining cells in an undifferentiated state (by silencing genes required for hematopoietic differentiation) (221). HDACi can induce the differentiation in haematological malignancies by interfering not only with the HDAC(s)-oncofusion proteins complex, but also in breast cancer (154), hepatoma cells (246), endometrial stromal sarcoma (96), glioblastoma (206), and thyroid carcinoma cells (253).
Metastasis and migration
HDACs (in particular, class I HDACs) play an important role in metastasis and migration pathways, as they repress the expression level of RECK and Rho-β, and decrease the level of Kangai-1 (a metastasis suppressor factor) through β-catenin pathway (55). HDACs work in multi-complexes with metastasis-associated protein 1 and 2 (247). HDACs have also been linked to extracellular matrix formation and its remodeling, thus increasing cancer cell invasion and migration (237). In human cancer tissues, reduced histone acetylation is significantly correlated with advanced tumor stage and the depth of tumor invasion (199, 216). Treatment with HDACi up-regulates a set of metastasis-suppressor genes and down-regulates a set of metastasis-promoting genes (107), as well as represses cancer cell invasion and metastasis both in vitro (120) and in vivo (148). Many cases are reported in literature (236), though the role played by HDACi remains controversial (136).
Angiogenesis
Solid tumor metastasis and progression require the formation of new blood vessels. One of the key initiators of angiogenesis is the protein HIF-1α, which is degraded once acetylated. Deacetylation of HIF-1α is mediated by HDACs (classes I, II, III) and induces activation of the angiogenic process (by the expression of vascular endothelial growth factor [VEGF]) (74). Anti-angiogenic effects seem to be related to the down-regulation of pro-angiogenic genes, such as VEGF and endothelial nitric oxide synthase (eNOS). The mechanism by which HDACi act is still unclear, but the destabilization of mRNA coding for these genes such as protein degradation may be involved. The degradation of VEGF and eNOS seems to increase after treatment with HDACi (182) as a consequence of HSP90 hyperacetylation (25, 170). The same effect is observable on the expression level and stability of the protein HIF-1α (255).
DNA damage and oxidative stress
Hyperacetylation of histones causes chromatin relaxation and, thus, increases TFs binding to DNA. HDACi have a negative impact on biological mechanisms, ensuring genome integrity and stability, such as DNA recognition and repair pathway and regulation of ROS concentration. In open chromatin, sites usually packaged and unexposed are not protected from damage (induced by radiation, drugs, and unbalanced ROS scavengers) and, therefore, act as signals for genome integrity failure, culminating in cell death pathway activation. Thus, targeting genome integrity in rapidly cycling cells (such as tumor cells), combining HDACi and radiotherapy, could be considered a preferred strategy in cancer (discussed in section “Examples of HDACi in Combination Treatments”). ROS, such as superoxide anion, hydrogen peroxide, and hydroxyl radicals, are a major source of endogenous DNA damage. Normal cells control ROS concentration through the action of enzymatic (thioredoxin 1/thioredoxin-binding receptor-2 cross-talk) and non-enzymatic ROS scavengers (220). The activity of thioredoxin-1 is modulated by its receptor, which binds the enzyme interfering with its activity. In response to HDACi, levels of thioredoxin-binding receptor-2 increase; while thioredoxin-1 is reduced (30). In particular, the inhibition of HDAC10 causes an accumulation of ROS, increased release of cytochrome C, and activation of proapoptotic molecules such as caspase 3, caspase 9, and Bid in SNU-620 gastric cancer cells (130).
HDACs and Sirtuins in Non-Cancer Human Diseases
The therapeutic potential of HDACi has also been investigated for a wide range of other diseases. Here, we focus on cardiac, cognitive, immunological, and metabolic disorders.
Cardiac disorders
Recent evidence has suggested that different HDACs are implicated in heart diseases such as arrhythmia, heart failure, acute coronary syndromes, and hypertrophy (117). Cardiac hypertrophy is characterized by increased cell size, enhanced protein synthesis, and heightened organization of the sarcomere. In this state, fetal genes such as natriuretic peptide precursor type A (Nppa), myosin heavy polypeptide 7 (Myh7), and skeletal α-actin are reactivated; whereas cardiac contractile proteins such as myosin heavy polypeptide 6 (Myh6) and calcium-handling proteins are repressed. In addition, immediate-early genes encoding c-fos, c-jun, and heat shock proteins are up-regulated. In many of these processes, HDACs play a critical role in changing the accessibility of chromatin to transcriptional machinery and deregulating levels of acetylation in non-histone proteins (thus interfering with intracellular cross-talk). One line of evidence (103) suggests that class IIa HDACs interact with MEF2, one of the early activators of hypertrophy. HDACi-based therapy could, thus, help prevent heart failure. The mechanism by which HDACi suppress cardiac hypertrophy is still being investigated, but overexpression of Krüppel-like factor 4 (KLF4) seems to be involved (116). Conversely, much evidence suggests that treatment with HDACi causes QT prolongation and heart diseases (see section “Side effects of HDACi”) (203).
Neurodegenerative disorders
Neurodegeneration is an umbrella term for progressive loss of structure and/or function of neurons. For an in-depth description of this topic, we refer the reader to Fischer et al. (60) and the references therein. Despite the relatively recent discovery of sirtuins, the role of SIRT1 (and marginally SIRT2) in neurodegenerations seems to be clearer than that played by other HDACs (52). In Alzheimer's disease (AD) models, SIRT1 performs a protective function, as demonstrated by the fact that its overexpression or activation by small molecules (SIRT activators) reduces β-amyloid peptide content, a hallmark of AD, and ameliorates cognitive capabilities and memory. In Huntington's disease, SIRT1, a mediator of the beneficial metabolic effects of calorie restriction, protects neurons against mutant htt toxicity. The mechanisms subtending these effects are still not fully understood. A possible underlying defect in transcriptional regulation has been described for the diseases cited earlier, strongly suggesting the use of HDACi. By way of an example, VPA is able to decrease A-β production plaques in AD transgenic mice by inhibiting GSK-3β-mediated γ-secretase cleavage of APP protein. At the cellular level, transcriptional regulatory proteins such as CBP, NF-κB, p53, brain-derived neurotrophic factor, and PGC1a, a key regulator of mitochondrial gene expression, are each implicated in these diseases. These proteins are potentially amenable to modulation by HDACi and conceivably with SIRT1 activators or inhibitors (222).
Inflammation and immune disorders
Recently, several HDACi have also been recognized as promising anti-inflammatory agents (135). The most exciting field of application is the treatment of rheumatoid arthritis (26, 185, 230). The anti-inflammatory capability of HDACi seems to be due to the inhibition of transcriptional functions of important proteins, such as NF-κB, leading to a reduction in the expression of pro-inflammatory cytokines (TNF-α, interleukin-1β, and IFN-γ). HDACi treatments are currently under investigation for other immunological disorders, including inflammatory bowel diseases, systemic lupus erythematosus, and psoriasis. Growing evidence shows that HDACi exert immune-modulatory effects, causing an up-regulation of surface antigens on tumoral cells (193), leading to their enhanced recognition. HDACi up-regulate the expression of major histocompatibility complex class I and II proteins (4, 197).
Metabolic syndromes
HDACs may be considered a promising target for insulin sensitization, as demonstrated by several studies (37) in which PPAR-γ ligands are compared with canonical HDACi. HDAC3, 4, 5, and 7 work together to enhance glucose production in hepatocytes by the activation of FOXO1. This pathway leads to an increase in hepatic gluconeogenesis by the expression of G6Pase. Thus, interfering with HDACi could be beneficial for controlling obesity and diabetes (162, 238). In the context of metabolic and energetic status, the involvement of SIRT1 is more clear, because many of its deacetylation targets are key metabolic regulators. By shuttling between the nucleus and the cytosol, SIRT1 can interact with eNOS (145) and Atgs (critical components of autophagy machinery) (129), as well as with the proteins involved in mitochondrial respiration (p53 and PGC-1α) (101), gluoconeogenesis and oxidative stress (FOXOs and CRTC2), and lipid anabolism (LXRs) (133) in the nucleus.
HDACi and SIRTi in Clinical Trials Against Cancer
Based on published reports, at least 20 structurally different HDACi are currently being used in clinical trials as mono- and combination therapy for hematological and solid cancers. In this review, the focus has been limited to FDA-approved HDACi or those at an advanced stage of experimentation. All information regarding clinical trials comes from clinicaltrials.gov.
Vorinostat
Vorinostat is at the most advanced stage of clinical development and is studied worldwide, as shown in Figure 4. A second-generation polar compound, Vorinostat binds to the catalytic domain of HDACs. This enables the hydroxamic moiety to chelate zinc ion located in the catalytic pockets of HDACs, thereby inhibiting deacetylation and leading to an accumulation of both hyperacetylated histones and TFs. Vorinostat was the first HDACi approved by FDA (2006) for clinical use in treating patients with advanced cutaneous T-cell lymphoma. Table 1 reports a list of trials (completed with and without results) having Vorinostat as a single drug. Although Vorinostat has shown strong antitumor activity against hematological malignancies, it is also currently being investigated for solid tumors. As an example, Vorinostat is most efficacious against glioblastoma, demonstrating the validity of HDACi-based therapies for brain tumor (66). Interestingly, four different trials employing Vorinostat against glioblastoma are currently being recruited (NCT00555399, NCT01378481, NCT01266031, and NCT01738646). Similar to many HDACi, Vorinostat has been used in a wide range of trials in combination with other drugs (demethylating agents and/or Bortezomib) and/or radiation (Table 2).
FIG. 4.

World map showing distribution of Vorinostat currently in clinical trials. Colors indicate the number of studies with locations in that region. Some trials are carried out simultaneously in different countries. Downloaded and modified from clinicaltrials.gov. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Table 1.
Clinical Trials with Vorinostat As Single Drug
| NCT number | Study results | Conditions | Phases | Funded by | Start data |
|---|---|---|---|---|---|
| NCT00305773 | Has results | AML (different Fab classifications) | 2 | NIH | January 2006 |
| NCT00128102 | No results available | Mesothelioma|Lung cancer | 3 | Industry | June 2005 |
| NCT00278330 | No results available | BLCML|Recurrent ALL|Recurrent ALL|RAEB|Relapsing CML|ALL|AML | 1 | NIH | January 2006 |
| NCT00132067 | No results available | Primary peritoneal cavity cancer|Recurrent ovarian epithelial cancer | 2 | NIH | October 2005 |
| NCT00330161 | No results available | Recurrent prostate cancer|Stage IV prostate cancer | 2 | NIH | March 2006 |
| NCT00363883 | No results available | Localized, metastatic, recurrent, regional TCC of the renal pelvis and ureter|TCC of the bladder | 2 | NIH | June 2006 |
| NCT00641706 | No results available | Adult giant cell glioblastoma|Adult glioblastoma|Adult gliosarcoma|Brain tumor | 2 | NIH | July 2008 |
| NCT00238303 | No results available | Adult giant cell glioblastoma|Adult glioblastoma|Adult gliosarcoma|Brain tumor | 2 | NIH | September 2005 |
| NCT00132002 | No results available | Male breast cancer|Recurrent breast cancer|Stage IV breast cancer | 2 | NIH | June 2005 |
| NCT00776503 | No results available | Myelodysplastic syndromes | 1/2 | Other|Industry | May 2008 |
| NCT00134043 | No results available | Thyroid cancer | 2 | NIH | December 2005 |
| NCT00479232 | Has results | Leukemia, myelocytic, acute MDS|MDS | 1 | Industry | June 2007 |
| NCT00632931 | Has results | Advanced cancer relapsed|Advanced cancer refractory | 1 | Industry | June 2007 |
| NCT00907738 | Has results | Advanced cancer | 2 | Industry | August 2005 |
| NCT00132028 | Has results | Lymphoma | 2 | NIH | September 2005 |
| NCT00771472 | Has results | Lymphoma | 1 | Industry | August 2008 |
| NCT00336141 | No results available | Colorectal cancer | 1 | Other|Industry | June 2006 |
| NCT00373490 | Has results | Tumors | 1 | Industry | July 2006 |
| NCT00127127 | No results available | Tumors | 1 | Industry | June 2005 |
| NCT00138203 | No results available | Lung cancer | 2 | Other|NIH | January 2006 |
| NCT00045006 | No results available | Cancer | 1 | Other|NIH | July 2001 |
| NCT00455351 | No results available | Pelvic cancer | 1 | Other | February 2007 |
| NCT00005634 | No results available | Leukemia|Lymphoma|MM and PCN|Prostate cancer|Small intestine cancer | 1 | Other|NIH | January 2000 |
| NCT00127140 | No results available | Lymphoma | 1 | Industry | June 2005 |
| NCT00097929 | No results available | B-cell lymphoma | 2 | Industry | May 2005 |
| NCT00091559 | No results available | Cutaneous T-cell lymphoma|Sezary syndrome|Mycosis fungoides | 2 | Industry | February 2005 |
| NCT00750178 | No results available | Cancer, Advanced | 1 | Industry | November 2004 |
| NCT00858234 | No results available | Multiple myeloma | 1 | Industry | March 2009 |
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; BLCML, blastic phase of chronic myeloid leukemia; CML, chronic myeloid leukemia; MDS, myelodysplastic syndrome; MM, multiple myeloma; PCN, plasma cell neoplasm; RAEB, refractory anemia with excess blasts; TCC, transitional cell carcinoma.
Table 2.
Clinical Trials with Vorinostat in Combination with Other Drugs
| NCT number | Recruitment | Study results | Conditions | Interventions | Phases | Funded by | Start date |
|---|---|---|---|---|---|---|---|
| NCT00875745 | Active, not recruiting | No results available | AML|APL|MDS | Sorafenib-Vorinostat | 1 | Other|Industry | April 2009 |
| NCT00838929 | Active, not recruiting | No results available | Brain metastases | Vorinostat|Radiation: radiation therapy | 1 | Other|Industry | March 2009 |
| NCT01023737 | Active, not recruiting | No results available | Malignant solid tumor | Hydroxychloroquine (HCQ)|Vorinostat | 1 | Other|Industry | November 2009 |
| NCT01110876 | Active, not recruiting | No results available | Brain cancer|Glioblastoma multiforme | Vorinostat|Erlotinib|Temozolomide | 1/2 | Other|Industry | June 2011 |
| NCT00821951 | Completed | Has results | NSCLC | Vorinostat|Radiotherapy | 1 | Other|Industry | May 2009 |
| NCT00948688 | Active, not recruiting | No results available | Pancreatic cancer|Adenocarcinoma of the pancreas | Radiation therapy|5-FU|Vorinostat | 1/2 | Other|Industry | August-2009 |
| NCT00937495 | Active, not recruiting | No results available | Tissue sarcoma | Vorinostat|Bortezomib | 2 | NIH | June 2009 |
| NCT00839956 | Active, not recruiting | No results available | Multiple myeloma | Bortezomib|Vorinostat | 2 | Other|NIH | February 2009 |
| NCT01038388 | Active, not recruiting | No results available | Multiple myeloma | Vorinostat, Lenalidomide, Bortezomib, Dexamethasone | 1 | Other|Industry | January 2010 |
| NCT00939991 | Active, not recruiting | No results available | Brain tumor|Glioblastoma | Vorinostat/Bevacizumab/Temozolomide | 1/2 | Other|Industry | October 2009 |
| NCT01132911 | Completed | No results available | Lymphoma|Sarcoma|Wilms Tumor|Neuroblastoma | Vorinostat Velcade (PS-341, Bortezomib) | 1 | NIH | May 2010 |
| NCT00858234 | Completed | No results available | Multiple myeloma | Vorinostat|Bortezomib | 1 | Industry | March 2009 |
Table reports a list of trials started in 2009 and now closed (2013).
APL, acute promyelocytic leukemia; NSCLC, non-small cell lung cancer.
Panobinostat
Panobinostat (LBH-589), developed by Novartis, is a non-selective HDACi that is currently being used in Phase I and II clinical trials as mono- and combination therapy for hematological tumors such as non-Hodgkin's lymphoma, acute leukemia myeloblasts, AML, MM, advanced solid tumors, and breast and lung cancer (46, 54, 208) (Table 3). Table 4 presents trials in which Panobinostat is used in combination with other drugs. Panobinostat is coupled with chemicals that are able to interfere with the methylated status of DNA (NCT00946647) and with tyrosine kinase inhibitor Lapatinib (NCT00632489) (124). No results are available for any of the trials listed in Table 4, many of which are recruiting or still active, highlighting scientific interest in the development of combinatorial therapies that improve the efficacy of HDACi. In many of these interventional studies, Panobinostat is being evaluated to determine the maximum tolerated dose in different pathologies and administration regimes (NCT00686218 and NCT01496118, not present in Table 4). Panobinostat is currently used in a Phase I/II clinical trial (NCT01680094), assessing its use in HIV-affected patients on highly active antiretroviral therapy (175).
Table 3.
Clinical Trials with Panobinostat (LBH-589) As Single Drug
| NCT number | Recruitment | Study results | Conditions | Phases | Funded by | Primary completion date |
|---|---|---|---|---|---|---|
| NCT01802879 | Not yet recruiting | No results available | Hematologic neoplasms | 2 | Industry | July 2018 |
| NCT01451268 | Recruiting | No results available | MDS|AML | 1/2 | Other | January 2016 |
| NCT00663832 | Completed | No results available | Prostate cancer | 1 | Industry | December 2011 |
| NCT01242774 | Recruiting | No results available | AML | 1 | Industry | November 2013 |
| NCT01245179 | Recruiting | No results available | Sickle cell disease | 1 | Other|Industry | December 2013 |
| NCT01034163 | Completed | No results available | Hodgkin's lymphoma | 3 | Industry | May 2012 |
| NCT01065467 | Active, not recruiting | No results available | Melanoma|Malignant melanoma | 1 | Other|Industry | December 2012 |
| NCT00840346 | Recruiting | No results available | AML | 1/2 | Other | September 2011 |
| NCT00878436 | Recruiting | No results available | Prostate cancer|Prostatic neoplasms | 1/2 | Other|Industry | December 2013 |
| NCT00985946 | Completed | No results available | Neuroendocrine tumors | 2 | Other|Industry | October 2012 |
| NCT01613976 | Recruiting | No results available | MDS|CMML|AML | 1 | Industry | September 2013 |
| NCT01136499 | Completed | No results available | Soft tissue sarcoma | 2 | Other | January 2012 |
| NCT00742027 | Active, not recruiting | No results available | Classical Hodgkin's lymphoma | 2 | Industry | July 2013 |
| NCT01680094 | Active, not recruiting | No results available | HIV infection | 1/2 | Other|Industry | March 2013 |
| NCT01007968 | Completed | No results available | Advanced solid tumors | 1 | Industry | November 2012 |
| NCT00670553 | Completed | No results available | Prostate cancer|Head and neck cancer|Esophageal cancer | 1 | Industry | January 2010 |
| NCT00532389 | Active, not recruiting | No results available | Multiple myeloma | 1 | Industry | December 2012 |
| NCT00739414 | Completed | No results available | Cancer|Advanced solid tumor | 1 | Industry | November 2009 |
| NCT00570284 | Completed | No results available | Cancer | 1 | Industry | February 2011 |
| NCT00550277 | Completed | Has results | Renal cell carcinoma | 2 | Other|Industry | March 2010 |
| NCT01055483 | Completed | No results available | Acute myeloid leukemia | 1 | Industry | February 2012 |
| NCT00412997 | Completed | No results available | Tumors|Cutaneous T-cell lymphoma | 1 | Industry | May 2008 |
| NCT00936611 | Active, not recruiting | No results available | Waldenstrom's macroglobulinemia | 2 | Other|Industry | December 2012 |
| NCT00667862 | Completed | No results available | Prostate cancer | 2 | Industry | November 2010 |
| NCT01032148 | Recruiting | No results available | Hodgkin's lymphoma|Non-Hodgkin's lymphoma | 1 | Other|Industry | July 2013 |
| NCT00490776 | Completed | No results available | Cutaneous T-cell lymphoma | 2/3 | Industry | September 2009 |
| NCT00594230 | Completed | Has results | MDS | 2 | Other|Industry | March 2011 |
| NCT00880269 | Completed | No results available | Refractory leukemia|AML | 2 | Industry | February 2012 |
| NCT01523834 | Recruiting | No results available | Diffuse large B-cell lymphoma | 2 | Other | January 2014 |
| NCT00535951 | Completed | No results available | Carcinoma, non-small-cell lung|Mesothelioma | 1 | Industry | January 2009 |
| NCT00690677 | Completed | No results available | Colorectal cancer | 2 | Other|Industry | December 2011 |
| NCT00449761 | Completed | No results available | Leukemia, myeloid, chronic | 2/3 | Industry | August 2008 |
| NCT00532675 | Active, not recruiting | No results available | Multiple myeloma | 1 | Industry | October 2013 |
| NCT00621244 | Completed | No results available | Lymphoma|Leukemia|Multiple myeloma | 1/2 | Industry | August 2009 |
| NCT01222936 | Completed | No results available | Small cell lung carcinoma | 2 | Other|Industry | June 2009 |
| NCT00451035 | Completed | No results available | Chronic myeloid leukemia in chronic phase | 2/3 | Industry | September 2008 |
| NCT00777049 | Active, not recruiting | No results available | Breast cancer | 2 | Other | November 2012 |
| NCT00939159 | Active, not recruiting | No results available | Myelodysplastic syndrome | 2 | Other|Industry | December 2014 |
| NCT00425555 | Active, not recruiting | No results available | Cutaneous T-cell lymphoma | 2/3 | Industry | June 2013 |
CMML, chronic myelomonocytic leukemia.
Table 4.
Clinical Trials with Panobinostat in Combination with Other Anticancer Drugs
| NCT number | Recruitment | Conditions | Interventions | Phases | Funded by | Start data |
|---|---|---|---|---|---|---|
| NCT01321346 | Recruiting | AML|ALL|Hodgkin's disease|Non-Hodgkin's lymphoma | Panobinostat|Cytarabine|Panobinostat | 1 | Other|Industry | March 2011 |
| NCT01169636 | Recruiting | Hodgkin's lymphoma | Panobinostat|Ifosfamide|Mesna|Carboplatin|Etoposide|Pegfilgrastim | 1/2 | Other|Industry | January 2011 |
| NCT01238692 | Recruiting | DLBCL | LBH589|Rituximab | 2 | Other|Industry | November 2010 |
| NCT01245179 | Recruiting | Sickle cell disease | Panobinostat | 1 | Other|Industry | November 2010 |
| NCT01651039 | Recruiting | MM | Panobinostat, lenalidomide and dexamethasone | 2 | Other|Industry | July 2012 |
| NCT01460940 | Recruiting | Hodgkin lymphoma | Panobinostat|Lenalidomide | 2 | Other|Industry | October 2011 |
| NCT00967044 | Active, not recruiting | Lymphoma | Panobinostat|Everolimus | 1/2 | Other|Industry | November 2009 |
| NCT00925132 | Recruiting | Metastatic melanoma | Temozolomide, decitabine, panobinostat | 1/2 | Other|Industry | January 2010 |
| NCT01549431 | Recruiting | MM | Panobinostat|Carfilzomib|Panobinostat|Carfilzomib|Dexamethasone | 1 | Other|Industry | January 2012 |
| NCT01336842 | Recruiting | Solid tumors|NSCLC | Panobinostat, cisplatin, pemetrexed | 1 | Other|Industry | April 2011 |
| NCT01463046 | Recruiting | AML|Advanced MDS | Panobinostat|Cytarabine|Daunorubicin | 1 | Other|Industry | January 2012 |
| NCT00788931 | Completed | HER-2 positive breast cancer|Metastatic breast cancer | LBH589|trastuzumab|paclitaxel | 1 | Industry | December 2008 |
| NCT01023308 | Active, not recruiting | MM | Panobinostat|Bortezomib | 3 | Industry | December 2009 |
| NCT00632489 | Active, not recruiting | Breast cancer | LBH589|Capecitabine|Lapatinib | 1 | Other|Industry | May 2008 |
| NCT01282476 | Recruiting | DLBCL | Panobinostat with Rituximab | 2 | Other | June 2011 |
| NCT01301807 | Recruiting | Myeloma | Carfilzomib|Panobinostat | 1 | Other|Industry | August 2011 |
| NCT00691938 | Active, not recruiting | AML|MDS | LBH589|Decitabine | 1/2 | Other | June 2008 |
| NCT00891033 | Active, not recruiting | MM | Bortezomib (Velcade)|Panobinostat | 1 | Other|Industry | April 2011 |
| NCT01440582 | Recruiting | Myeloma | Panobinostat|Bortezomib|Lenalidomide|Dexamethasone | 1/2 | Other|Industry | February 2013 |
| NCT01159418 | Active, not recruiting | Advanced solid tumors | Panobinostat (LBH589), carboplatin and paclitaxel | 1 | Other|Industry | June 2008 |
| NCT00946647 | Recruiting | MDS|CMML|AML | Panobinostat (LBH589) and 5-Azacytidine|5-Azacytidine | 2 | Industry | December 2009 |
| NCT01693601 | Recruiting | Myelofibrosis | Panobinostat|Ruxolitinib | 1/2 | Other | January 2013 |
| NCT01005797 | Recruiting | Renal cancer|NSCLC | Panobinostat (LBH589), sorafenib | 1 | Other|Industry | November 2009 |
| NCT00567879 | Completed | Breast cancer | Panobinostat, trastuzumab | 1 | Industry | April 2008 |
| NCT00859222 | Recruiting | Malignant glioma | LBH589|Bevacizumab | 1/2 | Other|Industry | March 2009 |
| NCT00907179 | Recruiting | NSCLC | LBH 589|Pemetrexed | 1/2 | Other|Industry | July 2009 |
| NCT01055795 | Completed | Advanced solid tumors | Bevacizumab, everolimus and LBH589 | 1 | Other|Industry | March 2010 |
| NCT01504776 | Recruiting | Mantle cell lymphoma | Panobinostat|Bortezomib | 1 | Other|Industry | April 2011 |
| NCT00556088 | Completed | Solid tumors | LBH589, paclitaxel, carboplatin, bevacizumab | 1 | Other|Industry | December 2007 |
| NCT00880269 | Completed | Refractory leukemia|AML | Panobinostat/LBH589 | 2 | Industry | August 2009 |
| NCT00738751 | Active, not recruiting | Lung cancer|Head and neck cancer | LBH589|Ertotinib | 1 | Other|Industry | November 2008 |
| NCT01341834 | Recruiting | Nasopharyngeal carcinoma, lymphomas, any EBV+solid tumor | LBH589 and RAD001 | 1 | Other|Industry | January 2011 |
| NCT01194908 | Recruiting | Breast cancer|Breast tumors|Breast neoplasms | Decitabine, LHB589, tamoxifen | 1/2 | Other|Industry | July 2010 |
| NCT00962507 | Completed | Lymphoma|MM and plasma cell neoplasm | Everolimus|Panobinostat | 1 | Other|NIH | July 2009 |
| NCT00978432 | Recruiting | DLBCL | RAD001|LBH589|RAD001 and LBH589 as a doublet | 2 | Other|Industry | February 2012 |
| NCT01175109 | Active, not recruiting | Chordoma | Imatinib+LBH589 | 1 | Other | October 2011 |
DLBCL, diffuse large-B cell lymphomas; EBV, Epstein-Barr virus.
Belinostat
Belinostat (PXD101) is a novel hydroxamic acid-type HDACi in late-stage clinical development with close to 1050 patients treated (March 2012). Belinostat is well tolerated, enabling combination with traditional chemotherapy without causing further bone marrow toxicity. Belinostat has been tested in a number of Phase I/II clinical trials in hematological cancers and solid tumors (Table 5). Data from these trials have provided evidence of the antitumor effect of Belinostat as monotherapy in peripheral T-cell lymphoma (PTCL) (95) and cutaneous T-cell lymphoma (CTCL), liver cancer (250), and thymoma. In addition, Belinostat has beneficial effects in combination with other anticancer drugs for the treatment of multiple types of cancer, including ovarian cancer (50), cancer of unknown primary, MM, AML, and bladder cancer (Table 6).
Table 5.
Clinical Trials with Belinostat (PXD101) As Single Drug
| NCT number | Recruitment | Study results | Conditions | Phases | Funded by |
|---|---|---|---|---|---|
| NCT00413075 | Completed | No results available | Solid tumor|Lymphoma | 1 | Industry |
| NCT00274651 | Completed | No results available | Cutaneous T-cell lymphoma|Peripheral T-cell lymphoma|Non-Hodgkin's lymphoma | 2 | Industry |
| NCT00865969 | Active, not recruiting | No results available | Peripheral T-cell lymphoma | 2 | Industry |
| NCT00301756 | Active, not recruiting | No results available | Fallopian tube cancer|Primary peritoneal cavity cancer|Ovarian tumor|Ovarian epithelial cancer | 2 | NIH |
| NCT01583777 | Not yet recruiting | No results available | Advanced cancer | 1 | Industry |
| NCT00321594 | Completed | No results available | Adult primary hepatocellular carcinoma|Liver cancer | 1/2 | NIH |
| NCT00589290 | Active, not recruiting | Has results | Thymoma|Thymic carcinoma | 2 | NIH |
| NCT01273155 | Recruiting | No results available | Neoplasms|Lymphomas | 1 | NIH |
| NCT00357162 | Completed | Has results | de Novo, previously, secondary MDS | 2 | NIH |
| NCT00357032 | Completed | No results available | AML | 2 | NIH |
| NCT00365053 | Completed | No results available | Mesothelioma | 2 | NIH |
| NCT00303953 | Completed | Has results | Recurrent adult Burkitt lymphoma|Recurrent adult DLCL | 2 | NIH |
DLCL, diffuse large cell lymphoma.
Table 6.
Clinical Trials with Belinostat in Combination with Other Drug/Regimens
| NCT number | Recruitment | Conditions | Interventions | Phases | Funded by |
|---|---|---|---|---|---|
| NCT01075425 | Recruiting | ALL|AML|MDS|CML | Bortezomib|Belinostat | 1 | Other|NIH |
| NCT00421889 | Completed | Ovarian cancer|Epithelial ovarian cancer|Fallopian tube cancer | Belinostat|Paclitaxel|Carboplatin | 1/2 | Industry |
| NCT00873119 | Completed | Occult primary | Belinostat, carboplatin, paclitaxel|Carboplatin, paclitaxel | 2 | Industry |
| NCT01317927 | Active, not recruiting | Solid tumor|Hematological malignancy | Belinostat, warfarin | 1 | Industry |
| NCT01310244 | Recruiting | NSCLC | Belinostat, carboplatin, paclitaxel | 1/2 | Industry |
| NCT01090830 | Terminated | NSCLC | Belinostat, carboplatin, paclitaxel and bevacizumab | 1/2 | Other |
| NCT01839097 | Recruiting | Peripheral T-cell lymphoma | Belinostat|CHOP | 1 | Industry |
| NCT00878800 | Completed | Soft tissue sarcomas | PXD101+doxorubicin | 1/2 | Industry |
| NCT01100944 | Active, not recruiting | Thymoma|Thymic carcinoma | Belinostat|Cisplatin|Cyclophosphamide|Doxorubicin | 1/2 | NIH |
| NCT00413322 | Completed | Tumor | Belinostat|5-Flurouracil (5-FU) | 1 | Industry |
| NCT01686165 | Recruiting | Large cell lymphoma | Belinostat|Biological: rituximab|Radiation: yttrium Y 90 ibritumomab tiuxetan | 2 | Other|NIH |
| NCT00334789 | Active, not recruiting | Unspecified adult solid tumor | Belinostat|Isotretinoin|Other: pharmacological study | 1 | NIH |
| NCT01188707 | Terminated | NSCLC | Belinostat and Erlotinib | 1/2 | Other|Industry |
| NCT00926640 | Recruiting | SCLC|Malignant epithelial neoplasms | (Belinostat) (PDX101)|Cisplatin|Etoposide | 1 | NIH |
| NCT00131261 | Completed | Multiple myeloma | PXD101|Dexamethasone | 2 | Industry |
Givinostat
Givinostat (originally ITF2357) was developed by Italfarmaco of Milan (Italy) patented in 1997 (WO 97/43251, US 6034096) and first described in 2005. It is in numerous Phase II clinical trials for relapsed leukemias and myelomas (NCT01761968) (79), and has been granted orphan drug designation in the European Union for the treatment of systemic juvenile idiopathic arthritis (NCT01261624, NCT01557452) and polycythaemia vera (NCT00928707). One interesting study involves assessing the use of Givinostat for Duchenne's disease (NCT01761292) (40).
PCI-24781
PCI-24781 (formerly CRA-024781) is currently being used in Phase I clinical trials for the treatment of neoplasia. The assessment of in vitro activity against tumor cell lines revealed the growth inhibition of multiple solid tumor lines, including colon, breast, lung, prostate, and ovarian cancers, as well as Hodgkin's and non-Hodgkin's lymphoma (15, 28, 210). Six different clinical interventional trials are now ongoing for PCI-24781, mainly for hematological diseases (NCT01149668, NCT00473577) and for testing the tolerability of the molecule in patients with hematological malignancies (NCT00562224, NCT00724984). A trial is now being recruited for the same drug (NCT01543763) in combination with PZP115891 (a tyrosine kinase inhibitor) (62).
Mocetinostat
Mocetinostat (MGCD0103) has shown clinical activity in several Phase I and II clinical trials both as a single agent (18) and in combination with Azacitidine and Gemcitabine (153) (Fig. 5). It has received orphan drug designation from FDA and has been designated an orphan medicinal product by the European Medicines Agency for the treatment of Hodgkin's lymphoma (NCT00543582) and AML (NCT00323934, NCT00324129).
FIG. 5.
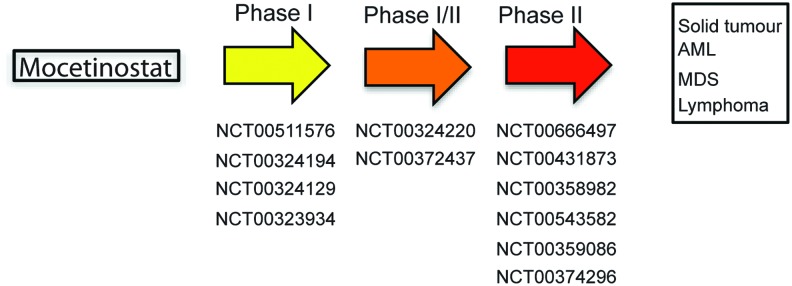
Clinical trials with Mocetinostat (MGCD0103). Studies are divided according to phase progression. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Entinostat
Entinostat (MS-275, SNDX-275) is a synthetic benzamide derivative shown to inhibit HDACs, and it displays antitumor activity in many preclinical models. Due to its relatively long half life, weekly and biweekly dosing schedules are explored in the clinic. A clinical trial with this agent was first performed in patients with advanced solid tumors or lymphoma in 2005. Since then, scientific interest in Entinostat has increased, and it is currently being used in Phase I/II clinical trials for recurrent advanced non-small cell lung cancer (NSCLC) combined with 5-azacytidine, or as a single drug for breast cancers (11, 109) (see Fig. 6 for disease-type classification).
FIG. 6.
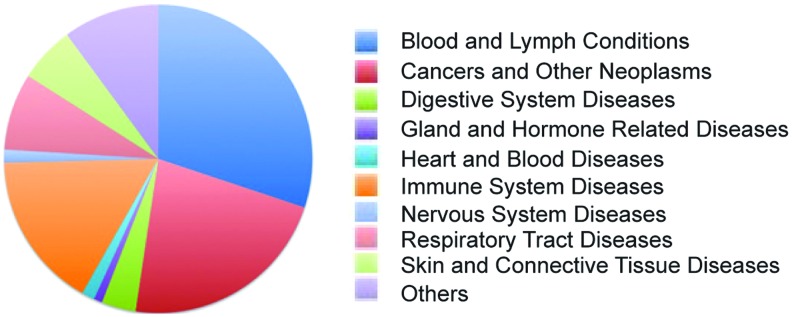
Clinical trials with Entinostat (MS-275) by topic classification. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Romidepsin
Romidepsin (INN, trade name Istodax) (90), a natural product obtained from the bacteria Chromobacterium violaceum, is an anticancer agent undergoing clinical trials for the treatment of CTCL (119), peripheral TCL, and other cancers (pancreatic, colorectal, renal, prostate, bladder, brain, thyroid, and ovarian) (106). In 2009, FDA granted approval for the use of Romidepsin in the treatment of CTCL in patients who had received at least one previous systemic therapy. Romidepsin is currently being used in more than 50 interventional trials, alone or in combination regimens (see clinicaltrials.gov). Romidepsin is often coupled with canonical chemotherapies (NCT00379639), demethylating agents (NCT01537744), and proteasome inhibitors (PIs; NCT00765102) (91, 194, 202).
Valproic acid
VPA has found clinical use as an anticonvulsant and mood-stabilizing drug, primarily in the treatment of epilepsy, bipolar disorder, and, less commonly, major depression. In addition, it is used to treat migraine headaches and schizophrenia (around 300 interventional and/or observational trials). VPA is also an HDACi and is under investigation for the treatment of HIV and various cancers (26 studies are reported for VPA used as HDACi) (43). Phase I and II clinical trials have tested VPA, alone or in combination, for lymphocytic leukemia, AML and myelodysplastic syndrome, melanoma, HIV infection, autoimmune lymphoproliferative syndrome, and human T-lymphotropic virus type-1-associated myelopathy/tropical spastic paraparesis. Some Phase II clinical trials have involved combination therapy with hydralazine, a DNA demethylating agent, and magnesium valproate for the treatment of cervical and breast cancers, and refractory solid tumors. Other clinical trials are ongoing for HD and amyotrophic lateral sclerosis.
Clinical trials with SIRT inhibitors or activators are still in early phases, and many of these involve resveratrol. For an in-depth description of this topic, we refer the reader to Nebbioso et al. (158) and references therein, such as trials NCT01521832, NCT01485952, NCT01485965, and NCT01521585.
Limits in the Current Use of HDACi
Tumor cells are extremely plastic, and one of the key aspects of the malignant transformation process is the capability to escape normal checkpoints. Consequently, tumor cells are generally more “independent” from the environment and they are also able to modify. In many cases, tumor cells develop mechanisms of resistance to HDACi caused by epigenetic and genetic characteristics, contributing to the generation and maintenance of a neoplastic phenotype. Cellular factors implicated in resistance phenomena to HDACi include drug efflux, target status (such as overexpression, mutation, and desensitization), chromatin alteration, up-regulation of oxidative stress response mechanism, defects in pro-apoptotic pathways, and up-regulation of anti-apoptotic signals/stimuli (Fig. 7).
FIG. 7.

Mechanisms leading to cell death and possible interfering causes in response to HDACi treatment in cancer cells. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Effect of HDACi efflux in cells
Multidrug resistance (MDR), mediated by multiple drug efflux ATP-binding cassette (ABC) transporters, is a critical issue in the treatment of many cancers (hematological and solid), with permeability (P)-glycoprotein (P-gp), multidrug resistance-associated protein 1 (MRP1), and breast cancer resistance protein ( ABCG2), which are consistently shown to be the key effectors of MDR in cell line studies (132, 241). Pumps conferring MDR are often overexpressed in many types of cancer, requiring the use of HDACi at a higher dose or in combination with inhibitors of transporter systems (132). Overexpression of these cellular pumps is also a side effect in treatment with HDACi. Hyperacetylation of the promoter regions encoding for these genes is often observed after HDACi treatment. To date, some information is available for Romidepsin, which has been found to interact with P-gp and MRP1 (168, 242, 243). The variability of HDACi structures can help overcome problems with cell intake. Changing permeability proprieties of the molecules using a combinatorial or rational approach could, therefore, improve the efficacy of available drugs; while further studies into pharmacokinetics and pharmacodynamics could reduce required doses and aspecific interactions.
HDAC status
As previously discussed (see section “HDACs and sirtuins in cancer: quantitative and functional abnormalities”), many HDACs are overexpressed in cancer. To date, various studies have investigated the aberrant expression of HDACs in tumorigenesis as well as the progression to metastatic/refractory phenotypes. Although overexpression and mutation of HDACs contributes to inducing tumorigenesis (41), this represents a serious drawback in HDACi-based therapies. In many cases, overexpression of HDAC1 alone is able to influence the outcome of epigenetic therapy (156), without considering the compensative effects always present when a member of a specific enzymatic class is targeted. In some cases, mutation of a specific HDAC causes resistance to treatment with specific inhibitors (HDAC2 is mutated in colon cancers) (140). Resistance acquired by alterations to HDACs requires further investigation with regard to the possibility of using pan- or selective inhibitors against the specific HDAC involved in a specific cancer (see next section).
Apoptotic/pro-survival mechanisms
Altered levels of anti-apoptotic proteins in cancer cells drive resistance against HDACi-mediated apoptosis. More specifically, it has been observed that by inhibiting the transcriptional activity of the JAK/STAT pathway, resistant CTCL cells can be re-sensitized to Vorinostat-induced apoptosis (53, 240). Consistently, overexpression of anti-apoptotic proteins such as Bcl-2, Bcl-XL is sufficient to render transformed cells resistant to HDACi. This observation is common in leukemias and lymphomas, in which the expression level of Bcl-2 protein is increased (211). The same is true for NF-κB, which is able to interfere with apoptosis in NSCL cancer lines, after treatment with HDACi. A different trend is observed for p21, which is overexpressed after treatment with HDACi (in particular, specific class I HDACi), acts as a negative regulator of cell-cycle progression in a p53-dependent or -independent manner, and has been implicated in the regulation of cell death. Overexpression of p21 also correlates with the start of biochemical processes, leading to DNA repair (12, 100). Since one of the mechanisms by which HDACi induce cell death is the creation of DNA damage (and genome instability), and since these phenomena contribute to the cytotoxic effects of HDACi, p21 may act as a resistance-inducing factor.
Chromatin and epigenetic modifications
DNA methylation contributes to malignant transformation (45), such as histone acetylation/deacetylation. The balance between the acetylated status of histones and the methylated status of DNA determines expression levels of onco-suppressors and oncogenic effectors. In many reports, the treatment of cells with HDACi alone is able to restore the expression of tumor suppressor genes, while other studies claim that a hierarchy in methylation and acetylation exists, with the dominance of DNA methylation as repressive marks in cancer cells. These findings suggest combining HDACi treatment with DNA demethylating agents (see section “Examples of HDACi in Combination Treatments”).
ROS scavengers as determinants of resistance
Redox pathways play an important role in the resistance of malignant transformed cells to HDACi. Many studies hypothesize a correlation between sensitivity to HDACi and levels of ROS scavengers (36, 72). A large body of information is available for thiol reductase (Trx), which is up-regulated in half of the tumors that are resistant to HDACi treatment (141, 220). Vorinostat is able to increase the level of Trx in normal cells (an important finding that explains HDACi selectivity for cancer cells) and, conversely, in transformed cells, Vorinostat induces up-regulation of the negative regulator of thioredoxin-binding protein. Increased levels of thioredoxin-binding protein lead to ROS accumulation and so, to the activation of pro-death pathways. In many pathological systems, such as myeloid leukemias, the level of Trx is up-regulated and normal HDACi-based therapies, consequently, fail. In this scenario, not only Trx but also other proteins participating in stress response and oxidative damage recognition could act as determinants of HDACi-resistance mechanisms.
Side Effects of HDACi
As with any class of anticancer agents, some HDACi in clinical trials, such as Romidepsin (2, 146, 164), Panobinostat (46), and Vorinostat (261), have been associated with serious cardio-toxicity in the treatment of solid cancers. The most important/frequent signal in cardio-toxicity status induced after HDACi administration is the prolongation of QT interval. The biological mechanism causing this side effect is not fully understood, but it may involve the aberrant expression/function of ion channels [e.g., (hERG)K+ channel]. (hERG)K+ channel is expressed in the nervous system and heart, where it determines the timing of electrical repolarization of the action potential in ventricular myocytes. Genetic mutations in and inhibition of (hERG)K+ channel can result in long QT syndrome. The function of (hERG)K+ channel in vivo is associated with other proteins such as MinK and MiRP1, and defects in their expression levels could, therefore, cause QT prolongation syndromes. To date, it is still unclear as to whether HDACi can react directly with K+ channels or whether their effect is due to an altered gene expression pathway. The former hypothesis is not corroborated by the fact that many HDACi induce QT prolongation independently of their structures. The greatest cardio-toxicity is, however, observed after treatment with hydroxamic-derivative HDACi. Again, it is unclear as to whether hydroxamic derivatives interact preferentially with (hERG)K+ channels or whether their broad-spectrum activity against all HDACs could exacerbate cardiac side effects (195).
Examples of HDACi in Combination Treatments
Although HDACi show promise as single anticancer agents, in particular with regard to their low-molecular range of activities and minimal toxicity, many studies are investigating the use of HDACi in combination with other drugs (21, 212). In this section, we present the most frequently used combinatorial therapeutic approaches. Figure 8 shows a comparison between the number of single and multiple agents in ongoing clinical trials.
FIG. 8.
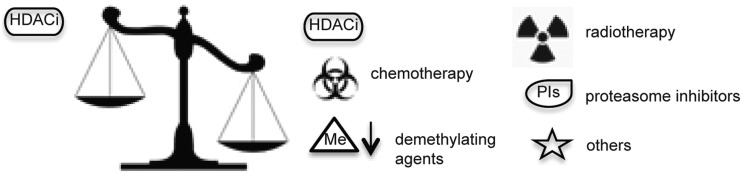
Schematic representation of the unbalanced number of trials with HDACi as a single drug and in combination regimens.
HDACi and death receptor agonists
HDACi can increase the expression and/or activity of proteins that directly transmit an apoptotic signal through death receptor pathways such as Fas and TRAIL ligands and death receptors, and downstream caspases (caspase-8 and −3), and can down-regulate proteins negatively regulating death-receptor signaling (e.g., c-FLIP, XIAP, survivin, IAP1/2). These findings provide a strong rationale for combining HDACi with death receptor stimuli. Since HDACi do not sensitize normal cells to TRAIL-mediated apoptosis, their combination with TRAIL is the most attractive option for cancer therapy (57, 99, 176, 226).
Combination of HDACi with irradiation
HDACi such as Vorinostat, TSA, VPA, and PCI-24781 enhance the radiosensitivity (γ-radiation) of tumoral cells. HDACi-mediated radiosensitization has been observed in several cancer cell lines such as lung, colon, prostate, breast, cervical, head, and neck. Preclinical studies have demonstrated that pre-treatment with HDACi followed by ionizing radiation is more effective (9, 83). Pre-treatment with Vorinostat in colorectal xenograft models produced a significant reduction in tumor volume compared with treatment with either therapy alone. Similar results have been observed in glioma and prostate tumors using other HDACi (34). At the molecular level, the synergistic antitumor effects of HDACi combined with radiotherapy enhance ATM, p53, and BRCA1 expression (114). Although the synergistic mechanisms are not fully understood, some findings suggest that two different biological pathways are involved. First, concomitant treatment with HDACi and radiotherapy induces the activation of ATM, culminating in the phosphorylation of p53 and, thus, triggering p53-mediated apoptotic cell death. ATM is activated by DNA damage induced by γ-radiation. Many authors report that treatment with HDACi exacerbates γ-radiation-mediated DNA damage, because chromatin is in a more open configuration (9, 122). HDACi also influence DNA repair mechanisms, with the down-regulation of proteins involved (such as Ku70, Ku86, and kinases). The capability of HDACi to induce a DNA damage response while suppressing the cellular repair mechanisms that are able to overcome it, therefore, provides a molecular rationale for combining HDACi and radiotherapy. This synergic approach could also improve the efficacy of anti-cancer therapies by controlling cutaneous radiation syndrome, a side effect of radiotherapy.
HDACi and regulators of proteasomal degradation
HDACi have recently been coupled with proteasome degradation regulators to enhance their efficacy against many cancers (92). Among all the PIs, Bortezomib is the best characterized. Proteasome inhibition results in the accumulation of mis-folded and damaged proteins, which, in turn, triggers a heat-shock protein response leading to apoptosis. The exact mechanism by which HDACi and PIs exert anticancer activity is not completely clear, but it is known to involve the regulation of Hsp90 and its client targets, such as the inhibition of NF-κB activity. ROS production and accumulation also very likely plays a role (59).
HDACi in combination with demethylating agents
Accumulating evidence has shown that the inhibition of aberrant epigenetic modifications caused by altered function/expression of HDACs could be more effective if the target strategy involving HDACi is coupled with demethylating agents. In many studies, HDACi treatment alone is insufficient to reactivate genes that are silenced by promoter hypermethylation, and is, therefore, used in combination with Decitabine, Azacitidine, and Zebularine. HDACi frequently used in combination regimens are VPA, sodium phenylbutyrate, and TSA for the treatment of AML, as well as colon, breast, endometrial, and thyroid cancers. Synergistic effects of HDACi and demethylating agents on gene expression might prolong the therapeutic time window. Although most clinical trials are still ongoing and concentrate predominantly on dosing schedules and safety of combination therapies, some studies have shown encouraging results. In patients with advanced hematological or solid malignancies, the combination of DNA-methyltransferase inhibitors and HDACi is well tolerated and induces biological effects such as DNA hypo-methylation and histone acetylation (33, 81, 102, 111, 188, 201).
HDACi with chemotherapeutic drugs
Combinatorial chemotherapeutic treatment is frontline therapy for many cancer types. Taxanes, which include paclitaxel and docetaxel, have also been used with HDACi such as TSA, Vorinostat, and PCI-24781. Since taxanes promote the stabilization of microtubules and interfere with transition from metaphase to anaphase during mitosis, the pre-treatment of cancer cells with HDACi enhances therapy by increasing tubulin acetylation. In particular, by combining class I/II HDACi with taxanes, a reduction in the proliferation of prostate, breast, ovarian, and gastric cancer cells has been observed (265). Other chemotherapeutic combinations, including DNA damage and microtubule-targeting agents, have also been used for cancer treatment. The addition of escalating doses of Belinostat in co-treatment with carboplatin and paclitaxel has been well tolerated for solid tumors (27, 50, 126). Vorinostat is also used in the carboplatin-paclitaxel chemotherapeutic regimen against NSCL. HDACi co-treatment with chemotherapeutic agents has also been employed in patients affected by anaplastic thyroid cancer. The combination of VPA and cisplatin-doxorubicin with radiotherapy decreases the volume of this tumor. However, clinical evaluation has, thus, far been limited to early-phase trials, making it difficult to draw conclusions regarding any added clinical benefit from the addition of HDACi. In conclusion, combining drugs that target different signaling pathways often lessens adverse side effects, while increasing the efficacy of treatment and reducing patient morbidity (187). In addition, multi-drug regimens using a variety of drugs and time schedules of drug administration may offer patients a more tailored therapy based on clinical status.
Open Questions: Specific or Pan-HDACi?
Unselectively inhibiting HDAC activity elicits not only therapeutic responses but also unexpected side effects. Considering the diversity of HDAC targets, selective HDACi (active against a specific HDAC class or a single isoform) may cause fewer adverse reactions and, thus, be more clinically useful. For example, class II HDACs are abundant in the heart and repress cardiac gene expression by direct or indirect association with TFs (7). As discussed in the section “Side effects of HDACi,” HDACi administration commonly causes QT interval prolongation. A selective HDACi, inactive against class II HDACs, could eliminate or reduce unwanted cardiac responses by preserving the activity of HDAC4, 5, 7 (class IIa), 6, and 10 (class IIb). To date, although considerable effort has been made toward their development, a few class-specific HDACi are available (for instance, MC1568, active on HDAC4 and 6 with IC50≈220 nM) (160). The difficulty in designing this kind of HDACi is essentially due to structural homology between HDACs and the way in which these inhibitors are developed. HDACi reported thus far are structurally diversified but, nevertheless, act as competitive inhibitors, targeting zinc ion in HDAC active sites. These compounds generally belong to the pan-inhibitor class. Sequence comparison indicates that the main differences between individual HDACs are found near the catalytic core, in the regions forming loops. Thus, inhibitors directed against this region, acting as non-competitive modulators, may display HDAC selectivity. Another way to develop non-competitive HDACi is to look at enzymatic regions that are responsible for interactions with DNA-binding proteins and/or transport proteins (for class IIa HDACs) (138). Non-competitive modulators have the advantage of being inactive (or less active) against other enzymes inside the cell requiring zinc ion as cofactor (such as RNA polymerase II, metalloprotease). The design of a new selective HDACi usually starts by taking a known active HDACi and modifying some of the structural features of the molecule without reducing its activity. Known inhibitors comprise a capping group, metal-binding moiety, and a linker region. While the capping group has been successfully modified for the design of several class-selective inhibitors (17), a combination of modifications at all three regions likely enhances their selective function. Modifications of the capping group are also useful in targeting HDACi to specific tissues. To confer tissue-selectivity properties to HDACi, they can be equipped with a surface recognition capping group that is capable of binding unique biological targets, such as expressed or overexpressed membrane receptor-binding molecules. One example of this strategy is compound CHR-2845, which is hydrolyzed by macrophage membrane receptor hCE-1 (carboxylesterase 1 receptor). The hydrolysis of CHR-2845 gives CHR-2847, the active compound accumulated only in cells expressing hCE-1 (84).
Concluding Remarks
In this review, we summarize studies on the classification, mechanisms of action, and chemistry of HDACs and their inhibitors. Several HDACi are currently being used in clinical trials for both solid and hematological malignancies. In combination with DNA-demethylating agents, chemopreventive, or classical chemotherapeutic drugs, HDACi are promising candidates for cancer therapy. Many questions regarding efficacy, tumor specificity, and gene regulation patterns still remain to be answered. A key issue is the identification of biomarkers that are predictive of positive outcome (i.e., are we aiming at the right “enemies” with the right “weapons”?). Is it possible to identify mRNAs and/or proteins that increase in response to a particular HDACi and which might also be used as indicators of drug efficacy? This is an ongoing discussion in epigenetics, and future research may help provide insights into response-based patient stratification. Due to the huge number of newly discovered epi-enzymes and their interconnection with tumorigenesis and cancer progression, a key task in future diagnostic, therapeutic, and prognostic strategies will focus on characterizing the interplay between genome and epigenome, and, thus, designing personalized epi-based single or combinatorial treatments.
Abbreviations Used
- ABC
ATP-binding cassette
- AD
Alzheimer's disease
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- APL
acute promyelocytic leukemia
- DLBCL
diffuse large B-cell lymphomas
- eNOS
endothelial nitric oxide synthase
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitors
- HIF-1α
hypoxia-inducible factor
- IFN- γ
interferon-γ
- MDR
multidrug resistance
- MDS
myelodysplastic syndrome
- MM
multiple myeloma
- MRP1
multidrug resistance-associated protein 1
- MSI
microsatellite instability
- NES
nuclear export signal
- NF-κB
nuclear factor-κB
- P-gp
permeability-glycoprotein
- PIs
proteasome inhibitors
- ROS
reactive oxygen species
- SIRT
sirtuin
- SIRTi
sirtuin inhibitors
- TF
transcription factors
- TNF
tumor necrosis factor
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- Trx
thiol reductase
- TSA
trichostatin A
- VEGF
vascular endothelial growth factor
- VPA
valproic acid
- YY1
Yin and Yang 1
Acknowledgments
This work was supported by EU: Blueprint (contract no. 282510), ATLAS (contract no. 221952); Epigenomics Flagship Project EPIGEN (MIUR-CNR); the Italian Association for Cancer Research (AIRC no. 11812); the Italian Ministry of University and Research (PRIN_2009PX2T2E_004); PON002782; and PON0101227. The authors apologize to the authors whose work could not be cited due to restrictions in the number of references. They are grateful to C. Fisher for linguistic editing of this article.
References
- 1.Adams H, Fritzsche FR, Dirnhofer S, Kristiansen G, and Tzankov A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin's lymphoma. Expert Opin Ther Targets 14: 577–584, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Amiri-Kordestani L, Luchenko V, Peer CJ, Ghafourian K, Reynolds J, Draper D, Frye R, Woo S, Venzon D, Wright J, Skarulis M, Figg WD, Fojo T, Bates SE, and Piekarz RL. Phase I trial of a new schedule of romidepsin in patients with advanced cancers. Clin Cancer Res 19: 4499–4507, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreoli F, Barbosa AJ, Parenti MD, and Del Rio A. Modulation of epigenetic targets for anticancer therapy: clinicopathological relevance, structural data and drug discovery perspectives. Curr Pharm Des 19: 578–613, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, Steinle A, and Salih HR. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res 65: 6321–6329, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett 280: 233–241, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Atadja P, Gao L, Kwon P, Trogani N, Walker H, Hsu M, Yeleswarapu L, Chandramouli N, Perez L, Versace R, Wu A, Sambucetti L, Lassota P, Cohen D, Bair K, Wood A, and Remiszewski S. Selective growth inhibition of tumor cells by a novel histone deacetylase inhibitor, NVP-LAQ824. Cancer Res 64: 689–695, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Backs J. and Olson EN. Control of cardiac growth by histone acetylation/deacetylation. Circ Res 98: 15–24, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Barber MF, Michishita-Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z, Tennen RI, Paredes S, Young NL, Chen K, Struhl K, Garcia BA, Gozani O, Li W, and Chua KF. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 487: 114–118, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barker CA, Bishop AJ, Chang M, Beal K, and Chan TA. Valproic acid use during radiation therapy for glioblastoma associated with improved survival. Int J Radiat Oncol Biol Phys 86: 504–509, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barneda-Zahonero B. and Parra M. Histone deacetylases and cancer. Mol Oncol 6: 579–589, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belinsky SA, Grimes MJ, Picchi MA, Mitchell HD, Stidley CA, Tesfaigzi Y, Channell MM, Liu Y, Casero RA, Jr., Baylin SB, Reed MD, Tellez CS, and March TH. Combination therapy with vidaza and entinostat suppresses tumor growth and reprograms the epigenome in an orthotopic lung cancer model. Cancer Res 71: 454–462, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, Fotedar A, and Fotedar R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 114: 599–610, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Berger SL. The complex language of chromatin regulation during transcription. Nature 447: 407–412, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Beumer JH. and Tawbi H. Role of histone deacetylases and their inhibitors in cancer biology and treatment. Curr Clin Pharmacol 5: 196–208, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Bhalla S, Balasubramanian S, David K, Sirisawad M, Buggy J, Mauro L, Prachand S, Miller R, Gordon LI, and Evens AM. PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NF-kappaB mechanisms and is synergistic with bortezomib in lymphoma cells. Clin Cancer Res 15: 3354–3365, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bi G. and Jiang G. The molecular mechanism of HDAC inhibitors in anticancer effects. Cell Mol Immunol 3: 285–290, 2006 [PubMed] [Google Scholar]
- 17.Bieliauskas AV. and Pflum MK. Isoform-selective histone deacetylase inhibitors. Chem Soc Rev 37: 1402–1413, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blum KA, Advani A, Fernandez L, Van Der Jagt R, Brandwein J, Kambhampati S, Kassis J, Davis M, Bonfils C, Dubay M, Dumouchel J, Drouin M, Lucas DM, Martell RE, and Byrd JC. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br J Haematol 147: 507–514, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolden JE, Shi W, Jankowski K, Kan CY, Cluse L, Martin BP, MacKenzie KL, Smyth GK, and Johnstone RW. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis 4: e519, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosch-Presegue L. and Vaquero A. The dual role of sirtuins in cancer. Genes Cancer 2: 648–662, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bots M. and Johnstone RW. Rational combinations using HDAC inhibitors. Clin Cancer Res 15: 3970–3977, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Boumber Y, Younes A, and Garcia-Manero G. Mocetinostat (MGCD0103): a review of an isotype-specific histone deacetylase inhibitor. Expert Opin Investig Drugs 20: 823–829, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, Matthias P, Muller CW, and Khochbin S. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J 25: 3357–3366, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, Garrido C, Yao TP, Vourc'h C, Matthias P, and Khochbin S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev 21: 2172–2181, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brouet A, Sonveaux P, Dessy C, Moniotte S, Balligand JL, and Feron O. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins. Circ Res 89: 866–873, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Buckland J. Rheumatoid arthritis: HDAC and HDACi: pathogenetic and mechanistic insights. Nat Rev Rheumatol 7: 682, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Buckley MT, Yoon J, Yee H, Chiriboga L, Liebes L, Ara G, Qian X, Bajorin DF, Sun TT, Wu XR, and Osman I. The histone deacetylase inhibitor belinostat (PXD101) suppresses bladder cancer cell growth in vitro and in vivo. J Transl Med 5: 49, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buggy JJ, Cao ZA, Bass KE, Verner E, Balasubramanian S, Liu L, Schultz BE, Young PR, and Dalrymple SA. CRA-024781: a novel synthetic inhibitor of histone deacetylase enzymes with antitumor activity in vitro and in vivo. Mol Cancer Ther 5: 1309–1317, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Buglio D, Khaskhely NM, Voo KS, Martinez-Valdez H, Liu YJ, and Younes A. HDAC11 plays an essential role in regulating OX40 ligand expression in Hodgkin lymphoma. Blood 117: 2910–2917, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, and Richon VM. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A 99: 11700–11705, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carafa V, Nebbioso A, and Altucci L. Sirtuins and disease: the road ahead. Front Pharmacol 3: 4, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cha TL, Chuang MJ, Wu ST, Sun GH, Chang SY, Yu DS, Huang SM, Huan SK, Cheng TC, Chen TT, Fan PL, and Hsiao PW. Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer Res 15: 840–850, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Chen M, Voeller D, Marquez VE, Kaye FJ, Steeg PS, Giaccone G, and Zajac-Kaye M. Enhanced growth inhibition by combined DNA methylation/HDAC inhibitors in lung tumor cells with silenced CDKN2A. Int J Oncol 37: 963–971, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chinnaiyan P, Vallabhaneni G, Armstrong E, Huang SM, and Harari PM. Modulation of radiation response by histone deacetylase inhibition. Int J Radiat Oncol Biol Phys 62: 223–229, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Choi JH, Kwon HJ, Yoon BI, Kim JH, Han SU, Joo HJ, and Kim DY. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn J Cancer Res 92: 1300–1304, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choudhary S. and Wang HC. Role of reactive oxygen species in proapoptotic ability of oncogenic H-Ras to increase human bladder cancer cell susceptibility to histone deacetylase inhibitor for caspase induction. J Cancer Res Clin Oncol 135: 1601–1613, 2009 [DOI] [PubMed] [Google Scholar]
- 37.Christensen DP, Dahllof M, Lundh M, Rasmussen DN, Nielsen MD, Billestrup N, Grunnet LG, and Mandrup-Poulsen T. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol Med 17: 378–390, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clarke N, Nebbioso A, Altucci L, and Gronemeyer H. TRAIL: at the center of drugable anti-tumor pathways. Cell Cycle 4: 914–918, 2005 [DOI] [PubMed] [Google Scholar]
- 39.Codd R, Braich N, Liu J, Soe CZ, and Pakchung AA. Zn(II)-dependent histone deacetylase inhibitors: suberoylanilide hydroxamic acid and trichostatin A. Int J Biochem Cell Biol 41: 736–739, 2009 [DOI] [PubMed] [Google Scholar]
- 40.Consalvi S, Mozzetta C, Bettica P, Germani M, Fiorentini F, Del Bene F, Rocchetti M, Leoni F, Monzani V, Mascagni P, Puri PL, and Saccone V. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol Med 19: 79–87, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conte M. and Altucci L. Molecular pathways: the complexity of the epigenome in cancer and recent clinical advances. Clin Cancer Res 18: 5526–5534, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, and Auwerx J. Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med 39: 335–345, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Davidson DC, Schifitto G, and Maggirwar SB. Valproic acid inhibits the release of soluble CD40L induced by non-nucleoside reverse transcriptase inhibitors in human immunodeficiency virus infected individuals. PLoS One 8: e59950, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr 133: 2485S–2493S, 2003 [DOI] [PubMed] [Google Scholar]
- 45.De S, Shaknovich R, Riester M, Elemento O, Geng H, Kormaksson M, Jiang Y, Woolcock B, Johnson N, Polo JM, Cerchietti L, Gascoyne RD, Melnick A, and Michor F. Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLoS Genet 9: e1003137, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Marinis F, Atmaca A, Tiseo M, Giuffreda L, Rossi A, Gebbia V, D'Antonio C, Dal Zotto L, Al-Batran SE, Marsoni S, and Wolf M. A phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in pretreated patients with small-cell lung cancer. J Thorac Oncol 8: 1091–1094, 2013 [DOI] [PubMed] [Google Scholar]
- 47.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, and van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370: 737–749, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dell'Aversana C, Lepore I, and Altucci L. HDAC modulation and cell death in the clinic. Exp Cell Res 318: 1229–1244, 2012 [DOI] [PubMed] [Google Scholar]
- 49.Deubzer HE, Schier MC, Oehme I, Lodrini M, Haendler B, Sommer A, and Witt O. HDAC11 is a novel drug target in carcinomas. Int J Cancer 132: 2200–2208, 2013 [DOI] [PubMed] [Google Scholar]
- 50.Dizon DS, Damstrup L, Finkler NJ, Lassen U, Celano P, Glasspool R, Crowley E, Lichenstein HS, Knoblach P, and Penson RT. Phase II activity of belinostat (PXD-101), carboplatin, and paclitaxel in women with previously treated ovarian cancer. Int J Gynecol Cancer 22: 979–986, 2012 [DOI] [PubMed] [Google Scholar]
- 51.Doetzlhofer A, Rotheneder H, Lagger G, Koranda M, Kurtev V, Brosch G, Wintersberger E, and Seiser C. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol Cell Biol 19: 5504–5511, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donmez G. and Outeiro TF. SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol Med 5: 344–352, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dupere-Richer D, Kinal M, Menasche V, Nielsen TH, Del Rincon S, Pettersson F, and Miller WH., Jr. Vorinostat-induced autophagy switches from a death-promoting to a cytoprotective signal to drive acquired resistance. Cell Death Dis 4: e486, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duvic M, Dummer R, Becker JC, Poulalhon N, Ortiz Romero P, Grazia Bernengo M, Lebbe C, Assaf C, Squier M, Williams D, Marshood M, Tai F, and Prince HM. Panobinostat activity in both bexarotene-exposed and -naive patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur J Cancer 49: 386–394, 2013 [DOI] [PubMed] [Google Scholar]
- 55.Ell B. and Kang Y. Transcriptional control of cancer metastasis. Trends Cell Biol 23: 603–611, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emiliani S, Fischle W, Van Lint C, Al-Abed Y, and Verdin E. Characterization of a human RPD3 ortholog, HDAC3. Proc Natl Acad Sci U S A 95: 2795–2800, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fandy TE, Shankar S, Ross DD, Sausville E, and Srivastava RK. Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia 7: 646–657, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farooq M, Hozzein WN, Elsayed EA, Taha NA, and Wadaan MA. Identification of histone deacetylase 1 protein complexes in liver cancer cells. Asian Pac J Cancer Prev 14: 915–921, 2013 [DOI] [PubMed] [Google Scholar]
- 59.Feng R, Oton A, Mapara MY, Anderson G, Belani C, and Lentzsch S. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br J Haematol 139: 385–397, 2007 [DOI] [PubMed] [Google Scholar]
- 60.Fischer A, Sananbenesi F, Mungenast A, and Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci 31: 605–617, 2010 [DOI] [PubMed] [Google Scholar]
- 61.Fischer DD, Cai R, Bhatia U, Asselbergs FA, Song C, Terry R, Trogani N, Widmer R, Atadja P, and Cohen D. Isolation and characterization of a novel class II histone deacetylase, HDAC10. J Biol Chem 277: 6656–6666, 2002 [DOI] [PubMed] [Google Scholar]
- 62.Fouliard S, Robert R, Jacquet-Bescond A, du Rieu QC, Balasubramanian S, Loury D, Loriot Y, Hollebecque A, Kloos I, Soria JC, Chenel M, and Depil S. Pharmacokinetic/pharmacodynamic modelling-based optimisation of administration schedule for the histone deacetylase inhibitor abexinostat (S78454/PCI-24781) in phase I. Eur J Cancer 49: 2791–2797, 2013 [DOI] [PubMed] [Google Scholar]
- 63.Fritzsche FR, Weichert W, Roske A, Gekeler V, Beckers T, Stephan C, Jung K, Scholman K, Denkert C, Dietel M, and Kristiansen G. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer 8: 381, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun 260: 273–279, 1999 [DOI] [PubMed] [Google Scholar]
- 65.Fulda S. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr Cancer Drug Targets 8: 132–140, 2008 [DOI] [PubMed] [Google Scholar]
- 66.Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, Reilly JF, Loboda A, Nebozhyn M, Fantin VR, Richon VM, Scheithauer B, Giannini C, Flynn PJ, Moore DF, Jr., Zwiebel J, and Buckner JC. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol 27: 2052–2058, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galasinski SC, Resing KA, Goodrich JA, and Ahn NG. Phosphatase inhibition leads to histone deacetylases 1 and 2 phosphorylation and disruption of corepressor interactions. J Biol Chem 277: 19618–19626, 2002 [DOI] [PubMed] [Google Scholar]
- 68.Gallinari P, Di Marco S, Jones P, Pallaoro M, and Steinkuhler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res 17: 195–211, 2007 [DOI] [PubMed] [Google Scholar]
- 69.Gantt SL, Gattis SG, and Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry 45: 6170–6178, 2006 [DOI] [PubMed] [Google Scholar]
- 70.Gao DJ, Xu M, Zhang YQ, Du YQ, Gao J, Gong YF, Man XH, Wu HY, Jin J, Xu GM, and Li ZS. Upregulated histone deacetylase 1 expression in pancreatic ductal adenocarcinoma and specific siRNA inhibits the growth of cancer cells. Pancreas 39: 994–1001, 2010 [DOI] [PubMed] [Google Scholar]
- 71.Gao L, Cueto MA, Asselbergs F, and Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem 277: 25748–25755, 2002 [DOI] [PubMed] [Google Scholar]
- 72.Gao S, Mobley A, Miller C, Boklan J, and Chandra J. Potentiation of reactive oxygen species is a marker for synergistic cytotoxicity of MS-275 and 5-azacytidine in leukemic cells. Leuk Res 32: 771–780, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, and Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol 18: 7185–7191, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Geng H, Harvey CT, Pittsenbarger J, Liu Q, Beer TM, Xue C, and Qian DZ. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem 286: 38095–38102, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghizzoni M, Haisma HJ, Maarsingh H, and Dekker FJ. Histone acetyltransferases are crucial regulators in NF-kappaB mediated inflammation. Drug Discov Today 16: 504–511, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Giandomenico V, Simonsson M, Gronroos E, and Ericsson J. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol Cell Biol 23: 2587–2599, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gimsing P. Belinostat: a new broad acting antineoplastic histone deacetylase inhibitor. Expert Opin Investig Drugs 18: 501–508, 2009 [DOI] [PubMed] [Google Scholar]
- 78.Glozak MA, Sengupta N, Zhang X, and Seto E. Acetylation and deacetylation of non-histone proteins. Gene 363: 15–23, 2005 [DOI] [PubMed] [Google Scholar]
- 79.Golay J, Cuppini L, Leoni F, Mico C, Barbui V, Domenghini M, Lombardi L, Neri A, Barbui AM, Salvi A, Pozzi P, Porro G, Pagani P, Fossati G, Mascagni P, Introna M, and Rambaldi A. The histone deacetylase inhibitor ITF2357 has anti-leukemic activity in vitro and in vivo and inhibits IL-6 and VEGF production by stromal cells. Leukemia 21: 1892–1900, 2007 [DOI] [PubMed] [Google Scholar]
- 80.Grant S. The novel histone deacetylase inhibitor NVP-LAQ824: an addition to the therapeutic armamentarium in leukemia? Leukemia 18: 1931–1933, 2004 [DOI] [PubMed] [Google Scholar]
- 81.Griffiths EA. and Gore SD. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin Hematol 45: 23–30, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gronroos E, Hellman U, Heldin CH, and Ericsson J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell 10: 483–493, 2002 [DOI] [PubMed] [Google Scholar]
- 83.Groselj B, Sharma NL, Hamdy FC, Kerr M, and Kiltie AE. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer 108: 748–754, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gryder BE, Sodji QH, and Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem 4: 505–524, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guardiola AR. and Yao TP. Molecular cloning and characterization of a novel histone deacetylase HDAC10. J Biol Chem 277: 3350–3356, 2002 [DOI] [PubMed] [Google Scholar]
- 86.Guenther MG, Barak O, and Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol 21: 6091–6101, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haberland M, Montgomery RL, and Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10: 32–42, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Han JW, Ahn SH, Park SH, Wang SY, Bae GU, Seo DW, Kwon HK, Hong S, Lee HY, Lee YW, and Lee HW. Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Res 60: 6068–6074, 2000 [PubMed] [Google Scholar]
- 89.Hanahan D. and Weinberg RA. Hallmarks of cancer: the next generation. Cell 144: 646–674, 2011 [DOI] [PubMed] [Google Scholar]
- 90.Harrison SJ, Bishton M, Bates SE, Grant S, Piekarz RL, Johnstone RW, Dai Y, Lee B, Araujo ME, and Prince HM. A focus on the preclinical development and clinical status of the histone deacetylase inhibitor, romidepsin (depsipeptide, Istodax((R))). Epigenomics 4: 571–589, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harrison SJ, Quach H, Link E, Seymour JF, Ritchie DS, Ruell S, Dean J, Januszewicz H, Johnstone R, Neeson P, Dickinson M, Nichols J, and Prince HM. A high rate of durable responses with romidepsin, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma. Blood 118: 6274–6283, 2011 [DOI] [PubMed] [Google Scholar]
- 92.Heider U, Rademacher J, Lamottke B, Mieth M, Moebs M, von Metzler I, Assaf C, and Sezer O. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur J Haematol 82: 440–449, 2009 [DOI] [PubMed] [Google Scholar]
- 93.Herceg Z, Lambert MP, van Veldhoven K, Demetriou C, Vineis P, Smith MT, Straif K, and Wild CP. Towards incorporating epigenetic mechanisms into carcinogen identification and evaluation. Carcinogenesis 34: 1955–1967, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hiratsuka M, Inoue T, Toda T, Kimura N, Shirayoshi Y, Kamitani H, Watanabe T, Ohama E, Tahimic CG, Kurimasa A, and Oshimura M. Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem Biophys Res Commun 309: 558–566, 2003 [DOI] [PubMed] [Google Scholar]
- 95.Howman RA. and Prince HM. New drug therapies in peripheral T-cell lymphoma. Expert Rev Anticancer Ther 11: 457–472, 2011 [DOI] [PubMed] [Google Scholar]
- 96.Hrzenjak A, Moinfar F, Kremser ML, Strohmeier B, Staber PB, Zatloukal K, and Denk H. Valproate inhibition of histone deacetylase 2 affects differentiation and decreases proliferation of endometrial stromal sarcoma cells. Mol Cancer Ther 5: 2203–2210, 2006 [DOI] [PubMed] [Google Scholar]
- 97.Huang EY, Zhang J, Miska EA, Guenther MG, Kouzarides T, and Lazar MA. Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev 14: 45–54, 2000 [PMC free article] [PubMed] [Google Scholar]
- 98.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, and Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature 417: 455–458, 2002 [DOI] [PubMed] [Google Scholar]
- 99.Iacomino G, Medici MC, and Russo GL. Valproic acid sensitizes K562 erythroleukemia cells to TRAIL/Apo2L-induced apoptosis. Anticancer Res 28: 855–864, 2008 [PubMed] [Google Scholar]
- 100.Insinga A, Cicalese A, Faretta M, Gallo B, Albano L, Ronzoni S, Furia L, Viale A, and Pelicci PG. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc Natl Acad Sci U S A 110: 3931–3936, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, and Kadowaki T. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 464: 1313–1319, 2010 [DOI] [PubMed] [Google Scholar]
- 102.Jackson M, Krassowska A, Gilbert N, Chevassut T, Forrester L, Ansell J, and Ramsahoye B. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol 24: 8862–8871, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jayathilaka N, Han A, Gaffney KJ, Dey R, Jarusiewicz JA, Noridomi K, Philips MA, Lei X, He J, Ye J, Gao T, Petasis NA, and Chen L. Inhibition of the function of class IIa HDACs by blocking their interaction with MEF2. Nucleic Acids Res 40: 5378–5388, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jin YH, Jeon EJ, Li QL, Lee YH, Choi JK, Kim WJ, Lee KY, and Bae SC. Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J Biol Chem 279: 29409–29417, 2004 [DOI] [PubMed] [Google Scholar]
- 105.Jones PA. and Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 3: 415–428, 2002 [DOI] [PubMed] [Google Scholar]
- 106.Jones SF, Infante JR, Spigel DR, Peacock NW, Thompson DS, Greco FA, McCulloch W, and Burris HA, 3rd. Phase 1 results from a study of romidepsin in combination with gemcitabine in patients with advanced solid tumors. Cancer Invest 30: 481–486, 2012 [DOI] [PubMed] [Google Scholar]
- 107.Joseph J, Mudduluru G, Antony S, Vashistha S, Ajitkumar P, and Somasundaram K. Expression profiling of sodium butyrate (NaB)-treated cells: identification of regulation of genes related to cytokine signaling and cancer metastasis by NaB. Oncogene 23: 6304–6315, 2004 [DOI] [PubMed] [Google Scholar]
- 108.Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, and Cristea IM. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol 9: 672, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, Lee B, Tsai S, Delgado IE, Rudek MA, Belinsky SA, Herman JG, Baylin SB, Brock MV, and Rudin CM. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov 1: 598–607, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jung M, Brosch G, Kolle D, Scherf H, Gerhauser C, and Loidl P. Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation. J Med Chem 42: 4669–4679, 1999 [DOI] [PubMed] [Google Scholar]
- 111.Kalac M, Scotto L, Marchi E, Amengual J, Seshan VE, Bhagat G, Ulahannan N, Leshchenko VV, Temkin AM, Parekh S, Tycko B, and O'Connor OA. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood 118: 5506–5516, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kao HY, Lee CH, Komarov A, Han CC, and Evans RM. Isolation and characterization of mammalian HDAC10, a novel histone deacetylase. J Biol Chem 277: 187–193, 2002 [DOI] [PubMed] [Google Scholar]
- 113.Kao HY, Verdel A, Tsai CC, Simon C, Juguilon H, and Khochbin S. Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J Biol Chem 276: 47496–47507, 2001 [DOI] [PubMed] [Google Scholar]
- 114.Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther 3: 513–519, 2004 [PubMed] [Google Scholar]
- 115.Kawai H, Li H, Avraham S, Jiang S, and Avraham HK. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int J Cancer 107: 353–358, 2003 [DOI] [PubMed] [Google Scholar]
- 116.Kee HJ. and Kook H. Kruppel-like factor 4 mediates histone deacetylase inhibitor-induced prevention of cardiac hypertrophy. J Mol Cell Cardiol 47: 770–780, 2009 [DOI] [PubMed] [Google Scholar]
- 117.Kee HJ. and Kook H. Roles and targets of class I and IIa histone deacetylases in cardiac hypertrophy. J Biomed Biotechnol 2011: 928326, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, Ji J, Wang XW, Park SH, Cha YI, Gius D, and Deng CX. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 20: 487–499, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim M, Thompson LA, Wenger SD, and O'Bryant CL. Romidepsin: a histone deacetylase inhibitor for refractory cutaneous T-cell lymphoma. Ann Pharmacother 46: 1340–1348, 2012 [DOI] [PubMed] [Google Scholar]
- 120.Kim SH, Ahn S, Han JW, Lee HW, Lee HY, Lee YW, Kim MR, Kim KW, Kim WB, and Hong S. Apicidin is a histone deacetylase inhibitor with anti-invasive and anti-angiogenic potentials. Biochem Biophys Res Commun 315: 964–970, 2004 [DOI] [PubMed] [Google Scholar]
- 121.Kops GJ, Weaver BA, and Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer 5: 773–785, 2005 [DOI] [PubMed] [Google Scholar]
- 122.Krishnan V, Liu B, and Zhou Z. ‘Relax and Repair’ to restrain aging. Aging (Albany NY) 3: 943–954, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kuzmichev A, Zhang Y, Erdjument-Bromage H, Tempst P, and Reinberg D. Role of the Sin3-histone deacetylase complex in growth regulation by the candidate tumor suppressor p33(ING1). Mol Cell Biol 22: 835–848, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.LaBonte MJ, Wilson PM, Fazzone W, Russell J, Louie SG, El-Khoueiry A, Lenz HJ, and Ladner RD. The dual EGFR/HER2 inhibitor lapatinib synergistically enhances the antitumor activity of the histone deacetylase inhibitor panobinostat in colorectal cancer models. Cancer Res 71: 3635–3648, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A, Minguez B, Tsai HW, Ward SC, Thung S, Friedman SL, and Llovet JM. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J Hepatol 56: 1343–1350, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lassen U, Molife LR, Sorensen M, Engelholm SA, Vidal L, Sinha R, Penson RT, Buhl-Jensen P, Crowley E, Tjornelund J, Knoblauch P, and de Bono JS. A phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br J Cancer 103: 12–17, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lavu S, Boss O, Elliott PJ, and Lambert PD. Sirtuins—novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov 7: 841–853, 2008 [DOI] [PubMed] [Google Scholar]
- 128.Lecker SH, Goldberg AL, and Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17: 1807–1819, 2006 [DOI] [PubMed] [Google Scholar]
- 129.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, and Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 105: 3374–3379, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee JH, Jeong EG, Choi MC, Kim SH, Park JH, Song SH, Park J, Bang YJ, and Kim TY. Inhibition of histone deacetylase 10 induces thioredoxin-interacting protein and causes accumulation of reactive oxygen species in SNU-620 human gastric cancer cells. Mol Cells 30: 107–112, 2010 [DOI] [PubMed] [Google Scholar]
- 131.Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, Wolfel C, Huber C, and Wolfel T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci U S A 102: 16013–16018, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Leonard GD, Fojo T, and Bates SE. The role of ABC transporters in clinical practice. Oncologist 8: 411–424, 2003 [DOI] [PubMed] [Google Scholar]
- 133.Li X, Zhang S, Blander G, Tse JG, Krieger M, and Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 28: 91–106, 2007 [DOI] [PubMed] [Google Scholar]
- 134.Li Y, Zhang X, Polakiewicz RD, Yao TP, and Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J Biol Chem 283: 12686–12690, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Licciardi PV, Ververis K, Tang ML, El-Osta A, and Karagiannis TC. Immunomodulatory effects of histone deacetylase inhibitors. Curr Mol Med 13: 640–647, 2013 [DOI] [PubMed] [Google Scholar]
- 136.Lin KT, Wang YW, Chen CT, Ho CM, Su WH, and Jou YS. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin Cancer Res 18: 4691–4701, 2012 [DOI] [PubMed] [Google Scholar]
- 137.Liu L, Li Y, and Tollefsbol TO. Gene-environment interactions and epigenetic basis of human diseases. Curr Issues Mol Biol 10: 25–36, 2008 [PMC free article] [PubMed] [Google Scholar]
- 138.Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, Baloglu E, Trump RP, Head MS, Hofmann GA, Murray-Thompson M, Schwartz B, Chakravorty S, Wu Z, Mander PK, Kruidenier L, Reid RA, Burkhart W, Turunen BJ, Rong JX, Wagner C, Moyer MB, Wells C, Hong X, Moore JT, Williams JD, Soler D, Ghosh S, and Nolan MA. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol 9: 319–325, 2013 [DOI] [PubMed] [Google Scholar]
- 139.Manzo F, Nebbioso A, Miceli M, Conte M, De Bellis F, Carafa V, Franci G, Tambaro FP, and Altucci L. TNF-related apoptosis-inducing ligand: signalling of a ‘smart’ molecule. Int J Biochem Cell Biol 41: 460–466, 2009 [DOI] [PubMed] [Google Scholar]
- 140.Mariadason JM. Making sense of HDAC2 mutations in colon cancer. Gastroenterology 135: 1457–1459, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Marks PA. Thioredoxin in cancer—role of histone deacetylase inhibitors. Semin Cancer Biol 16: 436–443, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Marquard L, Poulsen CB, Gjerdrum LM, de Nully Brown P, Christensen IJ, Jensen PB, Sehested M, Johansen P, and Ralfkiaer E. Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology 54: 688–698, 2009 [DOI] [PubMed] [Google Scholar]
- 143.Martin M, Kettmann R, and Dequiedt F. Class IIa histone deacetylases: regulating the regulators. Oncogene 26: 5450–5467, 2007 [DOI] [PubMed] [Google Scholar]
- 144.Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N, Khochbin S, Horinouchi S, and Yoshida M. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J 21: 6820–6831, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, and Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 104: 14855–14860, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McGraw AL. Romidepsin for the treatment of T-cell lymphomas. Am J Health Syst Pharm 70: 1115–1122, 2013 [DOI] [PubMed] [Google Scholar]
- 147.McKinsey TA, Zhang CL, Lu J, and Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408: 106–111, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Meehan WJ, Samant RS, Hopper JE, Carrozza MJ, Shevde LA, Workman JL, Eckert KA, Verderame MF, and Welch DR. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J Biol Chem 279: 1562–1569, 2004 [DOI] [PubMed] [Google Scholar]
- 149.Michishita E, Park JY, Burneskis JM, Barrett JC, and Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell 16: 4623–4635, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Milde T, Oehme I, Korshunov A, Kopp-Schneider A, Remke M, Northcott P, Deubzer HE, Lodrini M, Taylor MD, von Deimling A, Pfister S, and Witt O. HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res 16: 3240–3252, 2010 [DOI] [PubMed] [Google Scholar]
- 151.Minucci S. and Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6: 38–51, 2006 [DOI] [PubMed] [Google Scholar]
- 152.Moreno DA, Scrideli CA, Cortez MA, de Paula Queiroz R, Valera ET, da Silva Silveira V, Yunes JA, Brandalise SR, and Tone LG. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol 150: 665–673, 2010 [DOI] [PubMed] [Google Scholar]
- 153.Moreno-Bost A, Szmania S, Stone K, Garg T, Hoerring A, Szymonifka J, Shaughnessy J, Jr., Barlogie B, Prentice HG, and van Rhee F. Epigenetic modulation of MAGE-A3 antigen expression in multiple myeloma following treatment with the demethylation agent 5-azacitidine and the histone deacetlyase inhibitor MGCD0103. Cytotherapy 13: 618–628, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Munster PN, Troso-Sandoval T, Rosen N, Rifkind R, Marks PA, and Richon VM. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res 61: 8492–8497, 2001 [PubMed] [Google Scholar]
- 155.Mutze K, Langer R, Becker K, Ott K, Novotny A, Luber B, Hapfelmeier A, Gottlicher M, Hofler H, and Keller G. Histone deacetylase (HDAC) 1 and 2 expression and chemotherapy in gastric cancer. Ann Surg Oncol 17: 3336–3343, 2010 [DOI] [PubMed] [Google Scholar]
- 156.Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, and Tsuneyoshi M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep 18: 769–774, 2007 [PubMed] [Google Scholar]
- 157.Nakajima H, Kim YB, Terano H, Yoshida M, and Horinouchi S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp Cell Res 241: 126–133, 1998 [DOI] [PubMed] [Google Scholar]
- 158.Nebbioso A, Carafa V, Benedetti R, and Altucci L. Trials with ‘epigenetic’ drugs: an update. Mol Oncol 6: 657–682, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, Alvarez R, Schiavone EM, Ferrara F, Bresciani F, Weisz A, de Lera AR, Gronemeyer H, and Altucci L. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med 11: 77–84, 2005 [DOI] [PubMed] [Google Scholar]
- 160.Nebbioso A, Manzo F, Miceli M, Conte M, Manente L, Baldi A, De Luca A, Rotili D, Valente S, Mai A, Usiello A, Gronemeyer H, and Altucci L. Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep 10: 776–782, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Newbold A, Lindemann RK, Cluse LA, Whitecross KF, Dear AE, and Johnstone RW. Characterisation of the novel apoptotic and therapeutic activities of the histone deacetylase inhibitor romidepsin. Mol Cancer Ther 7: 1066–1079, 2008 [DOI] [PubMed] [Google Scholar]
- 162.Newman JC, He W, and Verdin E. Mitochondrial protein acylation and intermediary metabolism: regulation by sirtuins and implications for metabolic disease. J Biol Chem 287: 42436–42443, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nicolas E, Yamada T, Cam HP, Fitzgerald PC, Kobayashi R, and Grewal SI. Distinct roles of HDAC complexes in promoter silencing, antisense suppression and DNA damage protection. Nat Struct Mol Biol 14: 372–380, 2007 [DOI] [PubMed] [Google Scholar]
- 164.Noonan AM, Eisch RA, Liewehr DJ, Sissung TM, Venzon DJ, Flagg TP, Haigney MC, Steinberg SM, Figg WD, Piekarz RL, and Bates SE. Electrocardiographic studies of romidepsin demonstrate its safety and identify a potential role for K(ATP) channel. Clin Cancer Res 19: 3095–3104, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.North BJ. and Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol 5: 224, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Oehme I, Deubzer HE, Lodrini M, Milde T, and Witt O. Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opin Investig Drugs 18: 1605–1617, 2009 [DOI] [PubMed] [Google Scholar]
- 167.Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von Deimling A, Fischer M, and Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res 15: 91–99, 2009 [DOI] [PubMed] [Google Scholar]
- 168.Okada T, Tanaka K, Nakatani F, Sakimura R, Matsunobu T, Li X, Hanada M, Nakamura T, Oda Y, Tsuneyoshi M, and Iwamoto Y. Involvement of P-glycoprotein and MRP1 in resistance to cyclic tetrapeptide subfamily of histone deacetylase inhibitors in the drug-resistant osteosarcoma and Ewing's sarcoma cells. Int J Cancer 118: 90–97, 2006 [DOI] [PubMed] [Google Scholar]
- 169.Ouaissi M, Sielezneff I, Silvestre R, Sastre B, Bernard JP, Lafontaine JS, Payan MJ, Dahan L, Pirro N, Seitz JF, Mas E, Lombardo D, and Ouaissi A. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann Surg Oncol 15: 2318–2328, 2008 [DOI] [PubMed] [Google Scholar]
- 170.Park JH, Kim SH, Choi MC, Lee J, Oh DY, Im SA, Bang YJ, and Kim TY. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem Biophys Res Commun 368: 318–322, 2008 [DOI] [PubMed] [Google Scholar]
- 171.Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD, and Lam EW. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther 9: 844–855, 2010 [DOI] [PubMed] [Google Scholar]
- 172.Petrie K, Guidez F, Howell L, Healy L, Waxman S, Greaves M, and Zelent A. The histone deacetylase 9 gene encodes multiple protein isoforms. J Biol Chem 278: 16059–16072, 2003 [DOI] [PubMed] [Google Scholar]
- 173.Pflum MK, Tong JK, Lane WS, and Schreiber SL. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem 276: 47733–47741, 2001 [DOI] [PubMed] [Google Scholar]
- 174.Prince HM, Bishton MJ, and Johnstone RW. Panobinostat (LBH589): a potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol 5: 601–612, 2009 [DOI] [PubMed] [Google Scholar]
- 175.Rasmussen TA, Schmeltz Sogaard O, Brinkmann C, Wightman F, Lewin SR, Melchjorsen J, Dinarello C, Ostergaard L, and Tolstrup M. Comparison of HDAC inhibitors in clinical development: effect on HIV production in latently infected cells and T-cell activation. Hum Vaccin Immunother 9: 993–1001, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Reddy RM, Yeow WS, Chua A, Nguyen DM, Baras A, Ziauddin MF, Shamimi-Noori SM, Maxhimer JB, Schrump DS, and Nguyen DM. Rapid and profound potentiation of Apo2L/TRAIL-mediated cytotoxicity and apoptosis in thoracic cancer cells by the histone deacetylase inhibitor Trichostatin A: the essential role of the mitochondria-mediated caspase activation cascade. Apoptosis 12: 55–71, 2007 [DOI] [PubMed] [Google Scholar]
- 177.Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, and Marks PA. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci U S A 95: 3003–3007, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Riessland M, Brichta L, Hahnen E, and Wirth B. The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells. Hum Genet 120: 101–110, 2006 [DOI] [PubMed] [Google Scholar]
- 179.Ropero S. and Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol 1: 19–25, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rosato RR, Almenara JA, Dai Y, and Grant S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol Cancer Ther 2: 1273–1284, 2003 [PubMed] [Google Scholar]
- 181.Rosato RR, Almenara JA, and Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res 63: 3637–3645, 2003 [PubMed] [Google Scholar]
- 182.Rossig L, Li H, Fisslthaler B, Urbich C, Fleming I, Forstermann U, Zeiher AM, and Dimmeler S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res 91: 837–844, 2002 [DOI] [PubMed] [Google Scholar]
- 183.Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, and Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci U S A 96: 4592–4597, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Saji S, Kawakami M, Hayashi S, Yoshida N, Hirose M, Horiguchi S, Itoh A, Funata N, Schreiber SL, Yoshida M, and Toi M. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene 24: 4531–4539, 2005 [DOI] [PubMed] [Google Scholar]
- 185.Saouaf SJ, Li B, Zhang G, Shen Y, Furuuchi N, Hancock WW, and Greene MI. Deacetylase inhibition increases regulatory T cell function and decreases incidence and severity of collagen-induced arthritis. Exp Mol Pathol 87: 99–104, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sasaki H, Moriyama S, Nakashima Y, Kobayashi Y, Kiriyama M, Fukai I, Yamakawa Y, and Fujii Y. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer 46: 171–178, 2004 [DOI] [PubMed] [Google Scholar]
- 187.Sato T, Suzuki M, Sato Y, Echigo S, and Rikiishi H. Sequence-dependent interaction between cisplatin and histone deacetylase inhibitors in human oral squamous cell carcinoma cells. Int J Oncol 28: 1233–1241, 2006 [PubMed] [Google Scholar]
- 188.Savickiene J, Treigyte G, Borutinskaite VV, and Navakauskiene R. Antileukemic activity of combined epigenetic agents, DNMT inhibitors zebularine and RG108 with HDAC inhibitors, against promyelocytic leukemia HL-60 cells. Cell Mol Biol Lett 17: 501–525, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Segre CV. and Chiocca S. Regulating the regulators: the post-translational code of class I HDAC1 and HDAC2. J Biomed Biotechnol 2011: 690848, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shahbazian MD. and Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 76: 75–100, 2007 [DOI] [PubMed] [Google Scholar]
- 191.Shao Y, Gao Z, Marks PA, and Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A 101: 18030–18035, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Shen H. and Laird PW. Interplay between the cancer genome and epigenome. Cell 153: 38–55, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, Shrikant P, Fenstermaker R, and Pili R. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS One 7: e30815, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sherman EJ, Su YB, Lyall A, Schoder H, Fury MG, Ghossein RA, Haque S, Lisa D, Shaha AR, Tuttle RM, and Pfister DG. Evaluation of romidepsin for clinical activity and radioactive iodine reuptake in radioactive iodine-refractory thyroid carcinoma. Thyroid 23: 593–599, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Shultz MD, Cao X, Chen CH, Cho YS, Davis NR, Eckman J, Fan J, Fekete A, Firestone B, Flynn J, Green J, Growney JD, Holmqvist M, Hsu M, Jansson D, Jiang L, Kwon P, Liu G, Lombardo F, Lu Q, Majumdar D, Meta C, Perez L, Pu M, Ramsey T, Remiszewski S, Skolnik S, Traebert M, Urban L, Uttamsingh V, Wang P, Whitebread S, Whitehead L, Yan-Neale Y, Yao YM, Zhou L, and Atadja P. Optimization of the in vitro cardiac safety of hydroxamate-based histone deacetylase inhibitors. J Med Chem 54: 4752–4772, 2011 [DOI] [PubMed] [Google Scholar]
- 196.Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, and Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Rev Anticancer Ther 10: 935–954, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, and Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res 65: 11136–11145, 2005 [DOI] [PubMed] [Google Scholar]
- 198.Skov V, Larsen TS, Thomassen M, Riley CH, Jensen MK, Bjerrum OW, Kruse TA, and Hasselbalch HC. Increased gene expression of histone deacetylases in patients with Philadelphia-negative chronic myeloproliferative neoplasms. Leuk Lymphoma 53: 123–129, 2012 [DOI] [PubMed] [Google Scholar]
- 199.Song J, Noh JH, Lee JH, Eun JW, Ahn YM, Kim SY, Lee SH, Park WS, Yoo NJ, Lee JY, and Nam SW. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 113: 264–268, 2005 [DOI] [PubMed] [Google Scholar]
- 200.Song Y, Shiota M, Tamiya S, Kuroiwa K, Naito S, and Tsuneyoshi M. The significance of strong histone deacetylase 1 expression in the progression of prostate cancer. Histopathology 58: 773–780, 2011 [DOI] [PubMed] [Google Scholar]
- 201.Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, Cortes J, Wierda WG, Ouzounian S, Quezada A, Pierce S, Estey EH, Issa JP, Kantarjian HM, and Garcia-Manero G. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood 110: 2302–2308, 2007 [DOI] [PubMed] [Google Scholar]
- 202.Steiner FA, Hong JA, Fischette MR, Beer DG, Guo ZS, Chen GA, Weiser TS, Kassis ES, Nguyen DM, Lee S, Trepel JB, and Schrump DS. Sequential 5-Aza 2′-deoxycytidine/depsipeptide FK228 treatment induces tissue factor pathway inhibitor 2 (TFPI-2) expression in cancer cells. Oncogene 24: 2386–2397, 2005 [DOI] [PubMed] [Google Scholar]
- 203.Strevel EL, Ing DJ, and Siu LL. Molecularly targeted oncology therapeutics and prolongation of the QT interval. J Clin Oncol 25: 3362–3371, 2007 [DOI] [PubMed] [Google Scholar]
- 204.Sun X, Wei L, Chen Q, and Terek RM. HDAC4 represses vascular endothelial growth factor expression in chondrosarcoma by modulating RUNX2 activity. J Biol Chem 284: 21881–21890, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Suvarna BS. Sirtuins: the future insight. Kathmandu Univ Med J 10: 77–82, 2012 [DOI] [PubMed] [Google Scholar]
- 206.Svechnikova I, Almqvist PM, and Ekstrom TJ. HDAC inhibitors effectively induce cell type-specific differentiation in human glioblastoma cell lines of different origin. Int J Oncol 32: 821–827, 2008 [PubMed] [Google Scholar]
- 207.Tang Y, Zhao W, Chen Y, Zhao Y, and Gu W. Acetylation is indispensable for p53 activation. Cell 133: 612–626, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tate CR, Rhodes LV, Segar HC, Driver JL, Pounder FN, Burow ME, Collins-and Burow BM. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res 14: R79, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Taylor DM, Maxwell MM, Luthi-Carter R, and Kazantsev AG. Biological and potential therapeutic roles of sirtuin deacetylases. Cell Mol Life Sci 65: 4000–4018, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Thomas S, Thurn KT, Raha P, Chen S, and Munster PN. Efficacy of histone deacetylase and estrogen receptor inhibition in breast cancer cells due to concerted down regulation of Akt. PLoS One 8: e68973, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Thompson RC, Vardinogiannis I, and Gilmore TD. The sensitivity of diffuse large B-cell lymphoma cell lines to histone deacetylase inhibitor-induced apoptosis is modulated by BCL-2 family protein activity. PLoS One 8: e62822, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Thurn KT, Thomas S, Moore A, and Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol 7: 263–283, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Timp W. and Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 13: 497–510, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ting AH, McGarvey KM, and Baylin SB. The cancer epigenome—components and functional correlates. Genes Dev 20: 3215–3231, 2006 [DOI] [PubMed] [Google Scholar]
- 215.Toffolo E, Rusconi F, Paganini L, Tortorici M, Pilotto S, Heise C, Verpelli C, Tedeschi G, Maffioli E, Sala C, Mattevi A, and Battaglioli E. Phosphorylation of neuronal lysine-specific demethylase 1LSD1/KDM1A impairs transcriptional repression by regulating interaction with CoREST and histone deacetylases HDAC1/2. J Neurochem [Epub ahead of print]; DOI: 10.1111/jnc.12457, 2013 [DOI] [PubMed] [Google Scholar]
- 216.Toh Y, Ohga T, Endo K, Adachi E, Kusumoto H, Haraguchi M, Okamura T, and Nicolson GL. Expression of the metastasis-associated MTA1 protein and its relationship to deacetylation of the histone H4 in esophageal squamous cell carcinomas. Int J Cancer 110: 362–367, 2004 [DOI] [PubMed] [Google Scholar]
- 217.Tong JK, Hassig CA, Schnitzler GR, Kingston RE, and Schreiber SL. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 395: 917–921, 1998 [DOI] [PubMed] [Google Scholar]
- 218.Torti D. and Trusolino L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils. EMBO Mol Med 3: 623–636, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ueda T, Takai N, Nishida M, Nasu K, and Narahara H. Apicidin, a novel histone deacetylase inhibitor, has profound anti-growth activity in human endometrial and ovarian cancer cells. Int J Mol Med 19: 301–308, 2007 [PubMed] [Google Scholar]
- 220.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, and Marks PA. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A 102: 673–678, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Uribesalgo I. and Di Croce L. Dynamics of epigenetic modifications in leukemia. Brief Funct Genomics 10: 18–29, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Vaiserman AM. and Pasyukova EG. Epigenetic drugs: a novel anti-aging strategy? Front Genet 3: 224, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Valenzuela L, Dhillon N, and Kamakaka RT. Transcription independent insulation at TFIIIC-dependent insulators. Genetics 183: 131–148, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Van den Wyngaert I, de Vries W, Kremer A, Neefs J, Verhasselt P, Luyten WH, and Kass SU. Cloning and characterization of human histone deacetylase 8. FEBS Lett 478: 77–83, 2000 [DOI] [PubMed] [Google Scholar]
- 225.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, and Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U S A 101: 15064–15069, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.VanOosten RL, Earel JK, Jr., and Griffith TS. Histone deacetylase inhibitors enhance Ad5-TRAIL killing of TRAIL-resistant prostate tumor cells through increased caspase-2 activity. Apoptosis 12: 561–571, 2007 [DOI] [PubMed] [Google Scholar]
- 227.Verdel A, Curtet S, Brocard MP, Rousseaux S, Lemercier C, Yoshida M, and Khochbin S. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr Biol 10: 747–749, 2000 [DOI] [PubMed] [Google Scholar]
- 228.Verdel A. and Khochbin S. Identification of a new family of higher eukaryotic histone deacetylases. Coordinate expression of differentiation-dependent chromatin modifiers. J Biol Chem 274: 2440–2445, 1999 [DOI] [PubMed] [Google Scholar]
- 229.Verdin E, Dequiedt F, and Kasler HG. Class II histone deacetylases: versatile regulators. Trends Genet 19: 286–293, 2003 [DOI] [PubMed] [Google Scholar]
- 230.Vojinovic J. and Damjanov N. HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol Med 17: 397–403, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wade PA. Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum Mol Genet 10: 693–698, 2001 [DOI] [PubMed] [Google Scholar]
- 232.Wang GG, Allis CD, and Chi P. Chromatin remodeling and cancer, Part I: covalent histone modifications. Trends Mol Med 13: 363–372, 2007 [DOI] [PubMed] [Google Scholar]
- 233.Wang JC, Kafeel MI, Avezbakiyev B, Chen C, Sun Y, Rathnasabapathy C, Kalavar M, He Z, Burton J, and Lichter S. Histone deacetylase in chronic lymphocytic leukemia. Oncology 81: 325–329, 2011 [DOI] [PubMed] [Google Scholar]
- 234.Wang WH, Cheng LC, Pan FY, Xue B, Wang DY, Chen Z, and Li CJ. Intracellular trafficking of histone deacetylase 4 regulates long-term memory formation. Anat Rec (Hoboken) 294: 1025–1034, 2011 [DOI] [PubMed] [Google Scholar]
- 235.Weberpals JI, O'Brien AM, Niknejad N, Garbuio KD, Clark-Knowles KV, and Dimitroulakos J. The effect of the histone deacetylase inhibitor M344 on BRCA1 expression in breast and ovarian cancer cells. Cancer Cell Int 11: 29, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wedel S, Hudak L, Seibel JM, Makarevic J, Juengel E, Tsaur I, Wiesner C, Haferkamp A, and Blaheta RA. Impact of combined HDAC and mTOR inhibition on adhesion, migration and invasion of prostate cancer cells. Clin Exp Metastasis 28: 479–491, 2011 [DOI] [PubMed] [Google Scholar]
- 237.Whetstine JR, Ceron J, Ladd B, Dufourcq P, Reinke V, and Shi Y. Regulation of tissue-specific and extracellular matrix-related genes by a class I histone deacetylase. Mol Cell 18: 483–490, 2005 [DOI] [PubMed] [Google Scholar]
- 238.Wickman C. and Kramer H. Obesity and kidney disease: potential mechanisms. Semin Nephrol 33: 14–22, 2013 [DOI] [PubMed] [Google Scholar]
- 239.Witt O, Deubzer HE, Milde T, and Oehme I. HDAC family: what are the cancer relevant targets? Cancer Lett 277: 8–21, 2009 [DOI] [PubMed] [Google Scholar]
- 240.Wozniak MB, Villuendas R, Bischoff JR, Aparicio CB, Martinez Leal JF, de La Cueva P, Rodriguez ME, Herreros B, Martin-Perez D, Longo MI, Herrera M, Piris MA, and Ortiz-Romero PL. Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica 95: 613–621, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Xia CQ. and Smith PG. Drug efflux transporters and multidrug resistance in acute leukemia: therapeutic impact and novel approaches to mediation. Mol Pharmacol 82: 1008–1021, 2012 [DOI] [PubMed] [Google Scholar]
- 242.Xiao JJ, Foraker AB, Swaan PW, Liu S, Huang Y, Dai Z, Chen J, Sadee W, Byrd J, Marcucci G, and Chan KK. Efflux of depsipeptide FK228 (FR901228, NSC-630176) is mediated by P-glycoprotein and multidrug resistance-associated protein 1. J Pharmacol Exp Ther 313: 268–276, 2005 [DOI] [PubMed] [Google Scholar]
- 243.Xiao JJ, Huang Y, Dai Z, Sadee W, Chen J, Liu S, Marcucci G, Byrd J, Covey JM, Wright J, Grever M, and Chan KK. Chemoresistance to depsipeptide FK228 [(E)-(1S,4S,10S,21R)-7-[(Z)-ethylidene]-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,2 0,23-tetraazabicyclo[8,7,6]-tricos-16-ene-3,6,9,22-pentanone] is mediated by reversible MDR1 induction in human cancer cell lines. J Pharmacol Exp Ther 314: 467–475, 2005 [DOI] [PubMed] [Google Scholar]
- 244.Xie HJ, Noh JH, Kim JK, Jung KH, Eun JW, Bae HJ, Kim MG, Chang YG, Lee JY, Park H, and Nam SW. HDAC1 inactivation induces mitotic defect and caspase-independent autophagic cell death in liver cancer. PLoS One 7: e34265, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Xue Y, Wong J, Moreno GT, Young MK, Cote J, and Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell 2: 851–861, 1998 [DOI] [PubMed] [Google Scholar]
- 246.Yamashita Y, Shimada M, Harimoto N, Rikimaru T, Shirabe K, Tanaka S, and Sugimachi K. Histone deacetylase inhibitor trichostatin A induces cell-cycle arrest/apoptosis and hepatocyte differentiation in human hepatoma cells. Int J Cancer 103: 572–576, 2003 [DOI] [PubMed] [Google Scholar]
- 247.Yao YL. and Yang WM. The metastasis-associated proteins 1 and 2 form distinct protein complexes with histone deacetylase activity. J Biol Chem 278: 42560–42568, 2003 [DOI] [PubMed] [Google Scholar]
- 248.Yao YL. and Yang WM. Beyond histone and deacetylase: an overview of cytoplasmic histone deacetylases and their nonhistone substrates. J Biomed Biotechnol 2011: 146493, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Yao YL, Yang WM, and Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol Cell Biol 21: 5979–5991, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Yeo W, Chung HC, Chan SL, Wang LZ, Lim R, Picus J, Boyer M, Mo FK, Koh J, Rha SY, Hui EP, Jeung HC, Roh JK, Yu SC, To KF, Tao Q, Ma BB, Chan AW, Tong JH, Erlichman C, Chan AT, and Goh BC. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: a multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J Clin Oncol 30: 3361–3367, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.You A, Tong JK, Grozinger CM, and Schreiber SL. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc Natl Acad Sci U S A 98: 1454–1458, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Yu SL, Lee DC, Son JW, Park CG, Lee HY, and Kang J. Histone deacetylase 4 mediates SMAD family member 4 deacetylation and induces 5-fluorouracil resistance in breast cancer cells. Oncol Rep 30: 1293–1300, 2013 [DOI] [PubMed] [Google Scholar]
- 253.Yuan GB, Kuang AR, Fan Q, Yu LB, and Mi YX. Combined effects of all-trans-retinoic acid and trichostatin A on the induction of differentiation of thyroid carcinoma cells. Chin J Cancer 29: 379–384, 2010 [DOI] [PubMed] [Google Scholar]
- 254.Zhang B, Qin L, Zhou CJ, Liu YL, Qian HX, and He SB. SIRT3 expression in hepatocellular carcinoma and its impact on proliferation and invasion of hepatoma cells. Asian Pac J Trop Med 6: 649–652, 2013 [DOI] [PubMed] [Google Scholar]
- 255.Zhang D, Li J, Costa M, Gao J, and Huang C. JNK1 mediates degradation HIF-1alpha by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer Res 70: 813–823, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Zhang Y, Iratni R, Erdjument-Bromage H, Tempst P, and Reinberg D. Histone deacetylases and SAP18, a novel polypeptide, are components of a human Sin3 complex. Cell 89: 357–364, 1997 [DOI] [PubMed] [Google Scholar]
- 257.Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, and Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J 22: 1168–1179, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, and Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev 13: 1924–1935, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Zhang Y, Sun ZW, Iratni R, Erdjument-Bromage H, Tempst P, Hampsey M, and Reinberg D. SAP30, a novel protein conserved between human and yeast, is a component of a histone deacetylase complex. Mol Cell 1: 1021–1031, 1998 [DOI] [PubMed] [Google Scholar]
- 260.Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y, Sun J, Yu Y, Zhou W, Zheng Q, Wu M, Otterson GA, and Zhu WG. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol Cell Biol 26: 2782–2790, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Zhong HM, Ding QH, Chen WP, and Luo RB. Vorinostat, a HDAC inhibitor, showed anti-osteoarthritic activities through inhibition of iNOS and MMP expression, p38 and ERK phosphorylation and blocking NF-kappaB nuclear translocation. Int Immunopharmacol 17: 329–335, 2013 [DOI] [PubMed] [Google Scholar]
- 262.Zhou W, Liang IC, and Yee NS. Histone deacetylase 1 is required for exocrine pancreatic epithelial proliferation in development and cancer. Cancer Biol Ther 11: 659–670, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Zhou X, Marks PA, Rifkind RA, and Richon VM. Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci U S A 98: 10572–10577, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Zou H, Wu Y, Navre M, and Sang BC. Characterization of the two catalytic domains in histone deacetylase 6. Biochem Biophys Res Commun 341: 45–50, 2006 [DOI] [PubMed] [Google Scholar]
- 265.Zuco V, De Cesare M, Cincinelli R, Nannei R, Pisano C, Zaffaroni N, and Zunino F. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLoS One 6: e29085, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]