

Abstract
Site-specific protein-DNA photo-cross-linking was used to show that, when bound to its cognate site at various distances upstream of the TATA element, the chimeric transcriptional activator GAL4-VP16 can physically interact with a TATA box-binding protein (TBP)-transcription factor IIA (TFIIA)-TFIIB complex assembled on the TATA element. This result implies DNA bending and looping of promoter DNA as a result of the physical interaction between GAL4-VP16 and an interface of the TBP-TFIIA-TFIIB complex. This protein-protein interaction on promoter DNA minimally requires the presence of one GAL4 binding site and the formation of a quaternary complex containing TBP, TFIIB, and TFIIA on the TATA element. Notably, the topology of the TBP-TFIIA-TFIIB-promoter complex is not altered significantly by the interaction with DNA-bound activators. We also show that the ability of GAL4-VP16 to activate transcription through a single GAL4 binding site varies according to its precise location and orientation relative to the TATA element and that it can approach the efficiency obtained with multiple binding sites. Taken together, our results indicate that the spatial positioning of the DNA-bound activation domain is important for efficient activation, possibly by maximizing its interactions with the transcriptional machinery including the TBP-TFIIA-TFIIB-promoter quaternary complex.
Most models of transcriptional activation imply a physical interaction of DNA-bound transcriptional activators and the transcription machinery assembled on core promoters (1–3). This contact between the activation domain and components of the transcription machinery has been attributed diverse functions including: (i) the recruitment of key transcription factors (e.g. general transcription factors or co-activators) at the promoter, (ii) the stimulation of enzymatic activities involved in the transcription reaction (e.g. promoter melting, phosphorylation, initiation of RNA chain synthesis), and (iii) the relief of transcriptional blockades induced by various types of repressors including nucleosomes. In support of this view of transcriptional activation, a number of protein-protein interactions between various activation domains and members of the RNA polymerase II (RNAPII)1 transcription machinery have been characterized in solution and found to be important in mediating transcriptional activation (1–3). However, little is known about the formation of these protein-protein interactions when the interacting partners are bound to promoter DNA.
Ultimately, transcriptional activators regulate the activity of the basal RNAPII transcription machinery. The transcription reaction is a multi-step process in which a preinitiation complex containing TBP, TFIIB, TFIIE, TFIIF, TFIIH, and RNA-PII is first assembled onto promoter DNA (4, 5). TBP recognizes and binds the TATA element, inducing a bend of about 80° in the DNA helix (6, 7). TFIIB binds to and stabilizes the TBP-promoter complex (8). TFIIF, a factor composed of two subunits called RAP74 and RAP30, binds tightly to RNAPII (9, 10), recruits the enzyme to the TBP-TFIIB-promoter complex, and induces an isomerization of the preinitiation complex that includes wrapping of promoter DNA around RNAPII (11, 12). TFIIE, a factor also composed of two subunits called TFIIEα and TFIIEβ, stabilizes the preinitiation complex (13) and is involved in the melting of promoter DNA at the transcription initiation site through an ATP-independent mechanism (14, 15). TFIIH, a nine-subunit factor that has both kinase and helicase activities, mediates the ATP-dependent melting of promoter DNA and is involved in the transition between the initiation and elongation states of the complex (16–21) (also see Ref. 22 for a review). The initiation of chain synthesis proceeds through a cycle of abortive initiation events during which RNA-PII synthesizes short 2–10-nucleotide transcripts before escaping the promoter and entering a productive elongation mode (23). The activity of elongating RNAPII is modulated through the action of a number of elongation factors (24, 25).
The acidic activator GAL4-VP16 is a chimeric polypeptide composed of the DNA-binding domain of the yeast GAL4 protein (amino acids 1–147) and the transcriptional acidic activation domain of the viral protein VP16 (amino acids 412–490) (26). When GAL4 DNA-binding sites are located upstream of the TATA box of a promoter, this activator has been shown to stimulate transcription in a synergistic manner both in vivo and in vitro (see Ref. 27 for example). GAL4-VP16 can act on various steps of the transcription reaction, probably via its many interactions with components of the basal transcriptional machinery. These include interactions with TBP (28, 29), TFIIB (30, 31), TFIIA (32), TFIIH (33), and hTAFII32 (34). GAL4-VP16 was shown to stimulate three distinct events during the formation of the preinitiation complex: (i) formation of the TFIID-TFIIA-DNA complex (34); (ii) TFIIB recruitment (35); and, (iii) RAP30, TFIIEα, and RNAPII recruitment (36). A number of reports indicate that GAL4-VP16 can also act on open complex formation (37), transcript elongation (38), and reinitiation (39, 40).
Using site-specific protein-DNA photo-cross-linking, we have analyzed the physical interaction between GAL4-VP16 molecules bound to promoter sites various distances from the TATA element and a complex containing TBP, TFIIB, and TFIIA assembled onto the TATA element. Using templates with either one or five GAL4 DNA-binding sites, we show that DNA-bound GAL4-VP16 does indeed cross-link to many positions in the vicinity of the TATA element where a TBP-TFIIA-TFIIB complex is assembled. Interaction with DNA-bound GAL4-VP16 does not significantly modify the conformation of the TBP-TFIIA-TFIIB-promoter complex. We also show that a GAL4-VP16 dimer bound to one GAL4 binding site can stimulate transcription with an efficiency approaching that of GAL4-VP16 dimers bound to five sites if the single dimer is properly positioned upstream of the TATA box.
MATERIALS AND METHODS
Protein Factors
Recombinant yeast TBP (29), human TFIIB (41), human TFIIA (42), and GAL4-VP16 (43) were purified as described previously. The activity of all purified proteins was tested in various DNA binding assays and in in vitro transcription reactions.
In Vitro Transcription
In vitro transcriptions were performed as described previously (44). Promoters containing the adenovirus-2 major late (Ad2ML) promoter from nucleotides −50 to +10 fused to a G-less cassette and a varying number of GAL4 DNA-binding sites were used. Supercoiled template DNA (300 ng) and competitor DNA (300 ng, empty vector) were incubated with 1–5 μl of HeLa nuclear extract in both the absence and presence of 5–40 ng of GAL4-VP16. Transcript formation was monitored by primer extension and quantified using a PhosphorImager (Amersham Biosciences).
Primer Extension Analysis
Primer extension analyses were performed as previously described (45). Transcription reactions were incubated with a radiolabeled 30-mer complementary to nucleotides +37 to +67 of the coding strand of the Ad2ML promoter. This primer was labeled with [γ-32P]ATP using T4 polynucleotide kinase (United States Biochemical).
Protein-DNA Photo-cross-linking
The synthesis of the photoreactive nucleotide N3R-dUMP, the preparation of the probes, and the conditions for the binding reactions were performed as previously described (46). Photoprobes containing the photoreactive nucleotide at positions −45/−48, −39/−40, −34, −20, −19, and −15 were used. A typical reaction contained 100 ng of TBP, 100 ng of TFIIB, 200 ng TFIIA, and various amounts of GAL4-VP16 (as indicated in the figures). UV irradiation, nuclease treatment, and SDS-PAGE analysis of radio-labeled photo-cross-linking products were performed as described previously (46).
RESULTS AND DISCUSSION
DNA-bound GAL4-VP16 Specifically Cross-links to the Region of the TATA Box in the Presence of TBP, TFIIA, and TFIIB
Most models of transcriptional activation stipulate that an activator bound to its specific binding site upstream of the basal promoter could make contacts with the transcriptional machinery at the basal promoter through the formation of a loop in the promoter DNA (reviewed in Refs. 1–3). To obtain direct evidence of the interaction of activators with the transcriptional machinery on promoter DNA (as opposed to interactions between free molecules in solution), we asked whether or not GAL4-VP16 bound to its cognate sites upstream of the TATA box could cross-link to the region of TATA in the presence of TBP, TFIIB, and TFIIA. All three of these factors previously have been shown to bind to the activation domain of VP16 in solution (28 –32). To perform the cross-linking experiments with a template that supports transcriptional activation, we tested the ability of templates carrying between two and five GAL4 binding sites, placed in both possible orientations upstream of the TATA box, to support GAL4-VP16 activation in vitro from the Ad2ML promoter. As summarized in Fig. 1, the template with five GAL4 sites most efficiently supported transcriptional activation by GAL4-VP16 in a HeLa nuclear extract (11.9-fold). Notably, we were unable to obtain synergistic activation of this promoter under our experimental conditions. We then decided to use the template carrying five GAL4 sites (GML 33) in our photo-cross-linking experiments. As shown in Fig. 2A, GAL4-VP16 does cross-link to photoprobe − 39/−40 in the presence of TBP, TFIIB, and TFIIA (left panel). As expected, the p55 subunit of both recombinant TFIIA and TBP also cross-linked to photoprobe −39/−40 (Refs. 47–49; see below). The intensity of the GAL4-VP16 cross-linking signal in the presence of TBP, TFIIA, and TFIIB is about 30-fold higher than that obtained in the absence of TBP, TFIIA, and TFIIB (right panel). To ensure that the cross-linking of GAL4-VP16 to positions −39/−40 in the presence of TBP, TFIIA, and TFIIB requires binding of Gal-VP16 to the GAL4 sites located 37 bp upstream of TATA, we compared cross-linking signals obtained with photoprobes possessing or lacking GAL4 binding sites. Fig. 2B shows that the cross-linking of GAL4-VP16 to photo-probe −39/−40 is dependent upon the presence of the GAL4 binding sites. Together, these results indicate that a TBP-TFIIA-TFIIB-GAL4-VP16 complex can form on promoter DNA and that its formation requires the presence of both the TATA element and the GAL4 binding sites, thereby implying DNA bending and looping.
Fig. 1. Transcriptional activation using templates with multiple GAL4 binding sites.

Various numbers of tandemly repeated GAL4 binding sites (two sites, GML 272 and GML 274; three sites, GML 276 and GML 278; four sites, GML 280 and GML 282; five sites, GML 33) were placed in both possible orientations 37 bp upstream of the TATA box of the Ad2ML basal promoter fused to a G-less cassette. Maximal activation levels, expressed as the ratio of the number of transcripts produced in the presence GAL4-VP16 to that produced in its absence (fold activation), represent an average of 3–5 independent experiments.
Fig. 2. Photo-cross-linking of GAL4-VP16 to positions −39/−40.

A, photo-cross-linking experiments with photoprobe −39/−40 performed in the presence of increasing amounts of GAL4-VP16 either in the presence or absence of TBP, TFIIA, and TFIIB. The positions of TBP, TFIIA (p55), and GAL4-VP16 and the molecular mass markers (M) are indicated. B, photo-cross-linking experiments with photoprobe −39/−40 performed in the presence of TBP, TFIIA, and TFIIB with increasing amounts of GAL4-VP16 on templates carrying either five GAL4 binding sites or ones lacking a GAL4 binding site. The positions of TBP, TFIIA (p55), and GAL4-VP16 and the molecular mass markers (M) are indicated.
DNA-bound GAL4-VP16 Does Not Significantly Modify the Conformation of a TBP-TFIIA-TFIIB Complex on Promoter DNA
We and others have previously shown that TBP and TFIIA cross-link to a number of positions in the region of the TATA element of a promoter (47–49). We next compared the cross-linking of TBP and TFIIA at positions −45/−48, −34, −20, −19, and −15 in both the presence and the absence of GAL4-VP16 (Fig. 3). GAL4-VP16 specifically cross-linked to all of the photoprobes except −19. In the context of DNA looping, this finding is not surprising because five GAL4-VP16 dimers bound to a stretch of 100 bp of DNA (e.g. the five GAL4 binding sites) are expected to make multiple contacts with the TBP-TFIIA-TFIIB complex associated with the TATA box region.
Fig. 3. Photo-cross-linking of TBP, TFIIA, and GAL4-VP16 to different positions along promoter DNA.
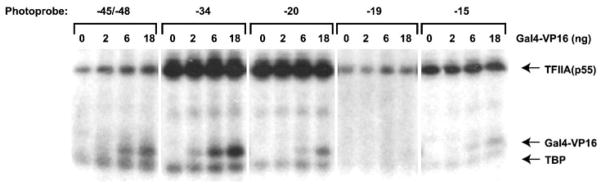
Photo-cross-linking experiments with photo-probes −45/−48, −34, −20, −19, and −15 were performed in the presence of TBP, TFIIB, and TFIIA with increasing amounts of GAL4-VP16 on templates containing five GAL4 binding sites. The positions of TBP, TFIIA (p55), and GAL4-VP16 are indicated.
Our results also show that the cross-linking of TBP and TFIIA to positions −39/−40 (Fig. 2), −34, −20, −19, and −15 (Fig. 3) is not significantly affected by the presence of GAL4-VP16. However, the cross-linking of TFIIA to photoprobe −45/−48 is weakly, but reproducibly, increased by increasing amounts of GAL4-VP16. Because this stimulation of the TFIIA cross-links by GAL4-VP16 is observed only at positions −45/−48, it is unlikely to be the result of either a general stabilization or a more efficient assembly of the TBP-TFIIA-TFIIB-promoter complex. We favor the alternative conclusion that DNA looping, induced by the contact of DNA-bound GAL4-VP16 with the TBP-TFIIA-TFIIB complex on the TATA box, tethers the DNA helix at −45/−48 to the bulk of TFIIA (see Fig. 6B for a schematic representation).
Fig. 6. Models for the interaction of DNA-bound GAL4-VP16 and the TBP-TFIIA-TFIIB-promoter (TATA) quaternary complex.

A, schematic representation of the expected location of GAL4-VP16 dimers on templates carrying a single GAL4 binding site. The figure shows a transversal view of the DNA with GAL4-VP16 dimers positioned on various faces of the helix. The color code represents the activation level obtained with each template: green, forward orientation; red, reverse orientation. Darker colors corresponds to stronger activation levels and lighter colors to lower activation levels. B, one GAL4-VP16 dimer bound to upstream promoter DNA (GML 258) can contact a TBP-TFIIA-TFIIB complex positioned at the TATA box through looping of the DNA helix. Although our results do not determine the interface of the TBP-TFIIA-TFIIB complex that is contacted by DNA-bound GAL4-VP16, one possibility is a domain of TFIIB (Val-135, Arg-137, Asn-139 and Asn-140; depicted in red) that previously has been shown to be essential for transcriptional activation (62). However, our results indicate that this TFIIB domain alone is not sufficient for the interaction of DNA-bound GAL4-VP16 with the transcriptional machinery bound to core promoter. As shown, DNA looping increases the contact of TFIIA with the DNA helix around positions −45/−48.
We have been unable to obtain cross-linking of DNA-bound GAL4-VP16 in the region of the TATA box in the presence of TBP alone, TBP and TFIIA, or TBP and TFIIB (data not shown), indicating that a TBP-TFIIA-TFIIB-promoter quaternary complex is minimally required to produce DNA looping and stable binding to DNA-bound GAL4-VP16. We did obtain cross-linking of DNA-bound GAL4-VP16 to the TATA box when we used photoprobes carrying a single GAL4 binding site (Fig. 4). This cross-linking signal is specific because most of it was lost in experiments performed in the absence of TBP, TFIIA, and TFIIB or when we used a template lacking GAL4 binding sites (data not shown). This result indicates that a single GAL4-VP16 dimer bound to DNA can interact with a TBP-TFIIB-TFIIA complex on the TATA box through DNA looping.
Fig. 4. Photo-cross-linking of GAL4-VP16 on templates containing a single GAL4 binding site.
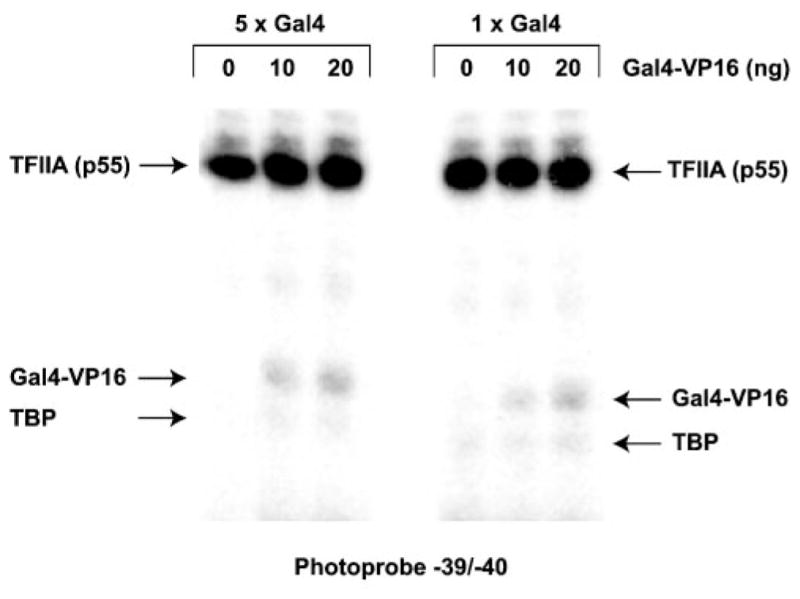
Cross-linking results using photoprobe −39/−40 carrying either five (GML 33) or only one (GML 258) GAL4 binding site(s) are compared. Experiments were performed with increasing amounts of GAL4-VP16 in the presence of TBP, TFIIA, and TFIIB.
The Location and Orientation of a Single GAL4 Binding Site Relative to the TATA Element Influences Activation Levels in Vitro
Our conclusion that GAL4-VP16 molecules bound to a single GAL4 binding site can efficiently contact a TBP-TFIIA-TFIIB-promoter complex suggests that a single GAL4 site could support efficient transcriptional activation if it is positioned properly upstream of the core promoter. To test the idea two series of templates with the Ad2ML basal promoter placed under the control of one GAL4 binding site (in both possible orientations) were constructed (Fig. 5A). In each series, the distance between the GAL4 site and the TATA box is different, being 37 (GML 248 and 250), 39 (GML 252 and 254), 41 (GML 256 and 258), 43 (GML 260 and 262), 45 (GML 264 and 266), or 47 (GML 268 and 270) bp. Because of the helical structure of DNA, GAL4-VP16 dimers bound to the various templates are localized on different faces of the DNA helix. Each 2-bp increment will move the activator molecule by 72° (a full helical turn being 360°). We used in vitro transcription assays in either the presence or absence of GAL4-VP16 to test activation with each of the constructs. The results are shown in Fig. 5B and are summarized in Fig. 5C. The different templates supported activation levels ranging from 2.3- to 7.5-fold. This result indicates that the precise location of the GAL4 binding site relative to the TATA element affects activation. We believe that activation depends on the precise face of the helix where the VP16 activation domain is positioned (see Fig. 6A for a schematic representation). As the site moves away from the TATA box, making one full turn around the DNA helix (in 72° increments), transcription activation increases up until the center of the site is located at nucleotide −80 and then decreases thereafter. This finding argues in favor of a preferred orientation, or side, of the helix for the GAL4-VP16 activation domain such that the interactions with the transcriptional machinery are maximized. In support of this conclusion, the templates that most efficiently support activation are those that place the GAL4-VP16 dimer on the same face of the DNA helix, whereas those that are least efficient place the GAL4-VP16 dimer on the opposite face of the helix (Fig. 6A).
Fig. 5. Transcriptional activation using templates with a single GAL4 binding site.

A, templates containing the Ad2ML basal promoter under the control of one GAL4 binding site. The GAL4 binding site was placed in both possible orientations (see arrows) at 37 (GML 248 and GML 250), 39 (GML 252 and GML 254), 41 (GML 256 and GML 258), 43 (GML 260 and GML 262), 45 (GML 264 and GML 266), and 47 (GML 268 and GML 270) bp from the TATA element. The maximum activation level (fold activation) of each template is indicated and is the average of five experiments. B, example of the primer extension experiments used to determine the activation levels. Experiments using the GML 33 template, which contains five GAL4 binding sites, are shown for purposes of comparison. C, summary of our in vitro transcription results.
As shown in Fig. 5, the activation level obtained with a template containing five GAL4 binding sites (11.7-fold) is only slightly higher than that obtained using a template with one GAL4 site at position −71 (7.5-fold). Templates containing two, 3, and four GAL4 binding sites produced equivalent or lower activation levels (Fig. 1). In contrast to the results of Lin et al. (27), we did not observe a synergistic effect of multiple GAL4 binding sites on transcriptional activation in vitro. Clearly, under our conditions, that is using non-chromatin templates, the adequate positioning of a single GAL4 binding site relative to TATA has an effect comparable with the use of multiple binding sites.
Together, our results support the notion that DNA-bound GAL4-VP16 can contact the transcription machinery around the TATA box, induce a looping of the DNA double helix, and stimulate transcription in a nonsynergistic manner. Although our results do not allow for the determination of the exact cause-and-effect relationship between these events, they indicate that the positioning of DNA-bound activation domains upstream of TATA is important in activation mechanisms and fully support the concept of a crucial role for direct physical contact between an activator and the RNAPII machinery through promoter DNA looping. An increasing body of evidence indicates that promoter DNA is wrapped around the transcription machinery during transcription initiation. By making contacts with the transcription machinery, a process that induces DNA bending and looping, transcriptional activators possibly stimulate the formation of a DNA wrap around RNAPII.
In recent years, many reports have indicated that transcriptional activators such as VP16 interact not only with the basal transcription machinery but also with the Mediator (TRAP-SMCC-ARC) complex (50 –52). In yeast, the VP16 activation domain can also modify the transcriptional status of chromatin by (i) relieving nucleosome-mediated repression through the recruitment of histone acetyltransferase complexes such as SAGA and NuA4 (53, 54) and by (ii) targeting the Swi/Snf complex, thus facilitating chromatin remodeling (55). In mammalian cells, VP16 interacts functionally with p300 (56, 57). The multiple interactions of a given activation domain with various transcriptional targets are required to produce both the high levels of activation obtained in vivo and in vitro on chromatin templates and the synergistic response obtained when iterative binding sites for a transcriptional activator drive the expression of a reporter gene on nucleosome-containing templates (1–5). Although previous reports have established that VP16 binds directly to TFIIA, TBP, and TFIIB, our results indicate that, separately, these interactions are not stable when the proteins are bound to promoter DNA and suggest that the TBP-TFIIA-TFIIB complex as a whole entity is the target of DNA-bound GAL4-VP16 dimers. More recently, a number of reports have shown that the ordered recruitment (and concomitant ordered release) of many transcription factors, including the general initiation factors, RNAPII, the Mediator complex, and diverse chromatin-modifying complexes, is required for gene activation in vivo (58). Here, we have provided biochemical evidence showing that specific protein-protein interactions between factors that are bound to distant positions on a DNA fragment can induce the formation of a loop that is important for transcriptional activation. The loop structure may be a key topological requirement for the association of some transcription complexes to promoter DNA. For example, and as we have suggested previously (22), the loop structure formed within promoter DNA as a result of the assembly of a TFIID-TFIIA-promoter complex (59) can provide the architectural requirement for the subsequent binding of the RNAPII complex. Rapid, tightly regulated association-dissociation of transcription complexes on promoter DNA could be facilitated by the formation of the loop structure. In support of this notion, topological constraints imposed to promoter DNA such as tight bending in the TATA box and initiation site regions were suggested to be involved in the displacement of a key repressive nucleosome during activation of the interferon-β gene (60, 61).
Acknowledgments
We thank members of our laboratory for helpful discussions, Diane Bourque for artwork, Vincent Trinh for the design of the molecular models in Fig. 6, and Will Home for critical reading of the manuscript.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research.
The abbreviations used are: RNAPII, RNA polymerase II; Ad2ML, adenovirus-2 major late; RAP, RNA polymerase II-associated protein; TBP, TATA box-binding protein; TF, transcription factor.
References
- 1.Ptashne M, Gann A. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- 2.Sauer F, Tjian R. Curr Opin Genet Dev. 1997;7:176–181. doi: 10.1016/s0959-437x(97)80126-8. [DOI] [PubMed] [Google Scholar]
- 3.Berk AJ. Curr Opin Cell Biol. 1999;11:330–335. doi: 10.1016/S0955-0674(99)80045-3. [DOI] [PubMed] [Google Scholar]
- 4.Orphanides G, Lagrange T, Reinberg D. Genes Dev. 1996;10:2657–2683. doi: 10.1101/gad.10.21.2657. [DOI] [PubMed] [Google Scholar]
- 5.Hampsey M. Microbiol Mol Biol Rev. 1998;62:465–503. doi: 10.1128/mmbr.62.2.465-503.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JL, Nikolov DB, Burley SK. Nature. 1993;365:520–527. doi: 10.1038/365520a0. [DOI] [PubMed] [Google Scholar]
- 7.Kim Y, Geiger JH, Hahn S, Sigler PB. Nature. 1993;365:512–520. doi: 10.1038/365512a0. [DOI] [PubMed] [Google Scholar]
- 8.Maldonado E, Ha I, Cortes P, Weis L, Reinberg D. Mol Cell Biol. 1990;10:6335–6347. doi: 10.1128/mcb.10.12.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flores O, Lu H, Killeen M, Greenblatt J, Burton ZF, Reinberg D. Proc Natl Acad Sci U S A. 1991;88:9999–10003. doi: 10.1073/pnas.88.22.9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conaway RC, Garrett KP, Hanley JP, Conaway JW. Proc Natl Acad Sci U S A. 1991;88:6205–6209. doi: 10.1073/pnas.88.14.6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forget D, Robert F, Grondin G, Burton ZF, Greenblatt J, Coulombe B. Proc Natl Acad Sci U S A. 1997;94:7150–7155. doi: 10.1073/pnas.94.14.7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robert F, Douziech M, Forget D, Egly JM, Greenblatt J, Burton ZF, Coulombe B. Mol Cell. 1998;2:341–351. doi: 10.1016/s1097-2765(00)80278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robert F, Forget D, Li J, Greenblatt J, Coulombe B. J Biol Chem. 1996;271:8517–8520. doi: 10.1074/jbc.271.15.8517. [DOI] [PubMed] [Google Scholar]
- 14.Flores O, Lu H, Reinberg D. J Biol Chem. 1992;267:2786–2793. [PubMed] [Google Scholar]
- 15.Holstege FC, Tantin D, Carey M, van der Vliet PC, Timmers HT. EMBO J. 1995;14:810–819. doi: 10.1002/j.1460-2075.1995.tb07059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaeffer L, Roy R, Humbert S, Moncollin V, Vermeulen W, Hoeijmakers JH, Chambon P, Egly JM. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- 17.Drapkin R, Reardon JT, Ansari A, Huang JC, Zawel L, Ahn K, Sancar A, Reinberg D. Nature. 1994;368:769–772. doi: 10.1038/368769a0. [DOI] [PubMed] [Google Scholar]
- 18.Lu H, Flores O, Weinmann R, Reinberg D. Proc Natl Acad Sci U S A. 1991;88:10004–10008. doi: 10.1073/pnas.88.22.10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serizawa H, Conaway RC, Conaway JW. Proc Natl Acad Sci U S A. 1992;89:7476–7480. doi: 10.1073/pnas.89.16.7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dvir A, Conaway RC, Conaway JW. Proc Natl Acad Sci U S A. 1997;94:9006–9010. doi: 10.1073/pnas.94.17.9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan Q, Moreland RJ, Conaway JW, Conaway RC. J Biol Chem. 1999;274:35668–35675. doi: 10.1074/jbc.274.50.35668. [DOI] [PubMed] [Google Scholar]
- 22.Coulombe B, Burton ZF. Microbiol Mol Biol Rev. 1999;63:457–478. doi: 10.1128/mmbr.63.2.457-478.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holstege FC, Fiedler U, Timmers HT. EMBO J. 1997;16:7468–7480. doi: 10.1093/emboj/16.24.7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conaway JW, Shilatifard A, Dvir A, Conaway RC. Trends Biochem Sci. 2000;25:375–380. doi: 10.1016/s0968-0004(00)01615-7. [DOI] [PubMed] [Google Scholar]
- 25.Shilatifard A. FASEB J. 1998;12:1437–1446. doi: 10.1096/fasebj.12.14.1437. [DOI] [PubMed] [Google Scholar]
- 26.Sadowski I, Ma J, Triezenberg S, Ptashne M. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 27.Lin YS, Carey MF, Ptashne M, Green MR. Cell. 1988;54:659–664. doi: 10.1016/s0092-8674(88)80010-2. [DOI] [PubMed] [Google Scholar]
- 28.Stringer KF, Ingles CJ, Greenblatt J. Nature. 1990;345:783–786. doi: 10.1038/345783a0. [DOI] [PubMed] [Google Scholar]
- 29.Ingles CJ, Shales M, Cress WD, Triezenberg SJ, Greenblatt J. Nature. 1991;351:588–590. doi: 10.1038/351588a0. [DOI] [PubMed] [Google Scholar]
- 30.Lin YS, Ha I, Maldonado E, Reinberg D, Green MR. Nature. 1991;353:569–571. doi: 10.1038/353569a0. [DOI] [PubMed] [Google Scholar]
- 31.Lin YS, Green MR. Cell. 1991;64:971–981. doi: 10.1016/0092-8674(91)90321-o. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi N, Boyer TG, Berk AJ. Mol Cell Biol. 1995;15:6465–6473. doi: 10.1128/mcb.15.11.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao H, Pearson A, Coulombe B, Truant R, Zhang S, Regier JL, Triezenberg SJ, Reinberg D, Flores O, Ingles CJ, Greenblatt J. Mol Cell Biol. 1994;14:7013–7024. doi: 10.1128/mcb.14.10.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klemm RD, Goodrich JA, Zhou S, Tjian R. Proc Natl Acad Sci U S A. 1995;92:5788–5792. doi: 10.1073/pnas.92.13.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W, Gralla JD, Carey M. Genes Dev. 1992;6:1716–1727. doi: 10.1101/gad.6.9.1716. [DOI] [PubMed] [Google Scholar]
- 36.Choy B, Green MR. Nature. 1993;366:531–536. doi: 10.1038/366531a0. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, Carey M, Gralla JD. Science. 1992;255:450–453. doi: 10.1126/science.1310361. [DOI] [PubMed] [Google Scholar]
- 38.Blau J, Xiao H, McCracken S, O’Hare P, Greenblatt J, Bentley D. Mol Cell Biol. 1996;16:2044–2055. doi: 10.1128/mcb.16.5.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zawel L, Kumar KP, Reinberg D. Genes Dev. 1995;9:1479–1490. doi: 10.1101/gad.9.12.1479. [DOI] [PubMed] [Google Scholar]
- 40.Yudkovsky N, Ranish JA, Hahn S. Nature. 2000;408:225–229. doi: 10.1038/35041603. [DOI] [PubMed] [Google Scholar]
- 41.Ha I, Lane WS, Reinberg D. Nature. 1991;352:689–695. doi: 10.1038/352689a0. [DOI] [PubMed] [Google Scholar]
- 42.Ozer J, Moore PA, Bolden AH, Lee A, Rosen CA, Lieberman PM. Genes Dev. 1994;8:2324–2335. doi: 10.1101/gad.8.19.2324. [DOI] [PubMed] [Google Scholar]
- 43.Chasman DI, Leatherwood J, Carey M, Ptashne M, Kornberg RD. Mol Cell Biol. 1989;9:4746–4749. doi: 10.1128/mcb.9.11.4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coulombe B, Killeen M, Liljelund P, Honda B, Xiao H, Ingles CJ, Greenblatt J. Gene Expr. 1992;2:99–110. [PMC free article] [PubMed] [Google Scholar]
- 45.Carey M, Lin YS, Green MR, Ptashne M. Nature. 1990;345:361–364. doi: 10.1038/345361a0. [DOI] [PubMed] [Google Scholar]
- 46.Robert F, Coulombe B. Methods Mol Biol. 2001;148:383–393. doi: 10.1385/1-59259-208-2:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coulombe B, Li J, Greenblatt J. J Biol Chem. 1994;269:19962–19967. [PubMed] [Google Scholar]
- 48.Lagrange T, Kim TK, Orphanides G, Ebright YW, Ebright RH, Reinberg D. Proc Natl Acad Sci U S A. 1996;93:10620–10625. doi: 10.1073/pnas.93.20.10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langelier MF, Forget D, Rojas A, Porlier Y, Burton ZF, Coulombe B. J Biol Chem. 2001;276:38652–38657. doi: 10.1074/jbc.M106422200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ito M, Yuan CX, Malik S, Gu W, Fondell JD, Yamamura S, Fu ZY, Zhang X, Qin J, Roeder RG. Mol Cell. 1999;3:361–370. doi: 10.1016/s1097-2765(00)80463-3. [DOI] [PubMed] [Google Scholar]
- 51.Naar AM, Beaurang PA, Zhou S, Abraham S, Solomon W, Tjian R. Nature. 1999;398:828–832. doi: 10.1038/19789. [DOI] [PubMed] [Google Scholar]
- 52.Malik S, Roeder RG. Trends Biochem Sci. 2000;25:277–283. doi: 10.1016/s0968-0004(00)01596-6. [DOI] [PubMed] [Google Scholar]
- 53.Utley RT, Ikeda K, Grant PA, Cote J, Steger DJ, Eberharter A, John S, Workman JL. Nature. 1998;394:498–502. doi: 10.1038/28886. [DOI] [PubMed] [Google Scholar]
- 54.Ikeda K, Steger DJ, Eberharter A, Workman JL. Mol Cell Biol. 1999;19:855–863. doi: 10.1128/mcb.19.1.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neely KE, Hassan AH, Wallberg AE, Steger DJ, Cairns BR, Wright AP, Workman JL. Mol Cell. 1999;4:649–655. doi: 10.1016/s1097-2765(00)80216-6. [DOI] [PubMed] [Google Scholar]
- 56.Kraus WL, Manning ET, Kadonaga JT. Mol Cell Biol. 1999;19:8123–8135. doi: 10.1128/mcb.19.12.8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kundu TK, Palhan VB, Wang Z, An W, Cole PA, Roeder RG. Mol Cell. 2000;6:551–561. doi: 10.1016/s1097-2765(00)00054-x. [DOI] [PubMed] [Google Scholar]
- 58.Cosma M. Mol Cell. 2002;10:227. doi: 10.1016/s1097-2765(02)00604-4. [DOI] [PubMed] [Google Scholar]
- 59.Oelgeschlager T, Chiang CM, Roeder RG. Nature. 1996;382:735–738. doi: 10.1038/382735a0. [DOI] [PubMed] [Google Scholar]
- 60.Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D. Cell. 2000;103:667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- 61.Lomvardas S, Thanos D. Cell. 2001;106:685–696. doi: 10.1016/s0092-8674(01)00490-1. [DOI] [PubMed] [Google Scholar]
- 62.Shaw SP, Carson DJ, Dorsey MJ, Ma J. Proc Natl Acad Sci U S A. 1997;94:2427–2432. doi: 10.1073/pnas.94.6.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]