

Abstract
Cystic fibrosis (CF), the most common recessive autosomal disease among Caucasians, is caused by mutations in the gene encoding the CF transmembrane conductance regulator (CFTR) protein. The most common mutation, F508del, leads to CFTR impaired plasma membrane trafficking. Therapies modulating CFTR basic defect are emerging, such as VX-809, a corrector of F508del-CFTR traffic which just succeeded in a Phase III clinical trial. We recently showed that VX-809 is additive to two other correctors (VRT-325 and compound 4a). Here, we aimed to determine whether the differential rescuing by these compounds results from cell-specific factors or rather from distinct effects at the early biogenesis and/or processing. The rescuing efficiencies of the above three correctors were first compared in different cellular models (primary respiratory cells, cystic fibrosis bronchial epithelial and baby hamster kidney [BHK] cell lines) by functional approaches: micro-Ussing chamber and iodide efflux. Next, biochemical methods (metabolic labeling, pulse-chase and immunoprecipitation) were used to determine their impact on CFTR biogenesis / processing. Functional analyses revealed that VX-809 has the greatest rescuing efficacy and that the relative efficiencies of the three compounds are essentially maintained in all three cellular models tested. Nevertheless, biochemical data show that VX-809 significantly stabilizes F508del-CFTR immature form, an effect that is not observed for C3 nor C4. VX-809 and C3 also significantly increase accumulation of immature CFTR. Our data suggest that VX-809 increases the stability of F508del-CFTR immature form at an early phase of its biogenesis, thus explaining its increased efficacy when inducing its rescue.
Keywords: Cellular systems, CFTR function, CFTR modulators, mechanism of action, novel therapies
Introduction
Cystic fibrosis (CF) is the most common severe autosomal recessive disease in Caucasians, caused by mutations in the gene (Riordan et al. 1989), that encodes for the CF transmembrane conductance regulator (CFTR) protein, a chloride (Cl−) channel expressed at the apical membrane of epithelial cells and regulating salt and water transport in epithelia (Collins 1992). Most CF mutations cause disease by disrupting either CFTR cell surface expression or its function as a Cl− channel.
The biosynthesis and maturation of CFTR is a complex and inefficient process, even for the wild-type (wt) protein. Correctly folded CFTR traffics from the ER to the plasma membrane (PM), passing through the Golgi where it undergoes full glycosylation. In contrast, when the protein is misfolded as occurs for the most frequent disease-causing mutant – F508del (Collins 1992), it is retained in the ER, thus remaining only core-glycosylated, and is rapidly targeted for ER-associated degradation via the ubiquitin-proteasomal pathway (UPP) (Jensen et al. 1995; Farinha et al. 2002; Farinha and Amaral 2005).
F508del mutation, associated with a severe clinical phenotype (Collins 1992), disrupts the function of CFTR in three ways: firstly, due to the protein folding defect, it prevents the trafficking to the PM (Cheng et al. 1990). Secondly, F508del greatly impairs channel regulation causing a major gating defect (Schultz et al. 1999). Thirdly, when rescued to the PM F508del-CFTR still presents a highly decreased half-life due to both accelerated endocytosis and fast ubiquitin-dependent turnover (Sharma et al. 2004; Swiatecka-Urban et al. 2005). Knowledge of how F508del and other CF mutations cause loss of Cl− channel function is leading to new, rational therapeutic approaches aimed at correcting such basic defects, through CFTR chemical modulators.
Rescue of F508del-CFTR was first shown for low temperature incubation (Denning et al. 1992) thus demonstrating that the folding defect associated with the F508del mutation is temperature-sensitive, but also that it is rescuable. Soon after, glycerol was also shown to achieve F508del-CFTR rescue in recombinant cells, albeit with some toxicity (Sato et al. 1996). These findings triggered a quest for small molecule compounds to rescue F508del-CFTR. These include both pharmacological/ chemical chaperones, directly promoting protein folding specifically/ unspecifically (Amaral 2004), or proteostasis modulators acting in the general cellular folding or quality control machineries (Balch et al. 2011).
Several high-throughput screening (HTS) efforts have produced a number of lead CFTR modulators, of two types, namely: (1) “correctors” (Van Goor et al. 2006) that rescue the trafficking defect of F508del-CFTR; and (2) “potentiators” (Pedemonte et al. 2005b) that restore Cl− channel gating. These compounds have become increasingly potent, more specific, and less toxic. Correctors include laboratory reagents like the quinazoline derivative VRT-325 (C3) (Van Goor et al. 2006) or compound 4a (C4), or preclinical correctors like VX-661 or VX-809 (Lumacaftor). VX-809, a predrug corrector showing the ability to rescue F508del-CFTR to the PM in primary human bronchial epithelial (HBE) cells in vitro (Van Goor et al. 2011), has undergone Phase II clinical trials with limited success under a monotherapy regime (Clancy et al. 2012), but more recently it was tested in Phase III clinical trials as combined therapy with the FDA/EMA-approved potentiator VX-770 (ivacaftor Kalydeco®), showing better results (Boyle et al. 2014).
Such screening initiatives have proven that F508del-CFTR correctors are much more difficult to identify than potentiators. Indeed, the effects of the best corrector (VX-809, lumacaftor) that has gone through Phase III clinical trial in combination with ivacaftor (VERTEX, 2014) are not as striking as those of the approved potentiator (Pedemonte et al. 2010; Van Goor et al. 2011).
Some of these correctors, such as VX-809 have been described to promote maturation of F508del-CFTR in a specific way (Wang et al. 2006; Van Goor et al. 2011), while others are unspecific. C3 for instance promotes maturation of both P-glycoprotein- and CFTR-processing mutants (Van Goor et al. 2006; Wang et al. 2007b). C4 also promotes the rescue of intracellularly retained mutants of melanocortin-4 receptor, a G-PCR related to juvenile obesity (Farinha et al. 2013). Moreover, it was shown that combinations of correctors may have additive effects on rescuing F508del-CFTR and other mutants, thus opening avenues for combination therapies (Wang et al. 2007a; Farinha et al. 2013).
However, the mechanism of action (MoA) of these compounds/drugs is still not completely understood. Recent studies have highlighted that not only cell background can influence the pharmacological rescue of mutant CFTR (Pedemonte et al. 2010; Amaral and Farinha 2013), but also the polarization status in the case of epithelial cells (Rowe et al. 2010). Furthermore, for CFTR modulators that are just laboratory reagents, there is only reduced knowledge on their comparative efficacy in heterologous expression systems and primary cells from CF patients, which may be critical to validate the respective MoA.
Regarding VX-809, several groups (including our own) have proposed that it acts through binding to the pocket created by the deletion of F508 at the interface between the first nucleotide-binding domain (NBD1) and the fourth intracellular loop (ICL4) of the second membrane-spanning domain (Farinha et al. 2013; He et al. 2013; Okiyoneda et al. 2013). VX-809 was also found not to confer to F508del-CFTR the long-term thermal stability of wt-CFTR (He et al. 2013). However, more studies need to address the efficacy of these compounds in native tissues from CF patients, so as to give a better prediction of their in vivo efficacy.
Our goal here was to compare the efficacy of VX-809 in functionally rescuing F508del-CFTR with that of currently used correctors C3 (VRT-325) and C4 in primary HBE cells and in other cell systems. Our data show that VX-809 always rescued F508del-CFTR more efficiently than the two other compounds in all cell types tested. In order to further investigate the step at which VX-809 may be acting, we determined the impact of the three compounds upon CFTR biosynthesis and turnover to find that VX-809 exerts its action at the early steps of CFTR biogenesis.
Materials and Methods
Cell culture and treatment with compounds
Access to non-CF and CF patient-derived tissue samples received approval from the Ethics Committee of the Hospitals involved. Primary non-CF and CF (derived from F508del CFTR homozygous patients) airway epithelial cells were isolated as previously (Moniz et al. 2013). Briefly, cells were expanded on collagen I/ fibronectin-coated plastic dishes before passage to the porous membrane inserts. Both primary and cystic fibrosis bronchial epithelial (CFBE) cells stably transduced with either wt- or F508del-CFTR (CFBE) (Bebok et al. 2005) were grown on collagen IV-coated porous membranes in air–liquid interface (ALI) for, respectively, 4–5 weeks and 6–9 days before experimentation. CFBE and baby hamster kidney (BHK) cells stably expressing wt- or F508del-CFTR were cultured as previously described (Farinha et al. 2002; Moniz et al. 2013). Cells were treated with the indicated concentrations of compound 4a (C4), VRT-325 (C3) (CFFT modulator library) and VX-809 (Selleck Chem, Munich, Germany) in 0.5% FBS (BHK) or 1% FBS (CFBE) supplemented medium. Concentrations of the compounds were selected as previously described (Farinha et al. 2013) or, in the case of functional assessment in CFBE cells, after testing the effectiveness of different concentrations relatively to its toxicity (see Results and Discussion and Conclusions), so as to obtain the maximal effect without a marked toxic effect. Stock solutions of forskolin (10 mmol/L) (Sigma, St.Louis, MO, USA), genistein (50 mmol/L) (Sigma) and CFTR channel blocker CFTRInh-172 (10 mmol/L) were prepared in either pure ethanol or dimethyl sulfoxide (DMSO). The incubation dose and time were selected to achieve maximal correction without toxicity for each cell line.
CFTR accumulation experiments
To assess the rate of CFTR biosynthetic accumulation, a modified pulse-labeling experiment was performed. Briefly, cells were starved for 30 min in methionine-free medium and then labeled for different periods of time (as indicated) with 150 μCi [35S]methionine at 37°C. Cells were then lysed and immunoprecipitation performed was performed with anti-CFTR 596 antibody (CFF, Bethesda, MD) as previously (Farinha et al. 2002). Rate of CFTR synthesis is measured by the incorporation of [35S]methionine and graphically represented at each time point as a fold increase relatively to the shortest incubation time (t = 10 min).
Pulse-chase
After incubation with compounds, cells expressing CFTR were starved for 30 min in methionine-free medium and then pulsed for 30 min at 37°C in the same medium supplemented with 150 μCi [35S]methionine. Pulse-chase experiments, followed by immunoprecipitation of CFTR were performed as above.
Iodide efflux
CFTR-mediated iodide efflux was measured at room temperature using the cAMP agonist forskolin (10 μmol/L) and the CFTR potentiator genistein (50 μmol/L; Sigma-Aldrich) as described (Lansdell et al. 1998; Roxo-Rosa et al. 2006).
Micro-Ussing chamber recordings
Transepithelial electrical resistance of the cells growing on Snapwell inserts was measured with the Chopstick Electrode (STX2 from WPI, Sarasota, FL, USA). Monolayers with resistance values above 450 Ω.cm2 were mounted in modified micro-Ussing chambers. Compounds (C3, C4 or VX-809) were not present during the recordings. Recordings were done as described previously (Tian et al. 2013). Briefly, the apical and basolateral surfaces of primary HBE cultures were continuously perfused with Ringer solution (in mmol/L: NaCl 145, KH2PO4 0.4, K2HPO4 1.6, D-glucose 5, MgCl2 1, Ca-gluconate 1.3). For CFBE41o-cells, the apical bath solution was replaced by a low Cl− Ringer solution containing (in mmol/L): NaCl 32; KH2PO4 0.4; K2HPO4 1.6; d-glucose 5; MgCl2 1; Ca-gluconate 5.7 and Na-gluconate 112. For primary HBE experiments, 20 μmol/L amiloride was added to apical bath solutions to exclude a potential interference by the epithelial sodium (Na+) channel (ENaC) on Vte recordings was excluded by luminal exposure to ENaC inhibitor amiloride. Following a 20 min equilibration period, baseline values were recorded. Subsequently, the cAMP agonist, forskolin (2 μmol/L), the CFTR potentiator genistein (50 μmol/L), and the CFTR channel blocker Inh172 (30 μmol/L) were added sequentially. Transepithelial resistance (Rte) was determined by applying short (1 s) current pulses (I = 0.5 μA) and the corresponding changes in transepithelial voltage (Vte) were recorded continuously. Equivalent cAMP-stimulated CFTR short-circuit currents (eqΔIsc) were calculated by Ohm’s law from Vte and Rte (eqIsc = Vte/Rte). Values for Vte were referred to the basolateral side of the monolayers.
Statistical analysis
Statistical analysis was made using two-tailed Student’s t-tests for simple (paired) comparisons (Fig. 6) and one-way Analysis of variance (ANOVA) followed by post hoc Tukey’s test for multiple comparisons (Figs.5). Statistical significance was considered for P < 0.05. The GraphPad Prism (La Jolla, CA, USA) version 5.0f software was used for statistical analysis.
Figure 6.

Turnover and processing of F508del-CFTR under treatment with C3 (A–C), C4 (D–F), and VX-809 correctors (G–I). (A, D, G) BHK cells expressing F508del-CFTR were labeled with 35S-methionine for 30 min and then chased for the indicated times (0, 1, 3, 5 h) before lysis. Immunoprecipitation was performed with the anti-CFTR M3A7 or 596 antibodies. After electrophoresis and fluorography, images were analyzed by densitometry. (B, E, H) Turnover of immature (band B) F508del-CFTR shown as the percentage of protein at a given time of chase (P) relative to the amount at t = 0 (P0). (C, F, I) Efficiency of processing of band B into band C shown as the percentage of band C at a given time of chase relative to the amount of band B at t = 0. Data are mean ± SEM at each point (n = 3–5). *P < 0.05 relative to DMSO.
Figure 5.
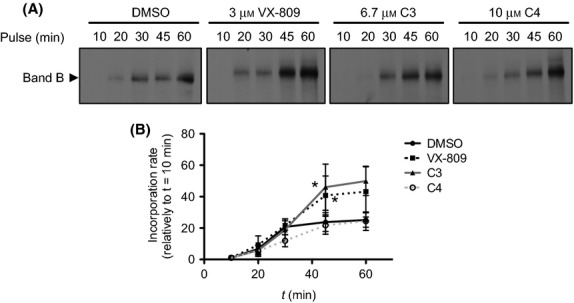
F508del-CFTR accumulation under treatment with VX-809, C3, and C4 correctors. BHK cells expressing F508del-CFTR (A) were incubated with radioactive methionine for the indicated periods of time at 37°C. (B) Rate of accumulation was measured by the increase in labeling relatively to the first time point (10 min). Data are mean ± SEM at each point (n = 4). *P < 0.05 relative to DMSO.
Results
Functional assessment of F508del-CFTR rescue by C3, C4, and VX-809 in BHK cells
We firstly functionally assessed the F508del-CFTR rescue by the three correctors in the three cellular models. To such end in BHK cells, since they do not polarize, we used the iodide efflux technique (instead of Ussing chamber measurements). BHK cells, although less relevant in the context of CFTR biology, are a simpler model that when transfected express high amounts of CFTR. This allows a more direct characterization of direct effects upon CFTR, independent of more sophisticated trafficking and regulated activity as in polarized cells. We were able to detect F508del-CFTR function after 12–24 h incubation with each of the three compounds plus potentiator genistein (Fig.1). Comparison of the respective activity peak levels with those of low-temperature rescued F508del-CFTR and of wt-CFTR, shows that for VX-809 the activity peak is ∼46% of wt-CFTR (Fig.1C), a value similar to that of low temperature rescued F508del-CFTR (∼55% of wt-CFTR), whereas C3 and C4 only restore the mutant function to ∼26% and ∼33% of wt-CFTR, respectively. Activity peaks for cells under all three correctors exhibit a delay of 2 min (VX-809) or 3 min (C3 and C4) compared to wt-CFTR (Fig.1B), whereas in low temperature rescue no delay is observed (Fig.1A).
Figure 1.
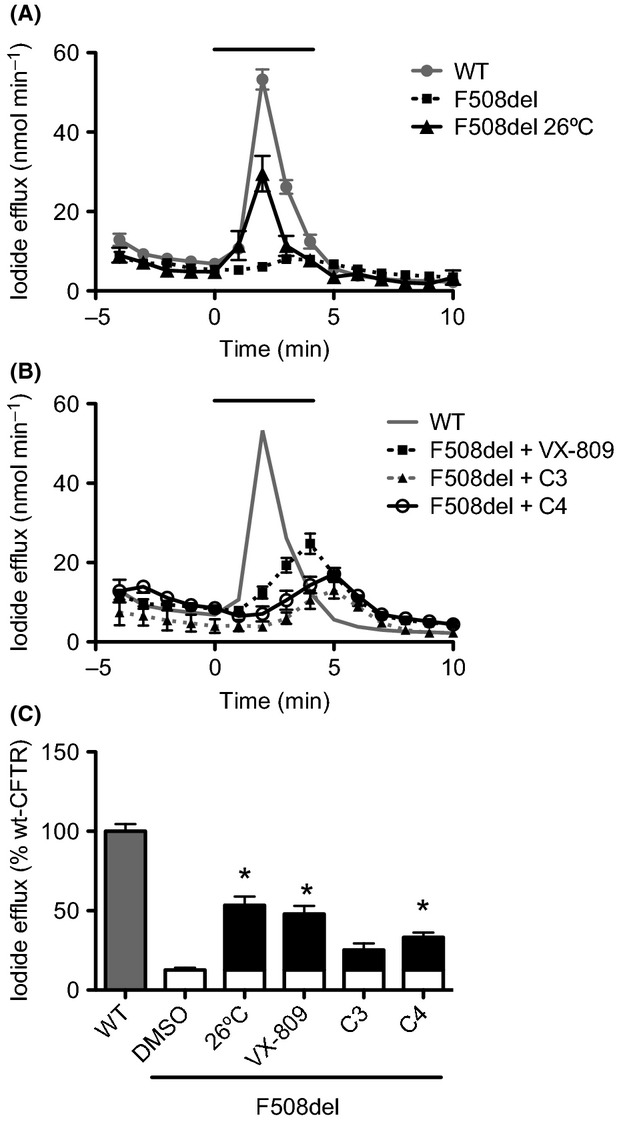
Functional assessment of F508del-CFTR rescue by C3, C4, and VX-809 in BHK cells. Iodide efflux from BHK cells stably expressing wt- or F508del-CFTR was measured directly or (F508del-CFTR cells) after low temperature (26°C, 48 h) incubation (A) or (F508del-CFTR cells) after treatment with 6.7 μmol/L C3, 10 μmol/L C4, 3 μmol/L VX-809 (B). Cells were stimulated with Forskolin (10 μmol/L) and Genistein (50 μmol/L) in the period indicated by the black solid line above graph of time course iodide efflux measurements. (C) Graph summarizing data of I− efflux peak magnitude generated by the different treatments of BHK F508del-CFTR cells expressed as a percentage of wt-CFTR activity. For F508del cells, the white bar corresponds to the efflux elicited by DMSO and the black bars to the increase promoted by each treatment. Data are mean ± SEM at each point (n = 4–6). Where error bars are not visible, the symbol has obscured them. *P < 0.05 relative to F508del-CFTR cells under DMSO (37°C).
Functional assessment of F508del-CFTR rescue by C3, C4, and VX-809 in polarized CFBE cells
We then investigated how the three correctors restore activity of F508del-CFTR stably expressed in polarized CFBE41o- cells. Variation in equivalent short-circuit currents (ΔIeq−sc) in cells treated with either VX-809 (∼2.2-fold increase vs. DMSO) or C4 (∼1.5-fold increase vs. DMSO) showed an enhancement of CFTR-mediated Cl− currents, whereas cells exposed to C3 revealed a much smaller response, consistent with reported inhibitory effect of this compound at higher concentrations on channel activity (Kim Chiaw et al. 2010) (Fig.2). Although the response of cells treated with VX-809 showed the greatest efficacy among all three correctors, low temperature restored F508del-CFTR function to even higher values (∼67% of wt-CFTR, ∼12-fold increase vs. DMSO). This observation is in agreement with previous studies in these cells (Rowe et al. 2010; Moniz et al. 2013).
Figure 2.
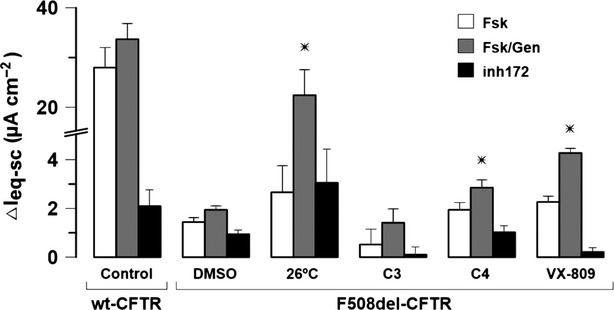
Functional assessment of F508del-CFTR rescue by C3, C4, and VX-809 in polarized CFBE41o-cells stably expressing wt- or F508del-CFTR. Ussing chamber experiments were carried out in cells incubated at low temperature (26°C, 24 h) or at 37°C and under treatment with DMSO (control) or correctors C3 (25 μmol/L, 24 h), C4 (15 μmol/L, 24 h), VX-809 (3 μmol/L, 48 h), as indicated. Summary of the induced short-circuit currents (ΔIeq−sc) of polarized CFBE41o- cells upon luminal stimulation with forskolin (2 μmol/L) and genistein (50 μmol/L) (Fsk/Gen) and blocked with CFTR inh172 (30 μmol/L). Data are means ± SEM. *P < 0.05 relative to DMSO-treated cells.
Functional assessment of endogenous F508del-CFTR rescue by C3, C4, and VX-809 in primary cultures of human bronchial epithelial cells
To test the rescuing effect of these CFTR correctors on patient-derived cells with endogenous F508del-CFTR, that is closer to in vivo conditions, we treated primary cultures of HBE cells derived from F508del-CFTR homozygous patients with compounds C3, C4, and VX-809. We used as a negative control equivalent cultures treated with the vehicle (DMSO). Response of DMSO-treated cultures cannot be distinguished from that of nontreated cultures, as we have shown previously (Moniz et al. 2013). Data from micro-Ussing chamber transepithelial voltage (Vte) measurements and respective ΔIeq−sc (Fig.3) show that VX-809 rescues F508del-CFTR function in response to either the cAMP agonist forskolin alone or in combination with potentiator genistein (Fig.3D). In contrast, the efficacy of both C3 and C4 is very modest, as previously shown (Van Goor et al. 2011). Importantly, these effects were specifically blocked by CFTR Inh172 (Fig.3C).
Figure 3.
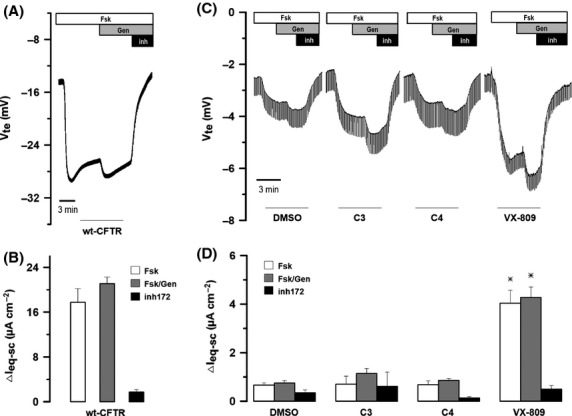
Functional assessment of F508del-CFTR Rescue by C3, C4, and VX-809 in primary cultures of CF (F508del/F508del) and non-CF human bronchial epithelial (HBE) polarized cells. (A) Original Ussing chamber recordings obtained for non-CF HBE cells (wt-CFTR) and (C) CF-HBE cells under treatments with DMSO (0.01%, 48 h), C3 (6.7 μmol/L, 24 h), C4 (10 μmol/L, 16 h) and VX-809 (3 μmol/L, 48 h). Note that, in primary F508del/F508del monolayers, VX-809 enhances the negative transepithelial voltage (Vte) deflection following the application of luminal forskolin (Fsk, 2 μmol/L), which is further potentiated by genistein (Gen, 50 μmol/L), and completely inhibited by CFTRinh-172 (inh, 30 μmol/L). (B) Summary of sensitive-ΔIeq−sc induced by CFTR agonists in wt-CFTR cells. (D) Summary of sensitive-ΔIeq−sc induced upon correctors and CFTR agonists exposure as referred in (C). Data are means ± SEM (triplicates from the same donor). *P < 0.05 relative to control (DMSO) cells.
In comparison to DMSO-treated control primary HBE cells, VX-809 treatment resulted in a ∼6-fold increase in Cl− transport in response to forskolin and genistein, whereas C3 or C4 resulted in only ∼1.5 and ∼1.2-fold increases, respectively (Fig.3D). This ∼6-fold increase in channel activity represented a recovery of activity equivalent to ∼20% of CFTR function in primary HBE cells from non-CF individuals (Fig.3A and B).
Comparison of functional correction of F508del-CFTR in different cellular models
To address the above-described cell specificity of correctors, we then compared the respective maximal effects (fold increase relatively to each DMSO-treated control). This analysis shows that VX-809 is the most effective of the three correctors (Fig.4A) in all cellular models tested. Importantly, VX-809 has its highest effect in primary cells (∼6-fold increase vs. DMSO), that is, significantly higher than observed in human bronchial epithelial (∼2.2-fold) or in hamster (∼3-fold) cell lines. The other correctors are not as effective in any of the three models tested. C3 is only effective in BHK cells whereas C4 has a modest effect both in CFBE and BHK cell lines. Curiously, neither of these laboratory correctors rescues F508del-CFTR in HBE primary cells (Figs. 3, 4A). When the rescuing efficacy of the three correctors was assessed by the percentage of wt-CFTR activity attained, once again VX-809 has the better results (Fig.4B). Interestingly, this analysis shows that the relative rescuing efficiencies for the three compounds are essentially maintained in all three cellular models tested (Fig.4B). Although the comparison shown joins together results from iodide efflux and Ussing chamber methods, both correspond, in the respective models (nonpolarized vs. epithelial polarized cells), to an assessment of global ion (Cl− or I−) transport (as opposed to a more thorough biophysical characterization of each CFTR channel), thus allowing a general inference on the overall effect of the compounds in the three models tested.
Figure 4.
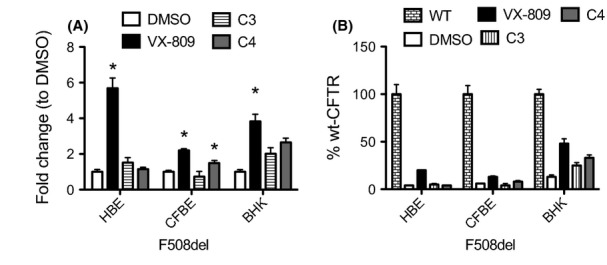
Comparison of functional correction of F508del-CFTR in different cellular models. (A) Summary of data of either induced short-circuit currents (ΔIeq−sc) upon stimulation with forskolin and genistein (for primary HBE cells or CFBE-F508del cell line) or I− efflux peak magnitude generated by the different treatments of BHK F508del-CFTR cells expressed as fold change relative to DMSO control. (B) Summary of data of either induced short-circuit currents (ΔIsc) upon stimulation with forskolin and genistein (for primary HBE cells or CFBE-F508del cell line) or I− efflux peak magnitude generated by the different treatments of BHK F508del-CFTR cells expressed as percentage of wt-CFTR control. Data are means ± SEM. *P < 0.05 relative to DMSO.
CFTR accumulation under treatment with C3, C4, and VX-809
Next, we investigated whether the higher efficiency of F508del-CFTR rescue by VX-809 is due to an effect at the early steps of biogenesis and folding of the mutant. We thus assessed the rates of protein accumulation under C3, C4, or VX-809. To this end, we exposed BHK cells expressing F508del-CFTR to each of the compounds for 24 h, then applied an [35S]methionine pulse at 37°C for different periods of time, lysed at the end of each labeling period, and CFTR was immunoprecipitated (see Materials and Methods). Due to its intrinsic properties, this cell line is a good model to biochemically assess direct effects upon CFTR biogenesis and turnover, when compared to polarized epithelial cells in which CFTR is very stable at the membrane and thus technically difficult to use in metabolic labeling experiments (Farinha et al. 2004). Results obtained for the yields of F508del-CFTR (Fig.5A) as an increase relative to the shortest incubation time (10 min) were plotted against time to determine the rate of immature form (band B) accumulation (Fig.5B). Data show that accumulation of labeled band B is increased in the presence of both VX-809 and C3, but not of C4, in comparison to the DMSO control. Indeed, under either VX-809 or C3 there is a statistically significant increase in the amount of immature full-length F508del-CFTR detected after 45 min of labeling (Fig.5B).
Turnover rate and processing efficiency of F508del-CFTR under C3, C4, and VX-809
To assess how the observed accumulation of immature F508del-CFTR (indicative of an early effect of the compounds) impacts on the stability of the immature form of CFTR and on its conversion to the mature form (band C), we analyzed the turnover of band B and the efficiency of its processing into band C by pulse-chase analysis, after 24 h-treatment with each of the three compounds (or DMSO control), and keeping the compounds present for all the duration of the experiment (see Materials and Methods).
Data in Figure6 provide evidence that the appearance of processed form of F508del-CFTR (band C) occurs earlier in VX-809-treated cells (Fig.6G, right panel) than for cells under C3 (Fig.6A, right panel) or C4 (Fig.6D, right panel). In contrast, for DMSO-treated cells (Fig.6A, D, G, left panels), only the immature form (band B) is detected. These results indicate that in compound treated cells, F508del-CFTR has exited the ER, and undergone full processing through the Golgi, plausibly reaching the cell surface, in agreement as previously reported (Pedemonte et al. 2005a; Van Goor et al. 2006, 2011). Moreover, VX-809 is statistically more efficient than C3 or C4 in doing so (as assessed by comparing dotted lines in Figs.6C, F, I by one-way ANOVA).
Quantification of band intensity over chase time to determine turnover rates (Fig.6B, E, H) and processing efficiency rates (Fig.6C, F, I) of the mutant protein under each corrector, shows that VX-809 significantly decreases the turnover rate of immature F508del-CFTR, being this accumulation statistically significant at 1, 3 and 5 h time points (Fig.6H). However, neither C3 (Fig.6B) nor C4 (Fig.6E) cause a similar effect.
This decrease in turnover rate by VX-809 also corresponds to an increase in its processing efficiency, that is, its conversion into the mature form. Indeed, while the amount of processed protein achieves ∼35% in VX-809-treated cells (Fig.6I), it only reaches ∼6% and ∼2% in cells treated with C3 (Fig.6C) or C4 (Fig.6F), respectively. These data are thus in agreement with functional data that show a higher rescuing efficacy for VX-809.
Discussion and Conclusions
In the last decade, multiple HTS initiatives identified several small molecules that partially rescue the trafficking defect of F508del-CFTR (correctors) like VRT-325 (C3), VRT-640, compound 4a (C4), as well as others with potential of becoming drugs: VX-661 and VX-809 (Pedemonte et al. 2005a; Loo et al. 2006; Van Goor et al. 2006, 2011).
Such screening initiatives have shown us so far that F508del-CFTR correctors are much more difficult to identify than potentiators. Indeed, the effects of the best one that has gone through clinical trial (VX-809, Lumacaftor) are not as spectacular as for the approved potentiator in primary airway epithelial cells from CF patients (Pedemonte et al. 2010; Van Goor et al. 2011). Its potency and efficacy is even more limited, in the clinical setting, as shown by the recent outcomes of the CFTR corrector VX-809 alone (Clancy et al. 2012) or in combination with VX-770 (VERTEX, 2014). Indeed, despite being encouraging these results are not comparable to the outcomes of CFTR potentiator VX-770 (Ivacaftor/Kalydeco) in G551D-homozygous patients (Van Goor et al. 2009; Accurso et al. 2010).
Because the MoA of correctors is not fully understood, more studies need to address the efficacy of these compounds in native tissues from CF patients, so as to give a better prediction of their in vivo efficacy.
Herein, we compared the efficacy of three well-known CFTR correctors (C3, C4) and prodrug VX-809 in different cellular models, including primary HBE cells, so as to understand whether their differential functional efficacy might be explained at the early steps of CFTR biogenesis.
Functional correction of F508del-CFTR
To address the cell specificity of correctors, our data show that the efficacy of C3, C4, and VX-809 in rescuing F508del-CFTR function depends on the cellular model tested. The rescuing efficiencies of C3, C4, and VX-809 (vs. wt-CFTR function) were (in %): 24, 32, and 46 (in BHK cells, Fig.1); 4, 8, and 13 (in CFBE41o- cells, Fig.2) and 5, 4, and 20 (in primary HBE cells, Fig.3).
These data confirm recent studies indicating that CFTR processing and activity are strongly influenced by the cellular model and evidencing enhanced maturation and cell surface stability demonstrated in polarized cells versus nonpolarizing systems (Pedemonte et al. 2010; Rowe et al. 2010). Although we used higher compound concentrations for both C3 (25 μmol/L) and C4 (15 μmol/L) in F508del-CFTR CFBE41o- cells, so as to observe functional rescue, our data demonstrate that C4 resulted in a modest ∼1.5-fold increase in ΔIeq−sc-Fsk/Gen relative to control (DMSO), smaller than that of VX-809 (∼2.2-fold) (Figs.4). On the other hand, high concentrations of C3 showed to be inhibitory on cAMP-induced channel activity (data not shown), as reported by others (Kim Chiaw et al. 2010). However, at these high concentrations of C3 and C4 some cell toxicity was elicited, as observed by decrease in Rte of the monolayers (Fig. S1) which, however, was not significant for VX-809.
Notwithstanding, these results do not warrant a full translation of efficiency into native tissues nor into the in vivo situation, as evidenced by results from clinical trials (Clancy et al. 2012). Supporting this notion, Inh172 effectively inhibits CFTR-mediated equivalent short-circuit currents (ΔIeq−sc) in primary HBE cell cultures (fig.3). However, this inhibitor only mildly inhibits CFTR-mediated currents in intact human native tissue, that is, rectal biopsies (data not shown).
Thus, differences in protein folding and biogenesis as well as in processing efficiency (as also shown here) may also contribute to the differences in the rescuing efficiencies of VX-809, C3, and C4 observed between polarized (20, 5, and 4% of wt-CFTR function in HBE cells, 13, 4, and 8% of wt-CFTR function in CFBE cells, respectively) and nonpolarized cells (46, 25, and 33% of wt-CFTR function in BHK cells, respectively). These differences compromise in fact good predictions of corrector efficacy (see also Pedemonte et al. 2010), based mainly on heterologous non-human systems. There is thus a need to use in screenings cellular models that recapitulate the compound efficacy in human bronchi. It is, nevertheless, interesting to note that the relative rescuing efficiencies by the three correctors are essentially maintained in all cellular models tested.
Insights into biogenesis and processing of correctors
To understand why VX-809 is more effective than the other two correctors, we assessed how these compounds affect the early steps of F508del-CFTR biogenesis.
C4 seems the only one of the three compounds that does not affect the accumulation of immature F508del-CFTR. Previously, C4 was described to partially correct the processing of another mutant ABC transporter, G268V-Pgp (Wang et al. 2006), and of a completely unrelated membrane protein, GPCR melanocortin-4 receptor (Farinha et al. 2013). It is thus plausible that C4 does not specifically correct a pocket of F508del-CFTR, as suggested (Farinha et al. 2013) but it may act unspecifically in secretory traffic, or as chemical chaperone.
Interestingly, C3 promotes an increase in F508del-CFTR accumulation but not a decrease in its turnover rate. This may reflect an ability to stabilize the early steps of cotranslational folding that, however, does not translate in the stabilization of full-length protein. This compound has been proposed to correct the F508del-induced defect at the NBD1:NBD2 dimer interface (Farinha et al. 2013), by promoting the interaction between the two halves of the CFTR molecule (Loo et al. 2009). Its ability to correct other misfolded variants of ABC transporters is, however, suggestive of a less specific interaction with F508del-CFTR. Nevertheless, it may just act on ABC transporters.
Some previous studies have suggested that VX-809 binds to CFTR at the NBD1:ICL4 interface (Farinha et al. 2013; He et al. 2013; Okiyoneda et al. 2013), whereas others have proposed that VX-809 may also correct the F508del-induced MSD1 misfolding (Ren et al. 2013). The direct binding of VX-809 to CFTR was later confirmed by biophysical methods (Eckford et al. 2014), that have also pointed out to mild potentiation for the compound when acutely applied to low temperature rescued CFTR. In addition, it was also shown that VX-809 ability to correct may be impaired by a chronic treatment with the potentiator VX-770 (Cholon et al. 2014; Veit et al. 2014). Our results show that VX-809 increases F508del-CFTR accumulation and decreases its turnover, so by stabilizing the mutant. Still consistent with a direct binding of the compound these data suggest that VX-809 increases the processing efficiency of F508del-CFTR as a result of an increased stabilization of the immature form. Accordingly, the lower processing efficiency of C3 and C4 may result from the absence of this stabilization which does not occur for these two molecules. It was previously shown that VX-809 does not lower the turnover of F508del-CFTR to a rate similar to its wt counterpart (He et al. 2013). However, our data shown here, demonstrate that there is a significant decrease in comparison to nontreated cell lines. Taken together, these observations are consistent with the concept that VX-809 is not able by itself to correct F508del-CFTR traffic to the levels of wt-CFTR (Amaral and Farinha 2013).
Conclusion
Our results here show that VX-809 is the most efficient of the three correctors studied in functionally rescuing F508del-CFTR in all three cellular models tested. Despite the utility of heterologous expression cells in the identification of correctors and good predictability in ranking the relative rescuing efficiencies of the compounds, data also evidence that testing them in relevant cells, that is, primary cultures of HBE cells from CF patients, is crucial to predict the effectiveness of compounds in clinical trials. On this regard, a very significant testing model would be primary cultures of human nasal epithelial cells (Beekman et al. 2014), with the additional advantage of allowing testing of modulators in patients with rare genotypes.
Our results on the MoA of VX-809 indicate that the increased efficacy of this compound seems to result from its effect on F508del-CFTR early biogenesis by stabilizing its immature form, likely by providing a kinetic advantage for the mutant to fold and thus exit the ER.
Acknowledgments
This work was supported by grants PTDC/SAU-GMG/122299/2010 (to M. D. A.), EXPL/BIM-MEC/1451/2013 (to C. M. F.) and centre grant (to BioISI, Centre Reference: UID/MULTI/04046/2013), from FCT/MCTES/PIDDAC, Portugal. MS, SC and IU are recipients of SFRH/BD/35936/2007, SFRH/BD/52491/2014 and SFRH/ BD/69180/2010 PhD fellowships (FCT, Portugal), respectively. Authors thank JP Clancy (University of Alabama, USA) for stably transduced CFBE41-o cell lines, B Bridges (RFUMS, USA) for CFTR modulators and JR Riordan (UNC, USA) for anti-CFTR antibody, both through CFF-USA. The authors thank CF patients and their families as well as José Fragata (Hospital Santa Marta, Lisboa) and Juan Pastor and Amparo Solè (Hospital Interuniversitario La Fe, Valencia) for access to explanted lungs. We are also grateful to Ines Pérez and Salvador Bejar Carbonell (Hospital la Fe (Valencia, Spain) for help with handling and shipping of patients’ materials and to Marta Palma and José Luis Múrias (FCUL, Lisboa) for help in the preparation of primary cultures.
Glossary
- ALI
air–liquid interface
- BHK
baby hamster kidney
- CFBE
cystic fibrosis bronchial epithelial
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- Cl−
chloride
- HBE
human bronchial epithelial
- HTS
high-throughput screening
- Isc
short-circuit current
- ICL4
fourth intracellular loop
- MoA
mechanism of action
- NBD1
first nucleotide-binding domain
- PM
plasma membrane
- Rte
transepithelial resistance
- Vte
transepithelial voltage
Disclosures
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Experiments carried out on polarized CFBE41o- at 37°C and treated with vehicle (DMSO, control) or correctors C3 (25 µmol/L), C4 (15 μmol/L) and VX-809 (3 μmol/L), as indicated. Summary of the transepithelial resistance (Rte) for the F508del-CFTR CFBE monolayers treated with DMSO or CFTR correctors. Data are means ± SEM (n = 3–4). *P < 0.05) relative to control (DMSO) cells.
References
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral MD. CFTR and chaperones: processing and degradation. J Mol Neurosci. 2004;23:41–48. doi: 10.1385/JMN:23:1-2:041. [DOI] [PubMed] [Google Scholar]
- Amaral MD, Farinha CM. Rescuing mutant CFTR: a multi-task approach to a better outcome in treating cystic fibrosis. Curr Pharm Des. 2013;19:3497–3508. doi: 10.2174/13816128113199990318. [DOI] [PubMed] [Google Scholar]
- Balch WE, Roth DM, Hutt DM. Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harb Perspect Biol. 2011;3:a004499. doi: 10.1101/cshperspect.a004499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebok Z, Collawn JF, Wakefield J, Parker W, Li Y, Varga K, et al. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J Physiol. 2005;569:601–615. doi: 10.1113/jphysiol.2005.096669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekman JM, Sermet-Gaudelus I, de Boeck K, Gonska T, Derichs N, Mall MA, et al. CFTR functional measurements in human models for diagnosis, prognosis and personalized therapy: report on the pre-conference meeting to the 11th ECFS Basic Science Conference, Malta, 26-29 March 2014. J Cyst Fibros. 2014;13:363–372. doi: 10.1016/j.jcf.2014.05.007. [DOI] [PubMed] [Google Scholar]
- Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2:527–538. doi: 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Cholon DM, Quinney NL, Fulcher ML, Esther CR, Jr, Das J, Dokholyan NV, et al. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci Transl Med. 2014;6:246ra296. doi: 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67:12–18. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS. Cystic fibrosis: molecular biology and therapeutic implications. Science. 1992;256:774–779. doi: 10.1126/science.1375392. [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Eckford PD, Ramjeesingh M, Molinski S, Pasyk S, Dekkers JF, Li C, et al. VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem Biol. 2014;21:666–678. doi: 10.1016/j.chembiol.2014.02.021. [DOI] [PubMed] [Google Scholar]
- Farinha CM, Amaral MD. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol Cell Biol. 2005;25:5242–5252. doi: 10.1128/MCB.25.12.5242-5252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha CM, Nogueira P, Mendes F, Penque D, Amaral MD. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem J. 2002;366:797–806. doi: 10.1042/BJ20011717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha CM, Mendes F, Roxo-Rosa M, Penque D, Amaral MD. A comparison of 14 antibodies for the biochemical detection of the cystic fibrosis transmembrane conductance regulator protein. Mol Cell Probes. 2004;18:235–242. doi: 10.1016/j.mcp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Farinha CM, King-Underwood J, Sousa M, Correia AR, Henriques BJ, Roxo-Rosa M, et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem Biol. 2013;20:943–955. doi: 10.1016/j.chembiol.2013.06.004. [DOI] [PubMed] [Google Scholar]
- He L, Kota P, Aleksandrov AA, Cui L, Jensen T, Dokholyan NV, et al. Correctors of {Delta}F508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013;27:536–545. doi: 10.1096/fj.12-216119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- Kim Chiaw P, Wellhauser L, Huan LJ, Ramjeesingh M, Bear CE. A chemical corrector modifies the channel function of F508del-CFTR. Mol Pharmacol. 2010;78:411–418. doi: 10.1124/mol.110.065862. [DOI] [PubMed] [Google Scholar]
- Lansdell KA, Kidd JF, Delaney SJ, Wainwright BJ, Sheppard DN. Regulation of murine cystic fibrosis transmembrane conductance regulator Cl- channels expressed in Chinese hamster ovary cells. J Physiol (Lon) 1998;512:751–764. doi: 10.1111/j.1469-7793.1998.751bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, Clarke DM. Correctors enhance maturation of DeltaF508 CFTR by promoting interactions between the two halves of the molecule. Biochemistry. 2009;48:9882–9890. doi: 10.1021/bi9004842. [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, Wang Y, Clarke DM. The chemical chaperone CFcor-325 repairs folding defects in the transmembrane domains of CFTR-processing mutants. Biochem J. 2006;395:537–542. doi: 10.1042/BJ20060013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniz S, Sousa M, Moraes BJ, Mendes AI, Palma M, Barreto C, et al. HGF stimulation of Rac1 signaling enhances pharmacological correction of the most prevalent cystic fibrosis mutant F508del-CFTR. ACS Chem Biol. 2013;8:432–442. doi: 10.1021/cb300484r. [DOI] [PubMed] [Google Scholar]
- Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat Chem Biol. 2013;9:444–454. doi: 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N, Tomati V, Sondo E, Galietta LJ. Influence of cell background on pharmacological rescue of mutant CFTR. Am J Physiol Cell Physiol. 2010;298:C866–C874. doi: 10.1152/ajpcell.00404.2009. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005a;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N, Sonawane ND, Taddei A, Hu J, Zegarra-Moran O, Suen YF, et al. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol Pharmacol. 2005b;67:1797–1807. doi: 10.1124/mol.105.010959. [DOI] [PubMed] [Google Scholar]
- Ren HY, Grove DE, De La Rosa O, Houck SA, Sopha P, Van Goor F, et al. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol Biol Cell. 2013;24:3016–3024. doi: 10.1091/mbc.E13-05-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rowe SM, Pyle LC, Jurkevante A, Varga K, Collawn J, Sloane PA, et al. DeltaF508 CFTR processing correction and activity in polarized airway and non-airway cell monolayers. Pulm Pharmacol Ther. 2010;23:268–278. doi: 10.1016/j.pupt.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roxo-Rosa M, Xu Z, Schmidt A, Neto M, Cai Z, Soares CM, et al. Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc Natl Acad Sci USA. 2006;103:17891–17896. doi: 10.1073/pnas.0608312103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem. 1996;271:635–638. doi: 10.1074/jbc.271.2.635. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Frizzell RA, Bridges RJ. Rescue of dysfunctional deltaF508-CFTR chloride channel activity by IBMX. J Membr Biol. 1999;170:51–66. doi: 10.1007/s002329900537. [DOI] [PubMed] [Google Scholar]
- Sharma M, Pampinella F, Nemes C, Benharouga M, So J, Du K, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164:923–933. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatecka-Urban A, Brown A, Moreau-Marquis S, Renuka J, Coutermarsh B, Barnaby R, et al. The short apical membrane half-life of rescued {Delta}F508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of {Delta}F508-CFTR in polarized human airway epithelial cells. J Biol Chem. 2005;280:36762–36772. doi: 10.1074/jbc.M508944200. [DOI] [PubMed] [Google Scholar]
- Tian Y, Schreiber R, Wanitchakool P, Kongsuphol P, Sousa M, Uliyakina I, et al. Control of TMEM16A by INO-4995 and other inositolphosphates. Br J Pharmacol. 2013;168:253–265. doi: 10.1111/j.1476-5381.2012.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, et al. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Sci Transl Med. 2014;6:246ra297. doi: 10.1126/scitranslmed.3008889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERTEX. 2014. Two 24-Week Phase 3 Studies of Lumacaftor in Combination with Ivacaftor Met Primary Endpoint with Statistically Significant Improvements in Lung Function (FEV1) in People with Cystic Fibrosis who have Two Copies of the F508del Mutation. Available at http://investors.vrtx.com/releasedetail.cfm?ReleaseID=856185 (Accessed 27 January 2015)
- Wang Y, Bartlett MC, Loo TW, Clarke DM. Specific rescue of cystic fibrosis transmembrane conductance regulator processing mutants using pharmacological chaperones. Mol Pharmacol. 2006;70:297–302. doi: 10.1124/mol.106.023994. [DOI] [PubMed] [Google Scholar]
- Wang Y, Loo TW, Bartlett MC, Clarke DM. Additive effect of multiple pharmacological chaperones on maturation of CFTR processing mutants. Biochem J. 2007a;406:257–263. doi: 10.1042/BJ20070478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Loo TW, Bartlett MC, Clarke DM. Modulating the folding of P-glycoprotein and cystic fibrosis transmembrane conductance regulator truncation mutants with pharmacological chaperones. Mol Pharmacol. 2007b;71:751–758. doi: 10.1124/mol.106.029926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Experiments carried out on polarized CFBE41o- at 37°C and treated with vehicle (DMSO, control) or correctors C3 (25 µmol/L), C4 (15 μmol/L) and VX-809 (3 μmol/L), as indicated. Summary of the transepithelial resistance (Rte) for the F508del-CFTR CFBE monolayers treated with DMSO or CFTR correctors. Data are means ± SEM (n = 3–4). *P < 0.05) relative to control (DMSO) cells.