

Abstract
We previously demonstrated that the intravenous anesthetic, propofol, restores the sensitivity of transient receptor potential vanilloid channel subtype-1 (TRPV1) receptors via a protein kinase C epsilon (PKCε)-dependent and transient receptor potential ankyrin channel subtype-1 (TRPA1)-dependent pathway in sensory neurons. The extent to which the two pathways are directly linked or operating in parallel has not been determined. Using a molecular approach, our objectives of the current study were to confirm that TRPA1 activation directly results in PKCε activation and to elucidate the cellular mechanism by which this occurs. F-11 cells were transfected with complimentary DNA (cDNA) for TRPV1 only or both TRPV1 and TRPA1. Intracellular Ca2+ concentration was measured in individual cells via fluorescence microscopy. An immunoblot analysis of the total and phosphorylated forms of PKCε, nitric oxide synthase (nNOS), and TRPV1 was also performed. In F-11 cells containing both channels, PKCε inhibition prevented the propofol- and allyl isothiocyanate (AITC)-induced restoration of TRPV1 sensitivity to agonist stimulation as well as increased phosphorylation of PKCε and TRPV1. In cells containing TRPV1 only, neither agonist induced PKCε or TRPV1 phosphorylation. Moreover, NOS inhibition blocked propofol-and AITC-induced restoration of TRPV1 sensitivity and PKCε phosphorylation, and PKCε inhibition prevented the nitric oxide donor, SNAP, from restoring TRPV1 sensitivity. Also, propofol-and AITC-induced phosphorylation of nNOS and nitric oxide (NO) production were blocked with the TRPA1-antagonist, HC-030031. These data indicate that the AITC- and propofol-induced restoration of TRPV1 sensitivity is mediated by a TRPA1-dependent, nitric oxide synthase-dependent activation of PKCε.
Keywords: PKCε and NOS, propofol, TRPA1, TRPV1
Introduction
Two prominent members of the transient receptor potential (TRP) family, the vanilloid subtype-1 (TRPV1) receptor and the ankyrin subtype-1 (TRPA1) receptor are extensively coexpressed in peripheral sensory neurons and function as sensory transducers of noxious stimuli (Caterina et al. 2001; Palazzo et al. 2008). The relative sensitivity of these receptors to their respective agonists can have a major impact on nociceptive signaling in health and disease. Several chemical agents are capable of acutely sensitizing TRPV1 receptors to thermal mediators resulting in the sensation of pain (Lu et al. 2006; Novakova-Tousova et al. 2007; Holzer 2008; Wang 2008), whereas desensitization of the receptor via repetitive stimulation plays an important role in blocking pain transmission (Mandadi et al. 2004; Larrucea et al. 2008). Recent evidence has suggested that cross talk between TRPV1 and TRPA1 receptors may serve to integrate distinct nociceptive signaling pathways and provide a novel mechanism for integration of nociceptive information (Akopian et al. 2007, 2008; Ruparel et al. 2008; Salas et al. 2009). Little is known about the cellular signaling pathways by which TRPV1 and TRPA1 channels communicate with each other in sensory neurons.
Propofol is one of the most commonly used intravenous anesthetics for the induction and maintenance of general anesthesia and sedation. Apart from its anesthetic properties, propofol has several nonanesthetic effects, one of which is the activation of TRPA1 ion channels to enhance pain and inflammation (Matta et al. 2007). Our laboratory has recently shown that propofol restores the sensitivity of TRPV1 receptors following agonist-induced desensitization via a TRPA1 dependent pathway in mouse dorsal root ganglion (DRG) sensory neurons (Zhang et al. 2011). Moreover, we also demonstrated in an earlier study that the propofol-induced restoration of TRPV1 sensitivity occurs via a protein kinase C epsilon (PKCε)-dependent phosphorylation of the TRPV1 receptor (Wickley et al. 2010). The extent to which the actions of propofol on TRPA1 and PKCε signaling pathways operate independently in parallel or are directly linked has not been determined. Moreover, the mechanism by which TRPA1 activation stimulates PKCε autophosphorylation and activation is also not known. We rationalized that activation of nitric oxide synthase (NOS) and production of nitric oxide (NO) following TRPA1 stimulation may play a role in mediating PKCε activation, at least in part, since TRPA1 activation has been associated with NO production in human lung carcinoma cells (Sun et al., 2014) and NO selectively activates PKCε in isolated adult rabbit hearts (Ping et al. 1999). Therefore, our objectives were to confirm that TRPA1 activation directly results in PKCε activation and to elucidate the cellular mechanism by which this occurs.
In the current study, we tested the hypothesis that inhibition of PKCε attenuates the restoration of TRPV1 sensitivity induced by the TRPA1 agonists, allyl isothiocyanate (AITC) and the general anesthetic, propofol. Moreover, we also tested the hypothesis that the presence of TRPA1 receptors is required for the propofol- and AITC-induced activation of PKCε as well as subsequent phosphorylation of TRPV1 receptors. Finally, we tested the hypothesis that TRPA1-dependent activation of NOS and NO production are involved in the autophosphorylation/activation of PKCε and restoration of TRPV1 sensitivity to agonist stimulation.
Materials and Methods
F-11 cell transfection with TRPV1 or TRPA1 cDNA
Cultured F-11 cells (DRG-like hybridoma cell line) were transfected with TRPV1 or TRPA1 cDNA via electroporation using the Neon Transfection System (Invitrogen, Carlsbad, CA) as per the manufacturer’s instructions for F-11 cells. Briefly, cultured F-11 cells at 80% confluence were harvested and washed with phosphate-buffered saline (PBS) without Ca2+ or Mg2+. The cells were resuspended in 100 μL of electrolytic Buffer R in a sterile eppendorf tube and 5 μg of either TRPV1 or TRPA1 cDNA was then added. Electroporation was performed using a pulse voltage of 1500 V, a pulse width of 35 msec and a pulse number of 2. Following electroporation, the cells were then suspended in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum at 37°C. A proportion of the cells were seeded onto coverslips in 6-well dishes and used for intracellular Ca2+ measurements, while the remainder were used for immunoblot analysis to confirm the presence and/or absence of TRPV1 and TRPA1.
Intracellular Ca2+ measurements
F-11 cells were incubated at room temperature (23°C) for 15 min with fura-2 acetoxy methylester (fura-2/AM; 2 μmol/L) in HEPES-buffered (2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid saline containing the following: 118 mmol/L NaCl, 4.8 mmol/L KCl, 1.2 mmol/L MgCl2, 1.25 mmol/L CaCl2, 11.0 mmol/L dextrose, 5 mmol/L pyruvate, and 25 mmol/L HEPES (pH 7.35). Coverslips containing the fura-2-loaded F-11 cells were placed in a temperature-regulated (30°C) chamber (Warner Instruments, Hamden, CT) mounted on the stage of an Olympus IX-81 inverted fluorescence microscope (Olympus America, Lake Success, NY). The cells were superfused continuously with HEPES-buffered saline at a flow rate of 2 mL/min. Drugs were delivered by switching from control buffer to drug-containing buffer for 20 sec unless noted otherwise. Intracellular free Ca2+ concentration ([Ca2+]i) measurements were simultaneously performed on multiple individual cells using a fluorescence imaging system (Easy Ratio Pro, Photon Technology International, Lawrenceville, NJ) equipped with a multi-wavelength spectrofluorometer (DeltaRAM X) and a QuantEM 512SC electron multiplying charge-coupled device camera (Photometrics, Tuscon, AZ). Images and photometric data were acquired by alternating excitation wavelengths between 340 and 380 nm (20 Hz) and monitoring an emission wavelength of 510 nm. Due to the fact that calibration procedures rely on a number of assumptions, the ratio of the light intensities at the two wavelengths was used to measure qualitative changes in [Ca2+]i. Just before data acquisition, background fluorescence was measured and automatically subtracted from the subsequent experimental measurement using Easy Ratio Pro.
Immunoblot analysis of PKCε, TRPV1, and nNOS (total and phosphorylated)
Immunoblot analysis was carried out on whole F-11 cell lysates (Zhang et al. 2011). Protein concentration was assessed using the Bradford method (Bradford 1976). All samples were adjusted to a protein concentration of 1–2 mg/mL in sample buffer, boiled for 5 min, and then kept at −20°C until use. Equal amounts of protein (50 μg) were separated by SDS-PAGE on 12% polyacrylamide gels and transferred to nitrocellulose membranes. Nonspecific binding was blocked with Tris-buffered saline solution (0.1% [vol/vol] Tween-20 in 20 mmol/L Tris base, 137 mmol/L NaCl adjusted to pH 7.6 with HCl, containing 3% [wt/vol] bovine serum albumin for 1 h at room temperature. Monoclonal antibodies against total PKCε (BD Transduction Laboratory, 1 Becton Drive, Franklin Lakes, NJ, 07417, USA) and polyclonal antibodies against serine 729 phosphorylated PKCε (Invitrogen, 5791 Van Alley Way, Carlsbad, CA 92008, USA), TRPV1 (Santa Cruz Biotechnology, 10410 Finnell Street, Dallas, TX, 75220, USA), serine 800 phosphorylated TRPV1 (Gift from Dr. Tominaga), serine 1417 phosphorylated nNOS (Abcam, 1 Kendall Square, Suite B2304, Cambridge, MA 02139, USA) or nNOS (Cell Signaling, 32 Tozer Road, Beverly MA, 01915, USA) were diluted 1:1000 in Tris-buffered saline containing 1% bovine serum albumin for immunoblotting (2 h). After washing in Tris-buffered saline three times (10 min each), membranes were incubated for 1 h at room temperature with horseradish-peroxidase-linked secondary antibody (goat anti-mouse and goat anti-rabbit; 1:5000 dilution in Tris-buffered saline containing 1% bovine serum albumin). Membranes were again washed and bound antibody was detected by enhanced chemiluminescence. Immunoreactivity was quantified by scanning densitometry and analyzed using software (ImageJ; National Institutes of Health, Washington, DC).
Selection criteria for determining subpopulations of transfected F-11 neurons
F-11 cells (Platika et al. 1985) that either transfected with only TRPV1 receptors or both TRPV1 and TRPA1 receptors were first identified by treating the cells with single applications of capsaicin and AITC. Cells responsive to only capsaicin were deemed to contain only functional TRPV1 receptors, while cells that were responsive to capsaicin and AITC were deemed to contain both TRPV1 and TRPA1 receptors. Cells that did not respond to capsaicin or AITC were excluded from the study and therefore were not used in the statistical analysis.
Measurement of NO production in TRPV1-TRPA1 cotransfected F-11 cells and fluorescence microscopy
NO detection kit for fluorescence microscopy was purchased from ENZO Life Sciences (10 Executive Boulevard, Farmingdale, NY, 11735, USA). All reagents were diluted in accordance to the product instructions. Cells were seeded the day before the experiment to ensure 50–70% confluency on the day of the experiment and were cultured in l-Arginine free media. On the day of the experiment, cells were pretreated with the NO scavenger c-PTIO and incubated with the NO detection reagent for 2 h. Cells were washed twice with wash buffer and then treated with experimental test agents including propofol (10 μmol/L), AITC (100 μmol/L), SNAP (100 μmol/L), and HC-030031 (0.5 μmol/L). Cells were treated with the NO-inducer, l-Arginine (100 μmol/L) and the NO-scavenger, 2-4-carboxyphenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (c-PTIO;10 μmol/L) as positive and negative controls, respectively. Posttreatment, cells were rinsed with wash buffer and examined under a fluorescence/confocal microscope (Olympus IX-70, FV5 PSU, Olympus America, 3500 Corporate Parkway, Center Valley, PA, 18034, USA) using the filter set Cyanine 5 (650/670 nm).
Composition of propofol
The Diprivan® lipid emulsion form of propofol (56 mmol/L [10 mg/mL] propofol, 10% soybean oil, 2.25% glycerol, 1.2% purified phospholipid) was used in all protocols and will be referred to as propofol throughout the manuscript. Intralipid vehicle was used as a control. The effect of Intralipid was examined at concentrations equivalent to those used for the propofol-Intralipid mixture.
Statistical analysis
All Ca2+ imaging experimental protocols were repeated in a minimum of five separate coverslips of F-11 cells. Results obtained from each coverslip (for F-11 cell experiments) were averaged so that all coverslips of cells were weighted equally. Gaussian distribution was examined by the Shapiro–Wilk normality test. Comparisons between the groups were made using repeated-measures one-way analysis of variance and the Bonferroni post hoc test. Differences were considered statistically significant at P < 0.05. All results are expressed as mean ± SEM. The error bars in the figures refer to the variability in the peak of the Ca2+ response (Ca2+ imaging) or to the variability in intensity of the bands (immunoblot analysis) compared to the untreated control or a specific intervention, as noted in the legends. Statistical analysis was conducted using Sigma Plot 12.5 software (Systat Software, San Jose, CA).
Results
Effect of PKCε inhibition on AITC- and propofol-induced restoration of TRPV1 sensitivity to agonist stimulation in cultured TRPA1-TRPV1 co-transfectedF-11 cells
Individual F-11 cells containing both functional TRPV1 and TRPA1 receptors were first identified by treating the cells with single applications of capsaicin (100 nmol/L) and AITC (100 μmol/L) a (Fig.1A). Repetitive stimulation of F-11 cells with capsaicin resulted in a progressive decrease (de-sensitization) in peak [Ca2+]i (Fig.1B). When AITC (100 μmol/L) was added to the bath during the 10 min pause in stimulation, subsequent reapplication of capsaicin resulted in a robust transient increase in [Ca2+]i (resensitization) (Fig.1B). A similar resensitizing effect was observed with propofol (Fig.1C). Pretreatment with the PKCε-specific inhibitory peptide εV1–2 (0.5 μmol/L) following desensitization inhibited the AITC- and propofol-induced restoration of TRPV1 sensitivity to capsaicin compared with F-11 cells treated only with propofol or AITC (Fig.1D and E). Summarized data depicting the effect of AITC and propofol alone or in the presence of εV1–2 on restoration of TRPV1 sensitivity to capsaicin in TRPV1-TRPA1 cotransfected F-11 cells are depicted in Figure1F. Summarized results are expressed as a percent of the response to the final application of capsaicin in untreated cells (control).
Figure 1.
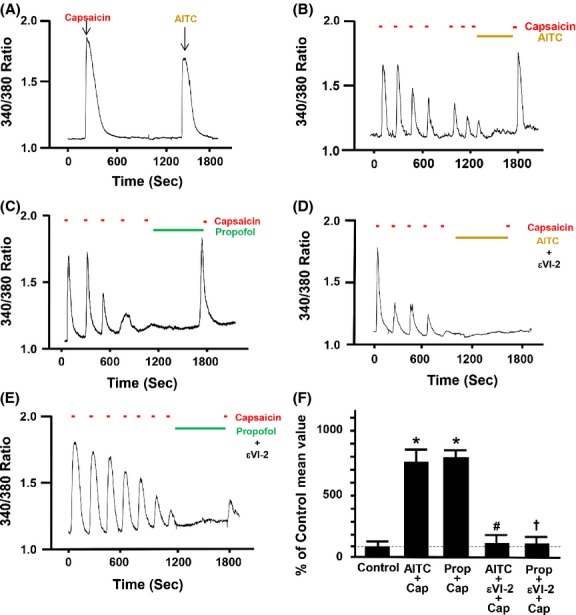
(A) Representative trace depicting the effect of a single application of the specific transient receptor potential vanilloid subtype-1 (TRPV1) agonist capsaicin (Cap; 100 nmol/L) and the specific transient receptor potential ankyrin receptor subtype-1 (TRPA1) activator allyl isothiocyanate (AITC, 100 μmol/L) on intracellular free calcium concentration in F11 cells transfected with both TRPA1 and TRPV1 complementary DNA (cDNA). Representative traces depicting the effect of AITC (100 μmol/L) alone, propofol (Prop, 10 μmol/L) alone, AITC in the presence of specific PKCε inhibitor εV1–2 (0.5 μmol/L), propofol in the presence of specific PKCε inhibitor εV1–2 after capsaicin-induced (100 nmol/L) desensitization on restoration of TRPV1 sensitivity in F-11 cells transfected with both TRPA1 and TRPV1 cDNA are depicted in Figure1B, C, D, and E, respectively. The εV1–2 inhibitor peptide was added alone for 5 min following desensitization and then AITC or propofol was brought on board in combination with the εV1–2 inhibitor peptide for an additional 5 min. Summarized data for Figure1B–E are depicted in Figure1F. Data are expressed as a percent of the response to the final application of capsaicin in the untreated control (% of control mean value ± SEM). *P < 0.05 compared to final capsaicin in the untreated control. #P < 0.05 compared to AITC plus capsaicin. †P < 0.05 compared to propofol plus capsaicin. n = seven separate coverslips of F-11 cells were used.
Effect of PKCε activation on restoration of TRPV1 sensitivity to agonist stimulation in TRPV1 transfected F-11 cells
F-11 cells containing only TRPV1 receptors were first identified by treating the cells with single applications of capsaicin (100 nmol/L) and AITC (100 μmol/L). Cells that responded with a transient increase in [Ca2+]i subsequent only to capsaicin were selected for the experiment (Fig.2A). Repetitive stimulation of F-11 cells with capsaicin resulted in a progressive decrease (desensitization) in peak [Ca2+]i that was maintained following a 10-min pause in capsaicin stimulation as indicated by the lack of any response to capsaicin after reapplication of capsaicin to the bath (Fig.2B). In contrast, when the PKCε activator peptide ψεRACK (0.5 μmol/L) was added to the bath during the 10-min pause in stimulation, subsequent reapplication of capsaicin resulted in a robust transient increase in [Ca2+]i (resensitization) (Fig.2C). Summarized data depicting the effect of ψεRACK (0.5 μmol/L) on TRPV1 resensitization are depicted in Figure2D. Summarized results are expressed as a percent of the response to the final application of capsaicin during the initial desensitization of TRPV1.
Figure 2.
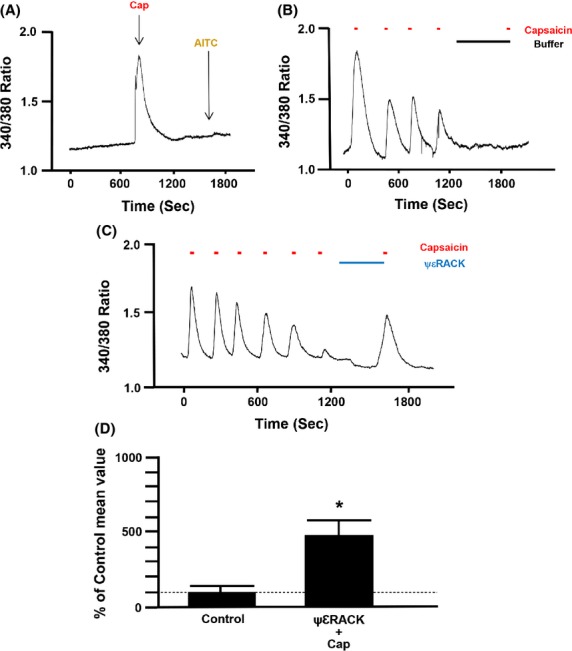
(A) Representative trace depicting the effect of a single application of capsaicin (100 nmol/L) and allyl isothiocyanate (AITC, 100 μmol/L) on intracellular free calcium concentration inF-11 cells transfected with only functional transient receptor potential vanilloid receptor type 1 (TRPV1) receptors c-DNA. Representative traces depicting the effect of time and the protein kinase C epsilon (PKCε) activator peptide (ΨεRACK, 0.5 μmol/L) on restoration of TRPV1 sensitivity in F-11 cells containing only functional TRPV1 receptors (B and C). Summarized data for Figure2C are depicted in Figure2D. *P < 0.05 compared to capsaicin-treated TRPV1-transfected F-11 cells. n = seven different cover slips containing TRPV1 transfected F-11 cells.
Effect of AITC and propofol on phosphorylation of PKCε at S729 in cultured F11 cells expressing TRPV1 only or both TRPV1 and TRPA1
To determine if the presence of TRPA1 receptors is required for propofol or AITC to activate PKCε at S729, immunoblot analysis of PKCεpS729 was performed before and after treatment of cells with either propofol (10 μmol/L; 10 min), AITC (100 μmol/L), or ψεRACK (0.5 μmol/L). We also assessed the effect of the PKCε inhibitor peptide, εV1–2 (0.5 μmol/L), on the propofol- and AITC-induced phosphorylation of S729. PKCεpS729 levels were normalized to total PKCε protein levels detected in the lysate. In TRPV1 transfectedF-11 cells, neither AITC (100 μmol/L) nor propofol (10 μmol/L) increased PKCεpS729 levels as compared to untreated control (Fig.3A). However, treatment with the PKCε activator peptide, ψεRACK (0.5 μmol/L), increased PKCεpS729 levels compared to control. In TRPV1-TRPA1 cotransfected F-11 cells, AITC, propofol, and ψεRACK all increased PKCεpS729 levels compared to control (Fig.3C). Moreover, the AITC- and propofol-induced increases in PKCεpS729 levels were prevented by pretreatment with the PKCε inhibitory peptide, εV1–2 in TRPV1-TRPA1 cotransfected F-11 cells (Fig.3C). In addition, following treatment with propofol (10 μmol/L) or AITC (100 μmol/L) in either cell type, the amount of immunodetectable PKCε in the membrane fraction was unchanged as compared to untreated control (Fig.3A and C). The Intralipid (amount equivalent to 10 μmol/L propofol) increased PKCεpS729 levels to 127 ± 8% of control similar to our previous findings (Wickley et al. 2010). Summarized data for Figure3A and C are depicted in Figure3B and D, respectively.
Figure 3.
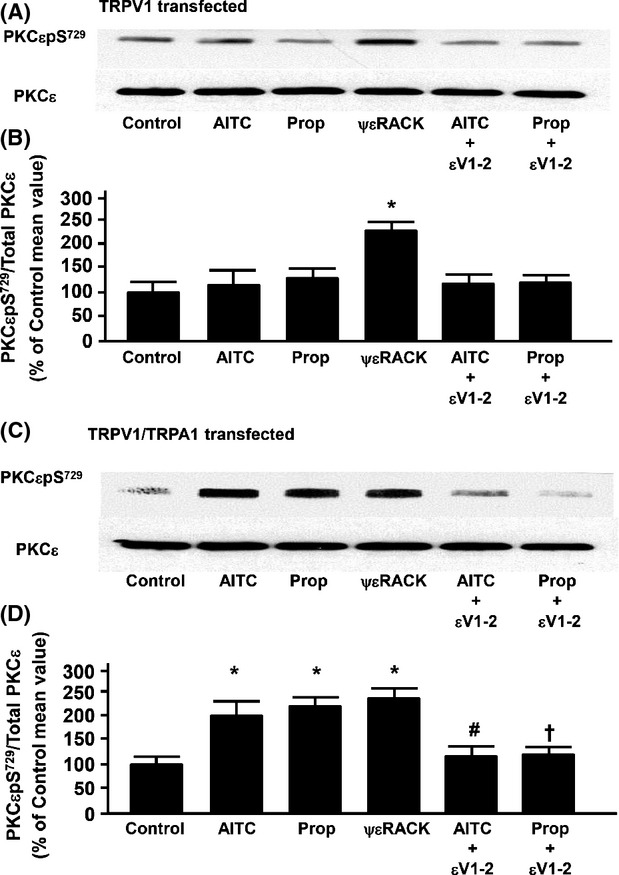
(A) Representative immunoblot depicting the effect of allyl isothiocyanate (AITC, 100 μmol/L) alone, propofol (Prop, 10 μmol/L) alone, the protein kinase C epsilon (PKCε) activator peptide ΨεRACK (0.5 μmol/L) alone, and AITC or propofol in the presence of the PKCε inhibitor peptide εV1–2 (0.5 μmol/L) on PKCε serine 729 phosphorylation (PKCεpS729) in F-11 cells transfected with transient receptor potential vanilloid receptor type 1 (TRPV1) channels only. Total PKCε was used as a loading control. (B) Summarized data for Figure3A. (C and D) Same as Figure3A and B, respectively, except in -11 cells transfected with both TRPV1 and transient receptor potential ankyrin receptor subtype-1 (TRPA1) channels. Data are expressed as a percent of the untreated control mean value ± SEM. *P < 0.05 compared to control. #P < 0.05 compared to AITC alone. †P < 0.05 compared to propofol alone. n = six different F-11 cell lysates.
Effect of propofol and AITC on TRPV1 phosphorylation at S800 in cultured F-11 cells expressing TRPV1 only or both TRPA1 and TRPV1
To determine if the presence of TRPA1 receptors is required for propofol or AITC to phosphorylate TRPV1 at S800, immunoblot analysis of TRPV1pS800 was performed. TRPV1pS800 levels were normalized to total TRPV1 protein detected in the lysate. In TRPV1 transfected F-11 cells, neither AITC (100 μmol/L) nor propofol (10 μmol/L) increased TRPV1pS800 levels as compared to untreated control (Fig.4A). However, treatment with the PKCε activator peptide, ψεRACK (0.5 μmol/L), increased TRPV1pS800 levels compared to control. In TRPV1-TRPA1 cotrasfected F-11 cells, AITC, propofol, and ψεRACK all increased TRPV1pS800 levels compared to control (Fig.4B). Moreover, the AITC- and propofol-induced increases in TRPV1pS800 levels were prevented by pretreatment with the PKCε inhibitory peptide, εV1–2. In addition, following treatment with propofol (10 μmol/L) or AITC (100 μmol/L) in either cell type, the amount of immunodetectable TRPV1 in the cell lysate was unchanged as compared to untreated control (Fig.4A and C). The Intralipid vehicle had no detectable effect on TRPV1pS800 levels (103 ± 5% of control). Summarized data for Figure4A and C are depicted in Figure4B and D, respectively.
Figure 4.
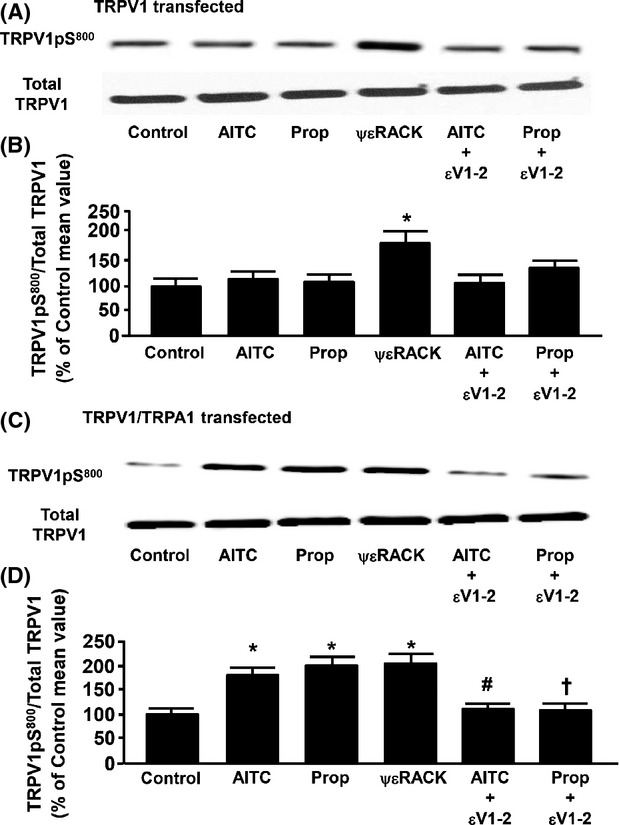
(A) Representative immunoblot depicting the effect of allyl isothiocyanate (AITC, 100 μmol/L) alone, propofol (Prop, 10 μmol/L) alone, the protein kinase C epsilon (PKCε) activator peptide ΨεRACK (0.5 μmol/L) alone, and AITC or propofol in the presence of the PKCε inhibitor peptide εV1–2 (0.5 μmol/L) on transient receptor potential vanilloid receptor type 1 (TRPV1) serine 800 phosphorylation (TRPV1pS800) in F-11 cells transfected with TRPV1 channels only. Total TRPV1 was used as a loading control. (B) Summarized data for Figure4A. (C and D) Same as Figure4A and B, respectively, except in F-11 cells transfected with both TRPV1 channels and transient receptor potential ankyrin receptor subtype-1 (TRPA1) channels. Data are expressed as a percent of the untreated control mean value ± SEM. *P < 0.05 compared to control. #P < 0.05 compared to AITC alone. †P < 0.05 compared to Prop alone. n = six different F-11 cell lysates.
Effect of NOS inhibition on propofol- and AITC-induced restoration of TRPV1 sensitivity
In F-11 cells transfected with both TRPV1 and TRPA1, inhibition of NOS with l-NG-nitroarginine methyl ester (l-NAME; 100 μmol/L) prevented the propofol-induced restoration of TRPV1 sensitivity by more than 90% (Fig.5B). Moreover, the NO donor SNAP (100 μmol/L) mimicked the effects of propofol on restoration of TRPV1 sensitivity (Fig.5C) and the restoring effect of SNAP was inhibited by the PKCε inhibitor peptide (0.5 μmol/L), εV1–2 (Fig.5D). Similarly, the PKCε activator peptide ψεRACK (0.5 μmol/L) restored TRPV1 sensitivity in the presence of NOS inhibition with l-NAME (Fig.5E). Pretreatment with l-NAME alone did not restore sensitivity of TRPV1 to capsaicin (102 ± 9% of control). Summarized data for Figure5A–E are depicted in Figure5F.
Figure 5.
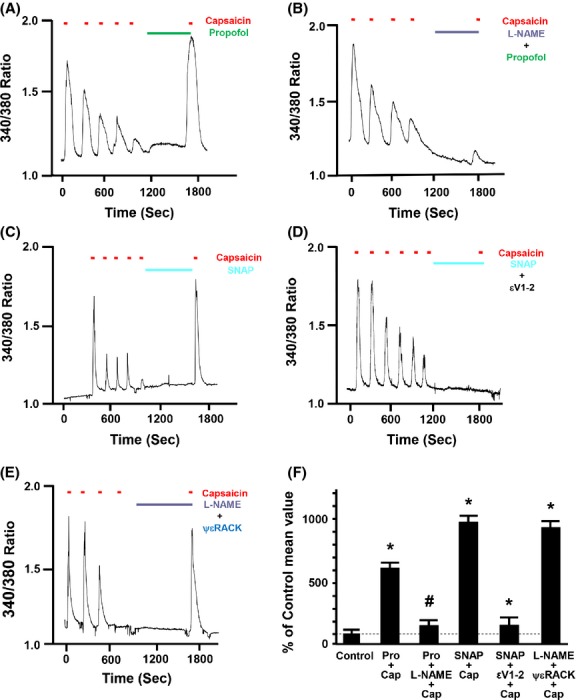
(A) Representative trace depicting the effect of propofol (Pro, 10 μmol/L) alone, l-NG-nitroarginine methyl ester (l-NAME; 100 μmol/L) plus propofol, S-nitroso-N-acetylpenicillamine (SNAP; 100 μmol/L) alone, SNAP plus protein kinase C epsilon (PKCε) inhibitor peptide εV1–2 (0.5 μmol/L), l-NAME plus the PKCε activator peptide ΨεRACK (0.5 μmol/L) after capsaicin-induced (100 nmol/L) desensitization on restoration of TRPV1 sensitivity to capsaicin (Cap) in mouse DRG neurons that contain functional TRPA1 and TRPV1 receptors are depicted in Figure5A, B, C and D and E, respectively. The NOS inhibitor l-NAME was added alone for 5 min following desensitization and then propofol or the PKCε activator peptide ΨεRACK were brought on board in combination with l-NAME for an additional 5 min. Similarly, the εV1–2 inhibitor peptide was added alone for 5 min following desensitization and then SNAP was brought on board in combination with the εV1–2 inhibitor peptide for an additional 5 min. Summarized data for Figure5A–E are depicted in Figure5F. Data are expressed as a percent of the response to the final application of capsaicin in the untreated control (% of control mean value ± SEM). *P < 0.05 compared to final capsaicin in the untreated control. #P < 0.05 compared to Pro plus capsaicin. †P < 0.05 compared to SNAP plus capsaicin. n = F-11 cells from seven separate cover slips.
Effect of NOS inhibition on propofol- and AITC-induced PKCε phosphorylation at S729 in cultured F-11 cells expressing both TRPA1 and TRPV1
In F-11 cells transfected with TRPA1 and TRPV1, pretreatment with the NO donor, SNAP, increased immunodetectable phosphorylation of PKCεpS729 (Fig.6A). Moreover, inhibition of NOS with l-NAME prevented the propofol- and AITC-induced increase in immunodetectable PKCεpS729. Incubation with l-NAME alone had no significant effect on basal levels of PKCεpS729 (98 ± 8% of control). Summarized data for Figure6A are depicted in Figure6B.
Figure 6.
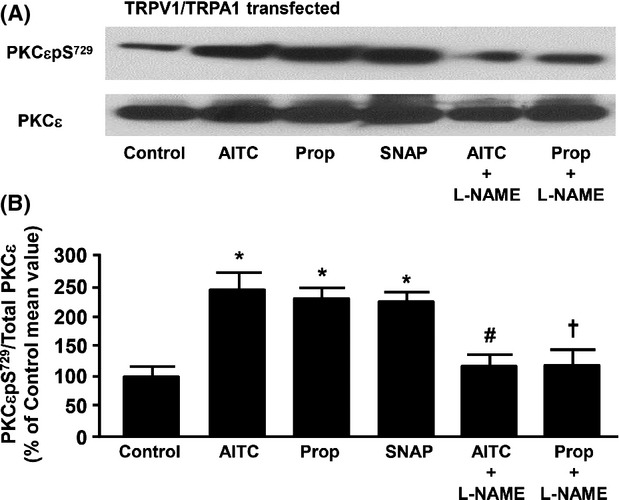
(A) Representative immunoblot depicting the effect of allyl isothiocyanate (AITC, 100 μmol/L) alone, propofol (Prop, 10 μmol/L) alone, S-nitroso-N-acetylpenicillamine (SNAP; 100 μmol/L), propofol in the presence of the NOS inhibitor, l-NG-nitroarginine methyl ester (l-NAME; 100 μmol/L) or allyl isothiocyanate (AITC, 100 μmol/L) in the presence of l-NAME on PKCε serine 729 phosphorylation (PKCεpS729) in F-11 cells transfected with both TRPV1 and TRPA1. Total PKCε was used as a loading control. (B) Summarized data for Figure6A. Data are expressed as a percent of the untreated control mean value ± SEM. *P < 0.05 compared to control. #P < 0.05 compared to AITC alone. †P < 0.05 compared to Prop alone. n = six different F-11 cell lysates.
Effect of propofol and AITC on phosphorylation of NOS at Ser1417 and on NO production in cultured F-11 cells expressing both TRPA1 and TRPV1
No significant change in the expression level of nNOS was observed in nontransfected, TRPV1-trasfected, TRPA1-transfected, and TRPV1-TRPA1 cotransfected F-11 cells (Fig.7A). However, TRPV1–TRPA1 cotransfected F-11 cells pretreated with propofol (10 μmol/L) and AITC (100 μmol/L) demonstrated increased phosphorylation of nNOS at Ser1417 compared to the nontreated cells lysate, that was significantly attenuated by the TRPA1 antagonist, HC-030031 (0.5 μmol/L; Fig.7B). Summarized data for Figure7B are depicted in Figure7C. AITC (100 μmol/L) - and propofol (10 μmol/L) -induced NO production was also detected and visualized in TRPV1-TRPA1 cotransfected F-11 cells, that was mimicked by SNAP (100 μmol/L). Moreover, propofol-induced NO production was attenuated in presence of HC-030031 (0.5 μmol/L). Changes in intracellular NO production were compared to cells pretreated with l-Arginine, an NO inducer (100 μmol/L), and c-PTIO, an NO scavenger (10 μmol/L) (Fig.7).
Figure 7.
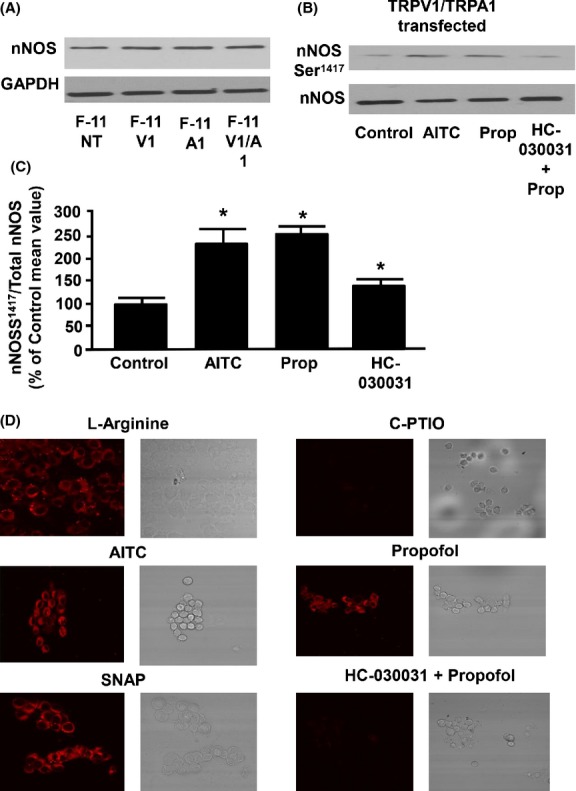
(A) Representative immunoblot depicting the expression of nNOS in nontransfected F-11 cells (F-11 NT), TRPV1-transfected F-11 cells (F-11 V1), TRPA1-transfected F-11 cells (F-11 A1), and TRPV1-TRPA1 co-transfected F-11 cells (F-11 V1/A1). GAPDH was used as a loading control. (B) Represents the effect of allyl isothiocyanate (AITC, 100 μmol/L) alone, propofol (Prop, 10 μmol/L) alone, and propofol in presence of HC-030031 (0.5 μmol/L) on nNOS serine 1417 phosphorylation (nNOSS1417) in F-11 cells transfected with both TRPV1 and TRPA1. Total nNOS was used as a loading control. (C) Summarized data for Figure7B. (D) Representative fluorescence images depicting the effect of l-Arginine (NO-inducer), c-PTIO (NO scavenger), AITC (100 μmol/L), propofol (10 μmol/L), SNAP (100 μmol/L), and propofol in the presence of HC-030031(0.5 μmol/L) on NO production in TRPA1 and TRPV1 cotransfected F-11 cells. Data are expressed as a percent of the untreated control mean value ± SEM. *P < 0.05 compared to control. n = six different F-11 cell lysates.
Discussion
This is the first study to delineate the role of PKCε in TRPA1-dependent restoration of TRPV1 sensitivity as well as to determine the mechanism by which TRPA1 activates PKCε. We previously demonstrated that propofol restores TRPV1 sensitivity via a PKCε-dependent phosphorylation of the channel (Wickley et al. 2010) and is dependent on the presence of functional TRPA1 receptors (Zhang et al. 2011). However, the extent to which propofol activates these two pathways independently and in parallel or whether they are directly linked to one another via an unknown signaling pathway has not been established. Our key findings are that the AITC and propofol-induced restoration of TRPV1 sensitivity are mediated by a TRPA1-dependent activation of PKCε Moreover, TRPA1-induced activation of PKCε is mediated via a NOS-dependent pathway.
Our first objective was to determine the whether activation of PKCε is directly linked to TRPA1-dependent restoration of TRPV1 sensitivity. Our current results substantiate our previous findings that inhibition of PKCε prevents the propofol-induced restoration of TRPV1 sensitivity (Wickley et al. 2010) and our current novel findings indicate that the PKCε inhibition also prevents the AITC-induced restoration of TRPV1 sensitivity to agonist stimulation. These data are also consistent with previous findings demonstrating increased sensitivity of desensitized TRPV1 by phorbol esters occurs through PKCε-mediated phosphorylation of TRPV1 (Mandadi et al. 2006).
Although our results indicate that PKCε is required for TRPA1 activation to restore TRPV1 sensitivity, indicating a TRPA1-dependent activation of PKCε, these results do not conclusively rule out the possibility that PKCε may activate TRPA1 and subsequently restore TRPV1 sensitivity. Therefore, we assessed the effect of the PKCε-specific activator peptide, ψεRACK, on restoration of TRPV1 sensitivity in F-11 cells expressing only TRPV1. Our findings indicate that direct activation of PKCε does not require the presence of TRPA1 receptors in order to restore TRPV1 sensitivity, and that TRPA1 stimulation is upstream of PKCε activation. Our current findings are similar to a previous study indicating that PKCε acts downstream of bradykinin and is principally responsible for sensitization of the heat response in nociceptors by bradykinin (Cesare et al. 1999).
As part of the activation process, PKCε is autophosphorylated at S729 rendering it catalytically competent. Our results indicate that in cells containing TRPV1 and TRPA1, propofol and AITC phosphorylate PKCε at S729. This effect was markedly attenuated in F-11 cells containing only TRPV1 indicating that propofol primarily activates PKCε via a TRPA1-dependent pathway. It should be noted that in addition to the important role of PKCε in TRPV1 regulation, there may also be a direct molecular interaction between TRPA1 and TRPV1 that operates in parallel (Ruparel et al. 2008; Salas et al. 2009; Staruschenko et al. 2010; Akopian 2011).
The PKCε-dependent phosphorylation of TRPV1 at S800 has been shown to increase the sensitivity of TRPV1 to agonist stimulation (Zhou et al. 2001; Mandadi et al. 2006; Srinivasan et al. 2008). In the current study, we now demonstrate that TRPA1 is required for propofol or AITC to phosphorylate TRPV1 at S800 indicating a TRPA1-dependent signaling cascade leading to activation of PKCε and subsequent phosphorylation of TRPV1 at S800. We previously demonstrated that the propofol increased TRPV1pS800 and that this increase was not observed in sensory neurons derived from PKCε−/− mice (Wickley et al. 2010).
Previous studies in other cell types have indicated that NO donors are capable of activating specific PKC isoforms (Burgstahler and Nathanson 1995; Liang and Knox 1999; Harada et al. 2004; Lee et al. 2006) including PKCε (Nishio and Watanabe 1997; Ping et al. 1999; Bolli 2001; Balafanova et al. 2002). In our study, NOS inhibition prevented the agonist-induced restoration of TRPV1 sensitivity. Moreover, the NO donor SNAP restored TRPV1 sensitivity, an effect that was abolished by PKCε inhibition and NOS inhibition failed to prevent restoration of TRPV1 sensitivity by the PKCε activator peptide. These data suggest that NO is a mediator of the TRPA1-dependent restoration of TRPV1 sensitivity and are consistent with a TRPA1-dependent induction of hyperthermia in rats which was dependent on NO production suggesting TRPA1 stimulates NOS activation (Ding et al. 2008).
Some studies have demonstrated that NO donors can directly activate TRP channels causing increases in intracellular Ca2+ (Yoshida et al. 2006; Miyamoto et al. 2009). In our study, we saw no increase in intracellular Ca2+ upon application of SNAP in cells transfected with both channels. This may be due to the significantly lower concentration of SNAP (100 μmol/L) used in our study compared to others using concentrations between 1 and 5 mmol/L (Miyamoto et al. 2009).
In order to biochemically determine the extent to which TRPA1 activation is linked to NO production and activation of PKCε, we utilized F-11 cells transfected with both channels. In these studies, NOS inhibition with l-NAME prevented the agonist-induced increase in immunodetectable PKCcpS729. Moreover, SNAP stimulated an increase in immunodetectable PKCεpS729. These data are consistent with the Ca2+ imaging data indicating a TRPA1-dependent activation of NOS leading to NO-dependent phosphorylation of PKCε and restoration of TRPV1 sensitivity. The precise mechanism by which NO causes PKCε activation is not known but several possibilities exist. NO reacts with superoxide anions to form peroxynitrite that can activate PKC (Reiter et al. 2000; Chakraborti et al. 2005; Li et al. 2011). Alternatively, the NO donor effect enhances PKC interaction with its specific anchor protein RACK2 via nitration of tyrosine residues of PKC, resulting in its activation (Balafanova et al. 2002).
This study also investigated the role of agonist-induced TRPA1 activation in activating nNOS and regulation of NO production in TRPV1 and TRPA1 coexpressing cells. Our data suggest that activation of TRPA1 leads to increased phosphorylation of nNOS at serine 1417 as well as NO production that is substantially attenuated in the presence of the TRPA1 antagonist, HC-030031. There were no significant changes in nNOS expression in any of the groups. These data further support the role of nNOS in TRPA1-mediated restoration of TRPV1 sensitivity and also confirms that nNOS is downstream of TRPA1.
Overall, our current findings are consistent with previous studies indicating cross talk between TRPV1 and TRPA1 receptors which regulate and modulate nociceptive ion channel function (Brand and Jacquot 2002; Akopian et al. 2007; Salas et al. 2009; Simons et al. 2010). Moreover, our data indicating TRPA1-dependent activation of NOS, leading to PKCε activation and subsequent restoration of TRPV1 sensitivity provides new evidence needed to elucidate the mechanisms by which TRPA1 and TRPV1 regulate each other’s function. A summary of the proposed cellular signaling pathway by which cross talk between TRPA1 and TRPV1 channels occurs is depicted in Figure8. The contribution of TRPV1 and TRPA1 to peripheral nociception supports the idea that one approach to control peripheral nociception is to better understand and manage cross regulation between TRPV1 and TRPA1 channels. A better understanding of these intricate mechanisms could prove vital in the search for therapeutic strategies to treat pathological conditions involving altered nociceptive processes.
Figure 8.

Schematic diagram depicting the proposed signaling pathway for cross talk between TRPA1 and TRPV1 ion channels (depicted by black arrows). Capsaicin (TRPV1 agonist); propofol and allyl isothiocyanate (AITC; TRPA1 agonists); HC-030031 (TRPA1 antagonist; l-NAME (neuronal nitric oxide synthase (nNOS) antagonist); NO (nitric oxide); SNAP (NO donor); protein kinase C epsilon (PKCε); ΨεRACK (PKCε activator peptide); εV1–2 (PKCε inhibitor peptide).
Acknowledgments
TRPA1 and TRPV1 cDNA were provided by David Julius, Ph.D., Professor, Department of Physiology, University of California at San Francisco. F-11 cells were gift from Probal Banerjee, Ph.D., Professor, Department of Chemistry, College of Staten Island, New York. Serine 800 phospho-TRPV1 antibody was provided by M.Tominaga, Division of Cell Signaling, Okazaki Institute for Integrative Biosciences (National Institute for Physiological Sciences), Japan.
Glossary
- AITC
allyl isothiocyanate
- c-PTIO
2-4-carboxyphenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide
- l-NAME
l-NG-nitroarginine methyl ester
- NO
nitric oxide
- NOS
nitric oxide synthase
- PKCε
protein kinase C epsilon
- RACK
receptor for activated C-kinase
- SNAP
S-nitroso-N-acetyl-d,l-penicillamine
- TRPA1
transient receptor potential ankyrin channel subtype-1
- TRPV1
transient receptor potential vanilloid channel subtype-1
Author Contributions
Zhang, Sinharoy, Sinha, Bratz, Damron participated in research design and performed the data analysis. Zhang, Sinharoy, Prudner conducted the experiments. Damron, Sinharoy, and Sinha wrote or contributed to the writing of the manuscript.
Disclosure
None declared.
References
- Akopian A. Regulation of nociceptive transmission at the periphery via TRPA1-TRPV1 interactions. Curr Pharm Biotechnol. 2011;12:89–94. doi: 10.2174/138920111793937952. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Ruparel NB, Jeske NA, Hargreaves KM. Transient receptor potential TRPA1 channel desensitization in sensory neurons is agonist dependent and regulated by TRPV1-directed internalization. J Physiol. 2007;583:175–193. doi: 10.1113/jphysiol.2007.133231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian A, Ruparel N, Patwardhan A, Hargreaves K. Cannabinoids desensitize capsaicin and mustard oil responses in sensory neurons via TRPA1 activation. J Neurosci. 2008;28:1064–1075. doi: 10.1523/JNEUROSCI.1565-06.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balafanova Z, Bolli R, Zhang J, Zheng Y, Pass JM, Bhatnagar A, et al. Nitric oxide (NO) induces nitration of protein kinase cepsilon (PKCepsilon), facilitating PKCe translocation via enhanced PKCepsilon-RACK2 interactions. J Biol Chem. 2002;277:15021–15027. doi: 10.1074/jbc.M112451200. [DOI] [PubMed] [Google Scholar]
- Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overveiw of a decade of research. J Mol Cell Cardiol. 2001;33:1897–1918. doi: 10.1006/jmcc.2001.1462. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brand G, Jacquot L. Sensitization and desensitization to allyl isothiocyanate (mustard oil) in the nasal cavity. Chem Sens. 2002;27:593–598. doi: 10.1093/chemse/27.7.593. [DOI] [PubMed] [Google Scholar]
- Burgstahler AD, Nathanson MH. NO modulates the apicolateral cytoskeleton of isolated hepatocytes by a PKC-dependent, CGMP-independent mechanism. Am J Physiol. 1995;269:G789–G799. doi: 10.1152/ajpgi.1995.269.5.G789. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 2001;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-e in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Chakraborti T, Das S, Chakraborti S. Proteolytic activation of protein kinase calpha by peroxynitrite in stimulating cytosolic phospholipase A2 in pulmonary endothelium: involvement of a pertussis toxin sensitive protein. Biochemistry. 2005;44:5246–5257. doi: 10.1021/bi0477889. [DOI] [PubMed] [Google Scholar]
- Ding Z, Gomez T, Werkheiser JL, Cowan A, Rawls SM. Icilin induces a hyperthermia in rats that is dependent on nitric oxide production and NMDA receptor activation. Eur J Phrmacol. 2008;578:201–208. doi: 10.1016/j.ejphar.2007.09.030. [DOI] [PubMed] [Google Scholar]
- Harada N, Miura T, Dairaku Y, Kametani R, Shibuya M, Wang R, et al. NO donor-activated PKC-d plays a pivitol role in ischemic myocardial protection through accelerated opening of mitochondrial K-ATP channels. J Cardiovasc Pharmacol. 2004;44:35–41. doi: 10.1097/00005344-200407000-00005. [DOI] [PubMed] [Google Scholar]
- Holzer P. The pharmacological challenge to tame the transient receptor potentail vanilloid-1 (TRPV1) nocisensor. Br J Pharmacol. 2008;155:1145–1162. doi: 10.1038/bjp.2008.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrucea C, Castro P, Sepulveda FJ, Wandersleben G, Roa J, Aguayo LG. Sustained increase of Ca2+ oscillations after chronic TRPV1 receptor activation with capsaicin in cultured spinal neurons. Brain Res. 2008;1218:70–76. doi: 10.1016/j.brainres.2008.04.035. [DOI] [PubMed] [Google Scholar]
- Lee S-J, Kim D-C, Choi B-H, Ha H, Kim K-T. Regulation of P53 by activated protein kinase C-d during nitric oxide-induced dopaminergic cell death. J Biol Chem. 2006;281:2215–2224. doi: 10.1074/jbc.M509509200. [DOI] [PubMed] [Google Scholar]
- Li S, Lin W, Tchantchou F, Lai R, Wen J, Zhang Y. Protein kinase C mediates peroxynitrite toxicty to oligodendrocytes. Mol Cell Neurosci. 2011;48:61–71. doi: 10.1016/j.mcn.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Liang M, Knox FG. Nitric oxide activates PKCa and inhibits Na+-K+-ATPase in opossum kidney cells. Am J Physiol. 1999;277:F859–F865. doi: 10.1152/ajprenal.1999.277.6.F859. [DOI] [PubMed] [Google Scholar]
- Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol. 2006;577:169–190. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandadi S, Numazaki M, Tominaga M, Bhat MB, Armati PJ, Roufogalis BD. Activation of protein kinase C reverses capsaicin-induced calcium-dependent desensitization of TRPV1 ion channels. Cell Calcium. 2004;35:471–478. doi: 10.1016/j.ceca.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Mandadi S, Tominaga T, Numazaki M, Murayama N, Saito N, Armati PJ, et al. Increased sensitivity of desensitized TRPV1 by PMA occurs through PKCe-mediated phosphorylation at S800. Pain. 2006;123:106–116. doi: 10.1016/j.pain.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Matta JA, Cornett PM, Miyares RL, Abe K, Sahibzada N, Ahern GP. General anesthetics activate a nociceptive ion channel to enhance pain and inflammation. Proc Natl Acad Sci. 2007;105:8784–8789. doi: 10.1073/pnas.0711038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Dubin AE, Petrus MJ, Patapoutian A. TRPV1 and TRPA1 mediate peripheral nitric oxide-induced nociception in mice. PLoS ONE. 2009;4:1–11. doi: 10.1371/journal.pone.0007596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio E, Watanabe Y. Nitric oxide donor-induced apoptosis in smooth muscle cells is modulated by protein kinase C and protein kinase A. Eur J Pharmacol. 1997;339:245–251. doi: 10.1016/s0014-2999(97)01368-x. [DOI] [PubMed] [Google Scholar]
- Novakova-Tousova K, Vyklicky L, Susankova K, Benedikt J, Samad A, Teisinger J, et al. Functional changes in the vanilloid dreceptor subtype 1 channel during and after acute desensitization. Neuroscience. 2007;149:144–154. doi: 10.1016/j.neuroscience.2007.07.039. [DOI] [PubMed] [Google Scholar]
- Palazzo E, Rossi F, Maione S. Role of TRPV1 receptors in descending modulation of pain. Mol Cell Endocrinol. 2008;286:S79–S83. doi: 10.1016/j.mce.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Ping P, Takano H, Zhang J, Tang XL, Qui Y, Li RC, et al. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ Res. 1999;84:587–604. doi: 10.1161/01.res.84.5.587. [DOI] [PubMed] [Google Scholar]
- Platika D, Boulos MH, Baizer L, Fishman MC. Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglia neurons with neuroblastoma cells. Proc Natl Acad Sci USA. 1985;82:3499–3503. doi: 10.1073/pnas.82.10.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter CD, Teng RJ, Beckman JS. Superoxide reacts with nitric oxide to nitrate tyrosine at physiological PH via peroxynitrite. J Biol Chem. 2000;275:460–466. doi: 10.1074/jbc.M910433199. [DOI] [PubMed] [Google Scholar]
- Ruparel N, Patwardhan A, Akopian A, Hargreaves K. Homologous and heterologous desensitization of capsaicin and mustard oil responses utilize different cellular pathways in nociceptors. Pain. 2008;135:271–279. doi: 10.1016/j.pain.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas MM, Hargreaves KM, Akopian AN. TRPA1-mediated responses in trigeminal sensory neurons: interaction between TRPA1 and TRPV1. Eur J Neurosci. 2009;29:1568–1578. doi: 10.1111/j.1460-9568.2009.06702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons CT, Carstens MI, Carstens E. Oral irritation by mustard oil: self-desensitization and cross-desenstization with capsaicin. Chem Sens. 2010;28:459–465. doi: 10.1093/chemse/28.6.459. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Wolfe D, Goss J, Watkins S, de Groat WC, Sculptoreanu A, et al. Protein kinase C epsilon contributes to basal and sensitizing responses of TRPV1 to capsaicin in rat dorsal root ganglion neurons. Eur J Pharmacol. 2008;28:1241–1254. doi: 10.1111/j.1460-9568.2008.06438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A, Jeske N, Akopian A. Contribution of TRPV1-TRPA1 interaction to the single-channel properties of the TRPA1 channel. J Biol Chem. 2010;285:15167–15177. doi: 10.1074/jbc.M110.106153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Wang Z, Cao J, Wang X, Han Y, Ma Z. Enhanced production of nitric oxide in A549 cells through activation of TRPA1 ion channel by cold stress. Nitric Oxide. 2014;40:31–35. doi: 10.1016/j.niox.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Wang Y. The functional regulation of TRPV1 and its role in pain sensitization. Neurochem Res. 2008;33:2008–2012. doi: 10.1007/s11064-008-9750-5. [DOI] [PubMed] [Google Scholar]
- Wickley PJ, Yuge R, Russell MS, Zhang H, Sulak M, Damron DS. Propofol modulates agonist-induced transient receptor potential vanilloid subtype-1 receptor de-sensitization via a protein kinase C epsilon-dependent pathway in mouse dorsal root ganglion sensory neurons. Anesthesiology. 2010;113:833–844. doi: 10.1097/ALN.0b013e3181eaa9a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Inoue R, Morii T, Takahashi N, yamamoto S, Hara Y, et al. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wickley PJ, Sinha S, Bratz IN, Damron DS. Propofol restores transient receptor potential vanilloid receptor subtype-1 sensitivity Via activation of transient receptor potential ankyrin receptor subtype-1 in sensory neurons. Anesthesiology. 2011;114:1169–1179. doi: 10.1097/ALN.0b013e31820dee67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou Z-S, Zhao Z-Q. PKC regulates capsaicin-induced currents in dorsal root ganglion neurons in rats. Neuropharmacology. 2001;41:601–608. doi: 10.1016/s0028-3908(01)00106-x. [DOI] [PubMed] [Google Scholar]