

Abstract
Ibrutinib (PCI-32765), a potent covalent inhibitor of Bruton’s tyrosine kinase, has shown efficacy against a variety of B-cell malignancies. Given the prominent role of CYP3A in ibrutinib metabolism, effect of coadministration of CYP3A perpetrators with ibrutinib was evaluated in healthy adults. Ibrutinib (120 mg [Study 1, fasted], 560 mg [studies 2 (fasted), and 3 (nonfasted)]) was given alone and with ketoconazole [Study 1; 400 mg q.d.], rifampin [Study 2; 600 mg q.d.], and grapefruit juice [GFJ, Study 3]. Lower doses of ibrutinib were used together with CYP3A inhibitors [Study 1: 40 mg; Study 3: 140 mg], as safety precaution. Under fasted condition, ketoconazole increased ibrutinib dose-normalized (DN) exposure [DN-AUClast: 24-fold; DN-Cmax: 29-fold], rifampin decreased ibrutinib exposure [Cmax: 13-fold; AUClast: 10-fold]. Under nonfasted condition, GFJ caused a moderate increase [DN-Cmax: 3.5-fold; DN-AUC: 2.2-fold], most likely through inhibition of intestinal CYP3A. Half-life was not affected by CYP perpetrators indicating the interaction was mainly on first-pass extraction. All treatments were well-tolerated.
Keywords: Bioavailability, CYP3A perpetrators, grapefruit juice, ibrutinib, ketoconazole, rifampin
Introduction
B-cell antigen receptor (BCR) signaling is implicated as a pivotal pathway in tumorigenesis in majority of B-cell malignancies. Antigen stimulation of normal B cells triggers dimerization of BCR initiating a downstream signaling kinase cascade, which in turn regulates multiple cellular processes, including proliferation, differentiation, apoptosis, and survival (Fuentes-Panana et al. 2004; Advani et al. 2013). Bruton’s tyrosine kinase (BTK), a critical terminal kinase enzyme in the BCR signaling pathway, is a promising target for therapeutic intervention in human malignancies. This downstream signal transduction protein plays a key role in the activation of pathways necessary for B-cell trafficking, chemotaxis, and adhesion, and has also been implicated in initiation, survival, and progression of mature B-cell lymphoproliferative disorders (Kuppers 2005).
Ibrutinib (Imbruvica®, PCI–32765), an orally active, BTK-targeting inhibitor, has been recently approved for the treatment of patients with chronic lymphocytic leukemia and mantle cell lymphoma who have received at least one prior therapy; Ibrutinib being the first covalent inhibitor of BTK to be advanced into human clinical trials. It forms a stable covalent bond with cysteine-481 on the active site of BTK and irreversibly inhibits BTK phosphorylation on Tyr223, impairing BCR signaling, and disrupting the proliferation and survival of malignant B-cells (IC50: 0.39 nM) (Honigberg et al. 2010). Ibrutinib is almost exclusively metabolized by cytochrome P450 (CYP) CYP3A. Absolute oral bioavailability (F) is low, ranging from 3.9% in the fasted state to 8.4% following a standard breakfast without grapefruit juice (GFJ) and 15.9% with GFJ (de Vries et al. 2015). A major metabolite of ibrutinib, PCI-45227, is a dihydrodiol metabolite that displays reversible binding with an inhibitory activity toward BTK approximately 15 times lower than that of ibrutinib (Parmar et al. 2014).
Ibrutinib has a mean peak plasma concentration observed at 1–2 h after administration. The mean terminal half-life is 4–13 h, with minimum drug accumulation after repeated dosing (<twofold) (Advani et al. 2013; Byrd et al. 2013; Imbruvica™., 2014). Population pharmacokinetic (PK) analysis indicated that ibrutinib clearance is independent of age (Marostica et al. 2015).
Multiple medications are administered in conjunction with ibrutinib for concurrent diseases including those for opportunistic infections. Given the prominent role of CYP3A in ibrutinib metabolism, drug–drug interactions (DDIs) that affect ibrutinib exposure and its metabolites, may occur when coadministered with potent CYP3A inhibitors or inducers. It is thus essential to understand the PK profile of ibrutinib when interacting with the CYP enzyme system, which can affect drug metabolism and clearance as well as, alter their safety and efficacy profile and/or of their active metabolites.
This paper discusses results from three phase 1 studies which were undertaken to obtain a comprehensive understanding of DDI between ibrutinib and CYP3A perpetrators ketoconazole (strong inhibitor) (Study 1), rifampin (strong inducer) (Study 2),and single-strength GFJ (classified as a moderate inhibitor, specific for intestinal CYP3A) (Study 3) and their effect on ibrutinib exposure. Because it became clear during clinical development that food by itself significantly increases the relative bioavailability, Study 3 was performed in nonfasted condition. In this way, the CYP3A DDI could be assessed under more relevant conditions, and data from both fasted and non-fasted condition could be used in building a robust physiologically based pharmacokinetic (PBPK) model.
Materials and methods
Study population
Healthy participants (nonsmokers) aged 18–55 years (inclusive) with body mass index 18–30 kg/m2 and body weight ≥50 kg were enrolled in all three studies. Study 1 enrolled only men, whereas studies 2 and 3 enrolled both men and women.
Participants with evidence of any clinically significant medical illness that could interfere with interpretation of study results or other abnormalities in physical examination, clinical laboratory parameters, vital signs, or ECG abnormalities etc. detected at screening, were excluded from all three studies. Women were required to be postmenopausal or surgically sterile. In all three studies, participants were to refrain from taking any over-the-counter or prescribed medications except acetaminophen (<3 g per day).
Protocols for each study were approved by an Independent Ethics Committee or Institutional Review Board at each study site and the studies were conducted in accordance with the ethical principles originating in the Declaration of Helsinki and in accordance with the ICH Good Clinical Practice guidelines, applicable regulatory requirements, and in compliance with the protocol. All participants provided written informed consent to participate in the studies.
Study design and treatment
Studies 1 and 2 were sequential design dedicated DDI studies of ibrutinib with ketoconazole and rifampin, respectively, versus Study 3, which was a two-way crossover DDI study with GFJ, combined with a formal absolute bioavailability study following single oral dose administration of ibrutinib in comparison with a single intravenous (i.v.) administration. All three studies were single center, and open-label.
Study 1 (clinicaltrials.gov identifier: NCT01626651) consisted of three phases: screening period (21 days), open-label treatment period (10 days), and follow-up period (10 ± 2 days). Participants received ibrutinib (120 mg, oral) on day 1 followed by blood sampling for PK analysis upto 72 h. Ketoconazole (Tara Pharmaceuticals), 400 mg oral, was given once-daily (q.d.) from days 4 to 9 (except on day 7); on day 7 they received ibrutinib (40 mg, oral) in combination with ketoconazole (400 mg, oral) 1 h before ibrutinib dosing.
Study 2 (clinicaltrials.gov identifier: NCT01763021) consisted of screening period (21 days), open-label treatment period (14 days) and follow-up period (10 ± 2 days). Participants received ibrutinib, 560 mg, oral, q.d. on day 1. Rifampin (VersaPharm) 600 mg oral, q.d. was given from days 4 to 13 (after the last ibrutinib sample collection for PK [over 72 h]). On day 11, a second single oral dose of ibrutinib 560 mg was administered, followed by the blood sample collection over 72 h.
In studies 1 and 2, ibrutinib was administered after overnight fast; food was withheld for 4 h after ibrutinib administration on dosing days.
Study 3 (clinicaltrials.gov identifier: NCT01866033) consisted of screening period (21 days), open-label treatment period (treatment A, B and C) (19 days), and follow-up period (10 ± 2 days). All participants received ibrutinib (560 mg) in treatment A and then randomized to either treatment B (ibrutinib [560 mg] administered 30 min after 240 mL of glucose in water) or treatment C (240 mL of GFJ [Albert Heijn pink] the evening before and 30 min before ibrutinib [140 mg]). In treatments B and C, participants had standard breakfast 30 min after ibrutinib dosing, as opposed to treatment A which was given in fasted condition. A single i.v. dose of 100 μg 13C6 PCI–32765 was administered 2 h after each ibrutinib oral dose.
In all three studies, participants took ibrutinib with 240 mL of water and lunch and subsequent standard meals were provided 4 h after oral ibrutinib dosing. Participants remained seated throughout morning (from 30 min before dosing until after lunch), in order to minimize inter and intrasubject intestinal blood flow differences.
Pharmacokinetic evaluations
Sample collection
Blood samples for all studies were collected by direct venipuncture or through an indwelling peripheral venous heparin lock catheter into heparin collection tubes. Samples were centrifuged at approximately 4°C (15 min at 1300 g); plasma was stored at ≤−70°C.
Blood samples for PK analysis were collected predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 48, and 72 h postdose on days 1 and 7 (Study 1 and 3) and days 1 and 11 (Study 2) for quantification of ibrutinib and PCI-45227. Additional blood samples were collected 2 h after ketoconazole dosing on day 7 (1 h after ibrutinib dosing) (Study 1) and 2 h after rifampin dosing on day 11 (Study 2) for ketoconazole and rifampin measurement, respectively. In Study 2, samples were collected and analyzed for determination of 4-β-hydroxycholesterol concentration on day -1, 12 h after ibrutinib administration on day 11 and 14.
Analytical methods
Plasma concentrations of ibrutinib, its metabolite, PCI–45227, ketoconazole, rifampin, and 13C6 ibrutinib, were determined using validated analytical liquid chromatography-tandem mass spectroscopy (LC-MS/MS) methods. Bioanalyses were conducted at Department of Bioanalysis, Janssen R&D and at Frontage Laboratories, Inc., Exton, PA. Quantification range was 0.100–25.0 ng/mL for ibrutinib and PCI-45227 (Study 1 and 2) and 0.5–100 ng/mL (Study 3) (de Vries et al. 2015). Quantification range for 13C6 PCI-32765 was 2–1000 pg/mL (de Vries et al. 2015).
Pharmacokinetic analysis
PK analyses were performed by noncompartmental methods using validated WinNonlin® software Version 5.2.1 (Certara USA, Inc. Princeton, NJ) and Phoenix WinNonlin 6.3. Key PK parameters included maximum observed plasma concentration (Cmax), time to reach maximum observed plasma concentration (tmax), elimination half-life associated with terminal slope (λz) of the semilogarithmic drug concentration-time curve (t1/2λ), area under the plasma concentration-time curve (AUC) from time 0 to 24 h (AUC24), AUC from time 0 to time of the last quantifiable concentration (AUClast), AUC from time 0 to infinity (AUC∞) and metabolite/parent ratios. In Study 1, apparent total clearance of drug after extravascular administration (CL/F), and apparent volume of distribution based on the terminal phase (Vdz) were also estimated. Additionally, in Study 3, absolute bioavailability (F) and total clearance of drug after i.v. administration (CL) were also estimated.
Safety evaluations
Safety evaluations included assessments of treatment-related adverse events (AE), vital signs, 12-lead electrocardiograms, clinical laboratory tests, and physical examinations. The AE severity was graded according to the National Cancer Institute - Common Terminology Criteria for Adverse Events (NCI-CTCAE) grading system version 4.03.
Analysis sets
Participants who had estimations of PK parameters of ibrutinib for both periods (ibrutinib administered alone and in combination with CYP3A perpetrators) were included in PK analysis set for statistical comparison. Participants who received at least one dose of study medication were included in safety analysis set.
Sample size determination
For all three studies, sample size determinations were based on statistical estimation. A sample size is considered adequate if the point estimates of GMR for PK parameters of primary interest (Cmax, AUCs) fall within the no-effect boundaries of clinical equivalence (90% CI) to the compound.
Statistical analyses
All individual and mean plasma concentrations and estimated PK parameters were presented by graphic and descriptive statistics methods for each treatment. Linear mixed-effect models were applied to evaluate potential DDI effect. Log-transformation was performed on PK parameters (Cmax, AUCs) prior to the analysis, and 90% confidence intervals for GMR (with/without coadministration of interacting drugs) were constructed on original scale.
Results
Subject disposition and demographics
All enrolled participants (Study 1: n = 18; Study 3: n = 8) completed studies 1 and 3. In Study 2, 17/18 enrolled participants completed the study. One participant was excluded from the PK-evaluable population due to a protocol deviation (use of prohibited concomitant pain medication). Demographics and baseline characteristics of the participants in three studies were consistent with the inclusion and exclusion criteria (Table1). All participants received scheduled doses of the study drugs.
Table 1.
Demographics and baseline characteristics (safety analysis set)
| Study 11 (n = 18) | Study 22 (n = 18) | Study 33 (n = 8) | |
|---|---|---|---|
| Sex, n | |||
| Women | – | 8 | 5 |
| Men | 18 | 10 | 3 |
| Race, n (%) | |||
| White | 4 (22) | 5 (28) | 8 (100) |
| Black or African American | 11 (61) | 11 (61) | – |
| Other/multiple | 2 (11) | 2 (11) | – |
| Ethnicity, n (%) | |||
| Not Hispanic or Latino | 16 (89) | 16 (89) | 8 (100) |
| Hispanic or Latino | 2 (11) | 2 (11) | 0 |
| Age (years) | |||
| Mean (SD) | 33.7 (8.8) | 41.1 (11.6) | 46.4 (8.1) |
| Baseline weight (kg) | |||
| Mean (SD) | 78.0 (7.4) | 77.6 (8.7) | 70.0 (13.2) |
| Baseline BMI (kg/m2) | |||
| Mean (SD) | 26.1 (2.3) | 26.4 (2.5) | 23.5 (2.7) |
BMI, body mass index; GFJ, grapefruit juice; SD, standard deviation.
Study 1: ibrutinib + ketoconazole.
Study 2: ibrutinib + rifampin.
Study 3: Ibrutinib + grapefruit juice.
Pharmacokinetic results
Study 1: effect of ketoconazole on pharmacokinetics of ibrutinib and its metabolite
Following coadministration of ibrutinib with ketoconazole under fasted condition, mean DN_Cmax (dose normalized to 120 mg) of ibrutinib increased from 11.8 to 325 ng/mL and mean DN_AUClast increased from 71.4 to 1599 ng·h/mL (Fig.1A). Although dose proportionality was not formally tested for ibrutinib, no deviations from linearity were observed neither in the Phase I escalating dose study nor population PK study (imbruvica™ 2014; Marostica et al. 2015), thus justifying the dose-normalization of ibrutinib exposure in this study. Intersubject variability in ibrutinib+ketoconazole treated participants for both Cmax and AUClast were >50% following ibrutinib administration alone and approximately 40% when coadministered with ketoconazole. The Vd/F and CL/F were both lower following ibrutinib + ketoconazole compared with ibrutinib alone (Vd/F: 885 L vs. 19049 L; CL/F: 92.0 L/h vs. 2014 L/h, whereas there was no change in mean tmax (2.00 h vs. 1.75 h) and t1/2 (6.32 h vs. 8.20 h) (Table S1).
Figure 1.
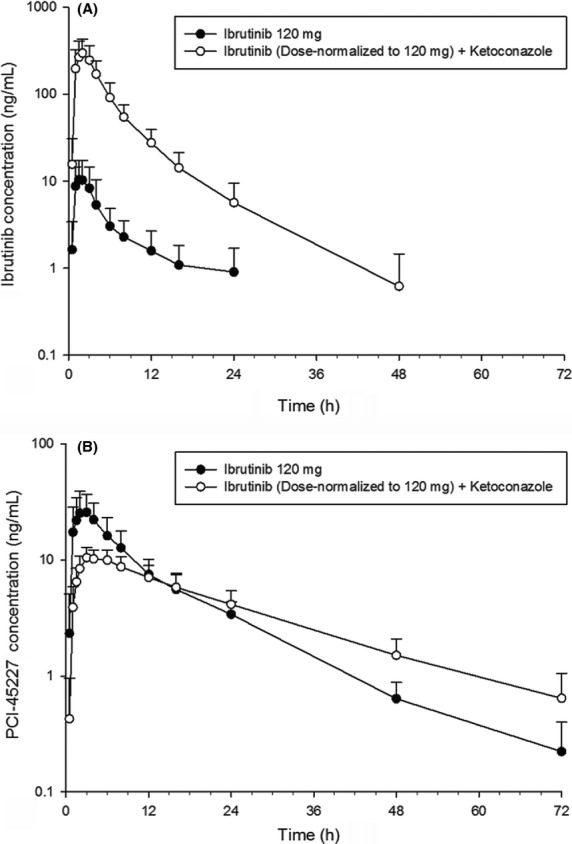
Dose-normalized mean (SD) logarithmic-linear plasma concentration-time profiles following oral administration of ibrutinib (120 mg) alone (day 1) and in combination with ketoconazole (40 mg Ibrutinib+400 mg Ketoconazole) (day 7) to healthy men. (A) Ibrutinib; (B) PCI-45227.
On the other hand, dose-normalized PCI-45227 exposure was lower following coadministration with ketoconazole compared with ibrutinib administration alone (Fig.1B). The DN_Cmax was 2.6 times lower (11.1 ng/mL vs. 29.1 ng/mL) and DN_AUClast was 1.2 times lower (256 ng·h/mL vs. 304 ng h/mL) following ibrutinib + ketoconazole treatment compared with ibrutinib alone. Intersubject variability for both Cmax and AUClast was approximately 40% following ibrutinib administration alone and approximately 20% following ibrutinib + ketoconazole. Tmax and t1/2 were both slightly longer following ibrutinib + ketoconazole (Tmax: 4.00 h vs. 2.00 h; t1/2 18.00 h vs. 11.41 h). Following coadministration with ketoconazole, ibrutinib mean DN_Cmax increased approximately 29-fold (geometric mean [GM]: 285.49 vs. 10.0 ng/mL), and mean DN_AUClast increased approximately 24-fold (GM: 1463.43 vs. 61.16 ng·h/mL) (Table2). Mean metabolite to parent ratios decreased from 2.64 to 0.05 for Cmax and from 5.03 to 0.19 for AUC24 following ibrutinib+ketoconazole coadministration.
Table 2.
Geometric mean ratio and the 90% CI of the combination treatment (studies 1, 2, and 3) over ibrutinib
| Parameter | Test treatment/reference treatment7 | N | Geometric mean | Ratio: (%)8 | 90% CI (%)8 | Intrasubject CV (%) |
|---|---|---|---|---|---|---|
| Ibrutinib + Ketoconazole4 | ||||||
| Cmax (ng/mL)1,2 | Ibrutinib + Ketoconazole | 18 | 286 | 2854.5 | 2397–3400 | 31 |
| Ibrutinib | 18 | 10 | ||||
| AUC24 (ng·h/mL)1,2 | Ibrutinib + Ketoconazole | 18 | 1390 | 2480.1 | 2002–3073 | 38 |
| Ibrutinib | 18 | 56 | ||||
| AUClast (ng·h/mL)1,2 | Ibrutinib + Ketoconazole | 18 | 1463 | 2392.8 | 1901–3012 | 41 |
| Ibrutinib | 18 | 61 | ||||
| AUC∞ (ng·h/mL)1,2 | Ibrutinib + Ketoconazole | 12 | 1860 | 2620.2 | 1996–3440 | 39 |
| Ibrutinib | 12 | 71 | ||||
| Ibrutinib + Rifampin5 | ||||||
| Cmax (ng/mL)2 | Ibrutinib + Rifampin | 17 | 3 | 7.9 | 6–12 | 69 |
| Ibrutinib | 17 | 32 | ||||
| AUC24h (ng·h/mL)2 | Ibrutinib + Rifampin | 17 | 23 | 10.9 | 8–15 | 61 |
| Ibrutinib | 17 | 214 | ||||
| AUClast (ng.h/mL)2 | Ibrutinib + Rifampin | 17 | 28 | 10.4 | 7–15 | 62 |
| Ibrutinib | 17 | 267 | ||||
| AUC∞ (ng·h/mL)2 | Ibrutinib + Rifampin | 4 | 46 | 15.2 | 5–46 | 74 |
| Ibrutinib | 4 | 300 | ||||
| Ibrutinib + Grapefruit Juice6 | ||||||
| Cmax (ng/mL)2,3 | Ibrutinib + GFJ | 8 | 437 | 360.4 | 269–483 | 31 |
| Ibrutinib | 8 | 121 | ||||
| AUC24 (ng·h/mL)2,3 | Ibrutinib + GFJ | 7 | 1337 | 252.9 | 219–293 | 13 |
| Ibrutinib | 7 | 529 | ||||
| AUClast (ng·h/mL)2,3 | Ibrutinib + GFJ | 8 | 1236 | 210.2 | 182–243 | 15 |
| Ibrutinib | 8 | 588 | ||||
| AUC∞ (ng·h/mL)2,3 | Ibrutinib + GFJ | 7 | 1378 | 214.5 | 184–250 | 14 |
| Ibrutinib | 7 | 643 | ||||
CV, coefficient of variation; GFJ, grapefruit juice.
Parameter values were natural log (ln) transformed and dose normalized to 120 mg ibrutinib before analysis.
A mixed-effect model with treatment as a fixed effect and participant as a random effect was used. Parameter values were natural log (ln) transformed before analysis.
The oral ibrutinib with grapefruit juice treatment group was dose normalized to 560 mg.
Ibrutinib: 40 mg and ketoconazole: 400 mg.
Ibrutinib: 560 mg and rifampin: 600 mg.
Ibrutinib: 560 mg and GFJ: 240 mL.
Test Treatment: ibrutinib + ketoconazole/ibrutinib + rifampin/ibrutinib + grapefruit juice, Reference Treatment: ibrutinib.
Ratio of parameter means (expressed as a percent) and 90% CIs were transformed back to the linear scale.
Plasma concentrations for ketoconazole at 1 h after drug intake on day 7 ranged from 231 to 15,800 ng/mL. Two participants had very low ketoconazole concentrations (352 ng/mL and 231 ng/mL); the interaction was still within the observed range.
Study 2: effect of rifampin on the pharmacokinetics of ibrutinib and its metabolite
Following coadministration with rifampin under fasted condition, ibrutinib mean Cmax and AUClast decreased (Cmax: 42.1 ng/mL to 3.38 ng/mL; AUClast: 335 ng·h/mL to 38.0 ng·h/mL) compared with ibrutinib administration alone (Fig.2A). Intersubject variability for both Cmax and AUClast was greater than 60% following ibrutinib administration alone and greater than 70% following ibrutinib+rifampin. Median tmax was delayed from 1.76 to 3.00 h. Terminal t1/2 was similar; due to multiple data points below the quantification limit in the elimination phase, it could only be calculated for 5 of 17 participants.
Figure 2.

Mean (SD) logarithmic-linear plasma concentration-time profiles of ibrutinib (560 mg) and metabolite PCI-45227 in absence and presence of CYP perpetrators: Rifampin (600 mg) (A and B: fasted condition) Grapefruit Juice (C and D: nonfasted condition).
Following coadministration with rifampin, PCI-45227 mean Cmax decreased from 70.0 ng/mL to 49.9 ng/mL and mean AUClast decreased from 946 ng·h/mL to 374 ng·h/mL (Fig.2B). Intersubject variability for both Cmax and AUClast was greater than 30% following ibrutinib administration alone and greater than 20% following ibrutinib + rifampin. Similar to the parent, median tmax of the metabolite was delayed following ibrutinib + rifampin coadministration compared with ibrutinib treatment alone (from 2.02 to 3.00 h). Terminal t1/2 trended shorter for the combination treatment. The GMR for Cmax and AUClast was 7.94% and 10.44% (or a 13- and 10-fold decrease), respectively, for ibrutinib + rifampin compared with ibrutinib alone (Table2). Metabolite to parent ratio for Cmax increased from 2.09 to 20.80, and the ratio for AUClast increased from 3.10 to 15.50.
Plasma concentrations for rifampin on day 11, 2 h after drug intake ranged from 289 ng/mL to 18400 ng/mL (mean ± SD = 9247 ± 4957 ng/mL). Low concentration of 289 ng/mL did not adversely affect induction, as decrease in ibrutinib exposure on day 11 in this participant was comparable with that observed in other participants. Compared with predose values (39.9 [15.50]) ng/mL), 4-β-hydroxycholesterol concentrations increased following multiple once-daily oral administrations of rifampin, providing evidence that sufficient induction of CYP3A had occurred when 560 mg ibrutinib (day 11) was administered after 1 week of rifampin 600 mg q.d., and that induction was maintained until the 72-h PK sample was collected on day 14 (day 11: 97.3 [29.4] ng/mL and day 14: 119 [39.6] ng/mL).
Study 3: effect of GFJ on ibrutinib exposure
Pretreatment with GFJ increased ibrutinib concentrations in plasma. DN_Cmax of ibrutinib increased by 3.5-fold, whereas DN_AUC increased by 2.2-fold in presence of single-strength GFJ compared with oral administration of 560 mg ibrutinib without GFJ under nonfasted condition (Fig.2C). On the other hand, the AUCs of ibrutinib following i.v. administration under nonfasted conditions with and without GFJ were the same. Mean Cmax was slightly higher with GFJ, but intrasubject variability was very high (84%) (Fig.3).
Figure 3.
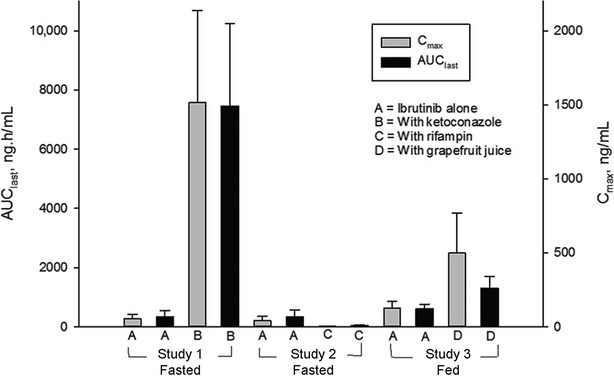
Mean (SD) AUClast and Cmax of ibrutinib following oral administration of ibrutinib alone and in combination with ketoconazole, rifampin, or grapefruit juice; data dose normalized to 560 mg.
There was no effect of GFJ on tmax of PCI-45227 (Fig.2D). Dose-normalized PCI-45227 concentrations and AUCs following oral administration of ibrutinib with or without GFJ were comparable. Cmax metabolite-to-parent ratio decreased from 1.03 to 0.32 with the addition of GFJ.
When ibrutinib was orally administered with GFJ, apparent clearance, CL/F and half-life both decreased by half; CL after i.v. administration, however, was unchanged with or without GFJ. The CL following i.v. administration under nonfasted condition was higher than that under fasted condition.
The magnitude of observed DDI was higher with higher baseline clearance. The higher the baseline CL/F, the larger the effect of ketoconazole. This trend was not observed clearly for the weaker (and intestine-specific) inhibitor GFJ, or for the inducer rifampin (Fig.4).
Figure 4.
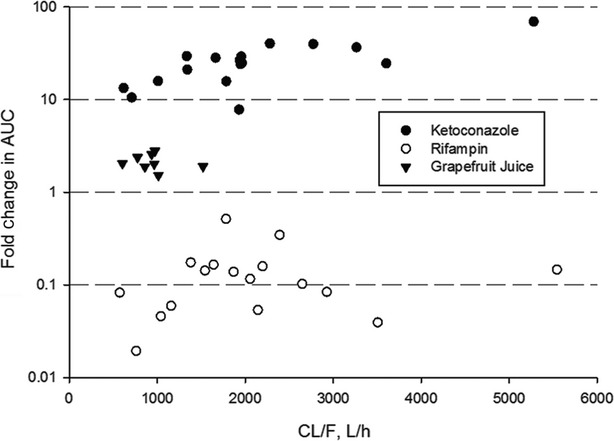
Fold-change in AUC versus baseline apparent clearance following oral administration of ibrutinib with ketoconazole or rifampin (both under fasted conditions) or with grapefruit juice (with standard meal).
Safety
Treatments were generally well-tolerated in all three studies. In Study 1, only one (6%) participant reported ≥1 AE (musculoskeletal discomfort) after ibrutinib administration alone, and six participants (33%) reported ≥1 AE following coadministration with ketoconazole. In Study 1, the most common AEs (>10%) following ibrutinib+ketoconazole coadministration were headache (n = 4, 22%), venipuncture-related hematoma and pain (n = 1, 6%), abdominal discomfort and dyspepsia (n = 2, 11%) (Table3). All AEs were mild in severity (grade 1), limited in duration, and, resolved without medical intervention. In Study 2, 5 participants (28%) reported ≥1AE after ibrutinib administration alone, compared with 22% (n = 4) following combination therapy with rifampin. Most common AEs following treatment with ibrutinib alone were musculoskeletal discomfort (n = 2, 11%) and following ibrutinib+rifampin included toothache (n = 2, 11%) and headache (n = 3, 17%). All AE were of grade 1 intensity except for two participants who experienced grade 2 AEs (headache and morbilliform skin rash) following rifampin treatment. In Study 3, the most common AEs reported by >1 subject included abdominal pain, diarrhea, and dizziness. All events were grade 1 in severity except for grade 2 abdominal pain in one participant. There were no serious AEs, AEs leading to discontinuation or AEs that did not resolve at end of the study, reported in the three studies. There were no relevant changes in vital signs or 12-lead ECG, laboratory safety or changes in physical examination findings in any of the studies.
Table 3.
Summary of treatment-emergent adverse events (AE) seen in more than 10% of participants in any study (safety analysis set)
| Study 11 n = 18 n (%) | Study 22 n = 18 n (%) | Study 33 n = 8 n (%) | |
|---|---|---|---|
| Number of participants with treatment-emergent AE | 6 (33) | 7 (39) | 3 (38) |
| Diarrhea | – | – | 3 (38) |
| Abdominal pain | – | – | 3 (38) |
| Dyspepsia | – | – | 1 (13) |
| Flatulence | – | – | 1 (13) |
| Vomiting | – | – | 1 (13) |
| Nausea | – | – | 1 (13) |
| Toothache | – | 2 (11) | |
| Musculoskeletal discomfort | – | 2 (11) | |
| Back injury | – | – | 1 (13) |
| Epicondylitis | – | – | 1 (13) |
| Dizziness | – | – | 2 (25) |
| Headache | 4 (22) | 3 (17) | 1 (13) |
| Hyperventilation | – | – | 1 (13) |
Study 1: ibrutinib + ketoconazole.
Study 2: ibrutinib + rifampin.
Study 3: ibrutinib + grapefruit juice.
Discussion
Ibrutinib is extensively metabolized by CYP3A and has low bioavailability due to extensive first-pass metabolism. Low bioavailability results in more variable PK and potential variability in desired therapeutic response as well as undesirable adverse effects. Studies reported here were therefore designed to understand the DDI potential between ibrutinib and CYP3A perpetrators (ketoconazole, rifampin and GFJ) and their probable effects on ibrutinib bioavailability in healthy participants.
In line with in vitro data that showed 96% of microsomal clearance could be attributed to CYP3A4 (Scheers et al. 2015), CYP3A perpetrators had a major effect on ibrutinib exposure, without affecting the terminal half-life. There was an increase in ibrutinib concentration following coadministration with ketoconazole; intersubject variability was lower compared to ibrutinib alone, which can be explained by close to complete bioavailability in this situation. Though ketoconazole concentration was within the observed range, a low value observed in two participants may be due to the over-expression of CYP3A in these two participants. These values correlated with ibrutinib Cmax and AUC that were approximately 80% and 70% lower, respectively, than the mean values, both with and without ketoconazole coadministration. Thus, the interaction did not differ significantly from the mean value, suggesting that complete inhibition was still obtained. In fasted condition, F was found to be 3.9% (90% CI = 3.06–5.02) in Study 3 (de Vries et al. 2015), implying that a 24-fold increase in ibrutinib AUC∞ with ketoconazole (Study 1 and hence a different cohort of participants) would result in F of approximately 73–121% (24 × 3.06 to 5.02%). In line with ketoconazole study observations, intersubject variability of ibrutinib exposure increased following rifampin coadministration, due to significantly decreased bioavailability.
There was a concomitant decrease in metabolite PCI-45227 concentration following coadministration of ibrutinib with both ketoconazole and rifampin. Decrease in exposure of PCI-45227 observed with rifampin may be attributed to further metabolism of PCI-45227 involving CYP3A or induction of P-gp, of which PCI-45227 is a substrate. Reductions in Cmax and AUC in Study 2 correlated with an increase in 4-β-hydroxycholesterol, an endogenous biomarker of CYP3A activity (Bjorkhem-Bergman et al. 2013; Marde Arrhen et al. 2013; Dutreix et al. 2014). The results were in agreement with previously reported data (Bjorkhem-Bergman et al. 2013).
Both studies 1 and 2 thus confirm that the metabolic pathway of ibrutinib is almost exclusively through CYP3A metabolism during first pass through intestine and liver. Results from this study verified the earlier in silico PBPK simulations (unpublished), which showed a 10- and 11-fold decrease (GMR) in simulated ibrutinib AUC and Cmax, respectively, following rifampin 600 mg q.d. Similarly, results were consistent with that obtained for the simulated ketoconazole interaction. As reported for other sensitive CYP3A substrates, it is thus recommended to avoid concomitant administration of strong CYP3A inhibitors and inducers in order to avoid potential ibrutinib over- and under-exposures, respectively.
Furanocoumarins found in GFJ have been shown to be potent inhibitors of CYP3A in humans contributing to GFJ-drug interactions in the gut (Guo et al. 2000; Guo and Yamazoe 2004). In Study 3, single-strength (250 mL) GFJ was administered at different time points before ibrutinib dosing, to enable the differentiation of the first-pass effect caused by the liver from that in the intestinal wall. Inhibition of mucosal metabolism results in reduced first-pass metabolism and increased bioavailability (Bailey et al. 1995; Ducharme et al. 1995; Veronese et al. 2003; Seidegard et al. 2012).
Pretreatment with GFJ increased ibrutinib concentrations and decreased apparent clearance by half. Intravenous clearance, however, remained unchanged. This is consistent with earlier PK studies performed using GFJ (Garg et al. 1998; Hanley et al. 2011; Shoaf et al. 2012). By comparing F of ibrutinib with and without GFJ, it was estimated that, under nonfasted condition, approximately 47% of ibrutinib is available after first-pass metabolism in the gut. Of this amount, the majority is cleared in the liver (de Vries et al. 2015).
It was known that ibrutinib is prone to a significant food effect (de Jong et al. 2015). Administration of ibrutinib in fasted condition resulted in approximately 60% of exposure (AUClast) to that observed either 30 min before or 2 h after a meal. In addition, a mass-balance study with ibrutinib conducted in fasted state indicated that oral absorption was complete (Scheers et al. 2015). This has led to the hypothesis that food, rather than via lipid-assisted solubilization, increases the oral bioavailability by diminishing hepatic and intestinal CYP3A first-pass elimination. Presumably, food ‘blunts’ the impact of hepatic and intestinal CYP3A metabolism by increasing the mesenteric and splanchnic blood flow, thus decreasing the residence time in GI and liver tissue and increasing transit time of the drug into central circulation, resulting in higher systemic bioavailability compared to fasted condition (e.g., caused by decreased residence time in hepatocytes and enterocytes). A similar observation was made for budesonide and other high extraction drugs (Marasanapalle et al. 2011; Seidegard et al. 2012). Thus, DDI on AUC of ibrutinib caused by GFJ in this study is less than that estimated following fasting condition. These data were subsequently used to expand the PBPK model to allow DDI simulations in presence of food [unpublished].
To minimize exposure to study drug, treatment dosages of 120 mg of ibrutinib alone and 40 mg in combination with ketoconazole were chosen in Study 1. Reduced dose of ibrutinib (40 mg) was chosen as a safety precaution as Study 1 was the first study conducted in healthy participants and also based on the assumption that ibrutinib exposure will increase following administration of ketoconazole. Furthermore, there were no reasons to suspect PK non-linearity. As ibrutinib exposures in Study 1 exceeded mean exposures at 560 mg in patients and were well-tolerated, therapeutic doses were applied in Study 2. To compensate for potential impact of CYP3A inhibition caused by GFJ at the level of absorption from intestinal lumen, a 140 mg ibrutinib dose was used rather than 560 mg dose in Study 3. Simulations using SimCyp® modeling and simulation software predicted that maximum increase in exposure caused by GFJ would be less than fourfold, thus warranting that exposures would not exceed those in the 560 mg treatment. The 400 mg for ketoconazole and 600 mg dose used for rifampin are the maximum recommended daily doses.
All three treatments were well-tolerated (regardless of meal or GFJ intake status) with no persisting treatment-emergent AEs. It should, however, be noted that these studies were conducted in healthy participants and the extent of interaction in patient setting, where they may be exposed to different drugs, needs to be ascertained.
In summary, ibrutinib showed an increased exposure following coadministration with a strong and moderate CYP3A inhibitor (ketoconazole and GFJ) and a decreased exposure in the presence of a strong CYP3A inducer (rifampin). Mean metabolite to parent ratios decreased following coadministration with CYP3A inhibitors (ketoconazole and GFJ), whereas it increased with a CYP3A inducer (rifampin). The magnitude of CYP3A inhibition and corresponding increase in ibrutinib exposure was higher in participants with a higher baseline clearance (i.e., lower AUC), likely because those participants had a higher baseline expression of CYP3A. Vice versa, this implies that participants with a high baseline exposure are not at risk of extreme increases when treated concomitantly with strong inhibitors, as complete bioavailability cannot be exceeded. No new safety signals were observed. Ibrutinib dose interruption or modification is warranted when treatment of a patient on ibrutinib requires administration of strong or moderate CYP3A inhibitors. PK and safety data collection in patients on concomitant CYP3A inhibitors is ongoing. From uncontrolled clinical trials with small and unequal sample group size, ibrutinib exposure in patients who received ibrutinib concomitantly with moderate or mild CYP3A inhibitors appears to be within twofold of the upper range of the exposure observed in patients who did not take CYP3A inhibitors. A DDI study of ibrutinib with CYP3A inhibitors in patients with a cancer diagnosis is planned to confirm the interaction potential in this patient population (Sukbuntherng et al. 2014).
Acknowledgments
Dr. Shalini Nair (SIRO Clinpharm Pvt. Ltd.) provided the writing assistance and Dr. Namit Ghildyal (Janssen Research & Development, LLC) provided additional editorial support for the development of this manuscript. The authors also thank the study participants, without whom this study would not have been accomplished, as well as the investigators for their participation in this study.
Glossary
- AE
adverse events
- BCR
B-cell antigen receptor
- BTK
Bruton’s tyrosine kinase
- DDI
drug–drug interaction
- GFJ
grapefruit juice
- LC-MS/MS
liquid chromatography-tandem mass spectroscopy
- PBPK
physiologically based pharmacokinetic
Author Contributions
De Jong, Skee, Sukbuntherng, Smit, Hellemans, Mannaert, Jiao, and Snoeys participated in research design of this study. De Vries conducted the experiments. Skee, Murphy, Snoeys, and Jiao performed data analysis or interpretation of this study. De Jong, Skee, Sukbuntherng, Smit, Mannaert, Jiao, Snoeys, de Vries, Hellemans, and Murphy reviewed or contributed to the development of the manuscript.
Disclosures
Authors de Jong, Skee, Smit, Mannaert, Jiao, Snoeys, de Vries, Hellemans, Murphy and Jiao are employees of Janssen Research & Development and hold stocks in the company. Author Sukbuntherng is an employee of Pharmacyclics Inc. All authors meet ICMJE criteria and all those who fulfilled those criteria are listed as authors. All authors had access to the study data and made the final decision about where to publish these data.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Summary of pharmacokinetic parameters of ibrutinib and PCI-45227.
References
- Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DG, Arnold JM, Bend JR, Tran LT, Spence JD. Grapefruit juice-felodipine interaction: reproducibility and characterization with the extended release drug formulation. Br J Clin Pharmacol. 1995;40:135–140. [PMC free article] [PubMed] [Google Scholar]
- Bjorkhem-Bergman L, Backstrom T, Nylen H, Ronquist-Nii Y, Bredberg E, Andersson TB, et al. Comparison of endogenous 4beta-hydroxycholesterol with midazolam as markers for CYP3A4 induction by rifampicin. Drug Metab Dispos. 2013;41:1488–1493. doi: 10.1124/dmd.113.052316. [DOI] [PubMed] [Google Scholar]
- Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducharme MP, Warbasse LH, Edwards DJ. Disposition of intravenous and oral cyclosporine after administration with grapefruit juice. Clin Pharmacol Ther. 1995;57:485–491. doi: 10.1016/0009-9236(95)90032-2. [DOI] [PubMed] [Google Scholar]
- Dutreix C, Lorenzo S, Wang Y. Comparison of two endogenous biomarkers of CYP3A4 activity in a drug-drug interaction study between midostaurin and rifampicin. Eur J Clin Pharmacol. 2014;70:915–920. doi: 10.1007/s00228-014-1675-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Panana EM, Bannish G, Monroe JG. Basal B-cell receptor signaling in B lymphocytes: mechanisms of regulation and role in positive selection, differentiation, and peripheral survival. Immunol Rev. 2004;197:26–40. doi: 10.1111/j.0105-2896.2004.0105.x. [DOI] [PubMed] [Google Scholar]
- Garg SK, Kumar N, Bhargava VK, Prabhakar SK. Effect of grapefruit juice on carbamazepine bioavailability in patients with epilepsy. Clin Pharmacol Ther. 1998;64:286–288. doi: 10.1016/S0009-9236(98)90177-1. [DOI] [PubMed] [Google Scholar]
- Guo LQ, Yamazoe Y. Inhibition of cytochrome P450 by furanocoumarins in grapefruit juice and herbal medicines. Acta Pharmacol Sin. 2004;25:129–136. [PubMed] [Google Scholar]
- Guo LQ, Taniguchi M, Xiao YQ, Baba K, Ohta T, Yamazoe Y. Inhibitory effect of natural furanocoumarins on human microsomal cytochrome P450 3A activity. Jpn J Pharmacol. 2000;82:122–129. doi: 10.1254/jjp.82.122. [DOI] [PubMed] [Google Scholar]
- Hanley MJ, Cancalon P, Widmer WW, Greenblatt DJ. The effect of grapefruit juice on drug disposition. Expert Opin Drug Metab Toxicol. 2011;7:267–286. doi: 10.1517/17425255.2011.553189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbruvica™. 2014. Summary of product information European Medicines Agency. Available at http://www.medicines.org.uk/emc/medicine/29383 (accessed January 9, 2015)
- de Jong J, Sukbuntherng J, Skee D, Murphy J, O’Brien S, Byrd JC, et al. The effect of food on the pharmacokinetics of oral ibrutinib in healthy participants and patients with chronic lymphocytic leukemia. Cancer Chemother Pharmacol. 2015;75:907–916. doi: 10.1007/s00280-015-2708-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukbuntherng J, de Jong J, Skee D, Pak Y, Fardis M, O’Brien S. 2014. Pharmacokinetics and safety of ibrutinib with concomitant use of CYP3A inhibitors in patients with B-cell malignancies. Presented at the American College of Clinical Pharmacy (ACCP) annual meeting, Austin, Texas, Oct 12–15.
- Kuppers R, et al. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5:251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- Marasanapalle VP, Boinpally RR, Zhu H, Grill A, Tang F. Correlation between the systemic clearance of drugs and their food effects in humans. Drug Dev Ind Pharm. 2011;37:1311–1317. doi: 10.3109/03639045.2011.571697. [DOI] [PubMed] [Google Scholar]
- Marde Arrhen Y, Nylen H, Lovgren-Sandblom A, Kanebratt KP, Wide K, Diczfalusy U. A comparison of 4beta-hydroxycholesterol: cholesterol and 6beta-hydroxycortisol: cortisol as markers of CYP3A4 induction. Br J Clin Pharmacol. 2013;75:1536–1540. doi: 10.1111/bcp.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marostica E, Sukbuntherng J, Loury D, de Jong J, de Trixhe XW, Vermeulen A, et al. Population pharmacokinetic model of ibrutinib, a Bruton tyrosine kinase inhibitor, in patients with B cell malignancies. Cancer Chemother Pharmacol. 2015;75:111–121. doi: 10.1007/s00280-014-2617-3. [DOI] [PubMed] [Google Scholar]
- Parmar S, Patel K, Pinilla-Ibarz J. Ibrutinib (imbruvica): a novel targeted therapy for chronic lymphocytic leukemia. P T. 2014;39:483–519. [PMC free article] [PubMed] [Google Scholar]
- de Vries R, Smit JW, Hellemans P, Jiao J, Murphy J, Skee D, et al. Stable Isotope-Intravenous Microdose for Absolute Bioavailability and Effect of Grapefruit Juice on Ibrutinib in Healthy Adults. Br J Clin Pharmacol. 2015 doi: 10.1111/bcp.12787. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheers E, Leclercq L, de Jong J, Bode N, Bockx M, Laenen A, et al. Absorption, metabolism, and excretion of oral 14C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos. 2015;43:289–297. doi: 10.1124/dmd.114.060061. [DOI] [PubMed] [Google Scholar]
- Seidegard J, Nyberg L, Borga O. Differentiating mucosal and hepatic metabolism of budesonide by local pretreatment with increasing doses of ketoconazole in the proximal jejunum. Eur J Pharm Sci. 2012;46:530–536. doi: 10.1016/j.ejps.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Shoaf SE, Mallikaarjun S, Bricmont P. Effect of grapefruit juice on the pharmacokinetics of tolvaptan, a non-peptide arginine vasopressin antagonist, in healthy subjects. Eur J Clin Pharmacol. 2012;68:207–211. doi: 10.1007/s00228-011-1106-4. [DOI] [PubMed] [Google Scholar]
- Veronese ML, Gillen LP, Burke JP, Dorval EP, Hauck WW, Pequignot E, et al. Exposure-dependent inhibition of intestinal and hepatic CYP3A4 in vivo by grapefruit juice. J Clin Pharmacol. 2003;43:831–839. doi: 10.1177/0091270003256059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of pharmacokinetic parameters of ibrutinib and PCI-45227.