

Abstract
Saroglitazar is a novel nonthiazolidinediones (TZD) and nonfibric acid derivative designed to act as dual regulator of lipids and glucose metabolism by activating peroxisome proliferator-activated receptors (PPAR). These studies evaluate the efficacy and safety profile of Saroglitazar in preclinical in vitro and in vivo models. The EC50 values of Saroglitazar assessed in HepG2 cells using PPAR transactivation assay for hPPARα and hPPARγ were 0.65 pmol/L and 3 nmol/L, respectively. In db/db mice, 12-day treatment with Saroglitazar (0.01–3 mg/kg per day, orally) caused dose-dependent reductions in serum triglycerides (TG), free fatty acids (FFA), and glucose. The ED50 for these effects was found to be 0.05, 0.19, and 0.19 mg/kg, respectively with highly significant (91%) reduction in serum insulin and AUC-glucose following oral glucose administration (59%) at 1 mg/kg dose. Significant reduction in serum TG (upto 90%) was also observed in Zucker fa/fa rats, Swiss albino mice, and in high fat -high cholesterol (HF-HC)-fed Golden Syrian hamsters. LDL cholesterol was significantly lowered in hApoB100/hCETP double transgenic mice and HF-HC diet fed Golden Syrian Hamsters. Hyperinsulinemic-Euglycemic clamp study in Zucker fa/fa rats demonstrated potent insulin-sensitizing activity. Saroglitazar also showed a significant decrease in SBP (22 mmHg) and increase (62.1%) in serum adiponectin levels in Zucker fa/fa rats. A 90-day repeated dose comparative study in Wistar rats and marmosets confirmed efficacy (TG lowering) potential of Saroglitazar and has indicated low risk of PPAR-associated side effects in humans. Based on efficacy and safety profile, Saroglitazar appears to have good potential as novel therapeutic agent for treatment of dyslipidemia and diabetes.
Keywords: Anti-diabetic, dual PPAR agonist, glitazar, hypertriglyceridemia, insulin sensitizer, lipaglyn
Introduction
Type 2 diabetes mellitus (T2DM), which is characterized by insulin resistance, hyperglycemia, and hyperinsulinemia, has become a major health problem across the world (Kiess et al. 2004; Wild et al. 2004). The morbidity and mortality due to type 2 diabetes are caused by micro- as well as by macrovascular damages as consequences of the complex metabolic dysfunctions. The microvascular complications are believed to be related to high plasma glucose levels, whereas the macrovascular damage and related cardiovascular mortalities are largely associated with an atherogenic lipid profile which is associated with arterial lipid deposition and inflammation (Kiess et al. 2004; Agrawal et al. 2006). Dyslipidemia in type 2 diabetes is usually characterized by high triglycerides, high proportion of dense low-density lipoprotein (LDL)-particles, and low levels of high-density lipoprotein cholesterol (HDL-C) (Valabhji et al. 2003; Rader 2007), a condition referred to as atherogenic diabetic dyslipidemia (Misra et al. 2004; Muačević-Katanec and Reiner 2011). Thus, an ideal therapy for T2DM should target both the glycemic and lipid abnormalities (Cziraky 2004; Rader 2007).
Peroxisome proliferator-activated receptors (PPARs) are the nuclear receptors that primarily modulate lipid metabolism and that activation has salutary effects on lipid and glucose metabolism. Thiazolidinediones (TZDs) are agonists of PPARγ that improve insulin sensitivity and lower blood glucose levels (Chakrabarti et al. 2003). TZDs have a variable ability to lower TG, increase HDL-C levels, and can decrease the levels of small dense LDL particles (Aronoff et al. 2000). The clinical use of PPARγ activators has fallen because of PPARγ-mediated side effects, such as the risk of fluid retention, heart failure, weight gain, and a potential increase in bone fractures (Patel et al. 2005; Berlie et al. 2007; Nissen and Wolski 2007).
Here, we report the efficacy of Saroglitazar a novel compound that has recently been granted marketing authorization in India (Brand Name-Lipaglyn™) with an indication for treatment of diabetic dyslipidemia and hypertriglyceridemia with T2DM not controlled by statins. In this report, we present the preclinical pharmacodynamics profile of Saroglitazar.
Materials and Methods
PPAR transactivation assay
HepG2 cells (National Centre of Cell Science) were maintained in growth medium composed of Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS (Hyclone, South Logan, Utah, USA), 1X MEM nonessential amino acid (Sigma, Aldrich, St. Louis, MO, USA), and 1 mmol/L sodium pyruvate and 1% penicillin/streptomycin. HepG2 cells were seeded in 12-well plates at a density of 400,000 cells/well in 1 mL of medium per well. Cells were transfected with PPRE3-TK-luc and 0.08 μg of the pCDNA (3.1) expression vector containing the cDNA of PPARα or 0.08 μg of the pSG5 expression vector containing the cDNA of PPARγ using Superfect (Qiagen, Hiden, Germany). Cells were incubated at 37°C, 5% CO2: 95% O2 and after 3 h 1.0 mL of the medium containing the respective test compounds were added to the respective wells. After the incubation for 20–22 h at 37°C under 5% CO2, cells were first washed with Phosphate Buffer Saline (pH 7.3-7.4), lysed and supernatant collected for luciferase and β-galactosidase activity. The luciferase activity was determined using commercial firefly luciferase assay according to the suppliers’ instructions (Promega, Madison, WI, USA) in white 96-well plate (Nunc, Roskilde, Denmark). The β-galactosidase activity was determined in ELISA reader at 415 nm.
Animals
The B6.BKS(D)-Leprdb/J(db/db) mice were obtained from Jackson Laboratory, Bar Harbor, ME, at 6 weeks of age. Zucker fa/fa rats, Swiss albino mice, hApoB100/hCETP double transgenic mice, Wistar rats, and Golden Syrian hamsters were bred at ZRC. All animals were housed in individually ventilated cages and given pelleted food (Standard Rodent diet, NIN, Hyderabad, India) and water ad libitum in a temperature (25 ± 3°C) and humidity (50–70%)-controlled environment with a 12/12-h dark–light cycle, in a facility accredited by AAALAC International (Association for Assessment and Accreditation of Laboratory Animal Care International).
Golden Syrian hamsters of 7–9 weeks age were put on a high-fat high-cholesterol (HF-HC) diet (so as to provide 35% total fat, and 24% total protein and 0.5% cholesterol) 21 days before initiation of the treatment and during the treatment period.
Female marmosets (Callithrix jacchus) were bred at NIRRH, Parel, Mumbai, India, and the study was conducted at the same animal facility.
All animal experimentations were carried out in accordance with the CPCSEA (Committee for the Purpose of Control and Supervision of Experiments on Animals) guidelines, using Institutional Animal Ethics Committee (IAEC) approved protocols.
Chemicals
Saroglitazar, [(S)-α-ethoxy-4-{2-[2-methyl-5-(4- methylthio) phenyl)]-1H-pyrrol-1-yl]-ethoxy})-benzenepropanoic acid magnesium salt] (Fig.1A), rosiglitazone and fenofibrate were synthesized by Cadila Healthcare Limited, Ahmedabad, India. WY14, 643 was procured from Sigma-Aldrich Co. LLC (St. Louis, MO, USA).
Figure 1.
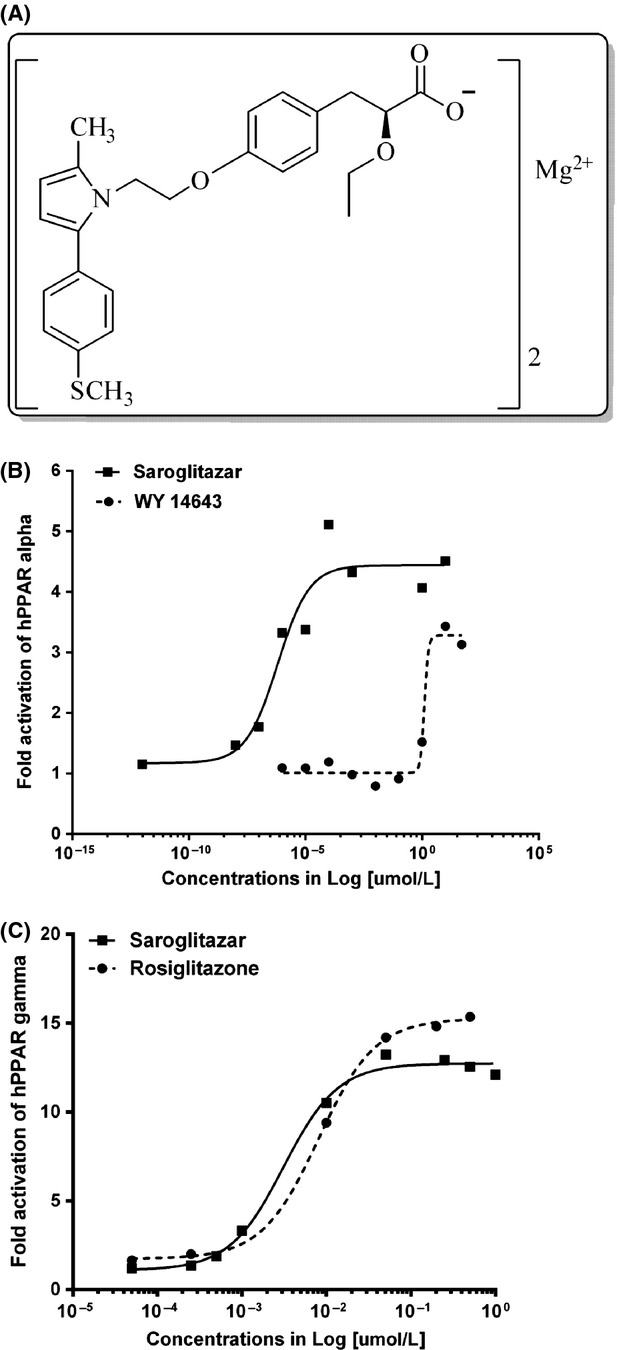
Structural formula of Saroglitazar and its in vitro activation. (A) Structural formula of Saroglitazar (B) Activation of PPARα and (C) activation of PPARγ by Saroglitazar. HepG2 cells were transfected with PPRE3-TK-luc and 0.08 μg of the cDNA (3.1) expression vector containing the cDNA of PPARα or 0.08 μg of the pSG5 expression vector containing the cDNA of PPARγ. The values are an average of duplicate experiments.
Experimental design, drug treatment, blood sampling, and parameters
Male C57BL/6J-db/db mice were bled on day 0 to determine pretreatment serum glucose and TG. Animals in defined serum glucose range were selected and randomized using MS office excel software and divided into different groups (n = 6) such that the mean serum glucose value of each group was not significantly different from that of the others. During next 12 days, each animal was dosed (by oral gavage) with vehicle (0.5% sodium carboxymethyl cellulose) or Saroglitazar (0.01, 0.03, 0.1, 0.3, 1, and 3 mg/kg per day) or pioglitazone (60 mg/kg per day) and on day 12 of the treatment, blood samples were collected (1 h after dosing) from orbital sinus under light ether anesthesia. The serum was isolated and analyzed for glucose, TG, free fatty acid (FFA), and insulin levels. On day 13, overnight fasted animals were weighed and subjected to Oral glucose tolerance test (OGTT). Briefly, after an overnight fast, 0 min blood collection was done and glucose load (1.5 g/kg in water) was administered orally. Blood samples were again collected at 30, 60, and 120 min after glucose load. Serum was separated and analyzed for glucose levels.
Sixteen-week-old Zucker fa/fa rats were bled and their serum TG levels were measured. Based on TG levels, animals were selected and randomized using MS office excel software and assigned to different treatment groups (n = 7) such that the average TG value of each group was not significantly different from other groups. For next 14 days, each animal was dosed (by oral gavage) daily with vehicle or Saroglitazar (0.01, 0.03, 0.1, 0.3, 1, 3, 10 mg/kg per day) or pioglitazone (50 mg/kg per day). On day 14, blood was collected 1 h post-dosing and serum was analyzed for TG, FFA, glucose, and insulin levels. On day 15, the animals were subjected to OGTT with glucose load (3 g/kg in water) and other procedures were similar as described in db/db mice OGTT study.
Hyperinsulinemic-euglycemic clamp study was done in a separate group of male Zucker fa/fa rats of 11–12 weeks old. Animals were randomized in such a way that their nonfasted serum glucose, body weight, and serum TG levels on day 0 of the treatment were not significantly different between groups. The rats were treated with vehicle or Saroglitazar (1 and 10 mg/kg per day) for 15 days by oral gavage. On day 8 of the treatment, left carotid artery and right jugular vein catheterization were performed in the rats using microrenathane (MRE)-40 tubing. On day 15, 1 h postdose administration, hyperinsulinemic-euglycemic clamp was performed in 5-h fasted animals. Briefly, 0 min blood collection was performed from the arterial line for insulin measurement and glucose was measured by glucometer (OneTouch Ultra 2) for basal glucose level. Regular Human insulin (Huminsulin, Eli Lilly and Company, Gurgaon, Haryana, India) was infused through the jugular vein at a constant rate of 2 μL/min with the help of an automated infusion pump to achieve hyperinsulinemia. Insulin was diluted in saline in such a way that the final infusion will be 10 mU/kg per min with a flow rate 2 μL/min. To maintain the blood glucose at target euglycemic level (100–120 mg/dL), 50% glucose was infused at a variable infusion rate by infusion pump. Blood glucose was measured at 10 min interval till the completion of the clamp period (0–120 min). Glucose infusion rate (GIR) was adjusted if necessary. When the blood glucose concentration was in steady state (euglycemic level 100–120 mg/dL) for 20 min, ∼1.5 h after starting the clamp, considered as clamped. Blood collection at 120 min was performed for insulin measurement. GIR (mg/kg per min) was calculated from concentration of infused glucose (50%), glucose pump rate, and body weight at each time point. The area under curve for GIR for each treatment group was calculated using GraphPad® software (La Jolla, CA, USA).
In another study, 15- to 16-week-old Zucker fa/fa rats were randomized and grouped (n = 7) based on serum TG and body weights and then for next 15 days each animal was dosed (by oral gavage) daily with vehicle or Saroglitazar (4 mg/kg per day) or pioglitazone (10 mg/kg per day) or fenofibrate (100 mg/kg per day). On day 14, blood was collected 1 h post-dosing and serum was analyzed for TG and adiponectin (Acrp30) levels. On day 15, the animals were subjected to noninvasive blood pressure measurement by using 8-channel noninvasive blood pressure monitor (NIBP-8, Columbus Instruments, Columbus, OH, USA).
Six- to eight-week-old Swiss albino mice in a defined serum TG range were selected and randomized using MS Office Excel software. During the next 6 days, each animal was dosed daily (by oral gavage) with vehicle or Saroglitazar (0.01, 0.03, 0.1, 0.3, 1, and 3 mg/kg per day) or fenofibrate (30 mg/kg per day). On day 6, animals were bled 1 h post-dosing and serum was analyzed for TG. On day 7, animals were subjected to intravenous lipid tolerance test (IVLTT). Briefly, Intralipid (3.3% v/v, from Pharmacia & Upjohn AB, Stockholm, Sweden) was administered through the intravenous route (10 mL/kg), and serum TG levels were measured at 0, 30, 60, and 120 min after injection.
Twenty seven- to thirty-three-week-old hApoB100/hCETP double transgenic mice were randomized and grouped (n = 6) such that their serum LDL-C levels on day 0 of the treatment were not significantly different between groups. The mice were treated with vehicle or Saroglitazar (0.1, 0.3, 1, 3, and 10 mg/kg per day) or fenofibrate (60 mg/kg per day) for 14 days by oral gavage. On day 14, at 1 h post dose, blood samples were collected for estimation of nonfasted serum LDL-C, TC, TG, and HDL-C levels.
Ten- to twelve-week-old adult female Golden Syrian hamsters were fed normal or HF-HC diet for 21 days. The animals were selected based on their body weights, randomized using MS Office Excel software and divided into six treatment groups (n = 7) such that the mean bodyweight value of each group was not significantly different from that of the others. During the next 14 days, each animal was dosed daily (by oral gavage) with vehicle, Saroglitazar (0.03, 0.1, 0.3, 1, 3, and 10 mg/kg per day) or pioglitazone (30 mg/kg per day). On day 14, animals were bled 1 h post-dosing and serum was analyzed for TC, TG, LDL-C, and HDL-C levels.
In order to understand the species differences, a three-month repeated dose efficacy and safety study was conducted in 12 female common marmosets (n = 4) and 30 female Wistar rats (n = 10). Animals randomized based on body weights and were divided into three equal groups and received the daily administration of vehicle (50% w/v honey for marmoset and 0.1% carboxymethylcellulose for Wistar rats) or Saroglitazar (1.5 and 15 mg/kg per day) for 90 days by oral gavage. Body weights and clinical signs were monitored throughout the study period. At the end of the treatment, blood was collected and animals were sacrificed. The blood samples were subjected to clinical pathological investigations, selected organs were weighed, and a detail histopathological evaluation was carried out.
Estimation mRNA in liver and white adipose tissue
Liver and epididymal adipose tissues were excised under aseptic condition, and total RNA was extracted by TRIzol reagent (Life Technologies, Carlsbad, CA, USA), following the manufacturer’s instructions. First-strand cDNA was generated from 1 μg of RNA in a 20 μL volume, by using the random primer in the first-strand cDNA synthesis kit. The reverse transcription reaction mixture (2.5 μL) was amplified in 100 mL volume, with primers specific for acyl-CoA oxidase (ACO), FATP, CD36, lipoprotein lipase (LPL), ApoCIII,aP2, ACRP30, and acidic ribosomal phosphoprotein (36B4) as control. Primer sequences were: ACO (F: 5′-TAAGTCTGTGTCTGTGGCATTCG-3′ and R: 5′-GCTGTGTACTGTCAATCTTAAGGG-3′); FATP (F: 5′-TGGTGGCTGCTCTTCTCAATG-3′ and R:5′-GTAGGAATGGTGGCCAAAGGC 3′); CD36 (F: 5′-GCCTGTGTATATTTCGCTTCCAC-3′ and R: 5′-GCCATCTCTACCATGCCAAGG-3′); LPL (F: 5′-GAACACCTACACACAAGCAAAGCC-3′ and R: 5′-CATAGACAGTACCAGGCTCGTTGC-3′); aP2 (F: 5′-GTGTGATGCCTTTGTGGGAA-3′ and R: 5′-TCCATCCCCTTCGCACCT-3′); ApoCIII (F: 5′-CCTCTTGGCTCTCCTGGCAT-3′ and R:5′-ATAGCTGGAGTTGGTTGGTCCTC-3′); 36B4 (F: 5′-AGGCCGTGGTGCTGATGG-3′ and R: 5′-TGGCCAACAGCATATCCCG-3′); ACRP (F: 5′-GCAGAGCCTGCAGAAGCCGAGTTCGGTGTAGGTCGTTCGCTC-3′ and R: 5′-CATACACCTGCAGCCAGACGAGCAGATACCAAATACTGTCC-3′).
Linearity of the PCR was tested by amplification of 200 ng of total RNA per reaction from 15 to 40 cycles. The linearity range was found to be between 15 and 35 cycles. In no case did the amount of RNA used for PCR exceed 200 ng per reaction. The samples were amplified for 30 cycles for ACO, FATP, and ACRP; 33 cycles for aP2, CD36, and LPL; and 30 cycles for 36B4, by using the following parameters: 94°C-3 min-1 cycle, followed by 94°C-30 sec, 55°C-30 sec, and 72°C-1 min.
PCR products (10 mL) were electrophoresed on 1.5% agarose gel. Band intensity was quantitated (under UV light). Levels of mRNA were expressed as the ratio of band intensity for the target gene relative to that of 36B4 gene.
Analytical methods
Serum glucose, TG, TC, LDL-C, HDL-C, and FFA were measured spectrophotometrically using commercially available kits (Pointe Scientific, Canton, Michigan, USA and Roche Diagnostics, Mannheim, Germany). Serum insulin was measured using ELISA kit from Crystal Chem., Downers Grove, IL, USA. Serum adiponectin levels were measured using Quantikine ELISA kit from R&D Systems, Minneapolis, MN, USA.
Compliance with design and statistical analysis requirements
All the studies conducted in rodent models consisted of at least six animals in each group. The marmoset study was conducted with n = 4 in each group. Less numbers were taken in primate studies due to ethical consideration. All groups, including controls, in each study consisted of equal number of animals. The animals were randomized based on various selection parameters as described in specific sections using MS Excel Software. The selection criterion was decided in advance. The animals were assigned to different groups in an unbiased manner. In vitro experiments were performed in duplicate for each concentration for EC50 calculations.
The in vivo study data are presented as Mean ± SEM. The percent reduction was calculated according to the formula:
where TT is the test day treated value, OT the zero day value treated, TC the test day control value, and OC the zero day control value. The difference among the experimental groups were analyzed by one-way analysis of variance (ANOVA), followed by Bonferroni/Dunnett’s test to evaluate the statistical difference between two groups. P < 0.05 was considered significant. All analyses were performed using GraphPad Prism software (GraphPad Software, La Jolla CA).
Results
Saroglitazar is a dual PPARα/γ activator with predominant PPARα activity
In vitro activity of Saroglitazar was evaluated using PPRE-luc transactivation assay in HepG2 cells transfected with hPPARα and hPPARγ receptors. The PPARα activation was compared with WY 14,643 and PPARγ activation was compared with rosiglitazone. As shown in Figure1B, Saroglitazar is a more potent activator of PPARα as compared to WY 14,643 (EC50 0.65 pmol/L vs. 1.2 μmol/L, respectively). Saroglitazar also showed PPARγ activity (Fig.1C) but at relatively higher concentrations (EC50 3 nmol/L) than that of PPARα. In this assay, rosiglitazone showed EC50 of 8 nmol/L for PPARγ activity. The results indicate that Saroglitazar is a potent and dual activator of PPARα and PPARγ, with predominant action on PPARα.
Antidiabetic and triglyceride-lowering activity in db/db mice
Saroglitazar treatment demonstrated a dose-dependent reduction in serum glucose, TG, and FFA in db/db mice after 12 days of treatment (Table1 and Fig.2A). Saroglitazar treatment also showed a significant improvement in oral glucose tolerance (59% reduction in AUCglucose, Fig.2B) and 91% reduction in serum insulin levels at 1 mg/kg dose as compared to control group. The ED50 for glucose reduction was found to be 0.19 mg/kg and reduction observed was 64.6% at 3 mg/kg. Saroglitazar caused TG reduction by upto 54.9% at 3 mg/kg and ED50 was found to be 0.05 mg/kg. The ED50 of for FFA lowering was 0.19 mg/kg and Saroglitazar caused upto 56.1% reduction at 3 mg/kg. Whereas pioglitazone caused 63% reduction in glucose and 59% reduction in TG at the dose of 60 mg/kg as compared to control group.
Table 1.
Effect of Saroglitazar in db/db mice.
| Group | Serum glucose (mg/dL) | Triglycerides (mg/dL) | Free fatty acids (mg/dL) | Body weight (g) |
|---|---|---|---|---|
| Control | 447.4 ± 25.3 | 104.30 ± 9.70 | 92.0 ± 6.40 | 40.9 ± 0.50 |
| Saroglitazar (0.01 mg/kg) | 457.2 ± 44.2 | 79.50 ± 2.90 | 95.8 ± 2.70 | 42.3 ± 1.36 |
| Saroglitazar (0.03 mg/kg) | 479.9 ± 20.6 | 75.30 ± 13.50 | 79.5 ± 5.80 | 41.4 ± 1.08 |
| Saroglitazar (0.1 mg/kg) | 359.5 ± 37.2 | 73.40 ± 9.9* | 75.9 ± 5.70 | 43.3 ± 0.42 |
| Saroglitazar (0.3 mg/kg) | 272.2 ± 22.4* | 61.00 ± 4.2* | 60.7 ± 3.6* | 44.2 ± 1.40 |
| Saroglitazar (1 mg/kg) | 183.9 ± 13.9* | 66.20 ± 1.8* | 49.3 ± 1.7* | 43.8 ± 0.65* |
| Saroglitazar (3 mg/kg) | 159.5 ± 5.6* | 50.50 ± 2.5* | 40.4 ± 1.1* | 44.3 ± 1.33* |
All values are measured at the end of treatment (12 day), and expressed as mean ± SEM (n = 6).
ED50Values: glucose reduction – 0.19 mg/kg, TG reduction – 0.05 mg/kg, and for FFA reduction – 0.19 mg/kg.
P < 0.05 as compared to control (ANOVA).
Figure 2.

Effect of Saroglitazar on (A) serum glucose and (B) AUC-glucose in oral glucose tolerance test in db/db mice treated for 12 days. Serum glucose was measured on pretreatment and on day 12 1 h post dose and on day 13 overnight fasted animals were subjected to oral glucose tolerance test with glucose load (1.5 g/kg) for AUC-glucose measurement. The values are calculated as percent change versus Control and expressed as mean ± SEM (n = 6). (C and D) Effect of Saroglitazar on expression of mRNA, in liver (C), and in white adipose tissue (D) of db/db mice treated with Saroglitazar for 12 days at 3 mg/kg dose determined by quantitative real-time PCR. The bars represent the fold change in the treatment groups compared with the vehicle control group, mean ± SEM (n = 6). *Indicates significantly different from vehicle-treated control group, P < 0.05 (ANOVA).
Gene expression study in db/db mice
In order to understand the mechanism of pharmacodynamic activity of Saroglitazar in db/db mice treated at 3 mg/kg dose for 12 days, the liver tissues were processed for the mRNA expression for ACO, FATP, CD36, LPL, and ApoCIII. Saroglitazar caused 2.4, 6.8, 1.7, and 2.9-fold increase in ACO, FATP, CD36, and LPL mRNA levels, respectively, and 70% downregulation in ApoCIII mRNA levels as shown in Figure2C. Simultaneously white Adipose tissues (WAT) were studied for aP2, FATP, CD36, LPL, and ACRP30 mRNA levels; Saroglitazar treatment showed 3.5, 1.9, 2.6, 3.1, and 1.5 fold increase in their expression as compared to control group (Fig.2D).
Lipid-lowering and insulin-sensitizing effect in Zucker fa/fa rats
In Zucker fa/fa rats, Saroglitazar treatment led to dose-dependent reduction in serum TG and improvement in oral glucose tolerance along with significant reduction in insulin. The 3 mg/kg dose showed reduction of 81.7% in serum TG, 69.3% in FFA, and 84.8% reduction in serum insulin and 51.5% improvement in AUCglucose (Table2 and Fig.3A and B) as compared to control group. The ED50 for TG-lowering effect was found to be 0.26 mg/kg and a reduction of 85.5% in serum TG was seen at 3 mg/kg dose. Pioglitazone showed comparable reduction up to 82% in serum TG, 84% in FFA, 95% in insulin, and 39% improvement in AUCglucose.
Table 2.
Effect of Saroglitazar in Zucker fa/fa rats.
| Group | Triglycerides (mg/dL) | OGTT:AUC glucose (mg/dL · min) |
|---|---|---|
| Control | 441.2 ± 81.7 | 28798.3 ± 3038.3 |
| Saroglitazar (0.01 mg/kg) | 453.1 ± 59.9 | 28970.9 ± 2045.1 |
| Saroglitazar (0.03 mg/kg) | 355.4 ± 29.5 | 28269.1 ± 1214.2 |
| Saroglitazar (0.1 mg/kg) | 316.1 ± 46.1 | 23709.1 ± 2683.8 |
| Saroglitazar (0.3 mg/kg) | 186.5 ± 13.7* | 18941.6 ± 1870.3* |
| Saroglitazar (1 mg/kg) | 93.6 ± 11.0* | 16637.5 ± 1591.5* |
| Saroglitazar (3 mg/kg) | 68.0 ± 3.0* | 13972.6 ± 436.1* |
| Saroglitazar (10 mg/kg) | 55.5 ± 4.1* | 13963.1 ± 548.9* |
Serum triglyceride levels were measured at the end of treatment (14th day) and AUC was measured on day 15th after oral glucose tolerance test, all values expressed as mean ± SEM. (n = 6).
ED50Value for TG reduction – 0.26 mg/kg.
P < 0.05 as compared to control (ANOVA).
Figure 3.
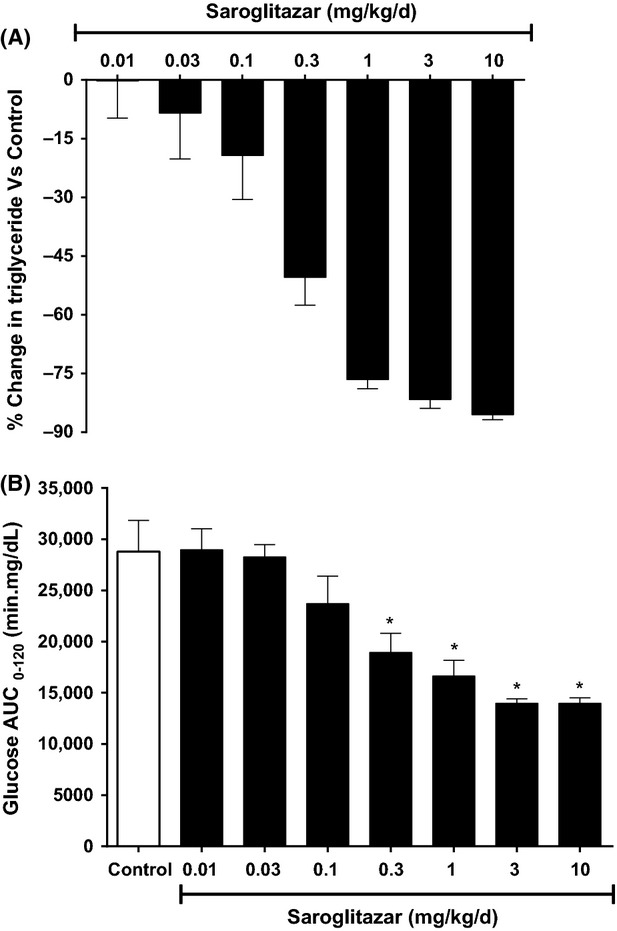
Effect of Saroglitazar on (A) serum triglyceride and (B) AUC- glucose in oral glucose tolerance test in Zucker fa/fa rats treated for 14 days. Serum triglyceride was measured on pretreatment and on day 14 1 h post dose and on day 15 overnight fasted animals were subjected to oral glucose tolerance test with glucose load (3 g/kg). The values are calculated as percent change versus control and expressed as mean ± SEM (n = 7). *Indicates significantly different from vehicle-treated control group P < 0.05 (ANOVA).
Insulin-sensitizing activity in Hyperinsulinemic-Euglycemic Clamp study
In Zucker fa/fa rats, blood glucose concentrations in steady state (100–120 mg/dL) were similar in Saroglitazar-treated Zucker fa/fa rats and controls (Table3). GIR, required to maintain this concentration in hyperinsulinemic condition was significantly higher in the Saroglitazar-treated Zucker fa/fa rats than in the controls (59% and 109% higher as compared to controls at 1 and 10 mg/kg dose, respectively) indicating that whole body insulin sensitivity was enhanced by Saroglitazar treatment (Fig.4A and B). When we calculated the AUC (0–120) for GIR it showed that Saroglitazar at 1 and 10 mg/kg showed 54% and 127% increase in AUC (0–120) GIR, respectively, in hyperinsulinemic clamp following once-daily oral treatment for 15 days. Insulin levels were decreased up to 42% at basal and there was no significant change when measured at the end of clamp period. It demonstrates that Saroglitazar has potent insulin-sensitizing activity.
Table 3.
Effect of saroglitazar on glucose infusion rate, blood glucose, and serum insulin levels at the end of the clamp experiments in Zucker fa/fa rats.
| Parameters | Control | Saroglitazar (1 mg/kg) | Saroglitazar (10 mg/kg) |
|---|---|---|---|
| Glucose infusion rate (mg/kg per min) | 11.6 ± 1.3 | 18.6 ± 1.6* | 24.5 ± 2.6* |
| Glucose (mg/dL) | 113.9 ± 4.5 | 111.3 ± 2.4 | 116.0 ± 1.9 |
| Insulin (ng/mL) | 17.6 ± 0.8 | 15.3 ± 0.8 | 15.0 ± 0.8 |
All values are measured at the end of clamp at 120 min after 15 days oral treatment and expressed as mean ± SEM. (n ≥ 6).
P < 0.05 as compared to control (ANOVA).
Figure 4.
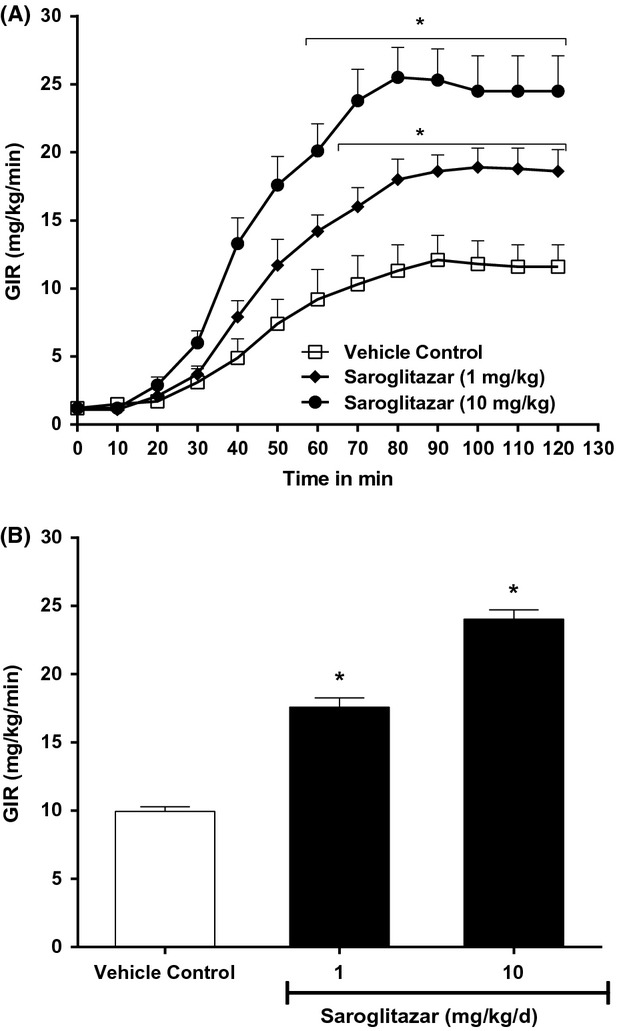
Effect of Saroglitazar on (A) glucose infusion rate (GIR) at 0–120 min (B) GIR at 60–120 min of hyperinsulinemic-euglycemic clamp done in Zucker fa/fa rats after 15 days oral administration. The values are calculated as percent change versus control and expressed as mean ± SEM (n ≥ 6). *Indicates significantly different from vehicle control P < 0.05 (ANOVA).
Effect on serum adiponectin levels and blood pressure in Zucker fa/fa rats
Once-daily treatment of Zucker fa/fa rats with Saroglitazar at 4 mg/kg dose, showed a significant decrease (P < 0.001) in Systolic blood pressure (SBP, 22 mmHg) and serum TG (80.9%) levels, whereas there was a significant increase (62.1%) in serum adiponectin levels (Table4). Pioglitazone (10 mg/kg) also showed a decrease in SBP (21 mmHg) and TG (48%). Effect of pioglitazone on serum adiponectin levels (24.1% increase) was less remarkable than Saroglitazar. On the other hand, fenofibrate (100 mg/kg) showed significant decrease in serum TG (54%), but had no significant effect on SBP and serum adiponectin levels.
Table 4.
Effect of Saroglitazar on serum TG, adiponectin levels, and SBP in Zucker fa/fa rats.
| Group | Serum triglycerides (mg/dL) | Serum adiponectin (μg/mL) | SBP (mmHg) |
|---|---|---|---|
| Control | 640.5 ± 91.8 | 5.3 ± 0.5 | 134.0 ± 2.8 |
| Saroglitazar (4 mg/kg) | 122.0 ± 8.1* | 8.2 ± 0.5* | 112.5 ± 2.6* |
| Pioglitazone (10 mg/kg) | 332.8 ± 64.0* | 7.5 ± 1.1 | 113.4 ± 2.4* |
| Fenofibrate (100 mg/kg) | 295.5 ± 85.6* | 4.3 ± 0.4 | 124.0 ± 5.3 |
Serum triglyceride and adiponectin levels were measured on (14th day) and SBP was measured on day 15th by noninvasive blood pressure measurement method, all values expressed as mean ± SEM. (n = 7).
P < 0.05 as compared to control (ANOVA).
Lipid-lowering effects in HF-HC diet fed syrian golden hamsters
Hamsters showed significant increase in TG, TC, and LDL-C after 21 days of HF-HC diet. When these hyperlipidemic hamsters were treated with Saroglitazar for 14 days, the animals showed dose-dependent reduction in serum TG and TC levels (Table5 and Fig.5A). The ED50 for TG-lowering effect was 0.37 mg/kg and a reduction of 89.8% was found at 10 mg/kg. Treatment with Saroglitazar at 10 mg/kg caused 52.7% reduction in TC. Saroglitazar also showed 61% reduction in LDL-C and 62.4% reduction in LDL-C/HDL-C ratio at 10 mg/kg dose. The TG/HDL-C ratio, which is often used as measure of atherogenic dyslipidemia (AD), was reduced by 91% after Saroglitazar treatment at 10 mg/kg as compared to control group (Fig.5B). Whereas, pioglitazone was less efficacious at 30 mg/kg with a 27% reduction in TG and 16% reduction in TC compared to control group.
Table 5.
Effect of Saroglitazar on high fat-high cholesterol (HF-HC) diet fed syrian golden hamsters.
| Group | Triglycerides (mg/dL) | Total cholesterol (mg/dL) | Body weight (g) |
|---|---|---|---|
| Normal diet control | 129.1 ± 9.3 | 78.3 ± 3.0 | 113.7 ± 5.1 |
| HF-HC diet Control | 493.2 ± 77.8 | 306.2 ± 27.7 | 105.3 ± 4.4 |
| Saroglitazar (0.03 mg/kg) | 634.4 ± 87.5 | 392.1 ± 13.5 | 106.4 ± 2.3 |
| Saroglitazar (0.1 mg/kg) | 354.3 ± 59.7 | 293.1 ± 16.7 | 112.6 ± 4.7 |
| Saroglitazar (0.3 mg/kg) | 371.0 ± 35.7 | 315.1 ± 13.1 | 107.1 ± 6.3 |
| Saroglitazar (1 mg/kg) | 272.5 ± 28.0 | 311.4 ± 30.8 | 103.0 ± 4.1 |
| Saroglitazar (3 mg/kg) | 109.9 ± 14.8* | 243.6 ± 24.7 | 100.1 ± 3.6 |
| Saroglitazar (10 mg/kg) | 50.5 ± 7.2* | 144.7 ± 6.6* | 81.6 ± 3.0* |
All values are measured at the end of treatment (14th day), and expressed as mean ± SEM (n = 6).
ED50Values: TG reduction – 0.37 mg/kg.
P < 0.05 as compared to control (ANOVA).
Figure 5.
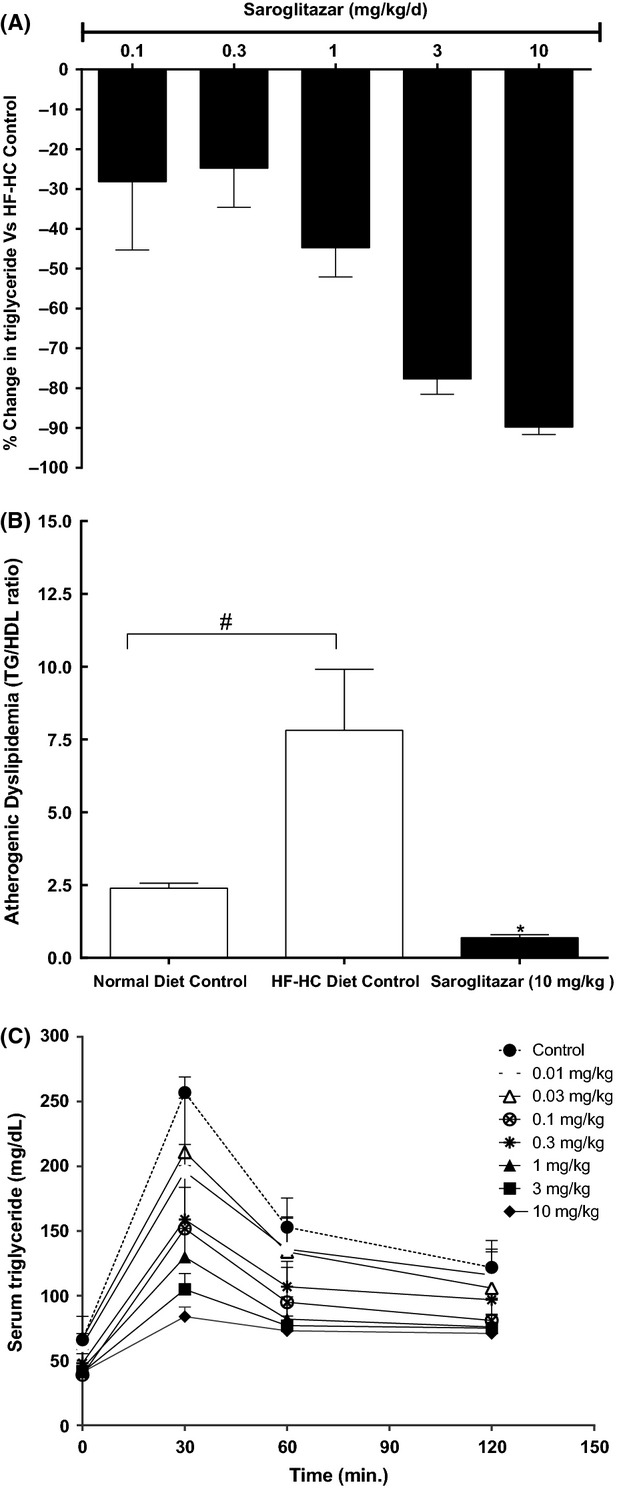
Effect of Saroglitazar on lipids in various animal models (A) effect on % change in serum triglyceride versus control and (B) effect on atherogenic dyslipidemia (TG/HDL-C) ratio in high fat-high cholesterol (HF-HC) diet fed Syrian Golden hamsters. Hamsters were kept on HF-HC diet and compound treatment was done for 14 days at 0.03, 0.1, 0.3, 1, 3, and 10 mg/kg. (C) Effect on serum triglyceride clearance in Swiss albino mice. Mice were treated with the compound for 6 days at 0.01, 0.03, 0.1, 0.3, 1, 3, and 10 mg/kg, and on day 7 overnight fasted animals were injected with 3.3% Intralipid as described in Methods. The values are calculated as percent change versus control and expressed as mean ± SEM (n = 7). #Indicates significantly different from normal diet control and *Indicates significantly different from HF-HC diet control P < 0.05 (ANOVA). In Figure (C) at 30 min serum triglyceride levels were significant (P < 0.05) at all doses of Saroglitazar, at 60 min time point 0.1–10 mg/kg dose levels and at 120 min time point 1–10 mg/kg dose levels showed significantly (P < 0.05) lowered triglyceride levels as compared to control group animals.
Effect on lipid levels and lipid tolerance test in swiss albino mice
Swiss albino mice showed dose-dependent reduction in TG after 6 days of Saroglitazar treatment (Table6). A reduction of 75.8% was observed in serum TG at 10 mg/kg dose and the ED50 for TG reduction was determined to be 0.09 mg/kg. In this study, fenofibrate showed only 28% reduction in TG at 30 mg/kg dose. When challenged intravenously with 10 mL/kg Intralipid (3.3%), Saroglitazar-treated animals showed dose-dependent improvement in lipid clearance (reduction in AUCtriglycerides). The enhanced lipolytic activity resulted in 68% reduction in AUCtriglycerides at 10 mg/kg dose (Fig.5C).
Table 6.
Effect of Saroglitazar in Swiss albino mice.
| Group | Triglycerides (mg/dL) | IVLTT-AUCtriglyceride (mg/dL · min) |
|---|---|---|
| Control | 100.3 ± 8.5 | 5918.0 ± 316.0 |
| Saroglitazar (0.01 mg/kg) | 69.6 ± 7.2* | 4654.0 ± 796.0 |
| Saroglitazar (0.03 mg/kg) | 73.8 ± 7.5* | 4447.0 ± 545.0 |
| Saroglitazar (0.1 mg/kg) | 45.9 ± 3.2* | 3739.0 ± 519* |
| Saroglitazar (0.3 mg/kg) | 59.5 ± 5.1* | 3739.0 ± 519* |
| Saroglitazar (1 mg/kg) | 36.6 ± 3.2* | 2722.0 ± 141* |
| Saroglitazar (3 mg/kg) | 31.8 ± 3.8* | 2379.0 ± 98* |
| Saroglitazar (10 mg/kg) | 24.1 ± 2.3* | 1909.0 ± 315* |
All values are measured at the end of treatment (6th day) serum TG, and IVLTT-AUC measured on day-7, values expressed as mean ± SEM. (n = 6).
ED50Values: TG reduction – 0.09 mg/kg.
P < 0.05 as compared to control (ANOVA).
Antidyslipidemic (LDL-C lowering) activity in hApoB100/hCETP double transgenic mice
Oral once-daily treatment of hApoB100/hCETP double transgenic mice with Saroglitazar for 14 days resulted in dose-dependent reductions in serum LDL-C with ED50 of 0.11 mg/kg (Table7). The reduction in LDL-C found was 72% on day 14 at 10 mg/kg dose. Saroglitazar also showed dose-dependent reduction in TC and TG levels on day 14 of the treatment. Saroglitazar at 1 mg/kg showed 67% reduction in LDL-C (Fig.6A), 50% reduction in TC (Fig.6B), 39% reduction in TG, and 61% reduction in LDL-C/HDL-C atherogenic ratio following once-daily oral treatment for 14 days.
Table 7.
Effect of Saroglitazar on total cholesterol, LDL-C, HDL-C, and triglycerides in hApoB100/hCETP double transgenic mice.
| Group | Total cholesterol (mg/dL) | LDL-C (mg/dL) | HDL-C (mg/dL) | Triglycerides (mg/dL) |
|---|---|---|---|---|
| Control | 151.2 ± 7.7 | 108.1 ± 10.7 | 43.6 ± 2.5 | 108.2 ± 14.7 |
| Saroglitazar (0.1 mg/kg) | 109.7 ± 6.2* | 74.1 ± 5.3* | 34.3 ± 3.5 | 86.8 ± 14.8 |
| Saroglitazar (0.3 mg/kg) | 83.9 ± 5.3* | 42.7 ± 2.9* | 38.4 ± 4.2 | 68.7 ± 13.4 |
| Saroglitazar (1 mg/kg) | 73.1 ± 2.8* | 35.3 ± 1.9* | 34.0 ± 2.7 | 52.3 ± 5.7* |
| Saroglitazar (3 mg/kg) | 66.5 ± 5.1* | 31.6 ± 3.3* | 30.7 ± 2.8* | 42.6 ± 5.8* |
| Saroglitazar (10 mg/kg) | 67.0 ± 5.2* | 29.9 ± 3.7* | 32.8 ± 3.7 | 41.6 ± 4.1* |
All values are measured at the end of treatment (14th day) and values expressed as mean ± SEM. (n = 6).
ED50Value: LDL reduction – 0.11 mg/kg.
P < 0.05 as compared to control (ANOVA).
Figure 6.
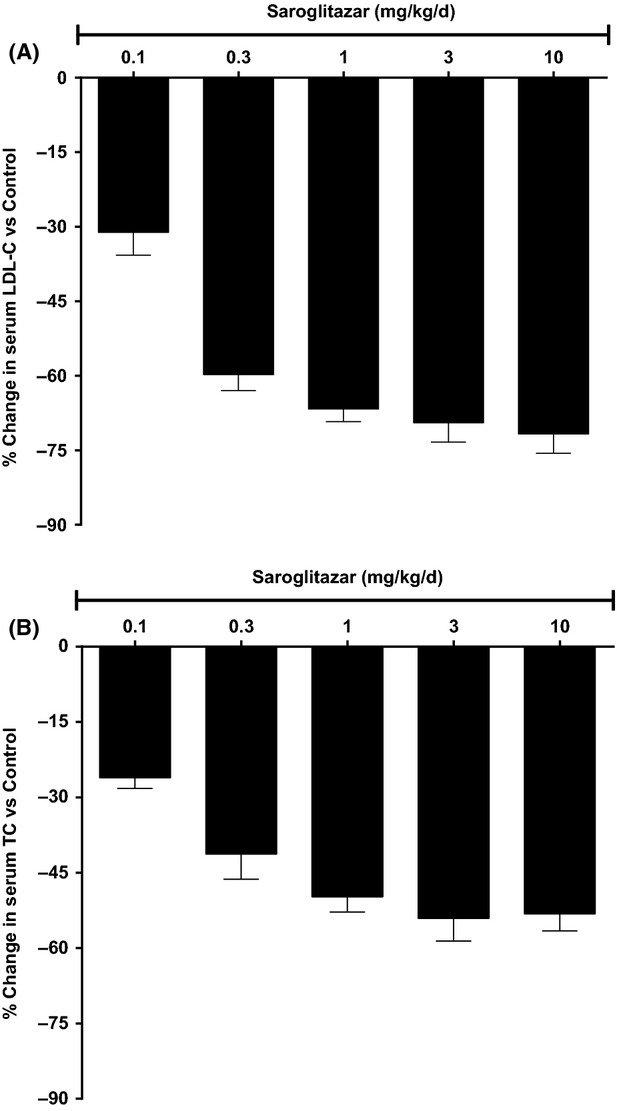
Effect of Saroglitazar on lipids in hApoB100/hCETP double transgenic mice (A) effect on % change in serum LDL-C versus control (B) effect on % change in serum total cholesterol versus control. Animals were treated with Saroglitazar for 14 days at 0.1, 0.3, 1, 3, and 10 mg/kg. The values are calculated as percent change versus control and expressed as mean ± SEM (n = 6).
Comparative efficacy and safety study in common marmoset and Wistar rats
Female Wistar rats and female marmosets were administered with Saroglitazar (1.5 and 15 mg/kg) or vehicle for 90 days. Treatment with Saroglitazar did not cause any unexpected changes in behavioral or clinical parameters in either species. There was no effect on body weights and feed consumption. As expected from PPARα agonists, Saroglitazar-treated Wistar rats showed higher liver weights and elevated serum ALT levels as compared to control group. Since Saroglitazar has relatively weaker PPARγ activity as compared to PPARα, the PPARγ mediated effects (reduction in hematocrit, body weight gain) were evident only at higher doses, which shows exposure about 66-fold higher than the exposure in human subjects observed at clinically effective dose of 4 mg. There were no other gross pathology findings associated with Saroglitazar treatments. Histopathological examination did not reveal any adverse lesions in vital organs except liver, which showed expected PPARα-mediated mild hepatocellular hypertrophy and minimal to mild single cell necrosis. Marmosets concurrently treated with similar doses of Saroglitazar also showed no behavioral changes and/or clinical signs, but showed significant (upto 53.4%) reduction in TG at 1.5 mg/kg (Fig.7) on day 90 as compared to control group which was expected efficacy end point. In contrast to rodents, organ weight data in marmosets did not reveal any changes in liver weight or gross morphology. Biochemical analysis showed no significant changes in serum alanine amino transferase (ALT) or serum electrolytes. Similarly, there were no gross pathological findings associated with the drug treatment. Histopathological examination revealed no adverse changes in any vital organs in the marmoset. Unlike rodents, livers of marmosets showed no changes in histomorphology. These clinical, pathological, and histopathological investigations revealed that Saroglitazar did not show any adverse toxic effects at doses upto at 15 mg/kg in marmosets.
Figure 7.
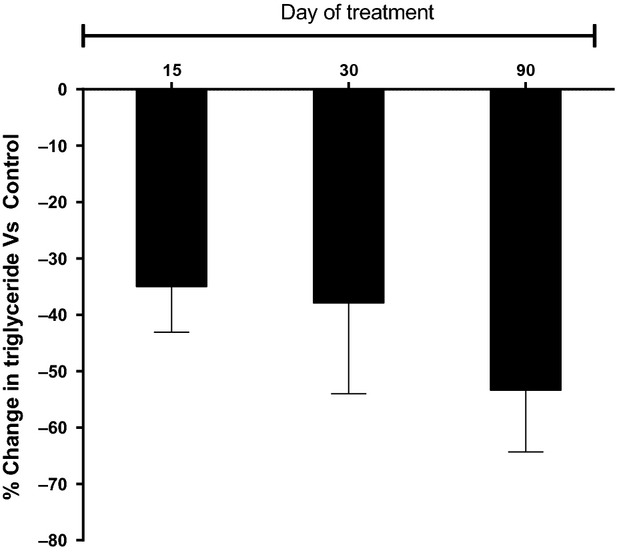
Effect of Saroglitazar treatment at 1.5 mg/kg dose on % change in serum triglyceride versus control on various treatment days in female common marmosets. Serum triglyceride was measured on pretreatment and on day 15, 30, and 90th of treatment at 1 h post dose. The values are calculated as percent change versus control and expressed as mean ± SEM (n = 4).
Discussion
Diabetic dyslipidemia is a complex cluster of metabolic abnormalities characterized by hypertriglyceridemia, high levels of LDL-C, low levels of HDL-C, and postprandial lipemia together with hyperglycemia and insulin resistance (Goldberg 2001; Taskinen 2002; Boullart et al. 2012). Since PPARα agonists are effective in managing lipids and PPARγ agonists have antihyperglycemic and insulin-sensitizing effects, using dual PPARα/γ agonist one can control both lipids and glucose simultaneously and fulfill the unmet medical need (Balakumar et al. 2007). In line with this hypothesis, Seber et al. (2006) and Boden et al. (2007) have demonstrated that combination of PPARα and PPARγ agonists, that is, rosiglitazone and fenofibrate was effective in controlling HbA1c and lipid levels. Interestingly in these studies they observed that edema and weight gain, the main side effects of PPARγ agonists were not observed. In fact PPARα activation has been found to also neutralize other PPARγ-related side effects (Samadfam et al. 2012). These observations suggest that a dual PPARα/γ agonist with optimum PPAR α and γ activity profile may show antidyslipidemic, antihyperglycemic efficacy with reduced side effects. The hypothesis that PPARα/γ dual agonism would provide complementary pharmacological effects has encouraged many research groups to develop these agents. Several dual PPAR agents were developed in past with varying ratios of PPARα and PPARγ activity (Rubenstrunk et al. 2007).
Saroglitazar is a novel alkoxy aryl propionic acid derivative, that showed dual PPARα/γ agonism with a predominant PPARα activity in an in vitro transactivation assay. Saroglitazar shows much higher PPARα transactivation potential as compared to the prototypical PPARα activator, WY14,643; its EC50 was found to be 0.65 pmol/L versus 1.2 μmol/L for WY 14,643. Saroglitazar showed potent PPARγ activation with an EC50 value of 3 nmol/L. Among the TZDs, rosiglitazone is known to be a full agonist and shows potent PPARγ activation (Balfour and Plosker 1999). In hPPARγ assay, rosiglitazone showed an EC50 of be 8 nmol/L. In this assay, Saroglitazar showed similar Emax as well as similar potency for PPARγ as rosiglitazone. Although, in vitro data indicate a very high selectivity of Saroglitazar for PPARα as compared to PPARγ, but difference in ED50 values for antihyertriglyceridemic and antihyperglycemic activities in animal models indicates that in vitro selectivity data may not accurately predict the actual efficacy profile observed in an in vivo system. This could be due to variable sensitivity of in vitro assays (Rubenstrunk et al. 2007), species difference, and differential role of coactivators and corepressors in PPAR mediated effects in different tissues.
The pharmacodynamics activity of Saroglitazar was extensively evaluated in various preclinical models of dyslipidemia and T2DM. The preclinical data confirmed that Saroglitazar has dual lipid-lowering and antihyperglycemic effects when evaluated in diabetic db/db mice, obese and insulin-resistant Zucker fa/fa rats; Swiss albino mice, hApoB100/hCETP double transgenic mice, HF-HC diet fed Golden Syrian hamsters, and non-human primates (Marmosets). The data obtained from in vivo models of T2DM and dyslipidemia indicate that Saroglitazar shows potent lipid- and glucose-lowering and insulin-sensitizing effects accompanied by expected PPAR-mediated changes in expression of target genes.
Regulation of blood glucose has been the cornerstone of both types 1 and type 2 diabetes treatment for decades. Saroglitazar was profiled in different animal models of type 2 diabetes. Saroglitazar showed significant reduction in serum glucose, FFA, and marked improvement in glucose tolerance. Hyperinsulinemic-euglycemic clamp study was done in Zucker fa/fa rats after 15 days of oral administration; Saroglitazar treatment showed significant and dose-dependent increase in GIR demonstrating increased whole body insulin sensitivity. Saroglitazar also showed significant increase in serum adiponectin levels and significant decrease in SBP in Zucker fa/fa rats. Its antihyperglycemic efficacy was comparable to pioglitazone when studied in diabetic and insulin-resistant animal models like db/db mice and Zucker fa/fa rats.
Although Saroglitazar is a predominantly PPARα agonist, it is known that PPARα agonist can contribute to antihyperglycemic effects due to improved lipid metabolism (Kobayashi et al. 1988; Ogawa et al. 2000). PPARα agonists are known to increase hepatic oxidation of fatty acids (FA), and thereby reduce the synthesis and secretion of TG (Desvergene and Wahli 1999; Minnich et al. 2001; Fruchart and Duriez 2006). By their actions on FA and TG, PPARα agonists may increase the insulin-stimulated glucose disposal in the skeletal muscle (Randle et al. 1963; Boden 1994; Goodpaster and Kelly 1998), and thereby ameliorate insulin resistance. Current guidelines recommend aggressive treatment of LDL-C in patients with T2DM. In hApoB100/hCETP double transgenic mice, a model that carries both human cholesteryl ester transfer protein (CETP) and human apolipoprotein B100 transgenes and exhibits human-like serum HDL/LDL distribution, Saroglitazar for 14 days resulted in dose-dependent reductions in LDL-C of upto 72% along with a 61% reduction in LDL-C/HDL-C atherogenic ratio.
It is also known that atherogenic dyslipidemia (AD) poses a cardiovascular risk (CV) risk due to elevated TG levels and accompanied by low HDL-C. AD is associated with insulin resistance (IR), and confers a marked increase in residual vascular risk, even when LDL-C is low (Hermans et al. 2010). It has been reported that TG/HDL-C ratio may be used as a surrogate marker for assessing CV risk (Hermans et al. 2010; Nicholls et al. 2011). In HF-HC fed hamster model, Saroglitazar treatment significantly reduced the TG/HDL-C ratio, which suggests that Saroglitazar may provide significant benefits to patients with atherogenic dyslipidemia.
Meal absorption is a complex phenomenon, postprandial hyperlipidemia and hyperglycemia are simultaneously present in the postabsorptive phase, particularly in diabetic patients and in subjects with impaired glucose tolerance (Ceriello et al. 2004). Furthermore, diabetic patients show significantly delayed postprandial TG clearance as compared to controls (Kumar et al. 2010). Persistent postprandial hypertriglyceridemia may not only result in a proatherogenic environment leading to atherosclerosis and macrovascular disease in type 2 diabetes subjects (Kumar et al. 2010), but also contribute in increased pancreatitis in T2DM patients (Berglund et al. 2012). In our study, Swiss albino mice administered with Saroglitazar for 6 days, showed dose-dependent improvement in lipid clearance (reduction in AUC triglycerides) up on intravenous Intralipid administration with up to 68% enhanced lipolytic activity at 10 mg/kg dose. It is known that LPL plays an important role in the removal of plasma TG, by hydrolyzing the TG of VLDL and chylomicron particles (Goldberg 1996). ApoCIII on the other hand appears to antagonize plasma TG metabolism, as it inhibits TG hydrolysis by LPL and hepatic lipase (Quarfordt et al. 1982; Ginsberg et al. 1986). Expression of LPL and ApoCIII mRNA was measured in liver tissues from db/db animals following treatment with Saroglitazar. In this study, 2.9-fold increase in LPL mRNA and 70% downregulation in ApoCIII expression was observed in Saroglitazar-treated animals as compared to vehicle-treated group. These observations suggest that improved lipid clearance after intravenous Intralipid challenge may be caused by increased LPL activity in Swiss albino mice. The data in HF-HC diet fed hamsters suggest that the efficacy of Saroglitazar is several folds better than that of fenofibrate and rosiglitazone.
PPARα agonists are known to cause rodent-specific liver proliferation and elevation of liver enzymes that are not seen in non-human primates and humans. To evaluate the species specificity, comparative efficacy and safety of Saroglitazar were studied in Wistar rats and marmosets, a non-human primate model. Saroglitazar was well tolerated and showed TG reduction effect in marmosets. It did not showed any adverse effect on behavior or toxicity parameters in marmosets. Elevation of liver enzymes (ALT) and hypertrophic/proliferative changes that are generally observed with PPARα agonists in rodents were observed in rats but were not seen in marmosets. These observations are in line with the reported literature on other PPARα agonists that rodent-specific adverse changes may not pose any risk to human (Green 1992; Heuvel 1999; Ammerschlaeger et al. 2004). Furthermore, Saroglitazar was found to have a good safety margins in chronic repeated dose toxicity in rats and dogs and in safety studies it was found to be safe in a battery of central nervous system (CNS), cardiovascular system (CVS), respiratory, and gastrointestinal (GI) parameters (unpublished data).
In phase-3 clinical trials, Saroglitazar appeared to be an effective and safe therapeutic option for improving hypertriglyceridemia in patients with T2DM. Saroglitazar (2 mg and 4 mg) significantly reduced plasma TG. It also lowered TC, LDL-C, VLDL, apolipoprotein-B, and fasting blood sugar levels. Saroglitazar treatment was generally safe and well tolerated. No serious adverse events were reported in Saroglitazar treatment arm and no persistent change in laboratory parameters. Clinical studies of Saroglitazar have also shown that it is a safe glitazar class compound (Jani et al. 2014; Pai et al. 2014).
In conclusion, Saroglitazar is a nonfibrate, non-TZD next generation PPAR agonist that has shown beneficial effects on lipids and glucose in various preclinical and clinical studies (Jani et al. 2014). It has shown good safety profile and may represent a novel therapeutic agent that will fulfill the unmet needs in T2DM and diabetic dyslipidemia.
Acknowledgments
This work was financially supported by Cadila Healthcare Limited, Ahmedabad, India. The authors are thankful to Dr. Shashank Joshi, Joshi Clinic, Mumbai, India, and Dr. Charles Burant, University of Michigan, for critical review of the manuscript. The authors are also thankful to the Director, National Institute for Research in Reproductive Health (NIRRH), Mumbai, and Vikas Dhige, Scientist at NIRRH, for supporting the work on Marmosets.
Glossary
- ACO
acyl-CoA oxidase
- ACRP30
adipocyte complement-related protein of 30 kDa
- aP2
adipocyte fatty acid-binding protein
- CETP
cholesteryl ester transfer protein
- FATP
fatty acid transporter protein
- GIR
glucose infusion rate
- HDL-C
high-density lipoprotein cholesterol
- HF-HC
high fat-high cholesterol
- IVLTT
intravenous lipid tolerance test
- LDL-C
low-density lipoprotein cholesterol
- LPL
lipoprotein lipase
- NIN
National institute of nutrition
- NIRRH
National institute for research in reproductive health
- OGTT
oral glucose tolerance test
- PPAR
peroxisome proliferator–activated receptors
- SAM
swiss albino mice
- SBP
systolic blood pressure
- TC
total cholesterol
- TG
triglycerides
- ZRC
Zydus research centre
Author Contribution
Mukul R. Jain, Suresh R. Giri, and Pankaj R. Patel conceptualized and designed the studies. Suresh R. Giri, Chitrang Trivedi, Bibhuti Bhoi, Akshyaya Rath, and Purvi Vyas performed the experiments. Mukul R. Jain, Geeta Vanage, Ramchandra Ranvir, and Suresh R. Giri contributed to study design and/or execution of marmoset study. Mukul R. Jain and Suresh R. Giri analyzed the data and drafted the manuscript. Mukul R. Jain critically reviewed the work.
Disclosures
MJ, SG, CT, BB, AR, PV, RR, and PP are employees of Cadila Healthcare Limited, Ahmedabad, India, the company that discovered, developed, and marketed Saroglitazar. GV is an employee of the National Institute for Research in Reproductive Health, Mumbai, a Government of India Institute and she has no conflicts of interest. All authors have approved the submission of the manuscript to this journal.
References
- Agrawal RP, Sharma P, Pal M, Kochar A, Kochar DK. Magnitude of dyslipedemia and its association with micro and macro vascular complications in type 2 diabetes: a hospital based study from Bikaner (Northwest India) Diabetes Res Clin Prac. 2006;73:211–214. doi: 10.1016/j.diabres.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Ammerschlaeger M, Beigel J, Klein KU, Mueller SO. Characterization of the species-specificity of peroxisome proliferators in rat and human hepatocytes. Toxicol Sci. 2004;78:229–240. doi: 10.1093/toxsci/kfh071. [DOI] [PubMed] [Google Scholar]
- Aronoff S, Rosenbalt S, Braithwaite S, Egan JW, Mathisen AL, Schneider RL. Pioglitazone hydrochloride monotherapy improves glycemic control in the treatment of patients with type 2 diabetes. Diabetes Care. 2000;23:1605–1611. doi: 10.2337/diacare.23.11.1605. [DOI] [PubMed] [Google Scholar]
- Balakumar P, Rose M, Ganti SS, Krishan P, Singh M. PPAR dual agonists: are they opening Pandora’s box? Pharmacological research. J Ital Pharmacol Soc. 2007;56:91–98. doi: 10.1016/j.phrs.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Balfour JB, Plosker GL. Rosiglitazone. Drugs. 1999;57:921–930. doi: 10.2165/00003495-199957060-00007. [DOI] [PubMed] [Google Scholar]
- Berglund L, Brunzell JD, Goldberg AC, Goldberg IJ, Sacks F, Murad MH, et al. Evaluation and treatment of hypertriglyceridemia: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:2969–2989. doi: 10.1210/jc.2011-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlie HD, Kalus JS, Jaber LA. Thiazolidinediones and the risk of edema: a meta-analysis. Diabetes Res Clin Pract. 2007;76:279–289. doi: 10.1016/j.diabres.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Boden G. Mechanism of fatty acid induced inhibition of glucose uptake. J. Clin. Invest. 1994;93:2438–2446. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden G, Homko C, Mozzoli M, Zhang M, Kresge K, Cheung P. Combined use of rosiglitazone and fenofibrate in patients with type 2 diabetes: prevention of fluid retention. Diabetes. 2007;56:248–255. doi: 10.2337/db06-0481. [DOI] [PubMed] [Google Scholar]
- Boullart ACI, Graaf JD, Stalenhoef AF. Serum triglycerides and risk of cardiovascular disease. Biochim Biophys Acta. 2012;1821:867–875. doi: 10.1016/j.bbalip.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Quagliaro L, Piconi L, Assaloni R, Da Ros R, Maier A, et al. Effect of postprandial hypertriglyceridemia and hyperglycemia on circulating adhesion molecules and oxidative stress generation and the possible role of simvastatin treatment. Diabetes. 2004;53:701–710. doi: 10.2337/diabetes.53.3.701. [DOI] [PubMed] [Google Scholar]
- Chakrabarti R, Vikramadithyan RK, Misra P, Hiriyan J, Raichur S, Damarla RK, et al. Ragaglitazar: a novel PPARα & PPARγ agonist with potent lipid lowering and insulin-sensitizing efficacy in animal models. Br J Pharmacol. 2003;140:527–537. doi: 10.1038/sj.bjp.0705463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cziraky MJ. Management of dyslipidemia in patients with metabolic syndrome. J Am Pharm Assoc. 2004;44:478–488. doi: 10.1331/1544345041475643. [DOI] [PubMed] [Google Scholar]
- Desvergene B, Wahli W. Peroxisome proliferator activated receptors: nuclear control of metabolism. Endocrinol. Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- Fruchart JC, Duriez P. Mode of action of fibrates in the regulation of triglyceride and HDL-cholesterol metabolism. Drugs Today (Barc) 2006;42:39–64. doi: 10.1358/dot.2006.42.1.963528. [DOI] [PubMed] [Google Scholar]
- Ginsberg HN, Le NA, Goldberg IJ, Gibson JC, Rubinstein A, Wangiverson P, et al. Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. J. Clin. Invest. 1986;78:1287–1295. doi: 10.1172/JCI112713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg IJ. Lipoprotein lipase and lipolysis. J. Lipids Res. 1996;37:693–707. [PubMed] [Google Scholar]
- Goldberg IJ. 2001. Diabetic dyslipidemia: statins versus fibrates in the treatment of diabetic dyslipidemia. Program and abstracts of the 61st Scientific Sessions of the American Diabetes Association; June: 22–26.
- Goodpaster BH, Kelly DE. Role of muscle intriglyceride metabolism. Curr Opin Lipidol. 1998;9:231–236. doi: 10.1097/00041433-199806000-00008. [DOI] [PubMed] [Google Scholar]
- Green S. Nuclear receptors and chemical carcinogenesis. TIPS. 1992;13:251–255. doi: 10.1016/0165-6147(92)90078-k. [DOI] [PubMed] [Google Scholar]
- Hermans MP, Ahn SA, Rousseau MF. log(TG)/HDL-C is related to both residual cardiometabolic risk and b-cell function loss in type 2 diabetes males. Cardiovasc Diabetol. 2010;9:88–97. doi: 10.1186/1475-2840-9-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuvel JPV. Peroxisome proliferator activated recepetor (PPAR) and carcinogenesis. Toxicol Sci. 1999;47:1–8. doi: 10.1093/toxsci/47.1.1. [DOI] [PubMed] [Google Scholar]
- Jani RH, Pai V, Jha P, Jariwala G, Mukhopadhyay S, Bhansali A, et al. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI) Diabetes Technol Ther. 2014;16:63–71. doi: 10.1089/dia.2013.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiess W, Bottner A, Bluher S, Raile K, Galler A, Kapellen TM. Type 2 diabetes mellitus in children and adolescents the beginning of a renal catastrophe? Nephrol Dial Transplant. 2004;19:2693–2696. doi: 10.1093/ndt/gfh455. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Shigeta Y, Hirata Y, Omori Y, SakamotoN NambuS, et al. Improvement of glucose tolerance in NIDDM by clofibrate. Randomised double-blind study. Diabe Care. 1988;11:495–499. doi: 10.2337/diacare.11.6.495. [DOI] [PubMed] [Google Scholar]
- Kumar V, Madhu SV, Singh G, Gambhir JK. Post-prandial hypertriglyceridemia in patients with type 2 diabetes mellitus with and without macrovascular disease. J Assoc Physicians India. 2010;58:603–607. [PubMed] [Google Scholar]
- Minnich A, Tian N, Byan L, Bilder G. A potent PPARα agonist stimulates mitochondrial fatty acid b-oxidation in liver and skeletal muscle. Am J Physiol Endocrinol Metab. 2001;280:E270–E279. doi: 10.1152/ajpendo.2001.280.2.E270. [DOI] [PubMed] [Google Scholar]
- Misra A, Luthra K, Vikram NK. Dyslipidemia in Asian Indians: determinants and significance. J Assoc Physicians India. 2004;52:137–142. [PubMed] [Google Scholar]
- Muačević-Katanec D, Reiner Z. Diabetic dyslipidemia or ‘diabetes lipidus’? Expert Rev Cardiovasc Ther. 2011;9(3):341–348. doi: 10.1586/erc.11.17. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Tuzcu M, Wolski K, Bayturan O, Lavoie A, Uno K. Lowering the triglyceride/high-density lipoprotein cholesterol ratio is associated with the beneficial impact of pioglitazone on progression of coronary atherosclerosis in diabetic patients. J Am Coll Cardiol. 2011;57:153–159. doi: 10.1016/j.jacc.2010.06.055. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Takeuchi K, Sugimura K, Fukuda M, Lee R, Ito S, et al. Bezafibrate reduces blood glucose in type 2 diabetes mellitus. Metabolism. 2000;49:331–334. doi: 10.1016/s0026-0495(00)90176-8. [DOI] [PubMed] [Google Scholar]
- Pai V, Paneerselvam A, Mukhopadhyay S, Bhansali A, Kamath D, Shankar V. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared to pioglitazone 45 mg in diabetic dyslipidemia (PRESS V) J Diabetes Sci Technol. 2014;8(1):132–141. doi: 10.1177/1932296813518680. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel C, Wyne KL, McGuire DK. Thiazolidinediones, peripheral oedema and congestive heart failure: what is the evidence? Diab Vasc Dis Res. 2005;2:61–66. doi: 10.3132/dvdr.2005.010. [DOI] [PubMed] [Google Scholar]
- Quarfordt SH, Michalopoulos G, Schirmer B. The effect of human C apolipoproteins on the in vitro hepatic metabolism of triglyceride emulsions in the rat. J Biol Chem. 1982;257:14642–14647. [PubMed] [Google Scholar]
- Rader DJ. Effect of insulin resistance, dyslipidemia, and intra-abdominal adiposity on the development of cardiovascular disease and diabetes mellitus. Am J Med. 2007;120(3):S12–S18. doi: 10.1016/j.amjmed.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Randle PJ, Garland PB, Hales CN, Newholme EA. The glucose fatty acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- Rubenstrunk A, Hanf R, Hum DW, FruchartJC StaelsB. Safety issues and prospects for future generations of PPAR modulators. Biochimicaet Biophysica Acta. 2007;1771:1065–1081. doi: 10.1016/j.bbalip.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Samadfam R, Awori M, Bénardeau A, Bauss F, Sebokova E, Wright M. Combination treatment with pioglitazone and fenofibrate attenuates pioglitazone-mediated acceleration of bone loss in ovariectomized rats. J Endocrinol. 2012;212(2):179–186. doi: 10.1530/JOE-11-0356. [DOI] [PubMed] [Google Scholar]
- Seber S, Ucak S, Ukcan B, Altuntas Y. The effect of dual PPAR αγ stimulation with combination of rosiglitazone and fenofibrate on metabolic parameters in type 2 diabetic patients. Diabetes Res Clin Pract. 2006;71:52–58. doi: 10.1016/j.diabres.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Taskinen MR. Diabetic dyslipidemia. Atheroscler Suppl. 2002;3:47–51. doi: 10.1016/s1567-5688(01)00006-x. [DOI] [PubMed] [Google Scholar]
- Valabhji J, Watson M, Cox J, Poulter C, Elwig C, Elkeles RS. Type 2 diabetes presenting as diabetic ketoacidosis in adolescence. Diabet Med. 2003;20:416–417. doi: 10.1046/j.1464-5491.2003.00942.x. [DOI] [PubMed] [Google Scholar]
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]