

Abstract
FFA2 is a receptor for short-chain fatty acids. Propionate (C3) and 4-chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide (4-CMTB), the prototypical synthetic FFA2 agonist, evoke calcium mobilization in neutrophils and inhibit lipolysis in adipocytes via this G-protein-coupled receptor. 4-CMTB contains an N-thiazolylamide motif but no acid group, and 4-CMTB and C3 bind to different sites on FFA2 and show allosteric cooperativity. Recently, FFA2 agonists have been described that contain both N-thiazolylamide and carboxylate groups, reminiscent of bitopic ligands. These are thought to engage the carboxylate-binding site on FFA2, but preliminary evidence suggests they do not bind to the same site as 4-CMTB even though both contain N-thiazolylamide. Here, we describe the characterization of four FFA2 ligands containing both N-thiazolylamide and carboxylate. (R)-3-benzyl-4-((4-(2-chlorophenyl)thiazol-2-yl)(methyl)amino)-4-oxobutanoic acid (compound 14) exhibits allosteric agonism with 4-CMTB but not C3. Three other compounds agonize FFA2 in [35S]GTPγS-incorporation or cAMP assays but behave as inverse agonists in yeast-based gene-reporter assays, showing orthosteric antagonism of C3 responses but allosteric antagonism of 4-CMTB responses. Thus, the bitopic-like FFA2 ligands engage the orthosteric site but do not compete at the site of 4-CMTB binding on an FFA2 receptor molecule. Compound 14 activates FFA2 on human neutrophils and mouse adipocytes, but appears not to inhibit lipolysis upon treatment of human primary adipocytes in spite of the presence of a functional FFA2 receptor in these cells. Hence, these new ligands may reveal differences in coupling of FFA2 between human and rodent adipose tissues.
Keywords: 4-CMTB, allosteric agonist, FFA2, GPR43, lipolysis, N-thiazolylamide
Introduction
Receptors with distinct roles in different cells or tissues present challenges for drug therapy, which aims to produce benefits in one tissue without provoking adverse effects in another. One proposed means to overcome this problem is allosterism, which describes the binding of ligands to a receptor at sites distinct from the binding site for the physiological ligand (the orthosteric site). Allosteric ligands which reduce signaling via orthosteric sites are classified as negative allosteric modulators or allosteric antagonists, and those which potentiate signaling via the orthosteric site are positive allosteric modulators. Allosteric agonists are ligands which potentiate signaling at the orthosteric site and themselves activate the receptor. One receptor family where allosterism is common and likely to have therapeutic relevance is the free fatty acid receptors (FFARs). Three of the five FFARs are phylogenetically related and tandemly encoded on human chromosome 19. They respond to long-chain fatty acids (LCFAs) in the case of FFA1 (GPR40), or short-chain fatty acids (SCFAs) in the case of FFA2 (GPR43) and FFA3 (GPR41) (Briscoe et al. 2003; Brown et al. 2003). A further LCFA receptor, FFA4 (GPR120), stimulates release of glucagon-like peptide-1 (GLP-1) (Hirasawa et al. 2005), and GPR84 is responsive to medium-chain fatty acids (Wang et al. 2006; Southern et al. 2013).
FFARs regulate metabolism and integrate the immune system with metabolic status. FFA2 is prominently expressed by neutrophils (Covington et al. 2006; Maslowski et al. 2009) and mediates chemotaxis toward agonists, especially in the gut where large quantities of SCFAs are produced by intestinal bacteria (Sina et al. 2009). Neutrophil FFA2 stimulates reactive-oxygen release, phagocytic activity, apoptosis, and release of intracellular Ca2+. However, FFA2−/− mice have more profound responses in models of colitis, arthritis and allergic asthma, and FFA2−/− neutrophils show increased chemotaxis toward fMLP and C5a and greater infiltration following peritoneal challenge. Hence, FFA2 appears to have an anti-inflammatory role (Maslowski et al. 2009; Sina et al. 2009). FFA2 is also expressed in adipose and coupled to inhibition of lipolysis (Brown et al. 2003), and FFA1, FFA2, and FFA3 are all expressed by pancreatic beta cells (Briscoe et al. 2003; Tang et al. 2015). These findings raised huge interest in FFARs as drug targets in metabolic and inflammatory disease, leading to the identification of orthosteric and allosteric ligands (Hudson et al. 2013b). Most synthetic agonists for FFA1 are structurally reminiscent of LCFAs and were presumed to have an orthosteric mode of binding, but a recent study showed interaction to occur through multiple allosteric sites (Lin et al. 2012).
Unlike FFA1, the first synthetic agonists of FFA2 did not resemble physiological agonists as they lacked carboxylate groups. The prototype synthetic FFA2 agonist is 4-chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide (4-CMTB), which contains a characteristic N-thiazolylamide group (Fig.1) and was identified by Amgen (Wang et al. 2010). 4-CMTB is highly selective for FFA2 over FFA3 and FFA1 (Lee et al. 2008) and acts as a chemoattractant for mouse bone marrow neutrophils (Maslowski et al. 2009). 4-CMTB activates FFA2 and also has a cooperative effect in combination with SCFAs such as propionate (C3) (Lee et al. 2008). The allosteric nature of its interaction was confirmed by showing that FFA2 residues are required for agonism by SCFAs, Arg(5.39) and Arg(7.35) (Stoddart et al. 2008) are not required for agonism by 4-CMTB (Smith et al. 2011). The 4-CMTB binding site has been modeled but not yet validated, although extracellular loop 2 is shown to support communication between SCFA and 4-CMTB sites (Smith et al. 2011). Recently patented FFA2 agonists are reminiscent of “bitopic” ligands: compounds containing structural elements of both orthosteric and allosteric ligands oriented to engage both sites simultaneously (Lane et al. 2013). These new FFA2 agonists contain both carboxylate (orthosteric) and N-thiazolylamide (allosteric), though there appears to be no “linker”, setting them apart from most rationally designed bitopic ligands. A key question, therefore, is whether acid N-thiazolylamides have a bitopic mode of action, binding simultaneously to FFA2 residues that bind SCFAs and those that bind allosteric agonists such as 4-CMTB. Previously they were shown to engage the same basic residues as C3, that is, they are orthosteric ligands (Hudson et al. 2013a). Preliminary evidence was presented that binding was not mutually competitive with 4-CMTB, which argued against a bitopic mode of action, however, in this study 4-CMTB did not show allosteric cooperativity with the particular acid N-thiazolylamides tested. Here, we have synthesized four further acid N-thiazolylamide FFA2 agonists. We show that each has an allosteric interaction with 4-CMTB, which we interpret in the context of potential bitopic and nonbitopic modes of action. Finally, we use these allosteric and orthosteric tools to characterize FFA2 signaling in immune and adipose primary human cells.
Figure 1.
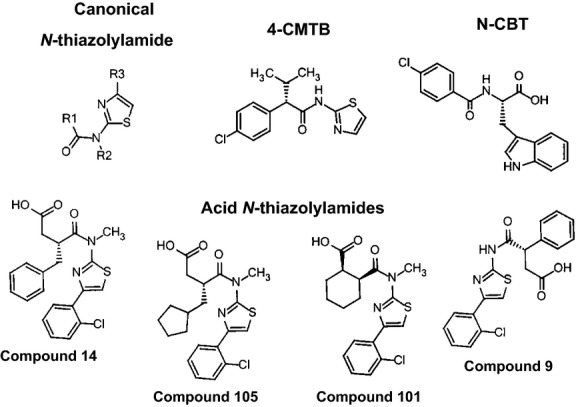
Structures of N-thiazolylamide and synthetic FFA2 ligands used in this study.
Materials and Methods
Forskolin, IBMX, BSA, and phorbol myristic acid were obtained from Sigma-Aldrich Ltd., Gillingham, UK.
Measurement of [35S]GTPγS binding
Scintillation proximity assay (SPA) was performed using LEADseeker™ Homogeneous Imaging (PerkinElmer Life and Analytical Sciences, Seer Green, UK) according to the manufacturer’s instructions. In brief, 109 HEK-F cells stably expressing rat Go in Gibco FreeStyle™ serum-free media (Life Technologies Ltd., Paisley, UK) were transduced for 24 h with BacMam baculovirusencoding human FFA2 (hFFA2) at multiplicity of infection (MOI) of 25. PBS-washed cell pellets were resuspended in nine volumes buffer (50 mmol/L 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) pH7.4, 1 mmol/L ethylenediamine tetraacetate (EDTA), 0.1 mmol/L Leupeptin, 25 mg/L Bacitracin). Pepstatin A (2 μmol/L) and freshly dissolved PMSF (1 mmol/L) were added and the suspension was homogenized in a chilled blender (Waring, Winsted, CT, USA; 20,000 rpm, 2 × 15 sec). Membranes were collected by sequential centrifugation (20 min at 500g, 20 min at 48,000g; 4°C), resuspended in 50 mmol/L HEPES pH7.4 with 1 mmol/L EDTA at 3–4 mg protein/mL, and frozen until use (−80°C). For assay, test compounds were dissolved (10 mmol/L) and diluted into dimethyl sulphoxide (DMSO), and SCFA standards were dissolved in dH2O (100 mmol/L) and serially diluted into assay buffer (HEPES 20 mmol/L pH 7.4, NaCl 100 mmol/L, MgCl2 10 mmol/L), before transfer of 0.5 or 1 μL/well to a Nunc 384 well plate. Membranes were thawed, passed through a 0.8-mm needle to ensure homogeneity, and incubated (1 h at 4°C with agitation) with beads in assay buffer supplemented with saponin (45 μg/mL). Guanosine diphosphate (GDP; 20 μmol/L) and [35S]GTPγS (0.27 nmol/L) were added and the mixture was dispensed (45 μL/well). After 60 min, [35S]GTPγS binding was determined by imaging (Viewlux; PerkinElmer) and expressed as % effect relative to the maximal effect of 4-CMTB (100%). Four-parameter curve fits were constrained to a minima of 0%.
Measurement of intracellular cAMP
LANCE™ competition immunoassay (PerkinElmer) was performed according to the manufacturer’s instructions. Briefly, U20S cells were transduced with hFFA2 BacMam using an MOI of 14, and stored as frozen aliquots until use. For assay, cells were thawed, washed, and resuspended in stimulation buffer (Hank’s buffered saline solution with 5 mmol/L HEPES pH 7.4, 0.01% bovine serum albumin (BSA) and 0.5 mmol/L 3-isobutyl-1-methylxanthine (IBMX)). Cell suspension was counted (Vi-CELL; Beckman Coulter Ltd., High Wycombe, UK), adjusted to 2 × 106/mL and dispensed (5 μL/well) to Nunc LV384-well plates containing 0.1 μL test compound in DMSO, followed by 5 μL/well 1:100 Alexa-fluor cAMP antibody in stimulation buffer containing forskolin (9 μmol/L). After 30 min, 10 μL/well detection solution containing EuSA and Biotin-cAMP was added and time-resolved fluorescence resonance energy transfer determined after 6 h by imaging (Viewlux).
Yeast growth bioassays
Yeast strain YIG69 was constructed by chromosomal integration of pRS306GPD-hFFA2 into the ura3 locus of MMY23. Strain YEG5 was produced by introducing episomal expression construct p426GPD-rFFA2 into MMY24 (Brown et al. 2003). MMY23 contains a chimeric Gpa1/Gαi1 subunit, whereas MMY24 contains Gpa1/Gαi3 (Brown et al. 2000), allowing functional coupling of hFFA2 or rFFA2 to the pheromone response pathway. Receptor activation was measured as described previously (Brown et al. 2011) by 24 h growth assays in 384-well microplates (50 μL/well; Greiner, Stonehouse, UK) with fluorescein di-β-d-galactopyranoside (10 μmol/L). Fluorescence was measured on an Envision plate reader (PerkinElmer). Signal:basal ratios were optimized by varying 3-aminotriazole (3AT), and final assay conditions were 5 mmol/L 3AT (YIG69) or 20 mmol/L 3AT (YEG5) except where otherwise indicated.
Measurement of intracellular calcium in human donor neutrophils
Healthy donor blood (50 mL; sex unknown) was collected into 1 mL 0.5 mol/L EDTA, and mixed with an equal volume of phosphate-buffered saline and half-volume 3% Dextran 200–500 kDa (Amersham Pharmacia Biotech, Uppsala, Sweden) in 0.85% NaCl. After 20 min, supernatant (20 mL) was layered onto 15 mL Lymphoprep and centrifuged (15 min at 700 g). Pellets were resuspended in 20 mL 0.2% NaCl, mixed for 1 min, and 30 mL 1.8% NaCl was added. Neutrophils were isolated by further centrifugation (5 min at 250 g) and loaded (60 min, 37°C, 106 cells/mL) in FLIPR buffer (10 mmol/L HEPES pH 7.4, 145 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 10 mmol/L glucose, 30 μmol/L probenecid, 2 mmol/L CaCl2) supplemented with 4 μmol/L Fluo-4AM. Cells were washed and transferred to Costar black 96-well plates (1.1 × 107 cells/mL; 90 μL/well), and 10 μL test compound (in FLIPR buffer/1% DMSO) was added with black tips in the FLIPR (Molecular Devices Ltd., Wokingham, UK), measuring fluorescence in real time. For antagonist experiments, cells were preincubated with 10 μmol/L N-(4-Chlorobenzoyl)-l-tryptophan (N-CBT) or vehicle for 30 min prior to treatment with agonist. Exposure of neutrophils to N-CBT caused a weak calcium transient via unknown mechanism; however, after 3–5 min intracellular calcium levels returned to basal and did not reduce responses to control agonists.
Lipolysis in mouse epididymal adipose tissue explants
FFA2-deficient (FFA2−/−) mice were obtained from Deltagen (San Mateo, CA). Genotypes of wild-type and FFA2−/− mice were identified by PCR analysis of genomic DNA from tail biopsies. Epididymal adipose tissues from male mice were harvested, cut into ∼15-mg sections and maintained in 96-well cell culture plates containing Krebs–Ringer buffer, 25 mmol/L glucose and 1% BSA for up to 1 h. For lipolysis assay, explants were transferred to fresh 96-well cell culture plates containing compounds, isoproterenol or insulin in Krebs–Ringer buffer supplemented with 25 mmol/L glucose and 1% BSA for 1 h at 37°C. Glycerol released into the supernatant during treatment was measured by free glycerol reagent (Randox, Crumlin, UK). All animal procedures were ethically reviewed and carried out in accordance with European Directive 86/609/EEC and the GSK Policy on the Care, Welfare and Treatment of Animals.
Measurement of GLP1 secretion
Mouse STC1 cells were cultured in DMEM containing 4.5 g/L d-glucose, 1% Penicillin–streptomycin–glutamine, 1 mmol/L sodium pyruvate, 25 mmol/L HEPES (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Biowest, Nuaille, France) at 37°C, 5% CO2 in a humidified atmosphere. Cells were serum starved overnight by omission of FBS. GLP1 secretion assay was performed over 30 min in HBSS media (Invitrogen) at 37°C. No DDP-IV inhibitor or diprotinin A was added to the incubation media. GLP1 level was measured using the Total GLP-1 Assay Kit (Mesoscale Discovery, Rockville, MD). Phorbol myristic acid was used as a control at 400 nmol/L.
Lipolysis in differentiated primary human adipocytes
Primary human subcutaneous preadipocytes (Zenbio, RTP, NC; sex unknown) were cultured and differentiated into adipocytes as per vendor instructions. Differentiated adipocytes were preincubated in Krebs–Ringer buffer (123 mmol/L NaCl, 5.4 mmol/L KCl, 20 mmol/L NaHCO3, 1 mmol/L NaH2PO4, 1 mmol/L CaCl2, 0.8 mmol/L MgCl2) supplemented with 25 mmol/L glucose and 0.5% (v/v) fatty-acid free BSA (Sigma Chemical Co., St. Louis, MO) for 2 h. Cells were then treated with compounds, pertussis toxin (PTX, 100 ng/mL), isoproterenol or insulin (both 200 nmol/L) in Krebs–Ringer buffer for 4 h. Glycerol released into the supernatant during treatment was measured by free glycerol reagent (Randox). Preadipocytes were sourced ethically and their research use was in accord with the terms of the informed consents.
Data analysis
All data presented were derived from a minimum of two independent experiments and are expressed as mean ± standard deviation, except where otherwise indicated. Initial curve-fitting was performed with four-parameter nonlinear regression isotherms in Prism 6.02 (GraphPad Software, San Diego, CA). Schild regressions for allosteric antagonists were performed in Prism using the equation (Kenakin 2009)
where α is the cooperativity factor. To estimate agonist and inverse agonist affinities in the yeast assay we used the operational model of (Slack and Hall 2012) using equation
where E is observed effect, A denotes agonist (or inverse agonist) with Ka the corresponding equilibrium dissociation constant, n is the Hill coefficient of the transducer function, χ is a measure of the coupling efficiency of the signal transduction system, ε is the efficacy parameter, and basal is the background signal observed in the absence of any activation of the system. This model can be applied to concentration–response curves with nonunit Hill coefficients and allows for receptor constitutive activity, as observed for hFFA2 expressed in yeast. Global nonlinear curve fitting was performed by minimizing the sum of squared residuals using the Excel solver add-in and accounting for likely log-normal distribution (Slack and Hall 2012), with E as the dependent variable and [A] as independent variable to give estimates of Ka, χ, ε, n, Emax, and basal. Statistical probability (P) was calculated in Microsoft Excel by one-way analysis of variance (ANOVA) followed by t-test.
Results
We synthesized four reported FFA2 agonists, designated 9, 14, 101, and 105 using the same numbering system as (Hoyveda et al. 2010). These compounds contain both carboxylic acid and N-thiazolylamide chemical moieties, as shown in Figure1. To confirm agonist activity, we measured [35S]-GTPγS incorporation in membranes prepared from HEKF-Go host cells transfected with hFFA2. Compounds 9, 14, 101, and 105 each activated hFFA2 resulting in similar maximum levels of [35S]-GTPγS incorporation to the standard FFA2 agonists C3 and 4-CMTB (Fig.2A and B). This suggests they all behave as full agonists under the conditions of this test. Half-maximal effective concentrations for compounds 9 and 101 were similar to those reported previously, whereas 14 and 105 appeared ∼1 log unit more potent in our experiments, compared with the previous report (Hoyveda et al. 2010). C3 and 4-CMTB were also more potent than in previous reports (Smith et al. 2011), by ∼0.4 log units (pEC50 values are presented in Table1). Next we tested hFFA2 activation by measuring cAMP production using LANCE™ competition immunoassays. hFFA2 was introduced into human U2 osteosarcoma (U2OS) cells by baculovirus-mediated transduction, and cells were treated with forskolin to elevate intracellular cAMP before exposure to test compound. As expected, C3 and 4-CMTB caused concentration-dependent decrease in cAMP (Fig.2C; Table1). Acetate (C2) also inhibited cAMP production (pEC50 = 3.9 ± 0.07; n = 2). 4-CMTB caused the greatest maximum decrease in cAMP of 118 ± 4% relative to C3, which was included in each experiment as normalizing standard (100%). (R)-3-benzyl-4-((4-(2-chlorophenyl)thiazol-2-yl)(methyl)amino)-4-oxobutanoic acid (Compound 14) was approximately equipotent to 4-CMTB (Fig.2D; Table1), which contrasts with the [35S]-GTPγS incorporation assay where compound 14 appeared more potent in activating hFFA2 than 4-CMTB. The trend of lower potency in cAMP compared with [35S]-GTPγS assays was also apparent for the other acid N-thiazolylamides tested. Compounds 105 and 101 were ∼2 log units less potent in the cAMP assay compared with [35S]-GTPγS incorporation. hFFA2 activation by 9 was only detected at the top concentrations tested (30 and 100 μmol/L) in the cAMP assay, and a pEC50 value for 9 was estimated at ∼4. The maximum cAMP reduction achieved by compounds 14 and 105 was close to that of 4-CMTB indicating that they behaved as full agonists. The maximum cAMP reduction achieved by compound 101 was significantly less (P < 0.05 compared with 4-CMTB, according to one-way ANOVA), showing behavior as a partial agonist.
Figure 2.
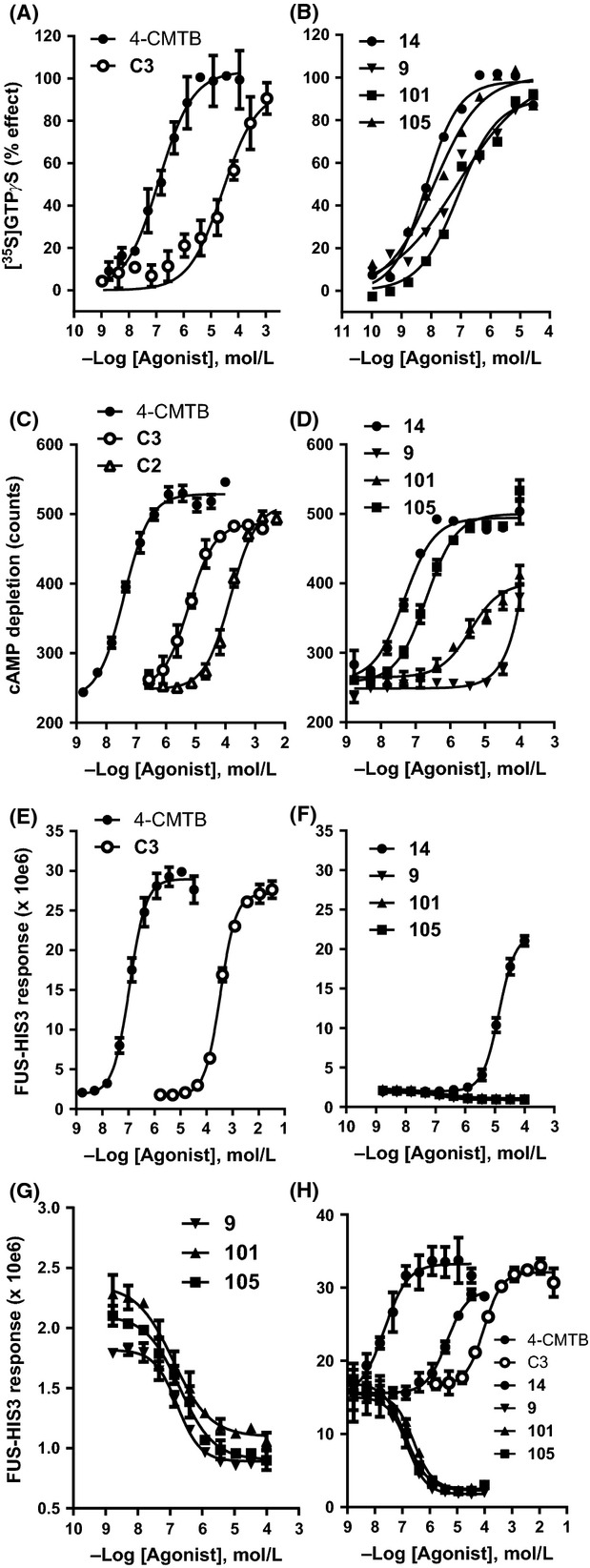
Agonist and inverse agonist effects of a panel of hFFA2 ligands. Propionate (C3), acetate (C2), 4-CMTB, and acid N-thiazolylamides 14, 9, 101, and 105 (free base) were tested for their ability to activate hFFA2. (A) and (B) Stimulation of hFFA2-mediated [35S]-GTPγS incorporation. (C) and (D) Inhibition of forskolin-stimulated cAMP production. (E) to (H) modulation of yeast growth via effects on the FUS1-HIS3 gene-reporter. Conditions for the yeast assay were 5 mmol/L 3AT (E), (F) and (G), which is the default used for hFFA2 yeast assays, or 1 mmol/L 3AT (H). Panels (F) and (G) present identical data but with an expanded y-axis in (G) to illustrate inverse agonism of compounds 9, 101, and 105. Panels show data from representative experiments; for clarity, error bars were omitted from panel (B). Mean pEC50 values and experimental replication are presented in Table1. Curves shown were separately fitted to four-parameter nonlinear regression isotherms but data in panels (E–H) were also subjected to simultaneous curve-fitting by application of an operational model (Figs. S1, S2). 4-CMTB, 4-chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide.
Table 1.
Agonist and antagonist potency at hFFA2 and rFFA2
| hFFA2 | rFFA2 | ||||
|---|---|---|---|---|---|
| [35S]-GTPγS pEC501 | [35S]-GTPγS pEC50 | LANCE-cAMP (pEC50 and %Effect) | Yeast pXC50 (5 mmol/L 3AT) | rFFA2 Yeast pEC50 | |
| C3 (propionate) | ND2 | 4.5 ± 0.08 n = 2 | 5.2 ± 0.05 (100%) n = 2 | 3.4 ± 0.11 (100%) n = 2 | 3.7 ± 0.20 (100%) n = 2 |
| 4-CMTB | ND2 | 6.9 ± 0.12 n = 3 | 7.1 ± 0.22 118 ± 4% n = 20 | 6.4 ± 0.25 105 ± 14% n = 45 | 7.1 ± 0.11 112 ± 16% n = 22 |
| Compound 14 | 7.0 | 8.2 ± 0.23 n = 4 | 7.7 ± 0.54 109 ± 7% n = 4 | 5.1 ± 0.163 65 ± 5%3 n = 4 | 5.0 ± 0.13 105 ± 18% n = 6 |
| Compound 9 | 7.2 | 7.0 ± 0.08 n = 2 | ∼4 n = 4 | 6.74 ± 0.154 n = 3 | Antagonist |
| Compound 101 | 7.2 | 7.1 ± 0.29 n = 3 | 5.4 ± 0.46 65 ± 10% n = 6 | 6.73 ± 0.234 n = 3 | Antagonist |
| Compound 105 | 7.1 | 8.1 ± 0.48 n = 4 | 7.1 ± 0.47 102 ± 3% n = 5 | 6.45 ± 0.204 n = 3 | Antagonist |
EC50 values for [35S]-GTPγS accumulation (Column 1) were taken from Hoyveda et al. (2010) and converted to pEC50 (-log10[EC50]) for comparison to pEC50 values generated herein (Column 2). Yeast pXC50 values indicate pEC50 values for compounds acting as agonists (C3, 4-CMTB and 14) and pIC50 values for compounds acting as inverse agonists (9, 101 and 105; underlined) in the yeast assay under the condition of 5 mmol/L 3AT. Compounds 9, 101, and 105 antagonized rFFA2 but accurate pIC50 values were not determined. 4-CMTB, 4-chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide
(Hoyveda et al. 2010)
ND: not disclosed
estimated (curve upper asymptote was not accurately defined)
pIC50 values.
Inhibition of forskolin-stimulated cAMP production and stimulation of [35S]-GTPγS incorporation result from activation of members of the Gi family of G-proteins (Gi and G0) by hFFA2 (Brown et al. 2003). We also investigated hFFA2 signaling to a chimeric G-protein containing the C-terminal residues of Gi1, in a yeast-based gene reporter assay (Brown et al. 2003). Host yeast cells used contain the FUS1-HIS3 reporter which encodes an enzyme for histidine biosynthesis, imidazoleglycerol-phosphate dehydratase (His3p). The FUS1 promoter is induced downstream of heterotrimeric G protein activation, and yeast growth in histidine-deficient media becomes dependent on G protein-coupled receptor (GPCR) activation when 3AT, which is an inhibitor of His3p enzyme, is supplemented into growth medium. As expected, standard agonists C3 and 4-CMTB stimulated growth of hFFA2-expressing yeast in the presence of 5 mmol/L 3AT. The maximum extent of activation was similar for each, suggesting they both behaved as full agonists (Fig.2E). However the acid N-thiazolylamide hFFA2 agonists showed contrasting effects in the yeast assay. Compound 14 activated hFFA2 but with ∼2.5 log units less potency than in the cAMP assay and ∼3.5 log units less potency than in the [35S]-GTPγS incorporation assay. The maximum extent of activation by 14 appeared less than for C3 and 4-CMTB suggesting it may behave as a partial agonist in this assay, though the maximum asymptote of the concentration-response to 14 was not accurately defined (Fig.2F). Compounds 9, 101, and 105 did not activate hFFA2 in the yeast assay, instead concentration-dependent inhibition of basal reporter gene activity was observed. This suggests hFFA2-expressing yeast exhibit agonist-independent growth, and 9, 101, and 105 act as inverse agonists. The extent of inhibition (at 5 mmol/L 3AT) was smaller than the agonist effect of compound 14, but could be observed by re-scaling the y-axis (Fig.2G).
Agonist-independent growth is observed for many GPCRs expressed in FUS1-HIS3 yeast cells, consistent with receptor constitutive activity (Bertheleme et al. 2013). Reducing 3AT concentration can amplify basal levels of constitutive GPCR signaling, since less FUS1-HIS3 induction is required before growth is observed. We used this approach to investigate further the action of compounds 9, 101, and 105. Reducing 3AT from 5 mmol/L to 1 mmol/L caused an increase in hFFA2 basal constitutive activity, as expected. This reduced the window for agonist activation but revealed characteristic inverse agonist activity of 9, 101, and 105 (Fig.2H; Table1). Compounds 14 and 105 in particular have closely related structures (Fig.1) and their differing activities in yeast (agonist and inverse agonist) result from the presence of phenyl in 14 compared to cyclopentyl in 105. This suggests that subtle conformational differences in ligand-bound FFA2 can lead to either agonist or inverse agonist behavior.
Next, we used the operational model of Slack and Hall (2012), which can be simultaneously applied to sets of agonists and inverse agonists to estimate ligand affinity. This model requires no prior knowledge of the orthosteric or allosteric nature of a ligand but does require data to be generated under conditions of varying receptor constitutive activity. Previously, this was achieved by altering [GDP] in [35S]-GTPγS incorporation assays (Slack and Hall 2012), but in yeast is readily achieved by varying [3AT]. The experiment shown in Figure2E–G was repeated at 5, 2, and 0.5 mmol/L 3AT corresponding to low, intermediate, and high constitutive activity, respectively. As the behavior of the three inverse agonists 9, 101, and 105 was similar, only compound 9 was included in modeling. Concentration–response data for C3, 4-CMTB, 14 and 9 were fitted simultaneously to common basal, Emax and n for all three 3AT concentrations (basal and Emax are respectively the minimal and maximal response that can be achieved in the yeast system; n is the Hill coefficient of the transducer function). Allowing the coupling efficiency χ to vary for each 3AT concentration and intrinsic efficacy ε to vary for each ligand gave curve fits shown in Figure S1. Affinity estimates were pKa = 2.9 for C3 and pKa = 5.9 for 4-CMTB, consistent with published pKa estimates from [35S]-GTPγS incorporation experiments (Smith et al. 2011). We tested the robustness of this approach by separately applying the operational model to data from Figure2E–G, but constraining pKa of C3 and 4-CMTB (Fig. S2). This gave values of n, χ (for each 3AT concentration), ε (for each ligand), and pKa estimates for 14 and 9 that were highly consistent. The relatively conservative structural differences between 14 and 9 (Fig.1) result in >2 log unit difference in apparent affinity for FFA2, in yeast: mean pKa = 4.29 for 14 (4.23 and 4.34) and pKa = 6.81 for 9 (6.93 and 6.69). Compounds that behave as either agonists or inverse agonists at a receptor have been classified as protean agonists (Kenakin 2001). We investigated whether 9, 101, and 105 could activate FFA2 in yeast under conditions of low receptor constitutive activity, by increasing [3AT]. At 20 mmol/L or 25 mmol/L 3AT, weak but significant positive efficacy of 9, 101, and 105 was observed (Fig. S3; P < 0.05). Hence, acid N-thiazolylamides can behave as protean agonists, exhibiting either partial agonism or inverse agonism depending on FFA2 constitutive activity.
In summary, acid N-thiazolylamides show different properties in different expression hosts and using different methods to measure hFFA2 activation. They range from potent agonists with positive, high efficacy in [35S]-GTPγS incorporation assays, to weak agonists with partial efficacy or inverse agonists with negative efficacy in yeast.
Next, we investigated allosteric cooperativity of hFFA2 agonists, using the yeast assay. As expected, 4-CMTB increased the potency of C3 (Fig.3A). The extent of shift of the C3 concentration–response curve (ΔpEC50) was 0.17 ± 0.06, 0.47 ± 0.08, 0.82 ± 0.02, and 0.94 ± 0.07 log units in the presence of 30, 100, 300 nmol/L, and 1 μmol/L (3e-8, 1e-7, 3e-7 and 1e-6 mol/L) 4-CMTB, respectively, comparing with the pEC50 for C3 alone (n = 2). This cooperativity of C3 and 4-CMTB at hFFA2 has not previously been shown in yeast, but has been published from studies of recombinant hFFA2 measuring Ca2+ mobilization, cAMP generation and [35S]GTPγS-incorporation (Lee et al. 2008; Hudson et al. 2013a), and lipolysis in FFA2-expressing mouse 3T3 cells (Lee et al. 2008). No cooperativity between C3 and compound 14 was observed, since increasing concentrations of 14 did not increase C3 potency (Fig.3B and D), consistent with 14 binding to the orthosteric (C3) site in hFFA2. This behavior is similar to a structurally related hFFA2 agonist, also containing both acid and N-thiazolylamide groups, which was shown to interact with the carboxylate-binding histidines within FFA2 (Hudson et al. 2013a). For the final pair of agonists, 14 and 4-CMTB, potency of 4-CMTB increased with increasing concentration of 14, indicating cooperativity (Fig.3C). ΔpEC50 values illustrate that whereas compound 14 caused no significant positive displacement of C3 pEC50, concentration-dependent positive displacement of 4-CMTB pEC50 was observed (Fig.3D). Allosteric modulation of 14 and 4-CMTB is consistent with binding of 4-CMTB and 14 to distinct hFFA2 sites. Hudson et al. (2013a) were unable to show allosteric cooperativity between 4-CMTB and a different acid N-thiazolylamide hFFA2 agonist (described as compound 2), which they attributed to probe dependence.
Figure 3.
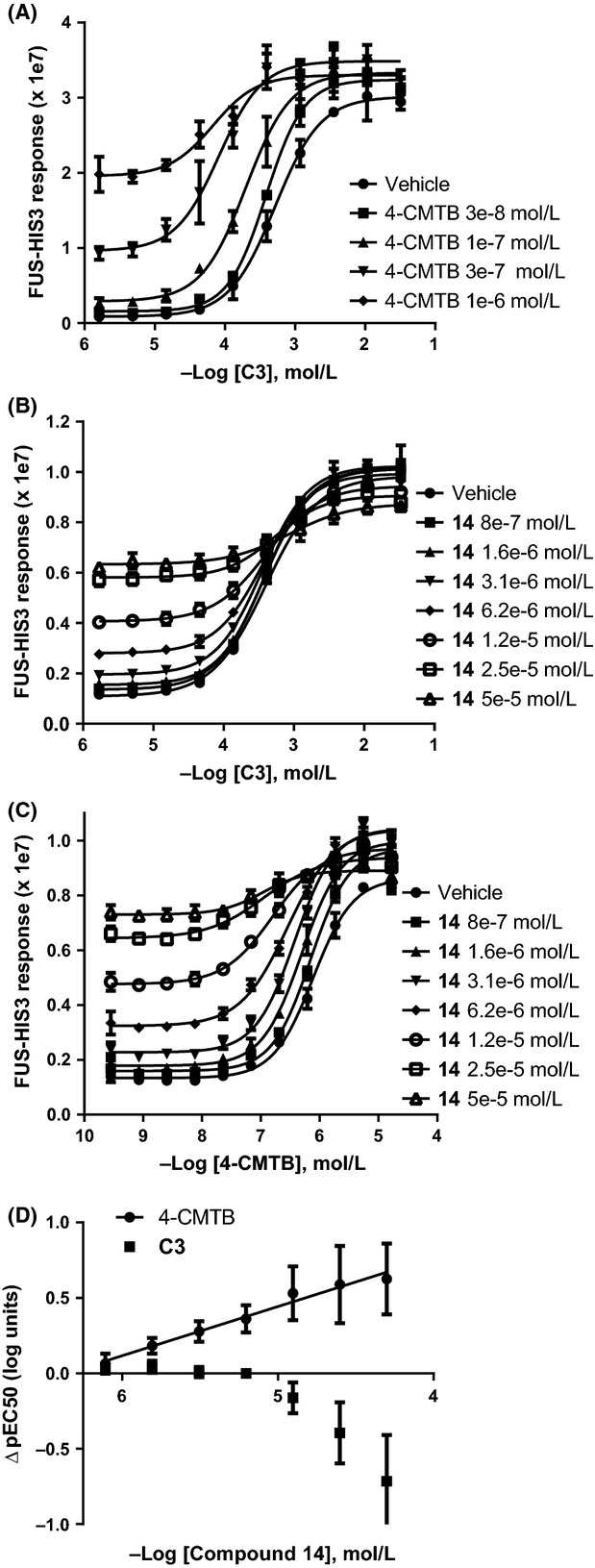
Positive modulation between hFFA2 agonists. Orthosteric agonist (Propionate; C3), allosteric agonist (4-CMTB) and the acid N-thiazolylamide compound 14 were combined in the hFFA2 yeast assay: (A) C3 plus 4-CMTB, (B) C3 plus compound 14, and (C) 4-CMTB plus compound 14 (representative experiments are shown). (D) Comparison of ΔpEC50 for agonist combinations C3 and 14 and 4-CMTB and 14 (n = 4 or 5 experiments). 4-CMTB, 4-chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide.
Next, we investigated whether hFFA2 inverse agonists 9, 101, and 105 could also antagonize C3 and 4-CMTB, in yeast. N-CBT (Fig.1; Table S1) is a tool hFFA2 antagonist that lacks the N-thiazolylamide group. N-CBT was originally described as a CCK antagonist (Bill 1990) and its activity at hFFA2 has not previously been published. However N-CBT is from the same chemotype as another hFFA2 antagonist, (S)-3-(2-(3-chlorophenyl)acetamido)-4-(4-(trifluoromethyl)phenyl) butanoic acid (CATPB) (Hudson et al. 2012). Increasing concentrations of N-CBT caused sequential shift of the C3 concentration–response curve (Fig.4A). Schild analysis showed a linear relationship across the full range of N-CBT concentrations tested with calculated pA2 = 6.3 (for slope of unity), agreeing with estimated pKB = 6.3 ± 0.16 (n = 8) and pKB = 6.3 ± 0.27 (n = 12) from yeast and [35S]-GTPγS incorporation assays, respectively (see Table S1). Antagonism of 4-CMTB by N-CBT differed in that lower concentrations of N-CBT caused sequential shift of the 4-CMTB concentration–response curve but at 16 and 50 μmol/L (1.6e-5 and 5e-5 mol/L) the extent of shift reduced and approached a maximum (Fig.4B). This observation is unlikely to be due to limiting solubility since N-CBT shows apparent competitive behavior with C3 at the same concentrations. Figure4B shows a linear unit-slope Schild plot fitted to lower N-CBT concentrations only, showing the deviation from linearity at higher concentrations which is characteristic of allosteric antagonism (Kenakin 2009). Using the Schild equation for allosteric antagonism gave estimated pKB = 6.5 ± 0.14, and α = 0.017 ± 0.003 (n = 2). hFFA2 antagonism by N-CBT in yeast is similar to the behavior of CATPB in ERK phosphorylation assays, in that CATPB shows orthosteric competitive antagonism of C3, but allosteric nonsurmountable antagonism of 4-CMTB (Hudson et al. 2013a). Together, this is consistent with N-CBT and CATPB (which both contain carboxylate groups) binding in the same site as C3 in hFFA2 and distinct from the 4-CMTB-binding site.
Figure 4.
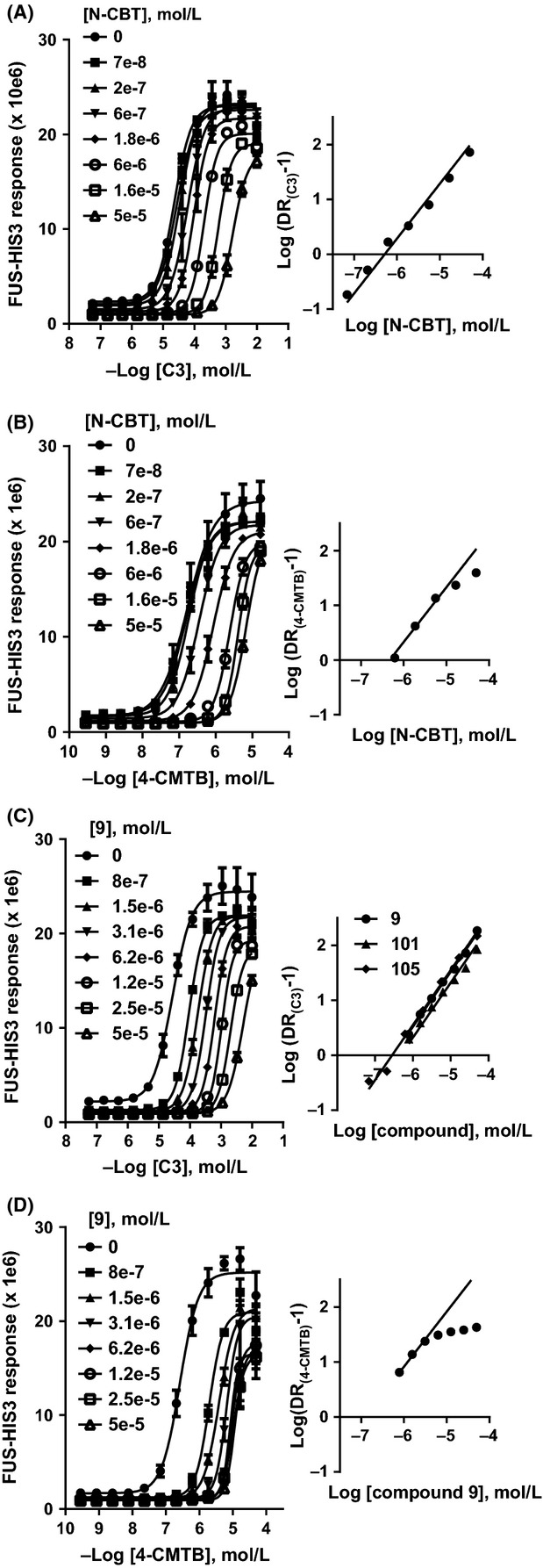
Orthosteric and allosteric antagonism of hFFA2. Responses to orthosteric agonist (Propionate; C3) and allosteric agonist (4-CMTB) were antagonized with N-CBT or acid N-thiazolylamide compounds 9, 101, and 105 in the hFFA2 yeast assay. Data show sets of concentration–response curves (left) and corresponding Schild plots with linear unit-slope fits (right) from the combinations (A): C3 and N-CBT; (B): 4-CMTB and N-CBT; (C): C3 and 9 and (D): 4-CMTB and 9. Schild analysis for antagonism of C3 by 101 and 105 is also shown in (C) but full sets of concentration–response curves for C3 and 101 and C3 and 105 are not presented. Unconstrained Schild slopes in (A) and (C) were not significantly different to unity. In (B) and (D), Schild plots were fitted with linear unit-slope lines to lower antagonist concentrations only, to illustrate the deviation from linearity at higher concentration characteristic of allosteric antagonists. Representative data are shown; for experimental replication (n) and estimated mean values of pA2, pKB, and α, see Results. 4-CMTB, 4-chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide; N-CBT, N-(4-Chlorobenzoyl)-l-tryptophan.
Inverse agonists 9, 101, and 105 antagonized hFFA2 in yeast with a similar profile to N-CBT. Increasing concentrations of 9 (Fig.4C) 101, and 105 caused sequential shift of the C3 concentration–response curve. Schild analysis for all three compounds (Fig.4C) showed linearity across the concentration range tested with slopes of unity (pA2 = 6.64 ± 0.17 for 9, pA2 = 6.40 ± 0.11 for 101, and pA2 = 6.54 ± 0.02 for 105; all n = 2). In contrast, shift of the concentration–response curve to 4-CMTB reached a maximum extent whereby further increases of 9 (Fig.4D), 101, and 105 caused no further inhibition. Schild analysis showed nonlinearity giving estimated pKB = 6.88 ± 0.17 and α = 0.01 ± 0.004 (n = 3) for 9, pKB = 6.69 ± 0.03 and α = 0.03 ± 0.02 (n = 3) for 101, and pKB = 6.68 ± 0.21 and α = 0.02 ± 0.009 (n = 4) for 105. As for N-CBT, the apparent competitive antagonism of C3 by 9, 101, and 105 is consistent with binding to the same site, whereas the apparent allosteric antagonism of 4-CMTB by 9, 101, and 105 is consistent with binding to a distinct site. Moderate depression of the maximum asymptote at higher antagonist concentrations may indicate insurmountable rather than surmountable effects (as have been modeled above) but these scenarios are difficult to differentiate in a live-cell assay since high organic load resulting in inhibition of cell-growth can lead to the same outcome. However, the primary conclusion, that antagonism of 4-CMTB by N-CBT, 9, 101, and 105 is allosteric, is unchanged.
We also tested agonist and inverse agonist effects at rat FFA2 (rFFA2) using the yeast assay. C3, 4-CMTB, and 14 behaved as full agonists at rFFA2 (Table1). Potency of 14 was not significantly different between the hFFA2 and rFFA2 yeast assays, whereas C3 and 4-CMTB were more potent at rFFA2 by 0.3 and 0.7 log units, respectively (P < 0.01; one-way ANOVA). Compounds 9, 101, and 105 all behaved as inverse agonists at rFFA2 and caused rightward shift of concentration–response curves of rFFA2 to C3 and 4-CMTB, in the yeast assay (data not shown). Therefore, the behavior of acid N-thiazolylamide ligands in the yeast assay is consistent between hFFA2 and rFFA2 orthologs.
Our data show that 14, 9, 101, and 105 are orthosteric ligands, binding to the same site in hFFA2 as C3, and having system-dependent efficacy. Compounds 14, 9, 101, and 105 do not compete for binding to the 4-CMTB site even though N-thiazolylamide is present in all these compounds. Since hFFA2 ligands are potential therapeutic agents in metabolic and immune diseases, we next studied ligand efficacy at FFA2 endogenously expressed in mouse and human cells. Neutrophils express high relative levels of FFA2 (Brown et al. 2003) and genetic deletion of mouse FFA2 abolishes both chemotaxis toward C3 and acetate-evoked intracellular Ca2+ release in bone marrow neutrophils (Maslowski et al. 2009; Vinolo et al. 2011). We measured intracellular Ca2+ mobilization in neutrophils purified from human donor blood, using FLIPR. C3 and 4-CMTB evoked robust concentration-dependent increases in intracellular Ca2+ (Fig.5A and B) of similar maximum magnitude to the control, leukotriene B4 (45 nmol/L; data not shown). Half-maximal effective concentrations were pEC50 = 4.0 ± 0.30 for C3 and pEC50 = 5.4 ± 0.28 for 4-CMTB (n = 8 donors across four separate occasions). Pretreatment with 10 μmol/L N-CBT blocked the Ca2+ response evoked by C3, confirming that hFFA2 mediates this effect (Fig.5A). N-CBT was also effective in reducing Ca2+ mobilization evoked by 4-CMTB, though to a lesser extent than with C3, possibly due to its allosteric antagonism of 4-CMTB (Fig.5B). Compound 14 appeared similarly effective to 4-CMTB in increasing neutrophil Ca2+, with the top four concentrations (1.2 to 33 μmol/L) causing significant Ca2+ mobilization (Fig.5C). However, 9, 101 and 105 were less effective, showing significant Ca2+ mobilization only at 33 and 10 μmol/L (101) or only at 33 μmol/L (9 and 105) (Fig.5C).
Figure 5.
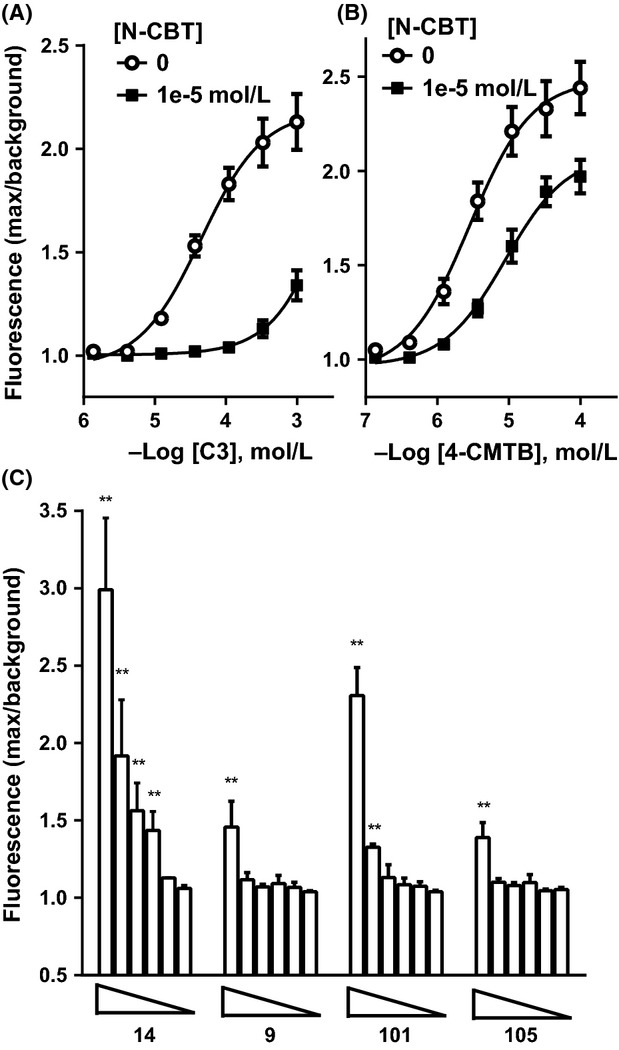
hFFA2-mediated calcium mobilization in human neutrophils. Human neutrophils from healthy volunteers were loaded with the calcium-sensitive dye Fluo4 and pretreated with either the hFF2 antagonist, N-CBT at 10 μmol/L (1e-5 mol/L), or vehicle. Transient intracellular calcium release evoked by the agonists C3 (A) or 4-CMTB (B) was measured by FLIPR. (C) Fluo4-loaded human neutrophils were challenged with N-thiazolylamide ligands 14, 9, 101, or 105: concentrations tested were 33, 11, 3.7, 1.2, 0.41, and 0.14 μmol/L. Data show mean ± SEM for n = 2 or 3 donors, (**P < 0.01; one-way ANOVA). 4-CMTB, 4-chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide; ANOVA, analysis of variance; N-CBT, N-(4-Chlorobenzoyl)-l-tryptophan.
Finally, we studied agonist efficacy at FFA2 in adipose tissue. Explants from epididymal adipose fat pads of wild-type and FFA2−/− mice were tested. Lipolysis resulted in glycerol release into the media, and was attenuated by insulin and by an agonist of hydroxycarboxylic acid receptor-2, and stimulated by isoproterenol, as expected. The effects of these agents were comparable between wild-type and FFA2-deficient explants (data not shown). Potency of 4-CMTB has been shown to be similar at mouse FFA2 (mFFA2) and hFFA2 (Lee et al. 2008). Moreover, 4-CMTB causes PTX-sensitive inhibition of lipolysis in differentiated mouse 3T3L1 adipocytes (Lee et al. 2008). Treatment of wild-type explants with either 4-CMTB, 14 (75 μmol/L) or C3 (20 mmol/L) resulted in significant inhibition of lipolysis, by approximately half in each case (Fig.6A). Treatment of explants from FFA2−/− animals caused no significant effects, confirming the involvement of FFA2 (Fig.6A). We also examined the capacity of 4-CMTB and 14 to stimulate GLP-1 secretion using mouse enteroendocrine STC-1 cells (Hudson et al. 2013a). Both 4-CMTB and 14 stimulated GLP-1 secretion but different potencies were observed. 4-CMTB (10 μmol/L) increased GLP-1 secretion ∼10-fold over basal, which was not further increased by higher concentrations of 4-CMTB, (25 and 50 μmol/L; Fig.6B). In contrast, 10 μmol/L 14 had no significant effect on GLP-1 secretion and higher concentrations were required to increase GLP-1. Even at the highest tested concentration (50 μmol/L), 14 increased GLP-1 secretion only to approximately sixfold over basal levels. Thus, 14 is less potent than 4-CMTB in evoking GLP-1 release from mouse STC-1 cells, possibly by fivefold or more.
Figure 6.
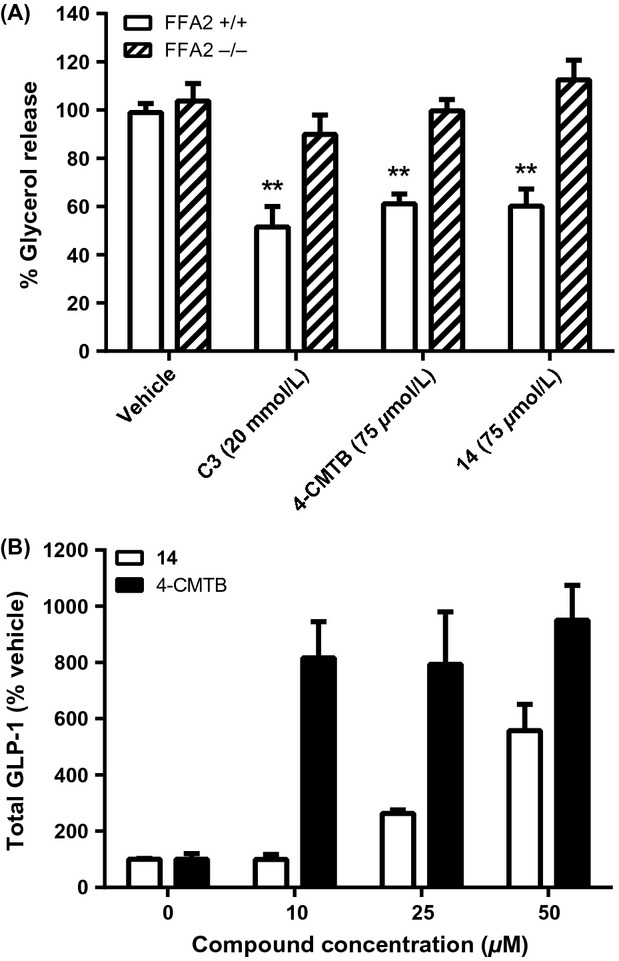
Effects of FFA2 agonists on lipolysis and GLP-1 release in mouse cells. (A) Adipose tissue explants from FFA2−/− mice and matched wild-type littermates were treated in vitro with vehicle, C3 (20 mmol/L), 4-CMTB, or 14 (75 μmol/L) and lipolysis determined by measurement of glycerol release. Data represent mean ± SEM (n = 6–11 across two experiment occasions; **P < 0.01). (B) Cultured mouse enteroendocrine STC-1 cells were treated with 4-CMTB or 14 (0–50 μmol/L) and GLP-1 secretion was measured by an ELISA-based method (MSD). Bars show mean ± SD; experiment repeated 2–5 times for each compound and representative experiment shown. 4-CMTB, 4-chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide.
On primary human adipocytes, 4-CMTB (50 μmol/L), and insulin significantly reduced lipolysis as expected (Fig.7A). The magnitude of this reduction was similar to the effect of 4-CMTB on mouse adipose explants (Fig.6A). Compounds 14, 9, 101, and 105 were each tested at 10, 25, 50, and 100 μmol/L, however, none significantly altered lipolysis at any concentration tested (only data for 50 μmol/L is shown; Fig.7A). To confirm the involvement of Gi-proteins, we showed that pretreatment with PTX prevented the inhibition of lipolysis by 4-CMTB (Fig.7B). Finally, to confirm involvement of hFFA2, we pretreated primary human adipocytes with N-CBT. N-CBT alone showed a trend toward increased basal lipolysis, which did not reach statistical significance (Fig.7C). The effect of 4-CMTB on lipolysis was significantly attenuated by 50 μmol/L N-CBT (Fig.7C). Considering 4-CMTB is a selective FFA2 agonist, and its effect on human adipocytes is PTX sensitive and is blocked by an FFA2-selective antagonist, this confirms the presence of functional hFFA2 in the primary human adipocytes. Hence, compound 14 is unable to inhibit lipolysis via hFFA2 on human adipocytes in this assay, even though it behaves as an agonist in recombinant assays of hFFA2, and is able to inhibit lipolysis via FFA2 in mouse adipose explants.
Figure 7.
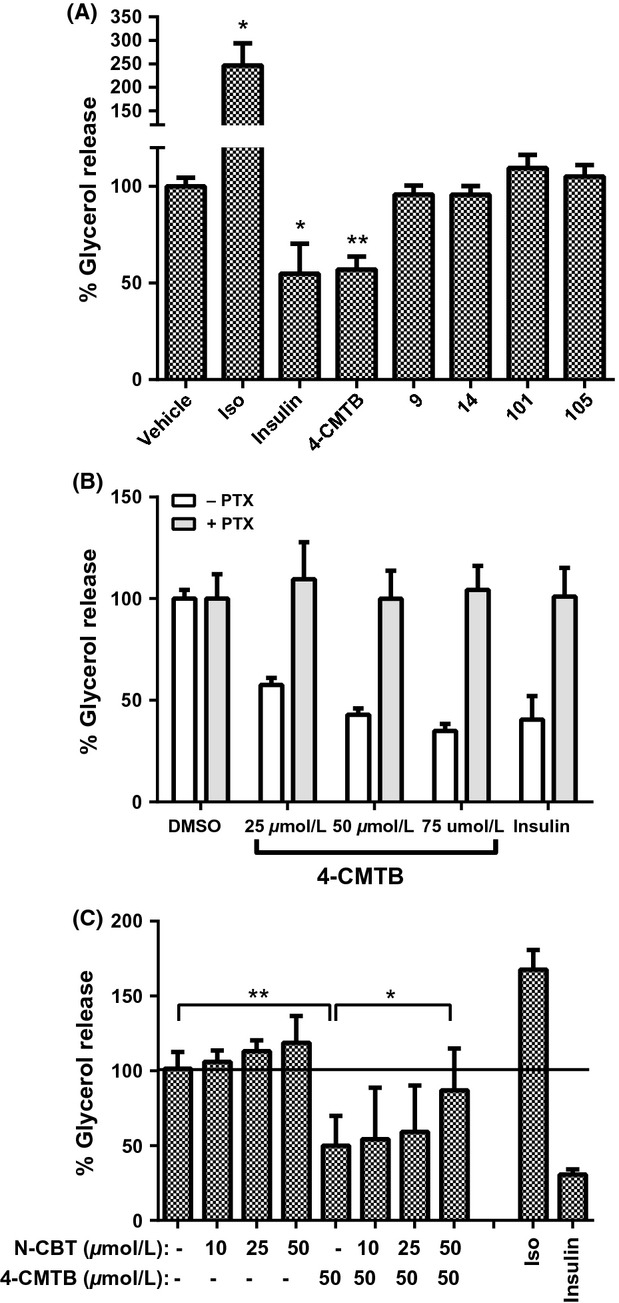
Inhibition of lipolysis by FFA2 agonists in primary human adipocytes. (A) Primary human adipocytes were treated with isoproterenol (Iso) or insulin (both at 200 nmol/L) or FFA2 agonists (10–100 μmol/L; only 50 μmol/L treatment is shown) and lipolysis determined as above. Bars represent mean ± SD (n = 4 determinations across two experiment occasions). (B) Primary human adipocytes were pretreated with PTX or vehicle before drug treatment. Bars represent mean ± SD from three experiments with each condition determined in duplicate in each experiment. (C) Primary human adipocytes were pretreated with the hFFA2 antagonist N-CBT (10–50 μmol/L) prior to 4-CMTB treatment. Data show mean ± SD from two experiments, each condition was determined n = 2–4 per experiment. *P < 0.05; **P < 0.01. 4-CMTB, 4-chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide; N-CBT, N-(4-Chlorobenzoyl)-l-tryptophan.
Discussion
In this study, we investigate the mode of binding of acid N-thiazolylamide ligands to the hFFA2 receptor. Drugs directed against hFFA2 have potential in treating metabolic conditions and inflammatory disorders, and the recent patent describing acid N-thiazolylamide FFA2 agonists could offer an alternative route to modulate FFA2 in vivo, distinct from allosteric hFFA2 agonists such as 4-CMTB. Acid N-thiazolylamides combine the critical feature of orthosteric agonists (carboxylate) with the N-thiazolylamide moiety found in 4-CMTB and related allosteric agonists, reminiscent of bitopic ligands (Lane et al. 2013). Acid N-thiazolylamides bind to the orthosteric (SCFA-binding) site of hFFA2, but only limited data have been available previously to indicate whether these ligands also overlap the binding site of 4-CMTB, though this seemed a likely hypothesis based on their chemical similarity. The previous report (Hudson et al. 2013a) described two acid N-thiazolylamides (compounds 1 and 2) structurally similar to the compounds used in this study. Compounds 1 and 2 were blocked competitively by the orthosteric hFFA2 antagonist CATPB, whereas blockade of 4-CMTB was noncompetitive (Hudson et al. 2013a). However, whilst this suggested that CATPB and 4-CMTB binding sites are distinct, it offered little information about the mutual binding of 4-CMTB with 1 or 2. In a β-arrestin assay where 4-CMTB behaved as a partial agonist with respect to 1, 4-CMTB could not antagonize a high concentration of 1, as might be expected if they were mutually competitive, and this was the evidence that 4-CMTB and 1 did not share a binding site (Hudson et al. 2013a). However, no allosteric modulation between 4-CMTB and 1 was observed, which was ascribed to probe dependence, the property of different pairs of allosteric and orthosteric ligands to exhibit different degrees of cooperativity (4-CMTB and 1 have neutral cooperativity). Here, we confirm that acid N-thiazolylamide ligands bind to the same site as SCFAs within hFFA2 by showing that 9, 101, and 105 competitively antagonize C3 in the yeast assay, comparable to another antagonist from a different chemical series, N-CBT. Furthermore, 14 does not show positive modulation of agonist responses to C3, also consistent with a common binding site. We provide direct evidence that acid N-thiazolylamides do not overlap the binding site of 4-CMTB by showing positive allosteric modulation between 4-CMTB and 14. Furthermore, antagonism of the response to 4-CMTB by 9, 101, and 105 is noncompetitive, exhibiting negative allosteric modulation. These data suggest two N-thiazolylamide-binding sites exist in the FFA2 structure, one accessed by 4-CMTB and the other (proximal to the SCFA-binding residues) accessed by 14, 9, 101, and 105. It seems somewhat paradoxical for two different FFA2 sites to bind the same conserved chemical structure (N-thiazolylamide), and an alternative possibility is that FFA2 has a single N-thiazolylamide site but FFA2 functions as a homodimer (or higher-order oligomer). According to this model, allosteric ligands can exert their effects by binding to one protomer and modulating affinity and/or efficacy at the partner protomer, within a dimer. Lane et al. (2014) recently proposed this model to explain allosteric behavior of the bitopic dopamine D2 receptor antagonist, SB269652. In their experimental system where D2 receptor dimers form from defined subunits, they converted the binding of SB269652 from allosteric to competitive by impairing binding to one protomer (Lane et al. 2014). A feature of this model is probe dependence, that is, binding of the allosteric ligand at the first protomer would have a high negative cooperativity for binding of the same ligand at the second protomer, allowing modulatory effects toward other ligands. To resolve these different models, it will be important to define the residues involved in the interaction with 4-CMTB and with the N-thiazolylamide moiety in compounds such as 14, by a combination of mutagenesis and structural approaches. Modeling suggested a hydrogen-bonding interaction of the N-thiazolylamide carbonyl in 4-CMTB with Leu173 in ECL2 of hFFA2 (Lee et al. 2008), but this is with a backbone nitrogen making it difficult to confirm through mutagenesis (Smith et al. 2011). Two mutations which reduce potency of compounds 1 and 2 without affecting C3 have been described (Q148EECL2 and V179A5,38), but whether these also affect 4-CMTB activity has not yet been published (Hudson et al. 2013a). Homology models of FFA2 (Lee et al. 2008; Smith et al. 2011) were based on the β2-adrenergic receptor and recent publication of the FFA1 structure in complex with TAK-875 (Srivastava et al. 2014) should permit more reliable models of FFA2 to be developed.
In general, the acid N-thiazolylamides show activity across human and rodent FFA2 orthologs, similar to 4-CMTB and in contrast to CATPB and N-CBT which are hFFA2-selective. Previously, compounds 1 and 2 were shown to activate hFFA2, rFFA2, and mFFA2 in three different assay formats (though 2 had somewhat reduced potency at mFFA2 and rFFA2) (Hudson et al. 2013a). Here, we have shown similar potencies of 14 at hFFA2 and rFFA2 in a common assay system (yeast). We have not tested 14 or other compounds at recombinant mFFA2, however, we show that 14 stimulates GLP-1 release by STC-1 cells, consistent with agonism of mFFA2. Also, we show that inhibition of explant lipolysis by both 4-CMTB and 14 (75 μmol/L) was mFFA2 dependent, though in this study we were not able to test ligands over a concentration range to assess relative potency due to limited availability of the mouse adipose explants. Hoyveda et al. (2010) showed that in mice, 14 (50 mg/kg) caused significant inhibition of lipolysis in vivo, and inhibited isoprenaline-stimulated lipolysis in isolated rat adipocytes (30 μmol/L). Hence, we corroborate here that acid N-thiazolylamides in general can inhibit lipolysis in rats and mice, and both in vitro and in vivo, via activation of FFA2 (Hoyveda et al. 2010).
Acid N-thiazolylamides exhibit system-dependent efficacy, illustrated by the switch from positive efficacy of 9, 101, and 105 in mammalian cells assays to negative efficacy in yeast. This switch may result from agonist bias, since our recombinant hFFA2 assays utilize different G-proteins, albeit all related to Gi (rat Go for [35S]-GTPγS incorporation, endogenous Gi proteins in U2OS cells for cAMP experiments, and Gpa1-Gi3 chimera in yeast). However, further experiments will be required to fully investigate this. Another potential difference is hFFA2 expression level. For [35S]-GTPγS incorporation and cAMP experiments, the same baculovirus transfection reagent was used but at different multiplicities of infection (MOI = 25 for [35S]-GTPγS incorporation and MOI = 14 for cAMP, consistent with the higher potency and efficacy in [35S]-GTPγS incorporation; Table1). Relative receptor levels on hFFA2-expressing yeast cannot presently be assessed due to the lack of radioligand tools. However, the lower potency of 14 and the switch from positive to negative efficacy of 9, 101, and 105 in yeast would be consistent with lower hFFA2 expression, as might result from inefficient production of the foreign protein. The switch from positive to negative efficacy of 9, 101, and 105 in yeast was also observed for rFFA2. We also observed system-dependent efficacy at endogenously expressed hFFA2, exemplified by compound 14 which induces calcium mobilization in human neutrophils but which we could not detect to inhibit lipolysis in primary human adipocytes (up to 100 μmol/L). This is in spite of the presence of functional hFFA2 in these cells, shown by N-CBT- and PTX-sensitive inhibition of lipolysis by 4-CMTB, and contrasts with the ability of 14 to inhibit lipolysis in rodent. If confirmed, the finding would indicate that adipocyte hFFA2 differs from the recombinant hFFA2 as well as rodent adipocyte FFA2. Differences between rodent and hFFA2 in adipose are precedented: agonists stimulate mouse adipocyte differentiation but not human (Dewulf et al. 2013). In vitro differentiated adipocytes from immortalized cell-lines SW872 and 3T3L1 (Lee et al. 2008; Hudson et al. 2013a) have commonly been used to understand whether agonist activity translates from recombinant assays to adipose FFA2, but our data suggest these may not present the same FFA2 pharmacology as primary human adipocytes. At present, full interpretation of the lack of activity of 14 to inhibit lipolysis of human adipocytes is not possible, but the data presented in this study illustrate potential influencing factors. First, FFA2 levels in the different assays and tissues should be compared, and there is preliminary evidence that radiolabeled CATBP may enable this (Ulven 2012). Second, the G protein mediating inhibition of lipolysis in human adipocytes needs to be defined, because the potentially bitopic nature of acid N-thiazolylamide ligands may cause a more constrained and G-protein selective conformation of FFA2 than C3. Finally, human adipocytes are complex cells expressing numerous additional factors that might confound extrapolation from isolated FFA2 in recombinant assays. FFA3 may also be expressed in adipose (Brown et al. 2003), and although the ligands tested herein are largely or wholly selective for FFA2 over FFA3, there is increasing evidence for GPCR cross-talk. Therefore, the possibility of cross-talk in human adipose between FFA2 and FFA3, or other GPCR, should be investigated.
One particular challenge in defining the physiological role of FFA2 has been the lack of a selective antagonist with affinity at rodent FFA2. CATPB and N-CBT are chemically similar and both are inactive at rodent FFA2 (Hudson et al. 2012) suggesting a shared mode of binding, distinct from that of the acid N-thiazolylamides which have affinity for both human and rodent orthologs. A recently described series of FFA2 antagonists structurally divergent to N-CBT and CATPB also lacks activity at rodent FFA2 (Pizzonero et al. 2014). Our data showing dissociation of efficacy from affinity within the acid N-thiazolylamide series suggest that identification of an example within this series having FFA2 affinity but lacking efficacy (at least in primary tissues) might be achieved, offering the prospect of a pan-species FFA2 antagonist to facilitate further validation of this target in metabolic and inflammatory conditions.
Acknowledgments
We thank Matilde Caivano and Sandrine Martin for practical assistance, David Hall for advice on use of the operational models, and Paul Martres, Ashley Barnes, and Martyn Foster for their scientific advice and support of these studies (all from GlaxoSmithKline).
Glossary
- 3AT
3-aminotriazole
- 4-CMTB
4-chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- C3
propionate
- CATPB
(S)-3-(2-(3-chlorophenyl)acetamido)-4-(4-(trifluoromethyl)phenyl) butanoic acid
- Compound 101
cis-2-((4-(2-chlorophenyl)thiazol-2-yl)(methyl)carbamoyl)cyclohexaneccarboxylic acid
- Compound 105
(R)-4-((4-(2-chlorophenyl)thiazol-2-yl)(methyl)amino)-3-(cyclopentymethyl)-4-oxobutanoic acid
- Compound 14
(R)-3-benzyl-4-((4-(2-chlorophenyl)thiazol-2-yl)(methyl)amino)-4-oxobutanoic acid
- Compound 9
(S)-4-(4-(2-chlorophenyl)thiazol-2-ylamino)-4-oxo-3-phenylbutanoic acid
- FFAR
free fatty acid receptor
- GLP-1
glucagon-like peptide-1
- GPCR
G protein-coupled receptor
- LCFAs
long-chain fatty acids
- MOI
multiplicity of infection
- N-CBT
N-(4-Chlorobenzoyl)-l-tryptophan
- PTX
pertussis toxin
- SCFA
short-chain fatty acid
- SPA
scintillation proximity assay
- U2OS
U2 osteosarcoma
Authorship Contributions
Participated in research design: Brown, Gower, Dean, Faucher, Gangar and Dowell. Conducted experiments: Brown, Tsoulou, Ward, Gower, Bhudia, and Chowdhury. Contributed new reagents or analytic tools: Ward, Bhudia, Chowdhury, and Faucher. Performed data analysis: Brown, Tsoulou, Ward, Gower, Bhudia, Chowdhury and Gangar. Wrote or contributed to the writing of the manuscript: Brown, Gangar and Dowell.
Disclosures
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Operational modelling of yeast hFFA2 agonist and inverse agonist concentration–response data.
Figure S2. Operational modelling of yeast hFFA2 agonist and inverse agonist concentration–response (data from Fig. 2E–G).
Figure S3. Protean agonist properties of acid N-thiazolylamide FFA2 ligands.
Table S1.N-CBT (N-(4-Chlorobenzoyl)-l-tryptophan) is an antagonist selective for human FFA2 with no activity at rat FFA2.
References
- Bertheleme N, Singh S, Dowell SJ, Hubbard J, Byrne B. Loss of constitutive activity is correlated with increased thermostability of the human adenosine A2A receptor. Br J Pharmacol. 2013;169:988–998. doi: 10.1111/bph.12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill A. 1990. Method and means for inducing, resp., preventing constriction of the pupil in the eye. Patent WO1990011773A1.
- Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278:11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Dyos SL, Whiteway MS, White JHM, Watson M-A, Marzioch M, et al. Functional coupling of mammalian receptors to the yeast mating pathway using novel yeast/mammalian G protein α-subunit chimeras. Yeast. 2000;16:11–22. doi: 10.1002/(SICI)1097-0061(20000115)16:1<11::AID-YEA502>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278:11312–11319. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Daniels DA, Kassim M, Brown S, Haslam CP, Terrell VR, et al. Pharmacology of GPR55 in yeast and identification of GSK494581A as a mixed-activity glycine transporter subtype 1 inhibitor and GPR55 agonist. J Pharmacol Exp Ther. 2011;337:236–246. doi: 10.1124/jpet.110.172650. [DOI] [PubMed] [Google Scholar]
- Covington DK, Briscoe CA, Brown AJ, Jayawickreme CK. The G-protein-coupled receptor 40 family (GPR40-GPR43) and its role in nutrient sensing. Biochem Soc Trans. 2006;34:770–773. doi: 10.1042/BST0340770. [DOI] [PubMed] [Google Scholar]
- Dewulf EM, Ge Q, Bindels LB, Sohet FM, Cani PD, Brichard SM, et al. Evaluation of the relationship between GPR43 and adiposity in human. Nutr Metab (Lond) 2013;10:11. doi: 10.1186/1743-7075-10-11. doi: 10.1186/1743-7075-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11:90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- Hoyveda H, Brantis CE, Dutheuil G, Zoute L, Schils D, Bernard J. 2010. Compounds, pharmaceutical composition and methods for use in treating metabolic disorders. Patent WO2010066682.
- Hudson BD, Tikhonova IG, Pandey SK, Ulven T, Milligan G. Extracellular ionic locks determine variation in constitutive activity and ligand potency between species orthologs of the free fatty acid receptors FFA2 and FFA3. J Biol Chem. 2012;287:41195–41209. doi: 10.1074/jbc.M112.396259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson BD, Due-Hansen ME, Christiansen E, Hansen AM, Mackenzie AE, Murdoch H, et al. Defining the molecular basis for the first potent and selective orthosteric agonists of the FFA2 free fatty acid receptor. J Biol Chem. 2013a;288:17296–17312. doi: 10.1074/jbc.M113.455337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson BD, Ulven T, Milligan G. The therapeutic potential of allosteric ligands for free fatty acid sensitive GPCRs. Curr Top Med Chem. 2013b;13:14–25. doi: 10.2174/1568026611313010004. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. A pharmacology primer: theory, applications and methods. Burlington, MA: Elsevier; 2009. Allosteric drug antagonism; pp. 129–146. . ???. ????, ed. [Google Scholar]
- Lane JR, Sexton PM, Christopoulos A. Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol Sci. 2013;34:59–66. doi: 10.1016/j.tips.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Lane JR, Donthamsetti P, Shonberg J, Draper-Joyce CJ, Dentry S, Michino M, et al. A new mechanism of allostery in a G protein-coupled receptor dimer. Nat Chem Biol. 2014;10:745–752. doi: 10.1038/nchembio.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Schwandner R, Swaminath G, Weiszmann J, Cardozo M, Greenberg J, et al. Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol Pharmacol. 2008;74:1599–1609. doi: 10.1124/mol.108.049536. [DOI] [PubMed] [Google Scholar]
- Lin DC, Guo Q, Luo J, Zhang J, Nguyen K, Chen M, et al. Identification and pharmacological characterization of multiple allosteric binding sites on the free fatty acid 1 receptor. Mol Pharmacol. 2012;82:843–859. doi: 10.1124/mol.112.079640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzonero M, Dupont S, Babel M, Beaumont S, Bienvenu N, Blanque R, et al. Discovery and optimization of an azetidine chemical series as a free fatty acid receptor 2 (FFA2) antagonist: from hit to clinic. J Med Chem. 2014;57:10044–10057. doi: 10.1021/jm5012885. [DOI] [PubMed] [Google Scholar]
- Sina C, Gavrilova O, Forster M, Till A, Derer S, Hildebrand F, et al. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J Immunol. 2009;183:7514–7522. doi: 10.4049/jimmunol.0900063. [DOI] [PubMed] [Google Scholar]
- Slack RJ, Hall DA. Development of operational models of receptor activation including constitutive receptor activity and their use to determine the efficacy of the chemokine CCL17 at the CC chemokine receptor CCR4. Br J Pharmacol. 2012;166:1774–1792. doi: 10.1111/j.1476-5381.2012.01901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NJ, Ward RJ, Stoddart LA, Hudson BD, Kostenis E, Ulven T, et al. Extracellular loop 2 of the free fatty acid receptor 2 mediates allosterism of a phenylacetamide ago-allosteric modulator. Mol Pharmacol. 2011;80:163–173. doi: 10.1124/mol.110.070789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern C, Cook JM, Neetoo-Isseljee Z, Taylor DL, Kettleborough CA, Merritt A, et al. Screening beta-arrestin recruitment for the identification of natural ligands for orphan G-protein-coupled receptors. J Biomol Screen. 2013;18:599–609. doi: 10.1177/1087057113475480. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Yano J, Hirozane Y, Kefala G, Gruswitz F, Snell G, et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature. 2014;513:124–127. doi: 10.1038/nature13494. [DOI] [PubMed] [Google Scholar]
- Stoddart LA, Smith NJ, Jenkins L, Brown AJ, Milligan G. Conserved polar residues in transmembrane domains V, VI, and VII of free fatty acid receptor 2 and free fatty acid receptor 3 are required for the binding and function of short chain fatty acids. J Biol Chem. 2008;283:32913–32924. doi: 10.1074/jbc.M805601200. [DOI] [PubMed] [Google Scholar]
- Tang C, Ahmed K, Gille A, Lu S, Grone HJ, Tunaru S, et al. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat Med. 2015;21:173–177. doi: 10.1038/nm.3779. [DOI] [PubMed] [Google Scholar]
- Ulven T. Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front Endocrinol (Lausanne) 2012;3:111. doi: 10.3389/fendo.2012.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinolo MA, Ferguson GJ, Kulkarni S, Damoulakis G, Anderson K, Bohlooly Y, et al. SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS One. 2011;6:e21205. doi: 10.1371/journal.pone.0021205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wu X, Simonavicius N, Tian H, Ling L. Medium-chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J Biol Chem. 2006;281:34457–34464. doi: 10.1074/jbc.M608019200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Jiao X, Kayser F, Liu J, Wang Z, Wanska M, et al. The first synthetic agonists of FFA2: discovery and SAR of phenylacetamides as allosteric modulators. Bioorg Med Chem Lett. 2010;20:493–498. doi: 10.1016/j.bmcl.2009.11.112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Operational modelling of yeast hFFA2 agonist and inverse agonist concentration–response data.
Figure S2. Operational modelling of yeast hFFA2 agonist and inverse agonist concentration–response (data from Fig. 2E–G).
Figure S3. Protean agonist properties of acid N-thiazolylamide FFA2 ligands.
Table S1.N-CBT (N-(4-Chlorobenzoyl)-l-tryptophan) is an antagonist selective for human FFA2 with no activity at rat FFA2.