

Abstract
G protein-coupled receptor desensitization is typically mediated by receptor phosphorylation by G protein receptor kinase (GRK) and subsequent arrestin binding; morphine, however, was previously found to activate a c-Jun N-Terminal Kinase (JNK)-dependent, GRK/arrestin-independent pathway to produce mu opioid receptor (MOR) inactivation in spinally-mediated, acute anti-nociceptive responses Melief, et. al., 2010 [1]. In the current study, we determined that JNK2 was also required for centrally-mediated analgesic tolerance to morphine using the hotplate assay. We compared JNK activation by morphine and fentanyl in JNK1−/−, JNK2−/−, JNK3−/−, and GRK3−/− mice and found that both compounds specifically activate JNK2 in vivo; however, fentanyl activation of JNK2 was GRK3-dependent, whereas morphine activation of JNK2 was GRK3-independent. In MOR-GFP expressing HEK293 cells, treatment with either arrestin siRNA, the Src family kinase inhibitor PP2, or the protein kinase C (PKC) inhibitor Gö6976 indicated that morphine activated JNK2 through an arrestin-independent Src- and PKC-dependent mechanism, whereas fentanyl activated JNK2 through a Src- GRK3/arrestin2-dependent and PKC-independent mechanism. This study resolves distinct ligand-directed mechanisms of JNK activation by mu opioid agonists and understanding ligand-directed signaling at MOR may improve opioid therapeutics.
Keywords: Arrestin, JNK, Tolerance, Opioid, Morphine, Fentanyl
Graphical abstract
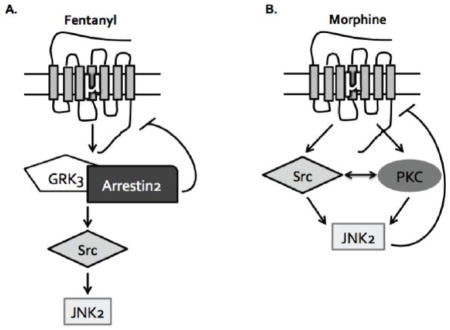
1. Introduction
The concept of ligand-directed signaling, also known as biased agonism or functional selectivity, has become increasingly important for understanding the different signaling events following G protein-coupled receptor (GPCR) ligand binding [2]. While the canonical mechanism of GPCR desensitization is GRK/arrestin mediated [3–8], alternative mechanisms of receptor desensitization have been documented. For instance, the development of spinally-mediated acute analgesic tolerance following treatment with the MOR agonist fentanyl requires the GRK3/arrestin pathway, whereas tolerance following administration of the MOR agonist morphine requires a distinctly different mechanism involving JNK2 [1]. This JNK-dependent mechanism of receptor inactivation has also been observed at the kappa opioid receptor (KOR), and the durations of action of a broad range of KOR antagonists positively correlates with the ability of the antagonist to activate JNK in mouse spinal cord [9, 10]. While JNK has been shown to be directly involved in morphine-induced MOR desensitization in the dorsal root ganglion [11], others have reported that JNK is not required for morphine-induced MOR desensitization in the locus ceruleus [12]. Thus MOR regulation mechanisms may be neuronal pathway selective, and further characterization of the role of JNK in different pain circuits and brain regions is required.
An additional issue is that although morphine and fentanyl desensitize MOR by different mechanisms, both compounds activate JNK [1]. How these ligands activate JNK and why one inactivates MOR, but the other does not, is not known. JNK activation requires phosphorylation of a threonine and tyrosine residue in the activation loop and the canonical mitogen-activated protein kinase kinases (MAPKK) MKK4 and MKK7 have been shown to phosphorylate these residues directly [13, 14]. Additional kinases have been implicated in opioid receptor mediated JNK activation upstream of the MAPKKs, including PKC and Src [1, 15–17]. In some instances, arrestin has been shown to act as a scaffold for JNK activation recruiting MKK4 and MKK7, in addition to ASK1, in close proximity with JNK [18–20]. It has also been demonstrated that arrestin can regulate the cellular localization of extracellular signal-regulated kinases (ERK) and other signaling molecules [21–23]. Based on our in vivo tolerance studies, we hypothesized that JNK activation by morphine and fentanyl required distinct upstream mechanisms, which can help explain the differential behavioral effect. A better understanding of the ligand-directed mechanisms that contribute to MOR mediated JNK activation and receptor desensitization will be necessary for the development of improved therapeutics that avoid tolerance or minimize arrestin-dependent responses. Additionally, this JNK mediated mechanism of receptor regulation may be more generally involved in other GPCR systems.
In this study, we examined the role of JNK in centrally mediated pain circuits and dissected the arrestin-dependent and arrestin-independent mechanisms of JNK activation by MOR in mouse spinal cord and MOR-GFP expressing HEK293 cells. Our study also suggests a role for additional kinases in JNK activation, including PKC and Src, which have been implicated in MOR desensitization following morphine [24, 25] and in [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO) mediated JNK activation [15], respectively. These results further elucidate mechanisms of arrestin-dependent and arrestin-independent JNK activation in an effort to better understand ligand-directed mechanisms of MOR desensitization.
2. Materials and Methods
2.1 Reagents
Morphine sulfate was provided by the NIDA Drug Supply (Bethesda, MD). Fentanyl citrate was purchased from Sigma (St Louis, MO). PP2 and Gö6976 were purchased from Calbiochem (Billerica, MA) and dissolved in DMSO. All compounds were diluted in 0.9% NaCl for animal studies and H20 for cell based assays.
2.2 Animals
Male C57Bl/6 wild type (WT) mice or GRK3−/−, JNK1−/−, and JNK2−/− mice (20–30g) on a C57Bl/6 background were generated and genotyped as previously described [1]. JNK3−/− mice were generated by Dr. RA Flavell (Yale University) [26] and provided by Dr. Zhengui Xia (UW Toxicology/Environmental Health). Mice were group housed and kept on a 12-h light/dark cycle with food and water available ad libitum. Animal procedures were approved by the Animal Care and Use Committee of the University of Washington and conform to the guidelines of the National Institutes of Health on the care and use of animals.
2.3 Analgesia
Nociceptive responses were measured using a 55°C hotplate (ICTC Life Sciences, model 39/336T) as previously described [27]. To make animals tolerant, mice were injected with morphine (20mg/kg, s.c.) or saline once per day in the morning for 3 days (Fig. 1A). Baseline response latencies were assessed 4 hr post injection on Day 1 and Day 3, then again 30 min following a second dose of morphine (10mg/kg, s.c.) on that day. Mice were removed from the hot plate after a nociceptive response (hindpaw withdrawal or shake) or before 30 sec to avoid tissue damage. The investigator was blinded to pretreatment and genotype
FIGURE 1.

JNK2 is Required for Hotplate Analgesic Tolerance to Morphine. A, Animals were treated with 20mg/kg morphine or saline as a control for 3 days to induce tolerance. On Day 1 and Day 3 all mice were challenged with 10mg/kg morphine 4 hr after their morning dose and tested on a 55°C hot plate pre and post 10mg/kg injection to assess analgesia, which was measured as the latency for hind paw withdrawal/flicks. Animals did not show robust analgesic tolerance on Day 1 (data not shown). B, WT animals pretreated with saline show a robust analgesic response to morphine (10mg/kg) (as indicated by an increased latency to withdraw) on Day 3 whereas animals pretreated with morphine (20mg/kg) do not. JNK2−/− animals show robust analgesia, as indicated by an increased latency to withdraw, to morphine regardless of pretreatment with saline or morphine, demonstrating that JNK2 is required for morphine analgesic tolerance on the hotplate. Data presented were calculated as the response latency 30 min post drug administration minus baseline withdrawal latency resulting in the post-pre value presented. Baseline responses did not vary across groups (data summarized in text). n=10–18; Data analyzed by two-way ANOVA with Bonferroni posttest.
2.4 Spinal Cord Dissection and Immunoblot
Mice were injected with saline, morphine (10mg/kg s.c.), or fentanyl (0.3mg/kg s.c.) at a volume of 10 mL/kg. Mice were decapitated 30–60 min post injection, and the lumbar region of the spinal cords were removed and homogenized in lysis buffer (50mM Tris-HCl, 300mM NaCl, 1mM EDTA, 1mM Na3VO4, 1 mM NaF, 10% Glycerol, 1% Triton-X 100) with phosphatase inhibitor cocktail (1:100, Calbiochem, Billerica, MA) and protease inhibitor cocktail (1:100, Calbiochem, Billerica, MA). Lysates were spun at 14,000×g for 30 min at 4°C and protein concentration of the supernatant was determined by BCA assay (Pierce). Lysates (15–20μg of protein) were heated at 100°C for 5 min and resolved on a 10% Bis-Tris polyacrylamide gel (Invitrogen Life Technologies, Grand City, NY) at 120V for 2 hr. Blots were transferred to nitrocellulose membranes (Whatman, Middlesex, UK) for 1.5–2 hr at 30V. Membranes were blocked with 5% BSA-TBST for 1 hr at 23°C and blotted for total-JNK (Cell Signaling Technology, Danvers, MA; 1:1000) or for phospho-JNK (Cell Signaling, Danvers, MA; 1:1000) and actin (Abcam, Cambridge, MA; 1:5000) immunoreactivities (-IR) in 5% BSA-TBST overnight at 4°C. Membranes were washed 3×5 min in TBST and incubated in IRdye secondary (Li-Cor Biosciences, Lincoln, NE; 1:10,000) in 1:1 5% milk-TBST and Odyssey buffer (Li-Cor Biosciences, Lincoln, NE) for 1 hr at 23°C. Membranes were washed for 3×5 min in TBST and scanned on the Odyssey Infrared Imaging System (Li-Cor Biosciences; Lincoln, NE). Relative fluorescent band intensity was measured using the Odyssey software and expressed as a ratio of total- or phospho-JNK-IR to actin-IR band intensity and then expressed as change over vehicle treatment. Both the 46 and 54kDa band were included for total- and phospho-JNK analysis. All westerns were run and analyzed blinded to treatment and genotype.
2.5 siRNA transfection
HEK293 cells stably expressing MOR-GFP were transiently transfected with siRNA against arrestin-2, arrestin-3, or a scrambled siRNA control (Dharmacon Research, Pittsburgh, PA, 50nM final concentration; 48 hr), by using Lipofectamine RNAiMAX (Life Technologies, Grand Island, NY) according to manufacturer’s recommendations. To assess expression knockdown, cells were homogenized in lysis buffer (50mM Tris-HCl, 300mM NaCl, 1mM EDTA, 1mM Na3VO4, 1mM NaF, 10% Glycerol, 1% Triton-X 100) with phosphatase inhibitor cocktail (1:100, Calbiochem, Billerica, MA) and protease inhibitor cocktail (1:100, Calbiochem, Billerica, MA). Lysates were spun at 14,000×g for 30 min at 4°C and protein concentration of the supernatant was determined by BCA assay (Pierce, Rockford, IL). Lysates (10μg of protein) were heated at 100°C for 5 min and resolved on a 10% Bis-Tris polyacrylamide gel as described above in section 2.4, then blotted for arrestin-2-IR or arrestin-3-IR (antibody provided by Dr. Jeffrey Benovic, Thomas Jefferson University, Philadelphia, PA) (1:3000) and actin (Abcam, Cambridge, MA; 1:5000) in 5% BSA-TBST overnight at 4°C. Relative fluorescent band intensity was measured using the Odyssey software and expressed as arrestin-2-IR or arrestin-3-IR intensity over actin-IR intensity and then expressed as change over scrambled siRNA treatment.
2.6 HEK293 Cell Treatments, Protein Extraction
Cells were incubated in serum-free media overnight and subsequently treated with opioid ligands and inhibitors as indicated. Fentanyl and morphine were tested at 10μM for 30 min. To inhibit Src family kinases, MOR-GFP HEK293 cells were treated with 5μM PP2 [28] 30 min before opioid administration. To inhibit Ca2+ sensitive PKC, MOR-GFP HEK293 cells were treated with 5μM Go6976 [29] 30 min before opioid; an equal volume of DMSO was employed as vehicle for PP2 and Gö6976.
2.7 HEK293 Immunoblots
To determine JNK phosphorylation levels in MOR-GFP expressing HEK293 cells, 40μg protein of each lysate was loaded onto 10% Bis-Tris polyacrylamide gel (Life Technologies, Grand Island, NY) and western blot was performed as described above in section 2.4 and blotted for phospho-JNK-IR (Cell Signaling Technology, Danvers, MA; 1:1000) and actin-IR (Abcam, Cambridge, MA; 1:5000). To determine Src phosphorylation levels in MOR-GFP expressing HEK293 cells, 30μg of each lysate was resolved then blotted for phospho-Tyr416-Src-IR (Cell Signaling Technology, Danvers, MA; 1:1000) and actin-IR (Abcam, Cambridge, MA; 1:5000).
2.8 Analysis
Analysis of Variance, Student’s t-tests, or one-sample t-tests were used to assess statistical significance of the data (α=0.05). Outliers were distinguished by the Grubb’s Test (α=0.05). In the figures shown, statistical significance is indicated by: * p<0.05, ** p<0.01, and *** p<0.001).
3. Results
3.1 JNK2−/− Mice Do Not Develop Centrally Regulated Analgesic Tolerance to Morphine
To assess whether JNK2 mechanisms contribute to acute analgesic tolerance to mu opioid agonists in the nociceptive circuits controlling hotplate responses as they do in the tail flick analgesia assay (Melief et al., 2010), wild-type (WT) and JNK2−/− (knockout) mice were treated with one 20mg/kg morphine injection every morning for three days or saline as a control (Fig. 1A). Tail flick analgesia is spinally mediated [30, 31], whereas the opioid-sensitive nociception controlling the hotplate response is centrally regulated in part by the periaqueductal gray and rostral ventromedial medulla [31–34]. On Day 1 and Day 3, nociceptive responses were assessed on the hotplate before and 30 min after an injection of 10mg/kg morphine, which was given 4 hr after a pretreatment of 20mg/kg morphine or saline (Fig. 1A). Animals did not show acute analgesic tolerance to morphine on Day 1 (data not shown) whereas on Day 3, WT animals pretreated with morphine (Fig. 1A) showed a significantly shorter latency to withdraw from the hotplate after a test dose of morphine (10mg/kg) compared to mice pretreated with saline, indicative of analgesic tolerance (Fig. 1B; p<0.01). This robust analgesic tolerance to morphine was not evident in JNK2−/− mice, which had equivalent latencies to withdraw regardless of pretreatment (Fig. 1B; p>0.05). Both genotypes showed equivalent baseline responses prior to the test dose of morphine (WT Sal=5.47 (±0.595) sec, WT Morphine=5.55 (±0.489) sec, JNK2−/− Sal=6.61 (±0.770) sec, JNK2−/− Morphine=7.27 (±0.812) sec). These results mirror previous results using the tail withdrawal assay [1], suggesting that similar JNK2 mechanisms regulate morphine tolerance in both spinal and central pain circuits. However, multiple morphine injections were required over the course of three days to observe the development of tolerance (Fig. 1) on the hotplate, suggesting that the mechanisms regulating centrally regulated analgesia differ temporally from those regulating spinal analgesia.
3.2 JNK2 is Specifically Activated by Morphine and Fentanyl in vivo
Both morphine and fentanyl increase phospho-JNK-immunoreactivity (phospho-JNK-IR), but produce acute analgesic tolerance by different mechanisms [1]. To understand the basis for this difference, we assessed the specific isoforms of JNK activated by morphine and fentanyl. JNK1−/−, JNK2−/−, and JNK3−/− male C57Bl/6 mice were treated with saline, morphine (10mg/kg s.c.), or fentanyl (0.3mg/kg s.c.) for 30 min followed by decapitation and dissection of spinal cords. This time point corresponds to the peak of the opioid analgesic response [1]. Although spinal cord contains a diversity of cell types that could respond to treatment either directly or indirectly, DAMGO stimulated [35S]GTPγS binding to spinal cord membranes was reduced by morphine pretreatment in wild type, but not mice pretreated with JNK inhibitor [1], suggesting that morphine activation of JNK does have direct effects on MOR signaling in spinal cord.
JNK1−/−, and JNK3−/− animals showed robust phospho-JNK-IR following morphine treatment (Fig. 2A,B; p<0.05 JNK1−/−, p<0.01 JNK3−/−). In contrast phospho-JNK-IR was not increased in JNK2−/− mice, indicating that the JNK2 isoform was specifically activated by morphine (Fig. 2A,B; p>0.05 JNK2−/−). Similarly, both JNK1−/− and JNK3−/− mice showed significant increases in phospho-JNK-IR in response to fentanyl, but this was absent in JNK2−/− mice (Fig. 2A,B; p<0.01 JNK1−/−, p>0.05 JNK2−/−, p<0.01 JNK3−/−). Together these data indicate that both morphine and fentanyl specifically activate spinal JNK2, but not JNK1 or JNK3.
FIGURE 2.

Morphine and Fentanyl Mediated Phospho-JNK Increase in the Spinal Cord is Selectively Abolished in JNK2−/− Mice, but not Significantly Affected by JNK1 or JNK3 Gene Knockout. A, Animals were treated with saline, morphine (10mg/kg, s.c.), or fentanyl (0.3mg/kg, s.c.) and their spinal cords were harvested 30 min post-treatment for phospho-JNK-IR analysis. Both MOR agonists increase phospho-JNK-IR in JNK1−/− and JNK3−/− mice but failed to increase phospho-JNK-IR over baseline in JNK2−/− mice. B, Representative images are shown for each phospho-JNK data set. C, Total-JNK-IR was also not altered by morphine or fentanyl in JNK2−/− mice, indicating that the effects on phospho-JNK were not a result of alterations in total protein levels. D, Representative images are shown for the total-JNK data set. n=3–10; Data analyzed by one-sample t-test; statistical outliers were determined by the Grubb’s Test with significance set to α=0.05.
Because phospho-JNK-IR was not increased in JNK2−/− mice, we confirmed that these mice did not show a change in total-JNK-IR following morphine or fentanyl (Fig. 2C,D; p>0.05), indicating that the lack of JNK activation in these mice is not due to alterations in total protein levels.
3.3 GRK3 is Required for Fentanyl, but not Morphine, Activation of JNK in vivo
Prior studies have demonstrated that JNK can be activated by an arrestin scaffold [19, 20]. To assess the role of arrestin in opioid-induced JNK activation, we measured the effect of GRK3 gene deletion (GRK3−/−) on phospho-JNK-IR in vivo. GRK3+/+ (wildtype littermates) and GRK3−/− male C57Bl/6 mice were treated with saline, morphine (10mg/kg s.c.), or fentanyl (0.3mg/kg s.c.), then spinal cords were harvested 60 min later. GRK3+/+ animals showed significant increases in phospho-JNK-IR following either morphine (p<0.01) or fentanyl (p<0.01) (Fig. 3A). GRK3−/− animals treated with morphine also showed an increase in phospho-JNK-IR over baseline (p<0.0001), whereas fentanyl did not change phospho-JNK-IR in GRK3−/− mice (p>0.05) (Fig. 3A,B). These results indicate that fentanyl and morphine activate JNK2 through distinct signaling cascades in vivo.
FIGURE 3.

Fentanyl, but not Morphine, Mediated Phospho-JNK Increase in the Spinal Cord is Selectively Abolished in GRK3−/− Mice. A, JNK activation following fentanyl administration requires GRK3 whereas morphine activation of JNK does not. Animals were treated with saline, morphine (10mg/kg, s.c.), or fentanyl (0.3mg/kg, s.c.) and their spinal cords were harvested 60 min post-treatment for phospho-JNK-IR analysis. Both MOR agonists activate JNK in GRK3+/+ mice. However, in GRK3−/− mice, morphine increased phospho-JNK-IR over baseline but fentanyl failed to do so. B, Representative images are shown for each data set. C, Total-JNK-IR was also not altered by morphine or fentanyl in GRK3−/− mice, indicating that the effects on phospho-JNK were not a result of alterations in total protein levels. D, Representative images are shown for the total-JNK data set. n=3–7; Data analyzed by one-sample t-test; statistical outliers were determined by the Grubb’s Test with significance set to α=0.05.
Additionally, GRK3−/− mice did not show a change in total-JNK-IR following morphine or fentanyl (Fig. 3C,D; p>0.05), indicating that the effects on phospho-JNK were not caused by alterations in total protein levels.
3.4 Arrestin-2 is Required for Fentanyl, but not Morphine, Activation of JNK
While the requirement of GRK3 for fentanyl activation of JNK in vivo implied an arrestin-dependent mechanism of JNK activation, to directly establish the role of arrestin in fentanyl activation of JNK, MOR-GFP expressing HEK293 cells were transfected with 50nM siRNA against either arrestin-2, arrestin-3, or a scrambled siRNA control for 48 hr. Cells were then lysed and analyzed for arrestin-2 and arrestin-3 expression using western blot. Arrestin-2 siRNA resulted in a 49 ± 5.6% reduction (p<0.01) in arrestin-2-IR without significantly affecting arrestin-3-IR compared to scrambled control (p>0.05) (Fig. 4A). Similarly, arrestin-3 siRNA reduced arrestin-3-IR by 54 ± 7.8% (p<0.01) without altering arrestin-2-IR compared to scrambled control (p>0.05) (Fig. 4B).
FIGURE 4.

Fentanyl Mediated Phospho-JNK Increase is Abolished Following Treatment with Arrestin-2 siRNA. MOR-GFP expressing HEK293 cells were transfected with siRNA against arrestin-2, arrestin-3, or a scrambled siRNA control. Cells were subsequently lysed for arrestin-2 and -3-IR (A) or treated with vehicle (H2O), 10μM morphine, or 10μM fentanyl for 30 min before lysis and analysis of phospho-JNK-IR for each sample, which was normalized as a percent of scrambled siRNA vehicle treated cells (C,D). A, Arrestin-2 siRNA reduced arrestin-2-IR by 49 ± 5.6%. Arrestin-3 siRNA did not significantly reduce arrestin-2-IR compared to a scrambled siRNA control (ctrl). B, Similarly, arrestin-2 siRNA did not significantly alter arrestin-3-IR compared to a scrambled siRNA control, whereas arrestin-3 siRNA reduced arrestin-3-IR by 54 ± 7.8%. C, Morphine treatment increased phospho-JNK-IR regardless of siRNA treatment, indicating that arrestin is not involved in morphine activation of JNK. Fentanyl, however, increased phospho-JNK-IR in control siRNA (ctrl) and arrestin-3 siRNA treated cells but this effect was abolished in arrestin-2 siRNA treated cells, indicating that arrestin-2 is required for fentanyl activation of JNK. D, Representative westerns are shown for each JNK dataset. n=10–13; Data analyzed by one-sample t-test (ctrl only) and student’s t-test with Welch’s correction when necessary.
To assess the role of arrestin in JNK activation, HEK293 cells were treated with vehicle, 10μM morphine, or 10μM fentanyl for 30 min, then lysed and processed for phospho-JNK-IR. Morphine treatment significantly increased phospho-JNK-IR, and the increase was not affected by either arrestin-2 or arrestin-3 siRNA (Fig. 4C,D). Fentanyl treatment also increased phospho-JNK-IR in scrambled siRNA and arrestin-3 siRNA treated cells (Fig. 4C,D). In contrast, fentanyl failed to increase phospho-JNK-IR in arrestin-2 siRNA treated cells (Fig. 4C,D; p>0.05). These results suggest that fentanyl activation of JNK required a GRK3/arrestin-2 dependent mechanism whereas morphine activation of JNK was arrestin-independent. Similar to the in vivo experiments previously described in section 3.2-3, morphine and fentanyl did not induce changes in total-JNK-IR in HEK293 cells (data not shown).
3.5 Src is Required for Fentanyl and Morphine Activation of JNK
In addition to an arrestin-dependent mechanism, Src kinase has also been implicated in JNK activation by opioid receptors [15–17]. To investigate the role of Src kinases, MOR-GFP expressing HEK293 cells were treated with vehicle, 10μM morphine, or 10μM fentanyl for 30 min. Both opioids significantly increased phospho-Src-IR (Fig. 5A,D). To determine the role of Src in JNK activation, MOR-GFP expressing HEK293 cells were pretreated for 30 min with 5μM PP2 [35–37], which inhibits Src family kinases [28], or vehicle prior to treatment with vehicle, 10μM morphine, or 10μM fentanyl for 30 min. Both morphine and fentanyl treatment significantly increased phospho-JNK-IR, and these effects were blocked by PP2 (Fig. 5B-D). These results suggest that Src mediates JNK phosphorylation either directly when activated by morphine or indirectly when activated by fentanyl.
FIGURE 5.

Src is Required for JNK Activation by Morphine and Fentanyl. MOR-GFP expressing HEK293 cells were treated with vehicle (H2O), 10μM morphine, or 10μM fentanyl for 30 min before lysis and analysis of phospho-Src-IR. Similar treatments were performed following a 30 min pretreatment with 5μM PP2 or an equivalent volume of DMSO vehicle followed by cell lysis and analysis of phospho-JNK-IR for each sample. A, phospho-Src-IR was increased over baseline following a 30 min morphine treatment or fentanyl treatment. B, Morphine increased phospho-JNK-IR in the presence of DMSO, but this effect was blocked by pre-incubation with PP2. PP2 did not increase phospho-JNK-IR on its own. C, Similar results were observed following fentanyl treatment. E, Representative westerns are shown for phospho-Src analysis and phospho-JNK analysis. n=4–13; Data analyzed by one-way ANOVA with Newman-Keuls post-test.
3.6 PKC is Required for Morphine, but not Fentanyl, Activation of JNK
Previous work has indicated that PKC is involved in morphine mediated activation of JNK [1] and morphine mediated receptor desensitization [25]. Therefore, MOR-GFP expressing HEK293 cells were treated with 5μM Go6976, which inhibits PKC [29], or DMSO prior to treatment with vehicle, 10μM morphine, or 10μM fentanyl for 30 min. Both morphine and fentanyl significantly increased phospho-JNK-IR (Fig. 6A-C). Pretreatment with Gö6976 also increased phospho-JNK-IR. Morphine did not further increase phospho-JNK-IR in the presence of Gö6976, whereas even in the presence of Gö6976, fentanyl treatment still elicited an increase in phospho-JNK-IR (Fig. 6B,C). These results suggest that PKC was required for morphine but not fentanyl induced JNK activation.
FIGURE 6.

PKC is Required Morphine Activation of JNK. MOR-GFP expressing HEK293 cells were treated with 5μM Go6976 or an equivalent volume of DMSO (vehicle) for 30 min. Cells were subsequently treated with vehicle (H2O), 10μM morphine, or 10μM fentanyl for 30 min before lysis and analysis of phospho-JNK-IR for each sample. A, phospho-JNK-IR was increased following morphine treatment in the presence of DMSO, but this effect was blocked by the inhibition of PKC with Gö6976 pretreatment, although Gö6976 elevated phospho-JNK-IR over baseline as well. B, Fentanyl treatment, however, lead to an increase in phospho-JNK-IR even in the presence of Gö6976, suggesting that PKC is not required for fentanyl mediated JNK activation. C, Representative westerns are shown for each phospho-JNK analysis. n=7–11; Data analyzed by one-way ANOVA with Newman-Keuls post-test.
4. Discussion
The principal findings of this study are that morphine and fentanyl activate JNK through distinct arrestin-independent and arrestin-dependent mechanisms, respectively, and this underlying ligand-directed signaling might explain the differential mechanisms of opioid tolerance observed in vivo. The concept of ligand-directed signaling has been applied to uncover multiple mechanisms of MOR desensitization, including GRK2 and GRK3 [5, 6], PKC [24, 38–40], JNK [1, 11], and ERK [41]. While GRK/arrestin mediated desensitization has been extensively characterized in multiple GPCR systems [4], less is known about JNK signaling and its role in receptor desensitization as only a few studies have directly tested the role of JNK on MOR desensitization directly using [35S]GTPγS and slice recordings in dorsal root ganglion neurons [1, 9, 11].
Previous studies have determined a role for JNK2 in spinally mediated morphine acute analgesic tolerance [1] and in this study we show that this mechanism of JNK2 mediated tolerance exists in additional centrally regulated pain circuits [31–34]. From this data, we conclude that JNK2 regulation of MOR is a more broadly applicable mechanism of morphine-induced MOR desensitization. How JNK2 activation causes MOR desensitization is not yet known; presumably a JNK-phosphorylated arrestin-like substrate binds and interferes with MOR signaling, but this hypothetical substrate has not yet been identified. Both morphine and fentanyl are able to activate JNK, however, and it was necessary to determine the signaling events that contribute to JNK activation in order to uncover a molecular basis for the differential JNK-dependent regulation of MOR following morphine and fentanyl stimulation. Fentanyl desensitization of MOR requires GRK3/arrestin, thus we presume that arrestin binding to MOR prevents the association of the putative JNK-phosphorylated substrate, but this hypothesis requires substantiation.
JNK was first characterized for its role in the stress response [42, 43]. Three genes encode JNK1, JNK2, and JNK3, and among these isoforms, ten splice variants have been identified [13, 26, 44]. In this study we determined that both morphine and fentanyl activate JNK2 specifically in vivo using knockout mice. While both compounds were able to activate the same isoform, which is involved in the development of morphine tolerance specifically, fentanyl activation of JNK requires GRK3, whereas morphine activation of JNK does not. This suggested arrestin-dependent and arrestin-independent mechanisms of JNK activation by MOR.
It has been shown that among the four arrestin subtypes, arrestin-3 can scaffold all three JNK isoforms along with the upstream kinases responsible for JNK phosphorylation and activation [18, 19, 45]. Surprisingly, we found that fentanyl activation of JNK required arrestin-2 and not arrestin-3, although it is possible that greater arrestin-3 knockdown might have suggested a requirement for arrestin-3 as well. Regardless, this suggests a previously unidentified mechanism of arrestin-2 mediated JNK2 activation. It is possible that arrestin-2 acts as a scaffold for JNK2, similar to arrestin-3, and based on studies with arrestin-3, it is likely that arrestin-2 is capable of scaffolding all three JNK isoforms. Further work will be required to confirm this proposed scaffolding activity of arrestin-2. Together, our data suggests that fentanyl activation of JNK2 requires GRK3/arrestin-2, whereas morphine activation of JNK2 is arrestin-independent.
As opiate tolerance is a multidimensional and complex process, we sought to uncover additional signaling molecules involved in JNK activation. Both morphine and fentanyl activate Src, and this Src activation was required for activation of JNK by both compounds at the time point tested. It has been previously shown that Src is responsible for MOR-mediated JNK activation following DAMGO through a PI3K-, Cdc42-, and Sos-dependent mechanism [15] and our study indicates that morphine-like compounds also require Src for JNK activation, although it is possible that upstream mechanisms of Src activation are distinct. Other studies would suggest that this is likely the case. For example, it has been shown that morphine mediated Src activation is arrestin-independent [36], whereas DAMGO mediated Src activation is likely arrestin-dependent whereby arrestin plays a critical role in maintaining Src at the cell membrane in an organized “lattice-like” distribution, which is critical for the ability of DAMGO to stimulate Src phosphorylation [46]. These studies in combination with our results form the basis for our model, where fentanyl mediated Src activation is downstream of arrestin recruitment whereas morphine mediated Src activation is arrestin-independent. While Src was not differentially involved in morphine and fentanyl mediated JNK activation, our study uncovered a differential role for PKC. These results indicate that PKC is specifically involved in morphine, but not fentanyl, activation of JNK at the time point tested. This demonstrates that although PKC has been shown to be involved in tolerance to both compounds in vivo [24, 25], it is differentially required for JNK activation in our studies. This study helps tie these previously separate findings together and provides the basis for future studies to further elucidate more comprehensive signaling mechanisms involved in MOR desensitization.
5. Conclusions
In conclusion, the present study further supports the role of JNK in morphine-induced MOR desensitization, and we propose distinct mechanisms of JNK activation by morphine and fentanyl. The arrestin-dependent and arrestin-independent mechanisms of JNK activation further our understanding of JNK regulation and provide an explanation for the differential mechanisms of tolerance observed in vivo. How morphine-activated JNK2 causes MOR desensitization is not clear. Further work will be required to confirm that arrestin-2 scaffolds JNK2 specifically and continues to explore the mechanism by which PKC and Src activate JNK. It still remains to be determined how this differential activation of JNK leads to distinct regulation of MOR downstream of JNK activation, although it is possible that differential upstream signaling cascades sequester JNK in distinct cellular compartments. This study provides the foundation for future work aimed at answering these questions and suggests that the arrestin-dependent and arrestin-independent mechanisms of JNK activation might be broadly applicable to additional GPCR systems and JNK signaling cascades.
Highlights.
JNK2 is required for morphine induced mu opioid receptor desensitization
Morphine and fentanyl activate JNK2 specifically in mouse spinal cord
Morphine activation of JNK2 is arrestin-independent and Src- and PKC-dependent
Fentanyl activation of JNK2 is Src- GRK3/arrestin2-dependent and PKC-independent
Mu agonists regulate MOR through different desensitization pathways
Acknowledgments
We thank Daniel I. Messinger who monitored the mouse breeding and genotyped the mice and Jessica Cao who helped with the hotplate analgesia experimental design.
Funding
This work was supported by USPHS grants R37-DA11672, T32-DA07278, KO5-DA020570, and PO1-DA035764 from the National Institute on Drug Abuse.
Abbreviations
- ASK1
Apoptosis signal-regulating kinase 1
- JNK
c-Jun N-terminal Kinase
- DAMGO
[D-Ala2, N-MePhe4, Gly-ol]-enkephalin
- ERK
Extracellular signal-regulated kinases
- GPCR
G protein-coupled receptor
- GRK
G protein receptor kinase
- IR
immunoreactivity
- KOR
kappa opioid receptor
- MAPKK
mitogen-activated protein kinase kinases
- MOR
mu opioid receptor
- PKC
protein kinase C
- WT
wild-type
Footnotes
Conflict of interests
None declared
Authorship Contributions
Participated in research design: Kuhar, Bedini, and Chavkin; Conducted experiments: Kuhar, Bedini, Melief, Chiu, and Striegel; Performed data analysis: Kuhar, Bedini, Chavkin; Wrote or contributed to the writing of the manuscript: Kuhar, Bedini, Chavkin
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Melief EJ, Miyatake M, Bruchas MR, Chavkin C. Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci. 2010;107:11608–11613. doi: 10.1073/pnas.1000751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- 3.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 4.Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- 5.Kovoor A, Celver JP, Wu A, Chavkin C. Agonist induced homologous desensitization of mu-opioid receptors mediated by G protein-coupled receptor kinases is dependent on agonist efficacy. Mol Pharmacol. 1998;54:704–711. [PubMed] [Google Scholar]
- 6.Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luttrell LM, Gesty-Palmer D. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev. 2010;62:305–330. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shenoy SK, Lefkowitz RJ. beta-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melief EJ, Miyatake M, Carroll FI, Beguin C, Carlezon WA, Jr, Cohen BM, Grimwood S, Mitch CH, Rorick-Kehn L, Chavkin C. Duration of action of a broad range of selective kappa-opioid receptor antagonists is positively correlated with c-Jun N-terminal kinase-1 activation. Mol Pharmacol. 2011;80:920–929. doi: 10.1124/mol.111.074195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruchas MR, Yang T, Schreiber S, DeFino M, Kwan SC, Li S, Chavkin C. Long-acting κ opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem. 2007;282:29803–29811. doi: 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mittal N, Tan M, Egbuta O, Desai N, Crawford C, Xie CW, Evans C, Walwyn W. Evidence that behavioral phenotypes of morphine in beta-arr2−/− mice are due to the unmasking of JNK signaling. Neuropsychopharmacology. 2012;37:1953–1962. doi: 10.1038/npp.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levitt ES, Williams JT. Morphine desensitization and cellular tolerance are distinguished in rat locus ceruleus neurons. Mol Pharmacol. 2012;82:983–992. doi: 10.1124/mol.112.081547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. Embo j. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 14.Lawler S, Fleming Y, Goedert M, Cohen P. Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Curr Biol. 1998;8:1387–1390. doi: 10.1016/S0960-9822(98)00019-0. [DOI] [PubMed] [Google Scholar]
- 15.Kam AY, Chan AS, Wong YH. Phosphatidylinositol-3 kinase is distinctively required for mu-, but not kappa-opioid receptor-induced activation of c-Jun N-terminal kinase. J Neurochem. 2004;89:391–402. doi: 10.1111/j.1471-4159.2004.02338.x. [DOI] [PubMed] [Google Scholar]
- 16.Kam AYF, Chan ASL, Wong YH. κ-opioid receptor signals through src and focal adhesion kinase to stimulate c-Jun N-terminal kinases in transfected COS-7 cells and human monocytic THP-1 cells. J Pharmacol Exp Ther. 2004;310:301–310. doi: 10.1124/jpet.104.065078. [DOI] [PubMed] [Google Scholar]
- 17.Kam AY, Chan AS, Wong YH. Rac and Cdc42-dependent regulation of c-Jun N-terminal kinases by the delta-opioid receptor. J Neurochem. 2003;84:503–513. doi: 10.1046/j.1471-4159.2003.01535.x. [DOI] [PubMed] [Google Scholar]
- 18.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 19.Zhan X, Kaoud TS, Kook S, Dalby KN, Gurevich VV. JNK3 enzyme binding to arrestin-3 differentially affects the recruitment of upstream mitogen-activated protein (MAP) kinase kinases. J Biol Chem. 2013;288:28535–28547. doi: 10.1074/jbc.M113.508085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kook S, Zhan X, Kaoud TS, Dalby KN, Gurevich VV, Gurevich EV. Arrestin-3 binds c-Jun N-terminal kinase 1 (JNK1) and JNK2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J Biol Chem. 2013;288:37332–37342. doi: 10.1074/jbc.M113.510412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi H, Narita Y, Nishida M, Kurose H. Beta-arrestin2 enhances beta2-adrenergic receptor-mediated nuclear translocation of ERK. Cell Signal. 2005;17:1248–1253. doi: 10.1016/j.cellsig.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 22.Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 23.Hanson SM, Cleghorn WM, Francis DJ, Vishnivetskiy SA, Raman D, Song X, Nair KS, Slepak VZ, Klug CS, Gurevich VV. Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J Mol Biol. 2007;368:375–387. doi: 10.1016/j.jmb.2007.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, Mundell SJ, Bailey CP, Kelly E, Henderson G. Agonist-selective mechanisms of mu-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- 25.Hull LC, Llorente J, Gabra BH, Smith FL, Kelly E, Bailey C, Henderson G, Dewey WL. The effect of protein kinase C and G protein-coupled receptor kinase inhibition on tolerance induced by mu-opioid agonists of different efficacy. J Pharmacol Exp Ther. 2010;332:1127–1135. doi: 10.1124/jpet.109.161455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the JNK3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 27.Terman GW, Jin W, Cheong YP, Lowe J, Caron MG, Lefkowitz RJ, Chavkin C. G-protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br J Pharmacol. 2004;141:55–64. doi: 10.1038/sj.bjp.0705595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 29.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 30.Irwin S, Houde RW, Bennett DR, Hendershot LC, Seevers MH. The effects of morphine methadone and meperidine on some reflex responses of spinal animals to nociceptive stimulation. J Pharmacol Exp Ther. 1951;101:132–143. [PubMed] [Google Scholar]
- 31.Le Bars D, Gozariu M, Cadden SW. Animal models of nociception. Pharmacol Rev. 2001;53:597–652. [PubMed] [Google Scholar]
- 32.Tsou K, Jang CS. Studies on the site of analgesic action of morphine by intracerebral micro-injection. Sci Sin. 1964;13:1099–1109. [PubMed] [Google Scholar]
- 33.Yaksh TL, Yeung JC, Rudy TA. Systematic examination in the rat of brain sites sensitive to the direct application of morphine: observation of differential effects within the periaqueductal gray. Brain Res. 1976;114:83–103. doi: 10.1016/0006-8993(76)91009-X. [DOI] [PubMed] [Google Scholar]
- 34.Tortorici V, Morgan MM, Vanegas H. Tolerance to repeated microinjection of morphine into the periaqueductal gray is associated with changes in the behavior of off- and on-cells in the rostral ventromedial medulla of rats. Pain. 2001;89:237–244. doi: 10.1016/S0304-3959(00)00367-5. [DOI] [PubMed] [Google Scholar]
- 35.Rey A, Manen D, Rizzoli R, Caverzasio J, Ferrari SL. Proline-rich motifs in the parathyroid hormone (PTH)/PTH-related protein receptor c terminus mediate scaffolding of c-Src with β-arrestin2 for ERK1/2 activation. J Biol Chem. 2006;281:38181–38188. doi: 10.1074/jbc.M606762200. [DOI] [PubMed] [Google Scholar]
- 36.Zhang L, Zhao H, Qiu Y, Loh HH, Law PY. Src phosphorylation of micro-receptor is responsible for the receptor switching from an inhibitory to a stimulatory signal. J Biol Chem. 2009;284:1990–2000. doi: 10.1074/jbc.M606762200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salazar EP, Rozengurt E. Bombesin and platelet-derived growth factor induce association of endogenous focal adhesion kinase with src in intact swiss 3T3 cells. J Biol Chem. 1999;274:28371–28378. doi: 10.1074/jbc.M807971200. [DOI] [PubMed] [Google Scholar]
- 38.Bailey CP, Kelly E, Henderson G. Protein kinase C activation enhances morphine-induced rapid desensitization of mu-opioid receptors in mature rat locus ceruleus neurons. Mol Pharmacol. 2004;66:1592–1598. doi: 10.1124/mol.104.004747. [DOI] [PubMed] [Google Scholar]
- 39.Bailey CP, Oldfield S, Llorente J, Caunt CJ, Teschemacher AG, Roberts L, McArdle CA, Smith FL, Dewey WL, Kelly E, Henderson G. Involvement of PKC alpha and G-protein-coupled receptor kinase 2 in agonist-selective desensitization of mu-opioid receptors in mature brain neurons. Br J Pharmacol. 2009;158:157–164. doi: 10.1111/j.1476-5381.2009.00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bailey CP, Llorente J, Gabra BH, Smith FL, Dewey WL, Kelly E, Henderson G. Role of protein kinase C and mu-opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur J Neurosci. 2009;29:307–318. doi: 10.1111/j.1460-9568.2008.06573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dang VC, Napier IA, Christie MJ. Two distinct mechanisms mediate acute mu-opioid receptor desensitization in native neurons. J Neurosci. 2009;29:3322–3327. doi: 10.1523/JNEUROSCI.4749-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 43.Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 44.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 45.Zhan X, Perez A, Gimenez LE, Vishnivetskiy SA, Gurevich VV. Arrestin-3 binds the MAP kinase JNK3alpha2 via multiple sites on both domains. Cell Signal. 2014;26:766–776. doi: 10.1016/S0092-8674(00)00116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walwyn W, Evans CJ, Hales TG. Beta-arrestin2 and c-Src regulate the constitutive activity and recycling of mu opioid receptors in dorsal root ganglion neurons. J Neurosci. 2007;27:5092–5104. doi: 10.1523/JNEUROSCI.1157-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]