

Abstract
Epidermal growth factor receptor (EGFR) inhibitors have demonstrated efficacy in squamous cell carcinoma of the head and neck (SCCHN). In addition to EGFR, other ErbB family members are expressed and activated in SCCHN. Afatinib is an ErbB family blocker that has been approved for treating patients with EGFR-mutated nonsmall cell lung cancer. We sought to determine the efficacy of afatinib in preclinical models and compare this to other EGFR-targeted agents. Afatinib efficacy was characterized in a panel of ten SCCHN cell lines and found to be most effective against cell lines amplified for EGFR. Afatinib had lower IC50 values than did gefitinib against the same panel. Two EGFR-amplified cell lines that are resistant to gefitinib are sensitive to afatinib. Cetuximab was not found to have a synergistic effect with afatinib either in vitro or in vivo. Both afatinib and cetuximab were effective in tumor xenograft model. Afatinib is an effective agent in SCCHN especially in models with EGFR amplification.
Keywords: Squamous cell carcinoma of the head and neck, Afatinib, Gefitinib, Cetuximab, Epidermal growth factor receptor
Introduction
Epidermal growth factor receptor (EGFR) and HER2 are both members of the ErbB tyrosine kinase receptor family which also comprises HER3 and HER4. When stimulated by the presence of a ligand, ErbB receptors homodimerize or heterodimerize with other members of the family, and through transphosphorylation, initiate intracellular signaling cascades [1]. Although HER2 has no known ligand, it is believed to be a preferred dimerization partner for EGFR and HER3 [2]. As members of the ErbB family are often deregulated in a number of malignancies [3], they have been an attractive option for targeted therapy. EGFR is thought to be highly expressed in as many as 90 % of the head and neck squamous cell carcinoma (SCCHN) tumors [4], while the expression of HER2 is more variable [5, 6].
One of the earliest agents to target the ErbB network system was cetuximab, a chimeric monoclonal antibody to EGFR. After demonstrating efficacy in preclinical models [7], cetuximab earned FDA approval for locally advanced as well as recurrent or metastatic SCCHN [8–11]. Another potential therapeutic target is the intracellular tyrosine kinase domain of ErbB receptors. First generation small molecule tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib, specifically target the tyrosine kinase of EGFR and are approved for clinical use in lung cancer. Gefitinib demonstrated excellent activity in preclinical SCCHN models [12, 13]. In phase 2 trials, gefitinib had a response rate of 9–10 % in patients with recurrent or metastatic SCCHN [14]. Erlotinib had similarly promising preclinical results [15] and slightly worse response rates in clinical studies [16].
Afatinib is an irreversible TKI that targets all kinase competent ErbB family members (EGFR, HER2, and HER4; HER3 lacks essential catalytic residues and displays weak to no activity) [17]. Early in vitro studies of afatinib in human tumor cell lines demonstrated significant activity as well as an increase in the proportion of cells in the sub-G0/G1 phase of the cell cycle [18, 19]. Effective antitumor activity of afatinib has also been shown in multiple xenograft models using human cell line-derived xenografts and transgenic mouse models [19, 20]. Phase I trials in patients with nonsmall cell lung cancer (NSCLC) and other solid tumors found afatinib to be well tolerated, with the most common adverse effects being diarrhea, rash, fatigue, and nausea [21, 22]. Although not designed to determine clinical response, there was a decrease in tumor size in 50 % of patients [22]. At this point, besides early evaluations assessing afatinib in combination with radiotherapy in one single model system (FaDu) [23], no work characterizing the effect of afatinib in a panel of SCCHN models has been reported.
Materials and methods
Viability assays
Cell Titer Blue assays (Promega, Madison, WI) were used to determine cell viability. Briefly, cells were seeded at 1000 cells/well on 96-well plates, incubated for 24 h in complete media, maintained in serum-free conditions for 24 h, and treated with afatinib or cetuximab for 72 h at 37 °C. Then, Cell Titer Blue reagent was added to each well and incubated for 2 h at 37 °C before 3 % SDS was added. Cell viability was quantified by scanning absorbance at 570 nm in a microplate reader (Bio-Tek, Winooski, VT).
Tumor xenografts treated with afatinib only or multiple ErbB-targeting agents
Mice were allowed to adjust to conditions at least for 5 days before they were used for experiments. They were housed in Macrolon type III cages in groups of ten under standardized specific pathogen-free (SPF) conditions at 21.5 ± 1.5 °C temperature and 55 ± 10 % humidity. Standardized diet (PROVIMI KLIBA) and autoclaved tap water were provided ad libitum. HN5 or FaDu cells were injected into the right flank of 6-week-old female BomTac:NMRI-Foxn1nu mice (Taconic, Denmark). Tumor take was monitored over time, and animals with established tumors (50–100 mm3) were randomized to the treatment groups (n = 10/group). Mice were treated with either vehicle or afatinib, daily or every other day or in a weekly alternating schedule (afatinib only experiment) or with vehicle or other agents daily (multiple agent experiment). Tumor volumes and body weights were recorded three times a week, and median tumor volumes as well as change in body weight were plotted over time.
Tumor xenografts treated with afatinib and cetuximab
HN5 and SCC25 cell line xenograft experiments were conducted as previously described [24]. Briefly, female athymic nude mice (Harlan, Indianapolis, IN) were maintained in a pathogen-free animal facility in accordance with the University of Chicago Animal Care and Use Committee. Mice received standard laboratory rodent food and water as desired. All handling procedures were conducted in a laminar flow biosafety hood. At 6–7 weeks of age, mice were injected subcutaneously in the right flank with HN5 or SCC35 cells. Drug treatment was initiated when mean tumor volumes reached 200 to 250 mm3. Mice were treated with cetuximab at 30-mg/kg body weight two times per week, via i.p. injection and/or afatinib at 12.5-mg/kg body weight once per day via oral gavage, or equal volume of diluents (control). For tumor growth analysis, tumor size was measured with a vernier caliper. Tumor volumes were calculated with the formula V = 0.52 × L × W2, where L and W represent the length and the width of the tumor (mm). The animals were monitored two times per week for body weight and tumor volume.
Quantification of TGFA, EGFR, and AREG mRNA expression and EGFR copy number by real-time PCR
The real-time PCR method for quantification of EGFR gene expression and copy number has been reported previously [25, 26].
FISH assays
FISH assay methods and analysis were previously described [24]. Briefly, for the evaluation of the EGFR gene copy number (GCN) alterations, dual-color FISH assays were conducted using an LSIEGFR SpectrumOrange:CEP7SpectrumGreen Probe mixture (Vysis/Abbott Molecular, Des Plaines, IL). HER2 amplification was studied using the Vysis PathVysion HER2 DNA Probe Kit according to manufacturer recommendations (Abbott Molecular, Des Plaines, IL). CEP7 or CEP17 probes were used to distinguish true gene amplification from EGFR or HER2 gene copy number gain (gene polysomy) and alterations in number of chromosome 7 or 17 homologs. The absolute number of each signal, the mean copy number of signal per cell, the ratios of EGFR to CEP7 or HER2 to CEP17, and the percentage of cells with given copy number of each signal per cell were calculated. Cells with a gene to chromosome signal ratio <2 were considered nonamplified, whereas those with a ratio greater than 2.0 (or ≥15 copies of EGFR per cells in ≥10 % of cells) were considered as having true amplification. Cells with ratios near cutoff points were equivocal or low amplified.
Western blotting
Western blots on cell lysates were performed as previously described [24]. Visualization and quantification were performed with Odyssey Infrared Imaging System (Li-Cor Biosciences). Experiments were repeated at least three times. PTEN antibody was purchased from Cell Signaling Technology, Inc. (Danvers, MA). Actin antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Secondary goat anti-rabbit IgG IRDye antibody was purchased from LI-COR Biosciences (Lincoln, NE). Secondary mouse IgM IRDye antibody was purchased from Rockland Immunochemicals Inc. (Gilbertsville, PA).
Results
Sensitivity to afatinib
Cell viability of ten SCCHN cell lines grown in monolayer cultures was determined over a range of afatinib concentrations (Fig. 1) and compared to IC50 ranges of the same cell lines to gefitinib (Table 1). In order to assess whether anti-proliferative activity could be improved upon with the addition of cetuximab, viability of cell lines with high IC50 values (SCC35 and Detroit 562) was tested at several doses (Fig. 2a, b). Treatment with cetuximab alone had little effect on cell viability, even at relatively high concentrations (100 nmol/L). The combination treatment resulted in CI values above 1, thus demonstrating no evidence of a synergistic or additive effect (data not shown). Treatment with afatinib and cetuximab was tried on additional cell lines with greater sensitivity to afatinib (SQ20B and SCC61) with similar results (Fig. 2c, d).
Fig. 1.

Viablility of ten SCCHN cell lines treated with a range of concentrations of afatinib. Results from Cell Titer Blue assays
Table 1.
Afatinib IC50s compared to gefitinib IC50s of SCCHN cell lines
| Cell line | Afatinib IC50 (μM) | Confidence interval | Gefitinib IC50 (μM) |
|---|---|---|---|
| SCC25 | 0.0102 | 9.796e-005 to 1.062 | 5–10 |
| HN5 | 0.06412 | 0.009348 to 0.4398 | 0.05–0.5 |
| SCC58 | 0.3405 | 0.07144 to 1.623 | >100 |
| SQ20B | 0.5176 | 0.08746 to 3.064 | 1–5 |
| MSK921 | 1.174 | 0.6654 to 2.071 | 1–5 |
| HN31 | 1.225 | 0.8320 to 1.805 | 1–5 |
| SCC61 | 1.903 | 0.6092 to 5.942 | 5 |
| SCC28 | 1.951 | 0.7886 to 4.827 | >5 |
| Detroit 562 | 2.485 | 0.9399 to 6.569 | 5–50 |
| SCC35 | 8.008 | 5.645 to 11.36 | 100 |
Fig. 2.
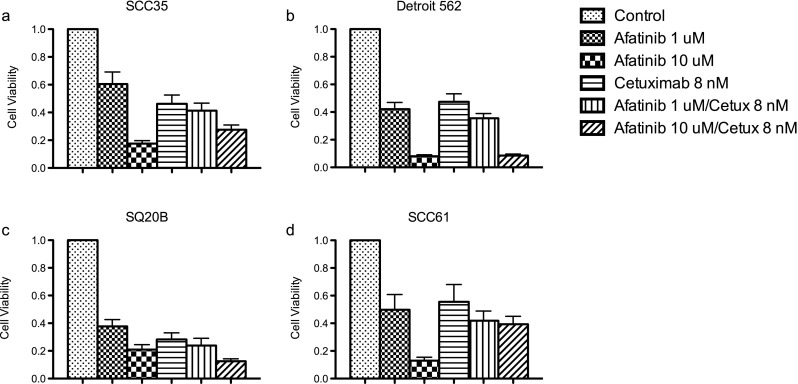
Viability of SCCHN cell lines treated with afatinib and cetuximab. Results from Cell Titer Blue assays. a SCC35, b Detroit 562, c SQ20B, d SCC61
In vivo tumor xenografts
In vivo activity of afatinib was first characterized against FaDu cells injected into the right flank of BomTac:NMRI-Foxn1nu mice. Treatment with afatinib at 10 mg/kg followed two regimens. Both daily and intermittent treatment regimens slowed tumor growth. Continuous dosing resulted in virtually no tumor growth, while intermittent dosing saw increase in tumor volume over time (Fig. 3a). Neither treatment regimen at the doses used had a significant effect on body weight (Fig. 3b).
Fig. 3.

In vivo activity of afatinib in FaDu cell line-derived xenografts. Mice were treated daily with either vehicle (black circle) or afatinib as described in (white triangle) or in a weekly alternating schedule (white square). Tumor volumes and body weights were recorded three times a week, and median tumor volumes (a) as well as change in body weight (b) were plotted over time
The anti-tumor activity of afatinib and several other ErbB targeting agents was further tested in mice bearing HN5 xenografts (Fig. 4). Daily treatment at 15 mg/kg resulted in tumor regressions in all treated animals. Four complete responses and six responses displaying 75 % or more reduction in tumor size compared to baseline were observed. Erlotinib, at 75 mg/kg, was less potent and induced six regressions and four tumors grew under treatment. Neratinib and lapatinib induced tumor growth delay, but no regressions were achieved.
Fig. 4.

In vivo activity of afatinib in HN5 cell line-derived xenografts. Mice were treated daily with either vehicle (gray), 2 × 100 mg/kg/day lapatinib (turquoise), 75 mg/kg/day erlotinib (green), 50 mg/kg/day neratinib (blue), or 15 mg/kg/day afatinib (orange) as described in “Materials and methods.” Tumor volumes were recorded three times a week. Median tumor volumes were plotted over time (a). On day 50, relative tumor volume changes were recorded b
The last set of in vivo experiments assessed the efficacy of afatinib in combination with cetuximab. Afatinib (12.5 mg/kg) and cetuximab (30 mg/kg), separately and in combination, were very effective at inhibiting in vivo tumor growth in both the HN5 and SCC35 mouse xenograft models (Fig. 5). At the doses tested, the combination treatment offered no advantage to single agent administration.
Fig. 5.

SCCHN cell line tumor xenografts respond to treatment by afatinib alone, or in combination with cetuximab. % tumor growth is shown. a HN5, b SCC35
PTEN expression
PTEN protein expression was examined by immunoblotting of SCCHN cell line lysates. We found that all lines express the protein at approximately equal levels (Fig. 6), eliminating PTEN loss as an explanation of primary EGFR inhibitor resistance in these lines.
Fig. 6.

PTEN expression in SCCHN cell lines. Western blotting shows that PTEN expression is consistent across cell lines. Actin serves as loading control
EGFR and HER2 amplification
Four of our SSCHN cell lines were EGFR amplified by FISH (Table 2, Fig. 7a and [26]). SCC58, HN5, and SQ20B exhibit high amplification (EGFR/CEP7 ratio >7), and SCC25 exhibits low amplification (EGFR/CEP7 ratio ~2). These same four lines show a gain of EGFR mRNA copies normalized to 18 s mRNA while the remaining cell lines do not (Table 2 and [26]). SCC28 cells do not show EGFR amplification (Table 2) but have high EGFR gene polysomy (Fig. 7b).
Table 2.
EGFR and HER2 gene copy number alterations and mRNA expression levels of SCCHN cell lines
| Cell line | FISH | qPCR | ||||||
|---|---|---|---|---|---|---|---|---|
| EGFR GCN1 | CEP7 GCN2 | EGFR/CEP7 3 | Interpretation | TGFa/18 s | AREG/18 s | EGFR/18 s | Interpretation | |
| SCC58 | 32.94 | 4.3 | 7.7 | High Ampl | 2.02 | 9.67 | 17.08 | Gain |
| SCC28 | 5.02 | 5.33 | 0.94 | No Ampl, | 0.39 | 1.86 | 1.77 | Normal |
| HER2 GCN1 | CEP17 GCN2 | HER2/CEP17 3 | Interpretation | |||||
| SCC25 | 3.25 | 3.05 | 1.07 | No Ampl, | ||||
| HN5 | 2.88 | 3.9 | 0.74 | No Ampl, | ||||
| SCC58 | 1.87 | 2.87 | 0.65 | No Ampl | ||||
| SQ20B | 2.06 | 4.20 | 0.49 | No Ampl | ||||
| SCC61 | 2.3 | 2.04 | 1.13 | No Ampl | ||||
| SCC28 | 2.85 | 2.93 | 0.97 | No Ampl, | ||||
FISH: 1Mean copy number EGFR or HER2 per cell; 2mean copy number of centromere enumeration probe (CEP7 or CEP17) per cell; 3mean gene to centromere ratio; Ampl, amplification, ≥15 copies of gene per cell in ≥10 % of cells or gene to CEP ratio ≥2.0. An average of 80 (range, 50 to 180) well-defined malignant nuclei were scored
qPCR: Mean values from three experiments were calculated relative to 18 s as control, arbitrarily set at 1. Gain, ≥7 copies of gene/18 s
Fig. 7.
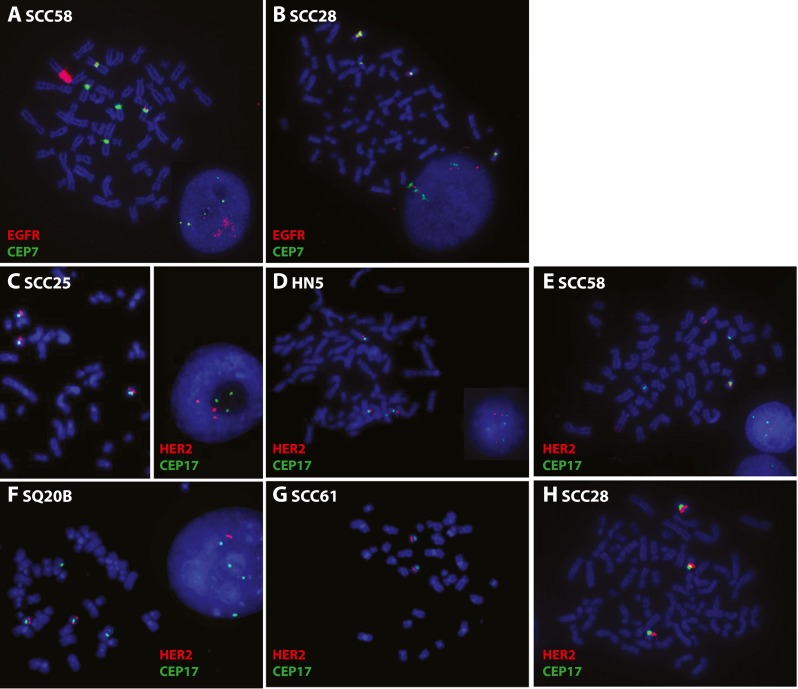
EGFR and HER2 FISH. Images of metaphase and interphase nuclei after FISH are presented. The EGFR and HER2 genes are localized by red fluorescent signals, and chromosome 7 and 17 centromeres (CEP7 and CEP17) are localized by green fluorescent signals. The cells were counterstained with DAPI (blue). EGFR/CEP7 staining of a SCC58 and b SCC28 cells. HER2/CEP17 staining of c SCC25, d HN5, e SCC58, f SQ20B, g SCC61, and h SCC28
SCC25, HN5, SCC58, SQ20B, SCC61, and SCC28 were also tested for HER2/CEP17 FISH. No HER2 amplification was found (Table 2 and Fig. 7c–h). SCC25, HN5, and SCC28 cells carried in average three copies of HER2 per cell due to trisomy for chromosome 17.
Discussion
In vitro, SCCHN cell lines show a range of sensitivities to afatinib. The four most sensitive cell lines, SCC58, SQ20B, SCC25, and HN5, show amplification of EGFR by FISH analysis and increased mRNA copy number by qPCR. This suggests that afatinib is most effective in cell lines where EGFR is amplified and possibly acts as a driver of cell growth. EGFR gene copy numbers have not been correlated with clinical activity of EGFR inhibitors in SCCHN; however, this has not been tested in a prospective study. Our data presented here and in the past [26] indicate that a prospective trial is warranted. When afatinib IC50 values are compared to those from gefitinib (Table 1), one sees that the order of increasing resistance is almost identical, the exceptions being SCC25 and SCC58, both of which are sensitive to afatinib but resistant to gefitinib. This suggests that afatinib may be a better therapeutic choice for cancers expressing high levels of EGFR and may relate to the broader ErbB inhibitory scope of afatinib as compared to gefitinib. Because gefitinib inhibits EGFR only, while afatinib targets EGFR, HER2, and HER4, one possibility could reside in differences due to HER2 signaling. However, none of the four most sensitive lines show an increased gene copy number for HER2 by FISH ruling out amplification as a possible mechanism. HER3 has been implicated as a mechanism of resistance to EGFR blockade in SCCHN in one paper [27]. As afatinib has been shown to block transphosphorylation of HER3 in vitro [19], it may be able to address this potential resistance mechanism. Indeed, afatinib activity was seen in cetuximab refractory patients in a phase II trial [28] suggesting a lack of cross-resistance in some instances.
PTEN expression was also not at play in afatinib resistance, as expression levels were similar in all cell lines. The main factors which could explain the differences in efficacy are the irreversible binding mode and the anticipated tighter blockade of the ErbB signaling network by afatinib. However, a measure of caution should be used in interpreting in vitro data, as SCC35, which exhibited the highest IC50 to afatinib (Table 1), was still sensitive to the drug in vivo (Fig. 5).
Gefitinib does block phosphorylation of AKT and ERK in SCC25 and SCC58 [26, data not shown], possibly pointing to signaling through STAT3 downstream. Elucidation of this mechanism could point to a more specific subset of cases in which afatinib is more effective than gefitinib. De Greve et al. [29] showed an objective response to afatinib in three patients with mutations in the tyrosine kinase domain of HER2 even after other ErbB targeting treatments had failed. Afatinib may be active in the presence of multiple genetic aberrations, which render other TKIs ineffective.
Indeed, afatinib was more effective than lapatinib, erlotinib, and neratinib in the HN5 tumor xenograft experiment. These three TKIs were only tested on one cell line, limiting broad conclusions about mechanism. However the combination of irreversible binding mode and broad ErbB signaling blockage is provided by afatinib alone among the TKIs tested here.
With these experiments, we were unable to show added benefit forcombination therapy with cetuximab in the most resistant cell lines to afatinib. Afatinib and cetuximab were very effective at arresting growth and decreasing tumor volume in vivo, both as single agents and in combination. In fact, these treatments worked so well that any benefit from the combination of drugs could not be seen.
Conclusion
Afatinib is most effective at arresting growth of SCCHN cell lines in vitro that are amplified and/or overexpress EGFR. Even EGFR-amplified cell lines which are resistant to gefitinib respond well to afatinib treatment. Afatinib was well tolerated in mice at the doses tested and was more effective with continuous dosing. Afatinib was more effective than other TKIs at arresting tumor xenograft growth, perhaps due to a combination of irreversible binding and broad inhibition of ErbB network signaling. Synergy between afatinib and cetuximab could not be observed in vitro due to the poor response to cetuximab and in vivo due to the excellent response to both drugs.
Acknowledgments
Funding
Boehringer-Ingelheim, NIDCR R21DE022454 to EEWC, NCI K12CA139160 to TG, ACS to AH.
Conflict of interest
Ms. Young has nothing to disclose. Dr. Soneru has nothing to disclose. Dr. Liu has nothing to disclose. Dr. Grushko has nothing to disclose. Miss Hardeman has nothing to disclose. Dr. Olopade has nothing to disclose. Dr. Baum has a patent WO 02/50043 A1 issued and is an employee of Boehringer Ingelheim RCV. Dr. Solca has a patent WO 02/50043 A1 issued and is an employee of Boehringer Ingelheim RCV. Dr. Cohen reports grants from Boehringer Ingelheim, grants from National Institutes of Health, during the conduct of the study.
References
- 1.Tzahar E, Waterman H, Chen X, Levkowitz G, KarunagaranD LS, Ratzkin BJ, Yarden Y. A hierarchical network or interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. 1996;16:5276–5287. doi: 10.1128/MCB.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klapper LN, Glathe S, Vaisman N, Hynes NE, Andrews GC, Sela M, Yarden Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc Natl Acad Sci U S A. 1999;96:4995–5000. doi: 10.1073/pnas.96.9.4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 4.Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579–3584. [PubMed] [Google Scholar]
- 5.Riviere A, Becker J, Loning T. Comparative investigation of c-erbB2/neu expression in head and neck tumors and mammary cancer. Cancer. 1991;67:2142–2149. doi: 10.1002/1097-0142(19910415)67:8<2142::AID-CNCR2820670823>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 6.Ibrahim SO, Vasstrand EN, Liavaag PG, Johannessen AC, Lillehaug JR. Expression of c-erbB proto-oncogene family member in squamous cell carcinoma of the head and neck. Anticancer Res. 1997;17:4539–4546. [PubMed] [Google Scholar]
- 7.Huang SM, Bock JM, Harari PM. Epidermal growth factor receptor blockage with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 1999;59(8):1935–1940. [PubMed] [Google Scholar]
- 8.Bonner JA, Harari PM, Girald J, Azarnia N, Shin D, Cohen R, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J, Youssoufian H, Amellal N, Rowinsky EK, Ang KK. Radiotherapy plus cetuximab for Squamous-Cell Carcinoma of the Head and Neck. NEJM. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 9.Burtness B, Goldwasser MA, Flood W, et al. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol. 2005;23:8646–8654. doi: 10.1200/JCO.2005.02.4646. [DOI] [PubMed] [Google Scholar]
- 10.Baselga J, Trigo JM, Bourhis J, et al. Phase II multicenter study of the antiepidermal growth factor receptor monoclonal antibody cetuximab in combination with platinum-based chemotherapy in patients with platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. J Clin Oncol. 2005;23:5568–5577. doi: 10.1200/JCO.2005.07.119. [DOI] [PubMed] [Google Scholar]
- 11.Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. NEJM. 2008;359:1116–1127. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 12.Wakeling AE, Guy SP, Woodburn JR, et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62:5749–5754. [PubMed] [Google Scholar]
- 13.Magne N, Fischel JL, Dubreuil A, et al. Influence of epidermal growth factor receptor (EGFR), p53, and intrinsic MAP kinase pathway status of tumour cells on the antiproliferative effect of ZD1839 (Iressa) Br J Cancer. 2002;86:1518–1523. doi: 10.1038/sj.bjc.6600299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen EE, Rosen F, Stadler WM, et al. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2003;21:1980–1987. doi: 10.1200/JCO.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 15.Pollack VA, Savage DM, Baker DA, et al. Inhibition of epidermal growth factor receptor-associated tyrosine phosphorylation in human carcinomas with CP-258,774: dynamics of receptor inhibition in situe and antitumor effects in athymic mice. J Pharmacol Exp Ther. 1999;291:739–748. [PubMed] [Google Scholar]
- 16.Soulieres D, Senzer N, Vokes E, Hidalgo M, Agarwala S, Siu L. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22:77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 17.Solca F, Dahl G, Zoephel A, Bader G, Sanerson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. JPET. 2012;343(2):342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 18.Khelwatty SA, Essapen S, Seddon AM, Modjtahedi H. Growth response of human colorectal tumour cell lines to treatment with afatinib (BIBW2992), an irreversible erbB family blocker, and its association with expression of HER family members. Int J Oncol. 2011;39(2):483–491. doi: 10.3892/ijo.2011.1054. [DOI] [PubMed] [Google Scholar]
- 19.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimamura T, Gewulich H, Solca F, Wong K. Efficacy of BIBW 2992, a potent irreversible inhibitor of EGFR and HER2 in human NSCLC xenografts and in a transgenic mouse lung-cancer model. J Thorac Oncol. 2007;2(8):S380. doi: 10.1097/01.JTO.0000283232.53465.68. [DOI] [Google Scholar]
- 21.Yap TA, Vidal L, Adam J, Stephens P, Spicer J, Shaw H, Ang J, Temple G, Bell S, Shahidi M, Uttenreuther-Fischer M, Stopfer P, Futreal A, Calvert H, de Bono JS, Plummer R. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J Clin Oncol. 2010;28:3965–3972. doi: 10.1200/JCO.2009.26.7278. [DOI] [PubMed] [Google Scholar]
- 22.Murakami H, Tamura T, Takahashi T, Nokihara H, Naito T, Nakamura Y, Nishio K, Seki Y, Sarashina A, Shahidi M, Yamamoto N. Phase I study of continuous afatinib (BIBW2992) in patients with advanced non-small cell lung cancer after prior chemotherapy/erlotinib/gefitinib (LUX-Lung 4) Cancer Chemother Pharmacol. 2012;69:891–899. doi: 10.1007/s00280-011-1738-1. [DOI] [PubMed] [Google Scholar]
- 23.Schutze C, Dorfler A, Eicheler W, Zips D, Hering S, Solca F, Baumann M, Krause M. Combination of EGFR/HER2 tyrosine kinase inhibition by BIBW 2992 and BIBW 2669 with irradiation in FaDu human squamous cell carcinoma. Strahlenther Onkol. 2006;182:256–264. doi: 10.1007/s00066-007-1696-z. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Kuo WL, Seiwert TY, Lingen M, Ciaccio MF, Jones RB, Rosner MR, Cohen EE. Effect of complementary pathway blockade on efficacy of combination enzastaurin and rapamycin. Head Neck. 2011;33:1774–1782. doi: 10.1002/hed.21701. [DOI] [PubMed] [Google Scholar]
- 25.Liu W, Wu X, Zhang W, Montenegro RC, Fackenthal DL, Spitz JA, Huff LM, Innocenti F, Das S, Cook EH, Jr, Cox NJ, Bates SE, Ratain MJ. Relationship of EGFR mutations, expression, amplification, and polymorphisms to epidermal growth factor receptor inhibitors in the NCI60 cell lines. Clin Cancer Res. 2007;13:6788–6795. doi: 10.1158/1078-0432.CCR-07-0547. [DOI] [PubMed] [Google Scholar]
- 26.Young NR, Liu J, Pierce C, Wei T-F, Grushko T, Olopade OI, Liu W, Shen C, Seiwert TY, Cohen EEW. Molecular phenotype predicts sensitivity of squamous cell carcinoma of the head and neck to epidermal growth factor receptor inhibition. Mol Oncol. 2013;7:359–368. doi: 10.1016/j.molonc.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Castanaro C, Luan B, Yang K, Fan L, Fairhurst JL, Rafique A, Potocky TB, Shan J, Delfino FJ, Shi E, Huang T, Martin JH, Chen G, Macdonald D, Rudge JS, Thurston G, Daly C. ERBB3/HER2 signaling promotes resistance to EGFR blockade in head and neck and colorectal cancer models. Mol Cancer Ther. 2014;13(5):1345–1355. doi: 10.1158/1535-7163.MCT-13-1033. [DOI] [PubMed] [Google Scholar]
- 28.Seiwert TY, Fayette J, Cupissol D, Del Campo JM, Clement PM, Hitt R, Degardin M, Zhang W, Blackman A, Ehrnrooth E, Cohen EE. A randomized, phase II study of afatinib versus cetuximab in metastatic or recurrent squamous cell carcinoma of the head and neck. Ann Oncol. 2014;25(9):1813–1820. doi: 10.1093/annonc/mdu216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Grève J, Teugels E, Geers C, Decoster L, Galdermans D, De Mey J, Everaert H, Umelo I, In't Veld P, Schallier D. Clinical activity of afatinib (BIBW 2992) in patients with lung adenocarcinoma with mutations in the kinase domain of HER2/neu. Lung Cancer. 2012;76:123–127. doi: 10.1016/j.lungcan.2012.01.008. [DOI] [PubMed] [Google Scholar]