

Abstract
Failure of neoplastic cells to respond to conventional chemotherapy is usually associated with factors that limit access of drugs to subcellular sites, differences in cell-cycle kinetics or mutations leading to loss of drug-activation pathways or other processes that govern response factors. For PDT, efficacy depends mainly on selective uptake of photosensitizers by neoplastic cells, oxygenation levels, the suitable direction of irradiation and the availability of pathways to cell death that are highly conserved among mammalian cell types. While it is possible to engineer PDT-resistant cell types, current evidence suggests that the major obstacles to cancer control relate to drug, light and oxygen distribution. This review discusses some of the factors that can govern PDT-induced cell death.
One of the major obstacles to the chemotherapy of cancer is the development of ‘resistance’ to whatever protocol is being employed. We are asking the malignant cell population to be more responsive to therapy than are the normal host cells from which cancer arose. This result can sometimes be achieved, but the pathways involved vary widely. In the treatment of childhood leukemia, advantage is taken of the relatively large number of leukemic cells in the S phase of the cell cycle at any given time. These leukemic cells are selectively targeted by drugs that antagonize DNA synthesis, e.g., the antifols.1 Resistance develops when a population of malignant cells arises that exhibit impaired drug accumulation, high levels of a target enzyme, enzyme deletions or turn on alternative pathways that prevent death. Since normal host cells do not mutate, host toxicity is never lost.
Some of the newer forms of chemotherapy take advantage of pathways elicited by malignant cells to permit uncontrolled growth. There are a variety of tyrosine kinases whose inhibition can markedly retard tumor proliferation.2 Once again, clones of malignant cells can develop routes to proliferation that bypass blockages. Photodynamic therapy appears to be relatively free from development of these ‘classic’ routes to drug resistance. In a recent review on this topic,3 several mechanisms were proposed as possible routes to PDT resistance: impaired uptake or accelerated loss of photosensitizers, altered intracellular sensitizer distribution, decreased activation (presumably meaning insufficient light, oxygen or both) and increased inactivation (which can mean anything from photobleaching to quenching of ROS by intracellular scavengers). How significant these factors are in clinical PDT responses is not entirely clear. The most common factors that contribute to incomplete PDT responses appear to be inadequate light distribution and hypoxia.
While these various processes might be demonstrated in cell-culture systems where it is possible to create PDT-resistant’ cell lines by long-term periodic exposure to sub-lethal PDT doses,4,5 clinical examples of such phenomena are rarely reported. Successes in the use of PDT for cancer control relate to the ability of reactive oxygen (and sometimes nitrogen) species to kill most cell types. Selectivity is governed by the preferential accumulation of the photosensitizer by malignant cell types together with the requirement for light.6 The latter can be directed away from organs, e.g., adrenals, pituitary gland, liver and spleen that tend to nonspecifically accumulate photosensitizers.
The ability of a relatively weak photon flux to successfully bring about cell death was at first puzzling. The power level required is far below what is needed for ablative therapy, e.g., with a carbon dioxide laser. Based on complex affinity relationships, the useful photosensitizing agents selectively partition to sites within cells where oxidative photodamage is especially lethal. The routes to cell death are obviously widely-conserved since PDT appeared to be effective against a wide variety of malignant cell types, assuming that sufficient levels of drug, light and oxygen are provided.
We now know that the most effective photosensitizing agents tend to localize in mitochondria, the ER or lysosomes, where irradiation brings about the initiation of apoptosis.7 This widely-conserved process irreversibly leads to cell death.8 Moreover, the process is autocatalytic, requiring release of only low levels of at trigger, e.g., cytochrome c, for its initiation.9 Since cytochrome c is a mitochondrial protein, it is not surprising that one example of PDT resistance involved the formation of ‘dense’ mitochondria.10 Other possible sources of resistance might be presence of high levels of quenchers of reactive oxygen species (ROS), e.g., glutathione. Highly-pigmented tumors will also tend to be resistant, since light penetration will be thereby limited.
As cells die, a series of events will occur as a response to apoptosis and other phenomena associated with cell death. The activation of executioner caspases during the process of apoptosis is known to result in the rapid activation of a large number of other enzymes and signaling pathways.11 All of this is downstream from the initial trigger for apoptosis, whatever that might be. Autophagy has been identified as an early response to photodamage. This is a process whereby cellular organelles and other components are engulfed by membranes that then fuse with lysosomes. The subsequent release of lysosomal proteases results in the contents of the autophagosome being degraded and recycled. Since autophagy is often cytoprotective,12,13 a mechanism can be proposed whereby photodamaged mitochondria or ER are sequestered and recycled before they can trigger apoptosis. Cells with an intrinsic overexpression of the autophagic process may have an enhanced ability to circumvent phototoxicity
The first report on apoptosis as a factor in PDT-induced cell death was provided by Oleinick's group.14 The field had advanced to the point that an electron microscope was no longer needed to detect apoptotic cells. Cleavage of DNA by endonucleases leads to a DNA fragmentation pattern that can be observed using gel electrophoresis. An explanation of the initiation of apoptosis after photodamage was provided when it was noted that one target for PDT was the anti-apoptotic protein Bcl-2.15 This protein was altered by photodamage, leaving pro-apoptotic proteins, e.g., Bad and Bax, unaffected and thereby capable of initiating apoptosis without interference. Oleinick's group later discovered that the interaction between PDT and Bcl-2 led to inactivation of this protein via formation of high molecular-weight polymers.16
The loss of functional Bcl-2 removes an inhibitory control on apoptosis and can also activate autophagy.17 Since Bcl-2 is found in both mitochondria and the ER, photodamage to the protein at either site can promote an apoptotic response. Cytochrome c, a trigger for apoptosis, is located in mitochondria and its release after mitochondrial photodamage is another efficient method for initiation of the apoptotic process. It is interesting to note that mutations leading to altered Bcl-2 levels are not necessarily correlated with PDT-resistance. Responses can be positively or negatively correlated with Bcl-2 content of malignant cell types.18,19
The route from lysosomal photodamage to apoptosis is less direct. When the level of lysosomal photodamage is sufficient to alter membrane permeability to large molecules, one result is the release of proteases into the cytoplasm. These proteases can then bring about cleavage of the pro-apoptotic protein Bid to form a ‘truncated’ protein termed t-Bid.20,21 This, in turn, interacts with the mitochondrial membrane so as to promote release of cytochrome c, and the inevitable triggering of apoptosis. Lesser levels of photodamage can result in a temporary loss of the pH gradient required to keep the lysosomal pH at a lower value than predominates in the cytoplasm, but this is reversible if the degree of photodamage is insufficient to cause protease release.22
Important targets for photodamage leading to apoptotic death are therefore mitochondria, lysosomes or the ER. One study indicated that when the plasma membrane is also a PDT target, this leads to decreased photokilling.23 The photosensitizer utilized was found to migrate to the cytoplasm during irradiation and cause photodamage to elements of the apoptotic process. It is interesting to note that, of the photosensitizing agents currently showing evidence of clinical efficacy, the plasma membrane is seldom if ever a primary target. A diagram outlining these various death pathways is provided in Fig. 1. The ability of autophagy to protect cells from photokilling is shown in 2. Wild-type 1c1c7 murine hepatoma cells and an ATG7 knockdown were photosensitized with benzoporphyrin derivative and irradiated with progressively greater light doses (690 nm). The ‘shoulder’ on the dose-response curve, representing the cytoprotective effect of autophagy is absent in the knockdown.
Figure 1.

Pathways whereby photodamage to mitochondria, lysosomes or the ER can initiate apoptotic death, along with possible routes to circumvention of death signals.
An autophagy-related protein termed ATG7 was found to be required for release of large molecules, i.e., proteases, from photodamaged lysosomes.22 In the absence of ATG7, cells became much less responsive to lysosomal photodamage. This could provide a mechanism for PDT resistance in malignant cell types with reduced ATG7 expression. It must be remembered, however, that a major anti-tumor effect of PDT is associated with vascular shutdown and it is unknown whether ATG7 is involved in this phenomenon. Among the agents that target lysosomes are N-aspartyl chlorin e6, commonly known as NPe6,24 and the palladium bacteriopheophorbide TOOKAD.25 Both also exert an antitumor effect associated with vascular shutdown. Neither sensitizer can readily enter tumor cells, so the efficacy of these agents appears to depend almost solely on the vascular effect. Photosensitizers without these permeability problems, capable of creating lysosomal photodamage, may be expected to be less effective in cells with reduced ATG7 levels.
The role of different ROS in the efficacy of PDT continues to be explored. One potential problem is that studies involving cell culture are often carried out under atmospheric conditions, e.g., ~20% oxygen levels. Most in vivo systems operate at 5% O2 and tumor loci may be <1%.26 This can affect the ROS production required for phototoxicity. There have been few studies designed to assess the effect of low O2 levels on PDT efficacy. In one series of experiments involving cell-free systems, it was found that the photochemistry of •OH formation was more sensitive to hypoxia than 1O2 formation.27 Whether or not this will translate into differences in in vivo efficacy remains to be determined.
We recently reported that low-dose lysosomal photodamage can markedly potentiate photokilling by a subsequent PDT dose directed at mitochondria.28 This appears to be a synergistic phenomenon that is unrelated to the discovery that promotion of lysosomal permeability, e.g., by bafilomycin, can also promote PDF efficacy.29 The latter effect is related to relocation of ferrous iron from permeabilized lysosomes to mitochondria where Fenton chemistry can produce the highly toxic OH radical from PDT-induced hydrogen peroxide in mitochondrial structures. While this approach may be effective in cell culture, the idea of poisoning every lysosome in the host in an effort to promote •OH production in malignant tissues is not persuasive.
A more selective approach might therefore involve targeting lysosomes to photodamage using any of the currently-available photosensitizing agents that specifically localize in these organelles. The list of such agents includes the chlorin derivative N-aspartyl chlorin e6 (NPe6), a galactose derivative of the pyropheophorbide HPPH30 and the phenothiazinium compound EtNBS.31 NPe6, with 4 carboxyl substituents, is too polar to readily penetrate cells and is mainly effective for vascular shutdown, but high extracellular levels can be used in cell culture for lysosomal targeting. In this regard, Cincotta et al. reported a synergistic effect when murine tumors were sequentially photodamaged by EtNBS followed by mitochondrial photodamage in vivo.32 At the time, there was no obvious mechanism to account for this finding.
With regard to possibilities for malignant cell types to escape photokilling, a few possibilities are obvious. A review cited above has summarized many of these,3 but newer information can now be taken into account. The vascular shut-down associated with PDT does not appear to be a likely site for PDT-resistance phenomena and is beyond the scope of this material. It has been postulated that PDT-resistant tumor cells tend to exhibit an elevated capacity for autophagy.33 This would lead to scavenging and recycling of damaged organelles, e.g., mitochondria, before pro-apoptotic molecules could be released. Cells defective in elements of the apoptotic process could exhibit PDT resistance. As noted above, ATG7 is required for release of apoptogenic proteases from lysosomes. Loss of ATG7 could therefore both promote PDT responses via the loss of the cytoprotective process of autophagy, but also reduce efficacy of PDT directed against lysosomes.
Agostinis’ group have identified a collection of signals that can influence the outcome of photodynamic processes.34-36 Oleinick's group had identified similar influences on PDT efficacy in cell culture in earlier studies.37 Girotti has explored the ability of nitric oxide, generated as a response to stress pathways, to have a cytoprotective effect.38 Many other such responses to PDT have been identified although their role as determinants of clinical responses is unknown.
The major detriments to PDT efficacy in the clinic may be related to lack of appropriate oxygenation levels, poor drug distribution or light penetration. While it is possible to generate ‘PDT-resistant’ cells, the level of resistance appears to be far less than what can be obtained with chemotherapy where lines resistant to 1000-fold increased levels of drugs have been reported.39 There may be rare instances of genetic factors involved in failures of PDT responses but to date the major factors remain hypoxia and/or inadequate numbers of photons of the appropriate wavelength reaching sites of photosensitizer accumulation. The most relevant factor in PDT responses may be the capacity for autophagic responses. Loss of autophagy-associated factors could affect photokilling involving lysosomal targets. Accelerated autophagic responses appear capable of limiting efficacy of photosensitizers. Since autophagy is dependent on functional lysosomes, the lysosome does become a tempting target for optimal PDT efficacy. In this context, it should not be forgotten that targeting of the tumor vasculature by PDT remains a potent route to cancer control, as is pointed out in Ref. 25.
Cell culture systems involving exploration of cell death processes do not always reflect what is pertinent in animal tumor models or in the clinic. There have recently been some studies in 3-dimensional models that more closely mimic the microenvironment of malignant cell lines and may offer additional advantages when attempting to translate studies done on 2D cultures to clinical relevance. For example, we had shown that a protocol involving a high drug dose, using the photosensitizer BPD could suppress the cytoprotective effects of autophagy; coupled with a correspondingly reduced light dose, this protocol promoted PDT efficacy on 2D culture.13 But this procedure was ineffective in a 3D model,40 a result likely related to the poor transmission of light through tissues containing a photosensitizer with a very substantial extinction coefficient.
The latter study provides an introduction to a newer approach to exploration of factors and determinants relating to PDT efficacy. While studies on monolayers of cells in 2D culture have been useful in assessing sub-cellular photosensitizer localization, modes of photokilling, protective effects than can be elicited and down-stream consequences, it is true that the microenvironment of the tumor cell is not well maintained in such models. Some approaches to more closely mimicking the in vivo situation have involved studies on 3D and microfluidic models.41-43 The goal is obtaining information by techniques that would be difficult to apply to whole-animal or clinical studies. While outcomes may suggest means for enhancing the anti-tumor effects of photodynamic therapy, an evaluation of relevance will come from translational research involving clinical experimentation.
Supplementary Material
{kind=link}
Figure 2.
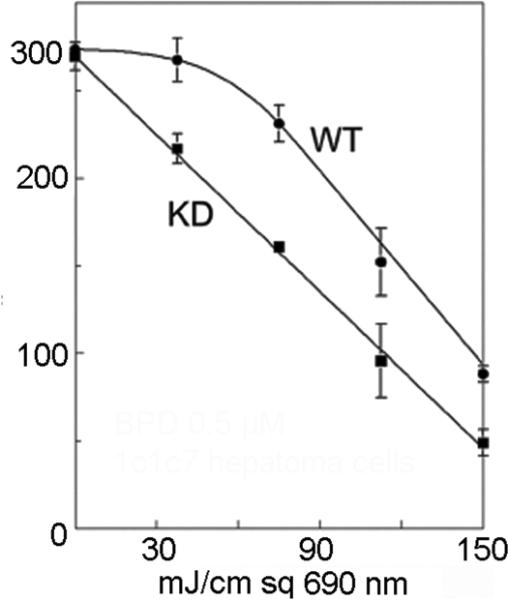
PDT dose-response curves for wild-type 1c1c7 cells and an ATG7 knockdown. This illustrates the cytoprotective effect of autophagy. The ‘shoulder’ on the dose-response curve is lost when autophagy is suppressed.
References
- 1.Visentin M, Zhao R, Goldman ID. The antifolates. Hematol. Oncol. Clin. North Am. 2012;26:629–648. doi: 10.1016/j.hoc.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drake JM, Lee JK, Witte ON. Clinical targeting of mutated and wild-type protein tyrosine kinases in cancer. Mol. Cell. Biol. 2014;34:1722–1732. doi: 10.1128/MCB.01592-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casas A, Di Venosa G G, Hasan T, Batlle A. Mechanisms of resistance to photodynamic therapy. Curr. Med. Chem. 2011;18:2486–2515. doi: 10.2174/092986711795843272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh G, Wilson S. Sharkey, Browman G, Deschamps P. Resistance to photodynamic therapy in radiation induced fibrosarcoma-1 and chinese hamster ovary-multi-drug resistant cells in vitro. Photochem. Photobiol. 1991;54:307–312. doi: 10.1111/j.1751-1097.1991.tb02021.x. [DOI] [PubMed] [Google Scholar]
- 5.Luna M, Gomer C. Isolation and initial characterization of mouse tumor cells resistant to porphyrin mediated photodynamic therapy. Cancer Res. 1991;51:4243–4249. [PubMed] [Google Scholar]
- 6.Dougherty TJ. Photodynamic Therapy-new approaches. Semin. Surg. Oncol. 1989;5:6–16. doi: 10.1002/ssu.2980050104. [DOI] [PubMed] [Google Scholar]
- 7.Agostinis P, Berg K, Cengel K, Foster T, Girotti A, Gollnick S, Hahn S, Hamblin M, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson B, Golab J. Photodynamic therapy of cancer: an update. CA Cancer J. Clin. 2011;61:250–281. doi: 10.3322/caac.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.WIlliams GT. Programmed cell death: apoptosis and oncogenesis. Cell. 1991;65:1097–1098. doi: 10.1016/0092-8674(91)90002-g. [DOI] [PubMed] [Google Scholar]
- 9.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 10.Sharkey S, Wilson B, Moorehead R, Singh G. Mitochondrial alterations in Photodynamic Therapy resistant cells. Cancer Res. 1993;53:4994–4999. [PubMed] [Google Scholar]
- 11.Dix MM, Simon GM, Cravatt BF. Verfaillie T, de Witte PA, Piette J, Agostinis P, editors. Global mapping of the topography and magnitude of proteolytic events in apoptosis, Autophagy pathways activated in response to PDT contribute to cell resistance against ROS damage. J. Cell Mol. Med. 2011;15:1402–1414. doi: 10.1111/j.1582-4934.2010.01118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inguscio V, Panzarini E, Dini L. Autophagy Contributes to the Death/Survival Balance in Cancer PhotoDynamic Therapy. Cells. 2012;1:464–491. doi: 10.3390/cells1030464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrzejak M, Price M, Kessel DH. Apoptotic and autophagic responses to photodynamic therapy in 1c1c7 murine hepatoma cells. Autophagy. 2011;7:979–984. doi: 10.4161/auto.7.9.15865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal ML, Clay ME, Harvey EJ, Oleinick NL. Photodynamic therapy induces rapid cell death by apoptosis in L5178Y mouse lymphoma cells. Cancer Res. 1991;51:5993–5936. [PubMed] [Google Scholar]
- 15.Kim H-RC, Luo Y, Li G, Kessel D. Enhanced apoptotic response to photodynamic therapy after bcl-2 transfection. Cancer Res. 1999;59:3429–3432. [PMC free article] [PubMed] [Google Scholar]
- 16.Xue L-Y, Chiu S-M, Oleinick NL. Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene. 2001;20:3420–3427. doi: 10.1038/sj.onc.1204441. [DOI] [PubMed] [Google Scholar]
- 17.Sinha S, Levine B. The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene. 2008;27(Suppl 1):S137–148. doi: 10.1038/onc.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He M. Agarwal J, Larkin H, Friedman L, Xue L L, Oleinick NL. The induction of partial resistance to photodynamic therapy by the protooncogene Bcl-2. Photochem. Photobiol. 1996;64:845–852. doi: 10.1111/j.1751-1097.1996.tb01845.x. [DOI] [PubMed] [Google Scholar]
- 19.Kawaguchi T, Yamamoto S, Naka N, Okishio K, Atagi S, Ogawara M, Hosoe S, Kawahara M, Furuse K. Immunohistochemical analysis of Bcl-2 protein in early squamous cell carcinoma of the bronchus treated with photodynamic therapy. Br. J. Cancer. 2000;82:418–423. doi: 10.1054/bjoc.1999.0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reiners J, Caruso J, Mathieu P, Chelladurai B, Yin X, Kessel D. Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves Bid cleavage. Cell Death Differ. 2002;9:934–944. doi: 10.1038/sj.cdd.4401048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu SM, Xue LY, Lam M, Rodriguez ME, Zhang P, Kenney ME, Nieminen AL, Oleinick NL. A requirement for bid for induction of apoptosis by photodynamic therapy with a lysosome- but not a mitochondrion-targeted photosensitizer. Photochem. Photobiol. 2010;86:1161–1173. doi: 10.1111/j.1751-1097.2010.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kessel D, Price M, Reiners J. ATG7 deficiency suppresses apoptosis and cell death induced by lysosomal photodamage. Autophagy. 2012;8:1333–1341. doi: 10.4161/auto.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kessel D, Luo Y, Deng Y, Chang CK. The role of subcellular localization in initiation of apoptosis by photodynamic therapy. Photochem. Photobiol. 1997;65:422–426. doi: 10.1111/j.1751-1097.1997.tb08581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamura H, Suzuki Y, Takeichi M, Saito T, Takayama M, Aizawa K. Morphologic evaluation of the antitumor activity of photodynamic therapy (PDT) using mono-L-aspartyl chlorin e6 (NPe6) against uterine cervical carcinoma cell lines. Int. J. Gynecol. Cancer. 2002;12:177–186. doi: 10.1046/j.1525-1438.2002.01087.x. [DOI] [PubMed] [Google Scholar]
- 25.Madar-Balakirski N, Tempel-Brami C, Kalchenko V, Brenner O, Varon D, Scherz A, Salomon Y. Permanent occlusion of feeding arteries and draining veins in solid mouse tumors by vascular targeted photodynamic therapy (VTP) with Tookad. PLoS One. 2010;5:e10282. doi: 10.1371/journal.pone.0010282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell. Biol. 2014;24:472–478. doi: 10.1016/j.tcb.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price M, Heilbrun L, Kessel D. Effects of the oxygenation level on formation of different reactive oxygen species during photodynamic therapy. Photochem. Photobiol. 2013;89:683–686. doi: 10.1111/php.12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kessel D, Reiners J. Enhanced Efficacy of Photodynamic Therapy via a Sequential Targeting Protocol. Photochem. Photobiol. 2014;90:889–895. doi: 10.1111/php.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saggu S, Hung H, Quiogue G, Lemasters J, Nieminen A. Lysosomal signaling enhances mitochondria-mediated photodynamic therapy in A431 cancer cells: role of iron. Photochem. Photobiol. 2012;88:461–468. doi: 10.1111/j.1751-1097.2012.01081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng X, Morgan J, Pandey SK, Chen Y, Tracy E, Baumann H, Missert JR, Batt C, Jackson J, Bellnier DA, Henderson BW, Pandey RK. Conjugation of 2-(1′-hexyloxyethyl)- 2-devinylpyropheophorbide-a (HPPH) to carbohydrates changes its subcellular distribution and enhances photodynamic activity in vivo. J. Med. Chem. 2009;52:4306–4318. doi: 10.1021/jm9001617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Georgakoudi I, Foster TH. Effects of the subcellular redistribution of two nile blue derivatives on photodynamic oxygen consumption. Photochem. Photobiol. 1998;68:115–122. [PubMed] [Google Scholar]
- 32.Cincotta L, Szeto D, Lampros E, Hasan T, Cincotta AH. Benzophenothiazine and benzoporphyrin derivative combination phototherapy effectively eradicates large murine sarcomas. Photochem. Photobiol. 1996;63:229–237. doi: 10.1111/j.1751-1097.1996.tb03019.x. [DOI] [PubMed] [Google Scholar]
- 33.Wei MF, Chen MW, Chen KC, Lou PJ, Lin SY, Hung SC, Hsiao M, Yao CJ, Shieh MJ. Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy. 2014;10:1179–1192. doi: 10.4161/auto.28679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dewaele M, Martinet W, Rubio N, Verfaillie T, de Witte PA, Piette J, Agostinis P. Autophagy pathways activated in response to PDT contribute to cell resistance against ROS damage. J. Cell Mol. Med. 2011;15:1402–1414. doi: 10.1111/j.1582-4934.2010.01118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garg AD, P, Agostinis P. ER stress, autophagy and immunogenic cell death in photodynamic therapy-induced anti-cancer immune responses. Photochem. Photobiol. Sci. 2014;13:474–487. doi: 10.1039/c3pp50333j. [DOI] [PubMed] [Google Scholar]
- 36.Rubio N, Verrax J, Dewaele M, Verfaillie T, Johansen T, Piette J, Agostinis P. p38(MAPK)-regulated induction of p62 and NBR1 after photodynamic therapy promotes autophagic clearance of ubiquitin aggregates and reduces reactive oxygen specieslevels by supporting Nrf2-antioxidant signaling. Free. Radic. Biol. Med. 2014;67:292–303. doi: 10.1016/j.freeradbiomed.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 37.Xue L, He J, Oleinick NL. Promotion of photodynamic therapy-induced apoptosis by stress kinases. Cell Death. Differ. 1999;6:855–864. doi: 10.1038/sj.cdd.4400558. [DOI] [PubMed] [Google Scholar]
- 38.Bhowmick R, Girotti AW. Cytoprotective signaling associated with nitric oxide upregulation in tumor cells subjected to photodynamic therapy-like oxidative stress, Free Radic. Biol. Med. 2013;57:39–48. doi: 10.1016/j.freeradbiomed.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inaba M, Johnson RK. Decreased retention of actinomycin D as the basis for cross-resistance in anthracycline- resistant sublines of P388 leukemia. Cancer Res. 1977;37:4629–4634. [PubMed] [Google Scholar]
- 40.Rizvi I, Anbil S, Alagic N, Celli J, Zheng LZ, Palanisami A, Glidden MD, Pogue BW, Hasan T. PDT dose parameters impact tumoricidal durability and cell death pathways in a 3D ovarian cancer model. Photochem. Photobiol. 2013;89:942–52. doi: 10.1111/php.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lou X, Kim G, Yoon H, Koo Y, Lee EK, Kopelmanb R, Yoon E. A high-throughput photodynamic therapy screening platform with on-chip control of multiple microenvironmental factors. Lap Chip. 2014;14:892–901. doi: 10.1039/c3lc51077h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y, Yang X, Zou J, Jia C, Hu Y, Du H, Wang H. Evaluation of photodynamic therapy efficiency using an in vitro three-dimensional microfluidic breast cancer tissue model. Lab Chip. 2014 doi: 10.1039/c4lc01065e. In Press DOI: 10.1039/c4lc01065e. [DOI] [PubMed] [Google Scholar]
- 43.Anbil S, Rizvi I, Celli JP, Alagic N, Pogue BW, Hasan T. Impact of treatment response metrics on photodynamic therapy planning and outcomes in a three-dimensional model of ovarian cancer. J. Biomed Opt. 2013;18:098004. doi: 10.1117/1.JBO.18.9.098004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.