

Abstract
Leber congenital amaurosis (LCA) and retinitis pigmentosa (RP) are retinal degenerative diseases which cause severe retinal dystrophy affecting the photoreceptors. LCA is predominantly inherited as an autosomal recessive trait and contributes to 5% of all retinal dystrophies; whereas RP is inherited by all the Mendelian pattern of inheritance and both are leading causes of visual impairment in children and young adults. Homozygosity mapping is an efficient strategy for mapping both known and novel disease loci in recessive conditions, especially in a consanguineous mating, exploiting the fact that the regions adjacent to the disease locus will also be homozygous by descent in such inbred children. Here we have studied eleven consanguineous LCA and one autosomal recessive RP (arRP) south Indian families to know the prevalence of mutations in known genes and also to know the involvement of novel loci, if any. Complete ophthalmic examination was done for all the affected individuals including electroretinogram, fundus photograph, fundus autofluorescence, and optical coherence tomography. Homozygosity mapping using Affymetrix 250K HMA GeneChip on eleven LCA families followed by screening of candidate gene(s) in the homozygous block identified mutations in ten families; AIPL1 – 3 families, RPE65- 2 families, GUCY2D, CRB1, RDH12, IQCB1 and SPATA7 in one family each, respectively. Six of the ten (60%) mutations identified are novel. Homozygosity mapping using Affymetrix 10K HMA GeneChip on the arRP family identified a novel nonsense mutation in MERTK. The mutations segregated within the family and was absent in 200 control chromosomes screened. In one of the eleven LCA families, the causative gene/mutation was not identified but many homozygous blocks were noted indicating that a possible novel locus/gene might be involved. The genotype and phenotype features, especially the fundus changes for AIPL1, RPE65, CRB1, RDH12 genes were as reported earlier.
Introduction
Retinal degenerations are the major cause of incurable blindness characterized by loss of photoreceptor cells and present with both genetic and phenotypic heterogeneity [1]. Leber congenital amaurosis (LCA; [OMIM] #204000) is an inherited retinal disease characterized by severe visual loss at birth, nystagmus, and minimal or absent recordable responses on the electroretinogram (ERG) before or by one year of age and accounting for 5% of all retinal dystrophies [2, 3]. LCA presents with variety of fundus changes e.g. the marbleized fundus appearance in CEP290 gene mutation, RPE atrophy, arteriolar narrowing, pigmentation in fundus in RPE65 gene involvement, granular pigmentation in GUCY2D and peripheral coats like vasculopathy in CRBI mutation etc [4]. Some of other clinical findings include high hypermetrophia, oculodigital signs, keratoconus and cataract [3]. The reported prevalence of the disease is 1:50,000–100,000 [5]. Retinitis pigmentosa (RP; [OMIM] #268000) also a retinal degenerative disease, is characterized by progressive degeneration of rods followed by cones, thereby affecting the night and peripheral vision initially and later central vision as well and the classic fundus appearance reveals dark pigmentary clumps in the mid-periphery (“Bone spicules”), attenuated retinal vessels, waxy optic disc pallor. Some of the secondary features of RP include cystoids macular edema, cataract, fine pigmented vitreous cells [6]. The age of onset of RP varies from very early childhood, to sixth decade of life [7] and affects 1 in 2500–7000 individuals in general population [8]. A recent epidemiological survey in Danish population has revealed the prevalence of generalized retinal dystrophies as 1:3454 [9].
LCA is usually inherited as an autosomal recessive disease, but few cases of dominant inheritance are also reported [10]. So far twenty-three genes are implicated in LCA. These candidate genes have been identified by using various methodoliges like, either by candidate gene screening, or linkage studies on large families or homozygosity mapping on nuclear families using either microsatellite markers or SNP microarrays, or screening genes which are involved in retinal function/tissue specific expression, or recently by whole exome seqeuncing [11–15]. These genes account for 70% of LCA cases but the mutation frequency vary among different ethnic populations [16]. The non-syndromic forms of RP are predominantly inherited as either autosomal dominant, autosomal recessive or X-linked recessive, but rarer forms such as X-linked dominant, mitochondrial, and digenic (due to mutations in two different genes) have also been reported [17]. Till date, seventy four loci and sixty seven causative genes have been identified for non-syndromic RP [18] and the prevalence of these known genes varies in different populations [16, 19]. The phenotypic and genetic heterogeneity of LCA, RP and various other inherited retinal dystrophies contribute to complexity of the clinical diagnosis and make molecular testing technically challenging. Also there is considerable overlap between the genotype and phenotype; the same gene may present with different phenotypes in the retinal degenerative disease (RDD) spectrum or the same gene may cause an isolated RDD or systemic disease along with RDD [20].
Identifying the causative gene would be very helpful in confirming the diagnosis at the molecular level, aiding in accurate genetic and reproductive counseling, carrier testing, prenatal testing and also predicting the prognosis of the disease [21]. Recent success in gene replacement therapy for RPE65 on patients with LCA resulting in slight improvement of vision has opened the possibility and holds promise as potential treatment modality for retinal dystrophies in the future [22–25]. These trials are proof-of-concept for gene transfer as a viable therapy for an entire family of eye diseases, thus proving the essentiality for molecular diagnosis and would be very vital for offering gene based therapies in future.
Homozygosity mapping using SNP arrays or microsatellite markers serves as a powerful tool, where the stretches of homozygous blocks indicate potential candidate gene/locus in autosomal recessive disease [26]. Consanguineous families, populations/communities practicing consanguinity for many generations and geographically isolated populations with inbreed marriages are good candidates for homozygosity mapping because there is an increased percentage of homozygosity in their genome due to identity by descent [27]. Homozygosity mapping has been extensively used to map disease loci for autosomal recessive diseases and there are reports of homozygosity mapping on autosomal recessive RP families from India. [28–30]. We have also previously reported identification of a novel missense mutation leading to activation of cryptic splice site in LCA5 in a consanguineous LCA family using homozygosity mapping [31]. There are reports from India on mutational screening of few genes for LCA by direct sequencing of the coding regions [32, 33], allele specific ligation assay [34], and APEX chip screening where reported mutations were screened [35]. Homozygosity mapping however adds on to the advantage that along with identifying the reported or novel mutations in the known candidate gene(s), novel loci/genes can also be mapped [36].
Here we have performed homozygosity mapping using Affymetrix 250K and 10K HMA GeneChip in eleven LCA and one arRP consanguineous south Indian families, respectively to know the prevalence of mutations in known genes and to know involvement of novel loci, if any.
Materials and Methods
Subjects
Eleven LCA and one arRP consanguineous families were enrolled in the study. Complete ophthalmic examination was carried out for all the affected individuals that included electroretinogram (ERG), fundus photograph, fundus auto fluorescence (FAF) in all patients and optical coherence tomography (OCT) where ever possible. Blood (10ml) was collected from all the affected individuals, unaffected siblings and parents after obtaining written informed consent. The study was approved by the Vision Research Foundation’s Institutional Review Board (IRB) and ethics committee and all the procedures were performed in accordance with institutional guidelines and the Declaration of Helsinki.
Homozygosity mapping
Genomic DNA was extracted using Nucleospin Blood XL kit (Macherey-Nagel, GmbH, Düren, Germany) according to the manufacturer’s instructions. The eleven LCA families (LCA1-LCA11) were genotyped for 262,000 SNPs using GeneChip Human Mapping 250K NspI Array (Affymetrix, Santa Clara, CA) and the individuals from the arRP family (arRP1) were genotyped for 11,555 SNPs using GeneChip Human Mapping 10K XbaI Array (Affymetrix, Santa Clara, CA) according to the manufacturer’s protocol.
SNP Genotyping was done on one or more affected family members along with an unaffected sibling. Following genotyping using 250K NspI GeneChip, the homozygous regions were analysed using Genotyping Console v4.0 (Affymetrix, Santa Clara, CA). The internal quality control check was set as 90%. Loss of heterozygosity (LOH) score is a measure for the likelihood of a stretch of SNPs to be homozygous based on the population SNP allele frequencies and a score of ≥15 is considered to be significant [37, 38]. Homozygous stretches between the affected and the unaffected were compared by LOH status. The homozygous blocks in the known LCA candidate genes loci and all other homozygous blocks were noted. We first screened the known LCA gene present in the largest homozygous block, followed by others, if required. When the causative mutation was identified, segregation analysis in the family members and control screening was performed to confirm its pathogenicity.
For the arRP1 family, that was genotyped using 10K HMA GeneChip, three affected and one unaffected members were taken for analysis. The internal quality control check was set as 90%. CEL files generated for each sample were analyzed using GTYPE software. The genotype generated was exported to excel sheet for further analysis. Here, the data was first sorted according to chromosome number and then by cytoband position (p arm and q arm). The sorted data was compared between the affected and unaffected for large continuous stretch of homozygous regions (consecutive SNP being homozygous). Chromosomal segments were considered to be homozygous if they had ≥39 consecutive SNPs homozygous since the likelihood of this to occur by chance is 1:100 in consanguineous families [39].
Mutation Analysis
Primers were designed using Primer 3 (v. 0.4.0) software [40] for the exons along with 50bp of flanking intronic regions for the LCA and arRP candidate genes. The exons were PCR amplified and sequenced using ABI 3100 Avant Genetic Analyser (Applied Biosystems, Foster City, CA). Segregation analysis within the family and control screening in 200 chromosomes was performed for the identified mutation(s) by direct sequencing.
Bioinformatics analysis
The intronic mutations were analysed by Human Splice Finder 2.4.1 [41, 42] and Mutation taster [43] for possible splicing defects and the missense mutations were anlaysed with PolyPhen-2 [44] and SIFT [45] to predict their possible effect on structure and function of the protein.
RNA extraction and cDNA analysis
Heparin blood samples were allowed to stand at room temperature for one hour and then the buffy coat collected separately. RNA was extracted using Nucleospin RNA II kit (Macherey-Nagel, GmbH, Düren, Germany). The RNA was converted to cDNA using Verso cDNA kit (Fischer Scientific, Surrey, U.K) and the cDNA amplified using specific primers encompassing exons 11, 12, and 13 of IQCB1 gene.
Results
Molecular Diagnosis
We performed the homozygosity mapping for the eleven LCA families with 250K HMA GeneChip on 32 individuals of which 23 were affected and 9 unaffected siblings and with 10K HMA GeneChip for three affected individuals and one unaffected individual in the arRP family. In each of the LCA family, we were able to identify on an average of about fifteen homozygous blocks ranging in size from 1Mb to 33 Mb. Out of eleven LCA families, we identified the causative mutation in ten families (90%), AIPL1 mutation in three, RPE65 mutation in two, and CRB1, GUCY2D, IQCB1, RDH12, SPATA7 mutation in one family each, respectively. In the arRP family, we identified a novel nonsense mutation in MERTK. Table 1 shows the list of known LCA candidate genes present within the homozygous blocks in each LCA and arRP family. Table 2 shows the total number of homozygous blocks >1Mb in the LCA and arRP families. Table 3 lists the mutations identified in LCA and arRP families. All the novel mutations are submitted in Leiden Open Variation Database (LOVD) v.3.0 [46].
Table 1. Size of homozygous blocks and the known LCA candidate genes identified in LCA families and the arRP family.
| S.No | Family ID | Number of Affected individuals taken for analysis | Size of the homozygous block in which known candidate gene(s) were present (Mb) | Chromosome location | Genes Screened | Gene reference ID |
|---|---|---|---|---|---|---|
| 1. | LCA-1 | 2 | 13 | 1p31.3 | RPE65 | NM_000329.2 |
| 2. | LCA-2 | 4 | 26 | 1q31.3 | CRB1 | NM_001257965.1 |
| 3. | LCA-3 | 2 | 3 | 17p31.1 | GUCY2D | NM_000180.3 |
| 4. | LCA-4 | 2 | 4.7 | 3q13.3 | IQCB1 | NM_001023570.2 |
| 5. | LCA-5 | 2 | 3.7 | 17p13.2 | AIPL1 | NM_014226.3 |
| 6. | LCA-6 | 2 | 4.05 | 14q11.2 | RPGRIP1 | NM_020366.3 |
| LCA-6 | 2 | 1 | 2q13 | MERTK | NM_006343.2 | |
| 7. | LCA-7 | 2 | 6 | 14q11.2 | RPGRIP1 | NM_020366.3 |
| LCA-7 | 2 | 1.3 | 14q24.1 | RDH12 | NM_152443.2 | |
| 8. | LCA-8 | 1 | 5 | 17p13.2 | AIPL1 | NM_014226.3 |
| 9. | LCA-9 | 2 | 30 | 1p31.3 | RPE65 | NM_000329.2 |
| 10. | LCA-10 | 2 | 4.9 | 17p13.2 | AIPL1 | NM_014226.3 |
| 11. | LCA-11 | 2 | 6 | 14q31.3 | SPATA7 | NM_018418.4 |
| 12. | arRP 1 | 3 | 1.1 | 2q13 | MERTK | NM_006343.2 |
Table 2. Total number of homozygous blocks ≥ 1Mb in the LCA and arRP families.
| S.No | Family ID | Number of ≥1Mb blocks |
|---|---|---|
| 1. | LCA-1 | 8 |
| 2. | LCA-2 | 7 |
| 3. | LCA-3 | 15 |
| 4. | LCA-4 | 8 |
| 5. | LCA-5 | 20 |
| 6. | LCA-6 | 16 |
| 7. | LCA-7 | 33 |
| 8. | LCA-8 | 28 |
| 9. | LCA-9 | 9 |
| 10. | LCA-10 | 15 |
| 11. | LCA-11 | 18 |
| 12. | arRP 1 | 1 |
Table 3. Mutations identified in LCA families and the arRP family.
| S.No | Family ID | Genes Screened | Exon/intron | Mutation identified | Predicted change in protein | Effect of identified sequence variations |
|---|---|---|---|---|---|---|
| 1. | LCA-1 | RPE65 | intron 8 | c.858+1G>T Reported [55] | (r.spl?) | Pathogenic |
| 2. | LCA-2 | CRB1 | Exon 9 | c.3307G>A Reported [47, 56] | p.(Gly1103Arg) | Pathogenic |
| 3. | LCA-3 | GUCY2D | Exon 3 | c.994delC Novel | p.(Arg332Alafs*63) | Pathogenic |
| 4. | LCA-4 | IQCB1 | intron 12 | c.1278+6T>A Novel | r.[1131_1278 del,1131_1278del] p.(Gln378Alafs*2) | Pathogenic |
| 5. | LCA-5 | AIPL1 | Exon 6 | c.834G>A Reported [35, 53, 57–60] | p.(Trp278*) | Pathogenic |
| 6. | LCA-6 | RPGRIP1 MERTK | - | Not identified | - | - |
| 7. | LCA-8 | AIPL1 | Exon 2 | c.247G>A Novel | p.(Glu83Lys) | Pathogenic |
| 8. | LCA-9 | RPE65 | Exon 13 | c.1409C>TReported [32] | p.(Pro470Leu) | Pathogenic |
| 9. | LCA-10 | AIPL1 | Exon 4 | c.613_622 delATCATCTGCC Novel | p.(Ile205*) | Pathogenic |
| 10. | LCA-11 | SPATA7 | intron 7 | c.913-2A>G Novel | (r.spl?) | Pathogenic |
| 11. | arRP1 | MERTK | Exon 4 | c.721C>T Novel | p.(Gln 241*) | Pathogenic |
| 12. | LCA-7 | RPGRIP1R DH12 | RDH12 intron 3 | c.344-8C>TNovel | (r.spl?) | Possibly pathogenic |
Segregation analysis (Fig 1a–1k) was performed in all the families and the mutation segregated within the family, with all the affected being homozygous for mutation, parent(s) being heterozygous carriers and the unaffected being either heterozygous for mutation or wild type. One hundred normal controls (200 chromosomes) were screened for the identified mutations and all were wild type.
Fig 1. Segregation analysis.

1a:arRP1 MERTK c.721C>T, 1b:LCA-1 RPE65 c.850+1G>T, 1c: LCA-2 CRB1 c.3307G>A, 1d:LCA-3 GUCY2D c.994delC, 1e:LCA-4 IQCB1 c.1278+6T>A, 1f:LCA-5 AIPL1 c.824G>A, 1g: LCA-7 RDH12 c.344-8C>T, 1h:LCA-8 AIPL1 c.247G>A, 1i:LCA-9 RPE65 c.1409C>T, 1j:LCA-10 AIPL1 c.613_622 delATCATCTGCC, 1k:LCA-11 SPATA7 c.913-2A>G. The arrow indicates the index case. The filled in circles and squares are affected females and males respectively. [M];[M]–affected with homozygous mutation, [M]; [=] –carries for any given mutation and [=]; [=] –wild type. Lines above the individual indicate availability of genotype.
The four intronic mutations; RPE65 c.858+1G>T, SPATA7 c.913-2A>G, IQCB1 c.1278+6T>A, RDH12 c.344-8C>T, are present either in the conserved splice acceptor or donor site or within ten bases of the intron following the exon. Analysis of cDNA prepared from lymphocytes of LCA-4 family members using specific primers encompassing IQCB1 exons 11–13, revealed a single transcript of 338bp in the affected and two transcripts of 338 and 487bp, respectively in the heretozygous carrier parents (Fig 2a). Direct sequencing revealed that in the proband, exon 12 has been completely deleted (338bp amplicon) whereas both the parents were heterozygous carriers (338bp deleted and 478 wild type amplicons, respectively; Fig 2b). This skipping of exon 12 in the affected is predicted to result in a truncated protein, p.(Gln378Alafs*2). The consequence of c.344-8C>T mutation on the splicing of the retinal-specific RDH12 gene could not been determined but the change was predicted to alter splicing. The two missense mutations, CRB1 c.3307G>A p.(Gly1103Arg) and AIPL1 c.247G>A p.(Glu83Lys) analysed with PolyPhen 2 [44] and SIFT [45] were both predicted to be probably damaging and deleterious (Table 4 details the results of the in-silico analysis).
Fig 2. 2% Agarose gel electrophoresis showing cDNA amplification of exon 11–13 of IQCB1.
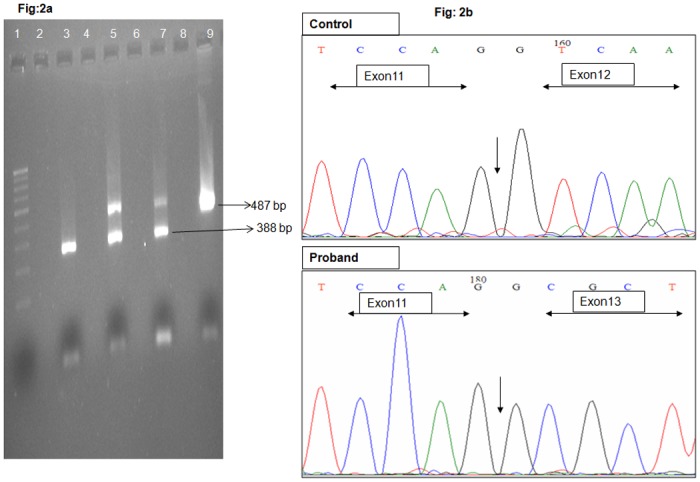
Lane 1-100bp ladder, Lane 3- Affected index case, Lane 5 & 7—Carrier parents, Lane 9—Control, Lane 2, 4, 6, 8—empty wells Fig 2b Eletrophoretogram trace showing the amplified cDNA of control and proband. In proband exon 11 is followed by exon 13 and exon 12 is completely deleted, whereas in control, exon 11, 12 and 13 is continuous. The end of exon 11 is marked in both the phoretograms.
Table 4. Probable effects of splice site mutations using HSF 2.4.1 and Mutation Taster and effects of missense mutations using PolyPhen and SIFT.
| S.No | Family ID | Gene | Mutation identified | HSF Wild type Consensous value (CV) | HSF Mutant consensous value (CV) | HSF delta CV (%) | MT Wild type scoring | MT Mutant scoring | MT Splice site change | cDNA Analysis | PolyPhen score | Polyphen predicted effect | SIFT score | SIFT Predicted effect |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | LCA-1 | RPE65 | c.858+1G>T | - | - | - | - | - | Likely to Disturb Normal Splicing, sequence motif lost | Not done | NA | NA | NA | NA |
| 2. | LCA-4 | IQCB1 | c.1278+6T>A | 79.28 | 75.78 | -4.42 | 0.36 | 0.95 | Donor increased | r.[1131_1278 del,1131_1278del]Exon12 skipping | NA | NA | NA | NA |
| 3. | LCA-7 | RDH12 | c.344-8C>T | 73.62 | 70.25 | -4.57 | 0.55 | 0.76 | Acceptor increased | Not done | NA | NA | NA | NA |
| 4. | LCA-11 | SPATA7 | c.913-2A>G | 86.72 | 57.09 | Site broken | 0.53 | 0.84 | Acceptor increased | Not done | NA | NA | NA | NA |
| 5. | LCA-2 | CRB1 | c.3307G>Ap.(Gly1103Arg) | NA | NA | NA | NA | NA | NA | NA | 0.91 | Probably damaging | 0 | Deleterious |
| 6. | LCA-8 | AIPL1 | c.247G>A p.(Glu83Lys) | NA | NA | NA | NA | NA | NA | NA | 1.0 | Probably damaging | 0 | Deleterious |
NA-not applicable
The human splice finder presents consensous value (CV) which indicate strength of the splice site range from 0 to 100. The splice sites of CV higher than 80 are considered as strong splice sites, 70–80 as less strong and 65–70 as weak, and a CV below 70 is considered to be non-functional [41]. The mutation taster (MT) scores the wild type and the mutant and a confidence score of >0.3 for the mutant indicates gain of completely new splice site [43].
Genotype-Phenotype correlation for the mutations identified in LCA
In this study, mutations in AIPL1 and RPE65 were identified in three and two families, respectively. Whereas mutations in other genes namely, GUCY2D, CRB1, IQCB1, RDH12, SPATA7 were identified in one family each.
Patients with AIPL1 mutations
In the three families, three different types of mutations were observed, a reported nonsense mutation, c.834G>A p.(Trp278*) in family LCA-5, a novel missense mutation, c.247G>A p.(Glu83Lys) in family LCA-8 and a novel 10-base pair deletion, c.613_622delATCATCTGCC p.(Ile205*) in family LCA-10. In LCA-5 family, both the affected siblings had normal disc, and mildly attenuated vessels. Yellowish atrophic patches were seen in the macular area in the younger sibling (10 yrs) (Fig 3a), while the elder sibling (14yrs) had atrophic macula with black pigments (Fig 3b). Both the siblings had salt and pepper fundus with bony spicules. In LCA-8 family there was only one affected person phenotyped at the age of 5 years, with normal disc, mildly attenuated vessels, and atrophic macula with peripheral RPE granularity. In LCA-10 family, the three affected siblings also had atrophic macular degeneration with bony spicules, attenuated vessels, all seen in their third decade of life. The cases in the genotyped families revealed three different mutations but were similar phenotypically with severity of the retinal changes increasing with age reflecting the progressive nature of the disease and macular degeneration being a characteristic feature in AILP1 mutation positive LCA cases.
Fig 3. Fundus photographs.
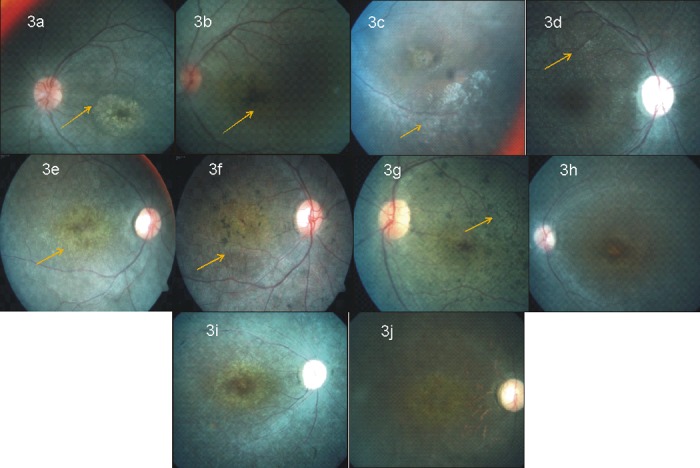
Fig 3a A 10yrs old female with c.824G>A p.(Trp278*) mutation in AIPL1 (LCA-5 family) showed normal disc, attenuated vessels, (arrow mark indicates) yellow patches in macula. Fig 3b A 14yrs old male with c.824G>A p.(Trp278*) mutation in AIPL1 (LCA-5 family, elder sibling) showed normal disc, attenuated vessels, (arrow mark indicates) black pigments in macula. Fig 3c A 18 yrs old female with c.850+1G>T (r.spl?) mutation in RPE65 (LCA-1 family) showed pallor disc, attenuated vessels with scar in the macula, peripheral RPE mottling (marked with arrow) Fig 3d A 28yrs old male with c.1409C>T p.(Pro470Leu) mutation in RPE65 (LCA-9 family) showed pallor disc, attenuated vessels, normal macula, with salt and pepper fundus. Arrow mark shows distinct pin head size yellow white dot like spots at the posterior pole. Fig 3e A 14 yrs old female with c.2971G>A p.(Gly991Arg) mutation in CRB1 (LCA-2 family) showed coin shaped pigment clumps and greyish atrophic changes seen in the macula, (arrow mark indicates the macula) Fig 3f A 18 yrs old female with c.2971G>A p.(Gly991Arg) mutation in CRB1 (LCA-2 family, elder sibling) showed pale disc, attenuated vessels, atrophic macula with nummular pigment clumps and greyish atrophic reflex (arrow mark indicates the macula) Fig 3g A 19yrs old male female with c.2971G>A p.(Gly991Arg) mutation in CRB1 (LCA-2 family, eldest sibling) showed coin shaped pigment clumps seen in the background (arrow mark indicates the coin shaped clumps) All the three affected siblings show progressive changes in macula with age for CRB1 mutation positive family. Fig 3h, 3i, 3j A 24 yrs old female, a 25 yrs old female and a 32 yrs old female with c.721C>T p.(Gln 241*) mutation in MERTK (arRP1 family) showing mild, milder and marked features of RP, respectively. Progressive changes with age in the macula are observed.
RPE65
In two families, we identified RPE65 mutation, a reported splice site mutation, c.858+1G>T (r.spl?) in family LCA-1 and a reported missense mutation, c.1409C>T p.(Pro470Leu) in family LCA-9. In LCA-1 family, both the affected siblings phenotyped in their second decade had pale disc with attenuated vessels, salt and pepper fundus with peripheral RPE mottling. The elder sibling also revealed macular scarring (Fig 3c) and the younger sibling had very few early alterations in the macula. In LCA-9 family, all the three affected siblings phenotyped in their third decade had pale disc, attenuated vessels, normal macula with salt and pepper appearance in the periphery. In both the families affected individuals had profound visual loss. The eldest sibling (28yrs) of LCA-9 family also showed presence of distinct pin head sized white spots at the posterior pole (Fig 3d).
CRB1
In family LCA-2 with four affected members, a reported missense mutation, c.2971G>A p.(Gly991Arg) was identified in CRB1. All the four affected members in their second decade had profound visual loss and all had a typical fundus picture of pale disc, para-arteriolar preservation of the retinal pigment epithelium (PPRPE), and atrophic macula with nummular pigment clumps and greyish atrophic reflex along with coin shaped pigment clumps seen at the background (Fig 3e, 3f and 3g).
GUCY2D
In family LCA3 with two affected members, a novel frameshift mutation, c.994delC p.(Arg332Alafs*63) was observed in GUCY2D. Both the siblings in their late teenage had profound visual loss and showed fundus picture of pale disc, minimal arteriolar attenuation and normal looking macula.
IQCB1
A novel IQCB1 splice mutation, c.1278+6T>A r.[1131_1278 del,1131_1278del] p. (Gln378Alafs*2) was seen in LCA-4 family. The two affected siblings showed pale disc, attenuated vessels, normal macula and plenty of hypo-pigmented lesion, tapetal reflex was seen at the background in the elder sibling (34y), whereas the younger sibling (27yrs) had little hypo-pigmentation.
RDH12
Family LCA-7 had a novel possible pathogenic variant in RDH12, c.344-8C>T (r.spl?). The two affected siblings, one aged 26yrs and other 10yrs showed normal disc, attenuated arteriolar vessels, and macula revealed small horizontal oval area (bull’s eye like lesion) along with metallic sheen in the background. Atrophic changes in the macula were seen in the first decade itself in the younger sibling.
SPATA7
A novel spice site mutation, c.913-2A>G (r.spl?) was seen in family LCA-11, with two affected siblings aged 8yrs and 2yrs. Fundus picture of both the siblings revealed presence of mild disc pallor, arteriolar attenuation and peripheral RPE mottling.
Phenotype of the MERTK mutation positive arRP family
In arRP1 family there were three affected siblings, two were in their second decade of life and the eldest sister was in her third decade of life. Fundus picture revealed pallor disc, marked attenuated vessels, atrophic macula, bone spicule pigment and widespread RPE atrophy. This family is marked by a progressive change in the fundus (Fig 3h, 3i and 3j).
The clinical details; refraction, visual acuity, nystagmus, ERG and fundus details of all the affected individuals are given in S1 Table.
Discussion
In our study, of eleven consanguineous LCA families analysed by homozygosity mapping followed by candidate gene screening, we identified the causative mutations in ten families (90%). Also we identified the causative gene and mutation in one arRP family studied. Homozygosity mapping involves detecting the disease loci by exploiting the fact that the adjacent region i.e short chromosomal segments surrounding the homozygous mutation had not been crossed over and the surrounding single nucleotide polymorphism (SNP) would also be in a homozygous state and these regions would be inherited by descent (Identical by descent) from a common ancestor [47]. For most of our families the largest homozygous blocks carrying the disease gene fell within the first three positions but there were also some exceptions, where the disease allele was not among largest homozygous blocks.
The significant homozygous blocks were in the average size from 1Mb to about 33Mb, differing for each family and harboring the candidate LCA gene(s). Also, when more number of affected members were genotyped, the number of homozygous blocks shared among the affected was less, enabling easier identification of the candidate locus/gene.
There were six novel mutations in six LCA families and one novel mutation in the arRP family identified in this study; AIPL1-2 mutations, and one each in GUCY2D, IQCB1, RDH12, SPATA7, and MERTK (in arRP family). Of the ten mutations identified in the eleven LCA families, two are splice site mutations, one each in RPE65, and SPATA7 and two intronic mutations within 10bp of the intron in IQCB1 and RDH12, respectively. These splice site mutations analysed with bioinformatics tools, HSF2.4.1 and Mutation taster 2 were predicted to result in loss of splicing, whereas the mutations within 10bp of the intron in RDH12 and IQCB1 were predicted to activate cryptic splice site. We performed cDNA analysis for the IQCB1 mutation, as this gene is expressed in lymphocyte as well. IQCB1 gene encodes for nephrocystin protein which interacts with calmodulin and retinitis GTPase regulator protein. Defects in this gene are reported for Senior-Loken Syndrome type 5 [48]. Splice site mutation has been previously reported in a nephronophthisis patient and also in two LCA families [49, 50]. In a study done by Estrada-Cuzcano et al [51], eleven IQCB1 mutations were identified in a cohort of 150 LCA patients. During revaluation, seven of the mutation positive cases were found to have developed renal complications, thus re-diagnosed to have Senior-Loken Syndrome, while rest of the four patients reported no kidney abnormalities but they had similar mutations found in nephronophthsis patients. In our cohort, the family LCA-4, with two affected siblings (sisters) was initially diagnosed with LCA and reported no renal abnormalities. However, when we recalled the family for cDNA analysis after identification of the mutation, the family reported that the proband now 34 years had a sudden onset of renal failure (both the kidneys) at the age of 31 years and is under treatment. We could perform the cDNA analysis in the younger affected sibling and the carrier parents only, and till now the younger sibling (29y) has no renal complications. cDNA analysis confirmed that the mutation, c.1278+6T>A activates cryptic splice site leading to complete skipping of exon 12 resulting in a predicted truncated protein p.(Gln378Alafs*2). IQCB1 mutation positive LCA patients may be at risk of developing renal abnormalities, however the onset of the renal failure is highly variable [51] and need to be counseled and managed appropriately. cDNA analysis for RDH12, c.344-8C>T mutation could not be done because the gene is not expressed in lymphocytes and has exclusive retinal expression. However, we consider this to be a possible pathogenic variant which might be causative for the disease phenotype; a) through homozygosity mapping we identified two large homozygous blocks spanning about 6Mb and 1.3 Mb containing two known LCA candidate genes, the larger block had RPGRIP1 and the smaller block had RDH12. Firstly, screening RPGRIP1 did not reveal any pathogenic variant, hence it was followed by screening RDH12, where we found the intronic variant, c.344-8 C>T, which segregated with disease phenotype in the family and was absent in 200 control chromosomes screened. b) in-silco analysis predicted the variant to activate the cryptic splice site affecting/altering the protein and c) phenotypically the fundus of both the affected sibs too showed maculopathy in the first decade of life, a feature previously observed in RDH12 mutation positive cases. [52].
Genotype-Phenotype correlation
In patients with AIPL1 mutations (three) atrophic macula and bony spicules were common features, as reported earlier [4, 53]. While fine pigments were seen in the periphery only in elder patients but these were not seen in younger patients. Patients with RPE65 mutation showed tapetal reflex, disc pallor, attenuated vessels, typical bony spicules with salt and pepper fundus and normal macula as described earlier [16]. Distinct yellow white dot like lesion appeared in eldest member of LCA-9 family as well as in the other family who had the same mutation from our previous study [32] (Fig 3d). Whether these particular RPE white dots are specific to this particular type of missense mutation or for mutations only in exon 13 is not known. In CRB1 mutation positive siblings showed typically described mild para-arteriolar preservation of the retinal pigment epithelium (PPRPE) in their fundus [54] along with coin shaped pigment clumps at the background. In RDH12 mutation positive patients too, pronounced maculopathy and bony spicules were observed as reported [52]. GUCY2D mutation positive patients showed normal macula and vessels and SPATA7 mutation positive patients showed mild disc pallor, arteriolar attenuation and peripheral RPE mottling.
Mutation negative family
In one family (LCA-6) we were unable to identify the causative gene/mutation in the known LCA candidate gene(s); there were two homozygous blocks with known LCA genes, RPGRIP1 and MERTK. These two did not harbor any pathogenic mutation, however there were fourteen other homozygous blocks shared between the affected and ranging in size from 1-7Mb with no known LCA candidate genes. The clinical details of the affected members of this family are given in S1 Table. Homozygosity mapping has revealed many homozygous blocks and the causative gene/mutation which may either be a novel gene or a gene involved in other retinal disease is most likely to be present in one of these blocks. We have however not screened the intronic and regulatory regions of the known candidate gene(s) in the family and hence cannot rule out the possibility of deep intronic mutations or mutations in regulatory regions that might be pathogenic. Nevertheless, homozygosity mapping has helped in indicating possible novel disease locus.
Conclusion
In our study, we performed the homozygosity mapping using 250K and 10K Affymetrix GeneChip for eleven consanguineous LCA families and one consanguineous arRP family, respectively. We were able to identify the mutations or likely disease-causing mutations in ten LCA and one arRP families (11 out of 12) (90%). Of the mutations identified 58% (7 out of 12) were novel involving seven different genes for LCA. The molecular diagnosis has not only confirmed the genetic heterogeneity and certain specific phenotypic features aiding in prognosis prediction but also helped in appropriate counseling. Absence of mutation in known candidate gene in one LCA family indicate involvement of further novel gene(s) in the disease.
Supporting Information
(XLS)
Acknowledgments
We thank all the patients and their family for their kind co-operation.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Indian Council of Medical Research, Government of India - 54/1/2010-BMS (icmr.nic.in/) to NS.
References
- 1. Chinchore Y, Mitra A, Dolph PJ. Accumulation of rhodopsin in late endosomes triggers photoreceptor cell degeneration. PLoS Genet. 2009;5(2):e1000377 10.1371/journal.pgen.1000377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dharmaraj SR, Silva ER, Pina AL, Li YY, Yang JM, Carter CR, et al. Mutational analysis and clinical correlation in Leber congenital amaurosis. Ophthalmic Genet. 2000;21(3):135–50. [PubMed] [Google Scholar]
- 3. Chung DC, Traboulsi EI. Leber congenital amaurosis: clinical correlations with genotypes, gene therapy trials update, and future directions. Journal of AAPOS: the official publication of the American Association for Pediatric Ophthalmology and Strabismus / American Association for Pediatric Ophthalmology and Strabismus. 2009;13(6):587–92. [DOI] [PubMed] [Google Scholar]
- 4. McKibbin M, Ali M, Mohamed MD, Booth AP, Bishop F, Pal B, et al. Genotype-phenotype correlation for leber congenital amaurosis in Northern Pakistan. Arch Ophthalmol. 2010;128(1):107–13. 10.1001/archophthalmol.2010.309 [DOI] [PubMed] [Google Scholar]
- 5. Allikmets R. Leber congenital amaurosis: a genetic paradigm. Ophthalmic genetics. 2004;25(2):67–79. [DOI] [PubMed] [Google Scholar]
- 6. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–809. [DOI] [PubMed] [Google Scholar]
- 7. Tsujikawa M, Wada Y, Sukegawa M, Sawa M, Gomi F, Nishida K, et al. Age at onset curves of retinitis pigmentosa. Archives of ophthalmology. 2008;126(3):337–40. 10.1001/archopht.126.3.337 [DOI] [PubMed] [Google Scholar]
- 8. Parmeggiani F. Clinics, epidemiology and genetics of retinitis pigmentosa. Current genomics. 2011;12(4):236–7. 10.2174/138920211795860080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bertelsen M, Jensen H, Bregnhoj JF, Rosenberg T. Prevalence of generalized retinal dystrophy in Denmark. Ophthalmic epidemiology. 2014;21(4):217–23. 10.3109/09286586.2014.929710 [DOI] [PubMed] [Google Scholar]
- 10. Nichols LL 2nd, Alur RP, Boobalan E, Sergeev YV, Caruso RC, Stone EM, et al. Two novel CRX mutant proteins causing autosomal dominant Leber congenital amaurosis interact differently with NRL. Hum Mutat. 2010;31(6):E1472–83. 10.1002/humu.21268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chiang PW, Wang J, Chen Y, Fu Q, Zhong J, Yi X, et al. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nature genetics. 2012;44(9):972–4. 10.1038/ng.2370 [DOI] [PubMed] [Google Scholar]
- 12. Mackay DS, Henderson RH, Sergouniotis PI, Li Z, Moradi P, Holder GE, et al. Novel mutations in MERTK associated with childhood onset rod-cone dystrophy. Mol Vis. 2010;16:369–77. [PMC free article] [PubMed] [Google Scholar]
- 13. Sergouniotis PI, Davidson AE, Mackay DS, Li Z, Yang X, Plagnol V, et al. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am J Hum Genet. 2011;89(1):183–90. 10.1016/j.ajhg.2011.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X, Wang H, Sun V, Tuan HF, Keser V, Wang K, et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. Journal of medical genetics. 2013;50(10):674–88. 10.1136/jmedgenet-2013-101558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Asai-Coakwell M, March L, Dai XH, Duval M, Lopez I, French CR, et al. Contribution of growth differentiation factor 6-dependent cell survival to early-onset retinal dystrophies. Human molecular genetics. 2013;22(7):1432–42. 10.1093/hmg/dds560 [DOI] [PubMed] [Google Scholar]
- 16. den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27(4):391–419. 10.1016/j.preteyeres.2008.05.003 [DOI] [PubMed] [Google Scholar]
- 17. Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12(4):238–49. 10.2174/138920211795860107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Available: https://sph.uth.edu/retnet/sum-dis.htm.
- 19. Kaplan J. Leber congenital amaurosis: from darkness to spotlight. Ophthalmic genetics. 2008;29(3):92–8. 10.1080/13816810802232768 [DOI] [PubMed] [Google Scholar]
- 20. Huang XF, Huang F, Wu KC, Wu J, Chen J, Pang CP, et al. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genetics in medicine: official journal of the American College of Medical Genetics. 2015;17(4):271–8. [DOI] [PubMed] [Google Scholar]
- 21. Millan JM, Aller E, Jaijo T, Grau E, Beneyto M, Najera C. [Genetic counselling in visual and auditory disorders]. Archivos de la Sociedad Espanola de Oftalmologia. 2008;83(12):689–702. [DOI] [PubMed] [Google Scholar]
- 22. Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, Roman AJ, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A. 2008;105(39):15112–7. 10.1073/pnas.0807027105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, et al. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther. 2009;20(9):999–1004. 10.1089/hum.2009.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, et al. Vision 1 year after gene therapy for Leber's congenital amaurosis. N Engl J Med. 2009;361(7):725–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cideciyan AV, Jacobson SG, Beltran WA, Sumaroka A, Swider M, Iwabe S, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci U S A. 2013;110(6):E517–25. 10.1073/pnas.1218933110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alkuraya FS. Homozygosity mapping: one more tool in the clinical geneticist's toolbox. Genetics in medicine: official journal of the American College of Medical Genetics. 2010;12(4):236–9. [DOI] [PubMed] [Google Scholar]
- 27. Woods CG, Cox J, Springell K, Hampshire DJ, Mohamed MD, McKibbin M, et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. American journal of human genetics. 2006;78(5):889–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lalitha K, Jalali S, Kadakia T, Kannabiran C. Screening for homozygosity by descent in families with autosomal recessive retinitis pigmentosa. Journal of genetics. 2002;81(2):59–63. [DOI] [PubMed] [Google Scholar]
- 29. Singh HP, Jalali S, Narayanan R, Kannabiran C. Genetic analysis of Indian families with autosomal recessive retinitis pigmentosa by homozygosity screening. Invest Ophthalmol Vis Sci. 2009;50(9):4065–71. 10.1167/iovs.09-3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kannabiran C, Singh H, Sahini N, Jalali S, Mohan G. Mutations in TULP1, NR2E3, and MFRP genes in Indian families with autosomal recessive retinitis pigmentosa. Molecular vision. 2012;18:1165–74. [PMC free article] [PubMed] [Google Scholar]
- 31. Ramprasad VL, Soumittra N, Nancarrow D, Sen P, McKibbin M, Williams GA, et al. Identification of a novel splice-site mutation in the Lebercilin (LCA5) gene causing Leber congenital amaurosis. Molecular vision. 2008;14:481–6. [PMC free article] [PubMed] [Google Scholar]
- 32. Mamatha G, Srilekha S, Meenakshi S, Kumaramanickavel G. Screening of the RPE65 gene in the Asian Indian patients with leber congenital amaurosis. Ophthalmic Genet. 2008;29(2):73–8. 10.1080/13816810802008259 [DOI] [PubMed] [Google Scholar]
- 33. Ramana Anandula Venkata RKCS, et al. RPE65 gene mutation: A rare event in lebers Congenital Amaurosis patients in Indian subcontinent. Research Journal of Biotechnology. May 2012;7 (2):18–21. [Google Scholar]
- 34. Sundaresan P, Vijayalakshmi P, Thompson S, Ko AC, Fingert JH, Stone EM. Mutations that are a common cause of Leber congenital amaurosis in northern America are rare in southern India. Mol Vis. 2009;15:1781–7. [PMC free article] [PubMed] [Google Scholar]
- 35. Verma A, Perumalsamy V, Shetty S, Kulm M, Sundaresan P. Mutational screening of LCA genes emphasizing RPE65 in South Indian cohort of patients. PloS one. 2013;8(9):e73172 10.1371/journal.pone.0073172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abu-Safieh L, Alrashed M, Anazi S, Alkuraya H, Khan AO, Al-Owain M, et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013;23(2):236–47. 10.1101/gr.144105.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, et al. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer research. 2005;65(14):6071–9. [DOI] [PubMed] [Google Scholar]
- 38. den Hollander AI, Lopez I, Yzer S, Zonneveld MN, Janssen IM, Strom TM, et al. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest Ophthalmol Vis Sci. 2007;48(12):5690–8. [DOI] [PubMed] [Google Scholar]
- 39. Woods CG, Valente EM, Bond J, Roberts E. A new method for autozygosity mapping using single nucleotide polymorphisms (SNPs) and EXCLUDEAR. Journal of medical genetics. 2004;41(8):e101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. [DOI] [PubMed] [Google Scholar]
- 41. Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic acids research. 2009;37(9):e67 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Skjorringe T, Tumer Z, Moller LB. Splice site mutations in the ATP7A gene. PloS one. 2011;6(4):e18599 10.1371/journal.pone.0018599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods. 2010;7(8):575–6. 10.1038/nmeth0810-575 [DOI] [PubMed] [Google Scholar]
- 44. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7(4):248–9. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- 46. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Human mutation. 2011;32(5):557–63. 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- 47. Hildebrandt F, Heeringa SF, Ruschendorf F, Attanasio M, Nurnberg G, Becker C, et al. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS genetics. 2009;5(1):e1000353 10.1371/journal.pgen.1000353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stone EM, Cideciyan AV, Aleman TS, Scheetz TE, Sumaroka A, Ehlinger MA, et al. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Archives of ophthalmology. 2011;129(1):81–7. 10.1001/archophthalmol.2010.330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Otto EA, Helou J, Allen SJ, O'Toole JF, Wise EL, Ashraf S, et al. Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Human mutation. 2008;29(3):418–26. [DOI] [PubMed] [Google Scholar]
- 50. Wang X, Wang H, Cao M, Li Z, Chen X, Patenia C, et al. Whole-exome sequencing identifies ALMS1, IQCB1, CNGA3, and MYO7A mutations in patients with Leber congenital amaurosis. Human mutation. 2011;32(12):1450–9. 10.1002/humu.21587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Estrada-Cuzcano A, Koenekoop RK, Coppieters F, Kohl S, Lopez I, Collin RW, et al. IQCB1 mutations in patients with leber congenital amaurosis. Investigative ophthalmology & visual science. 2011;52(2):834–9. [DOI] [PubMed] [Google Scholar]
- 52. Schuster A, Janecke AR, Wilke R, Schmid E, Thompson DA, Utermann G, et al. The phenotype of early-onset retinal degeneration in persons with RDH12 mutations. Invest Ophthalmol Vis Sci. 2007;48(4):1824–31. [DOI] [PubMed] [Google Scholar]
- 53. Dharmaraj S, Leroy BP, Sohocki MM, Koenekoop RK, Perrault I, Anwar K, et al. The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Archives of ophthalmology. 2004;122(7):1029–37. [DOI] [PubMed] [Google Scholar]
- 54. Ehrenberg M, Pierce EA, Cox GF, Fulton AB. CRB1: one gene, many phenotypes. Seminars in ophthalmology. 2013;28(5–6):397–405. 10.3109/08820538.2013.825277 [DOI] [PubMed] [Google Scholar]
- 55. Thompson DA, Gyurus P, Fleischer LL, Bingham EL, McHenry CL, Apfelstedt-Sylla E, et al. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Investigative ophthalmology & visual science. 2000;41(13):4293–9. [PubMed] [Google Scholar]
- 56. Beryozkin A, Zelinger L, Bandah-Rozenfeld D, Harel A, Strom TA, Merin S, et al. Mutations in CRB1 are a relatively common cause of autosomal recessive early-onset retinal degeneration in the Israeli and Palestinian populations. Investigative ophthalmology & visual science. 2013;54(3):2068–75. [DOI] [PubMed] [Google Scholar]
- 57. Sohocki MM, Bowne SJ, Sullivan LS, Blackshaw S, Cepko CL, Payne AM, et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nature genetics. 2000;24(1):79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sohocki MM, Perrault I, Leroy BP, Payne AM, Dharmaraj S, Bhattacharya SS, et al. Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Molecular genetics and metabolism. 2000;70(2):142–50. [DOI] [PubMed] [Google Scholar]
- 59. Gallon VA, Wilkie SE, Deery EC, Newbold RJ, Sohocki MM, Bhattacharya SS, et al. Purification, characterisation and intracellular localisation of aryl hydrocarbon interacting protein-like 1 (AIPL1) and effects of mutations associated with inherited retinal dystrophies. Biochimica et biophysica acta. 2004;1690(2):141–9. [DOI] [PubMed] [Google Scholar]
- 60. Testa F, Surace EM, Rossi S, Marrocco E, Gargiulo A, Di Iorio V, et al. Evaluation of Italian patients with leber congenital amaurosis due to AIPL1 mutations highlights the potential applicability of gene therapy. Invest Ophthalmol Vis Sci. 2011;52(8):5618–24. 10.1167/iovs.10-6543 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLS)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.