

Abstract
Survival rates for metastatic lung cancer, including non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), are poor with 5-year survivals of less than 5%. The immune system has an intricate and complex relationship with tumorigenesis; a groundswell of research on the immune system is leading to greater understanding of how cancer progresses and presenting new ways to halt disease progress. Due to the extraordinary power of the immune system—with its capacity for memory, exquisite specificity and central and universal role in human biology—immunotherapy has the potential to achieve complete, long-lasting remissions and cures, with few side effects for any cancer patient, regardless of cancer type. As a result, a range of cancer therapies are under development that work by turning our own immune cells against tumors. However deeper understanding of the complexity of immunomodulation by tumors is key to the development of effective immunotherapies, especially in lung cancer.
KEYWORDS : Lung cancer, immunotherapy, immune checkpoint, program death-ligand 1 (PD-L1), program death-1 (PD-1)
Introduction
Lung cancer is one of the leading causes of cancer-related death globally. Non-small cell lung cancer (NSCLC) is the most common type, accounting for nearly 85% of all newly diagnosed cases1. Most patients with NSCLC either present with metastatic disease or experience disease recurrence despite undergoing treatment for seemingly localized disease, underscoring the systemic nature of this disease. Cytotoxic chemotherapy regimens developed over the past few decades have produced only modest improvements in survival in metastatic NSCLC. A small subset of patients with tumors driven by activating mutations in the gene encoding epidermal growth factor receptor (EGFR) or rearrangements in the gene coding for anaplastic lymphoma kinase (ALK) benefit substantially from specific targeted therapies2-4. However, most of these patients eventually succumb to tumor progression within a few years of diagnosis. Thus therapies that obtain long lasting disease control are urgently needed.
The immune system plays an important role in controlling and eradicating cancer. Nevertheless, in the setting of malignancy, multiple mechanisms of immune suppression may exist that prevent effective antitumor immunity. Antibody therapy directed against several negative immunologic regulators is currently demonstrating significant success and is likely to become a major component of treatment for patients with a variety of malignancies. Therefore, this review focuses on the role of immune system in cancer and indeed lung cancer.
What is an immune checkpoint?
Thymus-derived lymphocytes (T-lymphocytes, T-cells) activation and expansion are necessary for an effective acquired immune response. Spontaneous lymphocytic infiltrates can be consistently observed in a variety of tumors. CD4 T-cells and CD8 T-cells make up the majority of T-lymphocytes. Interferon-γ producing CD8 T cells play an important role in inhibiting and killing tumor cells and impeding tumor growth. Interleukin-12 and granulocyte-macrophage colony-stimulating factor (GM-CSF) induce the activation of tumor-resident CD8 T effector/memory cells (Tem) followed by cytotoxic CD8 T effector cell expansion, a population that is a potent in situ resource for successful reactivation of systemic antitumor T cell immunity5. Amongst the many factors CD8 T cells produced, interferon-γ seems to be one of most significant cytokines in preventing and suppressing the development of cancers. In addition, the cytotoxic effects of CD8 T cells may also directly mediate death of tumor cells6.
After being activated and differentiated into distinct effector subtypes, CD4 T-cells play a major role in mediating immune response through the secretion of specific cytokines. These cells have multiple functions, ranging from activation of the cells of the innate immune system, B-lymphocytes, cytotoxic T-cells, as well as non-immune cells, and also play a critical role in suppression of immune reaction. Ongoing studies have identified new subsets of CD4 cells besides the classical T-helper 1 and 2 cells, like T-helper 17, follicular helper T-cell, induced T-regulatory cells (Treg), and the regulatory type 1 cells as well as the potentially distinct T-helper 97. Tregs, originally termed suppressive T-cells, were first described in the early 1970s as thymus-derived lymphocytes that tolerized bone marrow-derived lymphocytes to antigenic challenge8,9. Subsequent research demonstrated that T-cells expressing CD4 and CD25 [the alpha chain of interleukin-2 (IL-2) receptor] from tumor-bearing mice abrogated tumor rejection10-14. It was 10 years later that Sakaguchi and colleagues ascertained that CD25 could be used to identify these suppressive cells15. Later studies from the same laboratory established the forkhead box P3 (FoxP3) transcription factor as both a key intracellular marker of CD4+ CD25+ Tregs and a necessary factor for development and proper function of these cells16.
One of the key attributes is how the T-cells activate and distinguish “self” from “non-self” molecules. A series of positive and negative costimulatory receptors are expressed on a T-cell at variable levels according to the timing and circumstances of the immune response. The efficiency with which CD4 T-cells direct an immune response demands that proper regulatory measures are in place to prevent immune hyperactivation leading to autoimmune disease. This is very important especially for organs like the lungs that have large mucosal and gas-exchanging surfaces which are constantly exposed to the environment17. Such a critical process involves presentation of antigens to T-cells by antigen presenting cells (APC) and is highly regulated by molecules on T-cells and APC as well as tumor and stromal cells, known as immune checkpoints. Recognition of antigen- major histocompatibility complex (MHC) complexes by the T-cell antigen receptor is not sufficient for activation of naïve T-cells. Additional costimulatory signals are required and are provided by the engagement of CD28 on the T-cell surface with B7 molecules (CD80 and CD86) on the APC18,19 (Figure 1). The role of immune checkpoints is not only to trigger a sufficient immune response but also to inhibit stimulation to ensure the inductive immune response is not excessive. In fact, these immune checkpoints, usually referred to as molecules of inhibitory pathways in the immune system, are crucial for maintaining self-tolerance and modulating physiological immune responses in the periphery, in order to avoid or minimize tissue damage from excess reactions.
Figure 1.
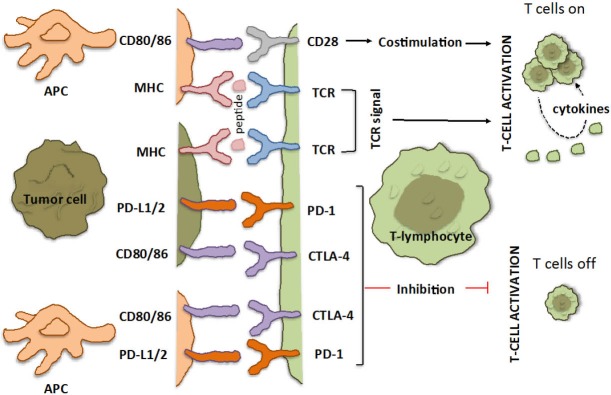
T-cell interaction with APC and tumor cells: the immune checkpoints CTLA-4 and PD-1/PD-L1. Depicted are various ligand-receptor interactions between T-cells, APCs and cancer cells that regulate the T-cell response to antigen. Activation of T-cells is a two-step process that requires recognition of specific peptides presented by MHC on the surface of cancer cells through their TCR, as well as a co-regulatory signal delivered by the CD28 family of receptors (the so-called immune checkpoints). The co-regulatory signal promotes T-cell clonal expansion, cytokine secretion, and functional activity of the T-cell. In the absence of this signal (even in the presence of a target peptide), T-cells fail to respond effectively and are functionally inactivated. This is designed as a fail-safe mechanism to ensure that the immune system is activated at the appropriate time in order to limit collateral damage to normal tissue and minimize the possibility of chronic autoimmune inflammation. Checkpoint pathways regulate these coregulatory signals and can be either stimulatory (switching T-cells on) or inhibitory (switching them off). CTLA-4 and PD-1 deliver inhibitor signals. CTLA-4 negatively regulates T-cell activation by binding to B7 molecules (CD80/86) on the surface of APC or tumor cell. Conversely, when these B7 molecules bind to CD28 they generate the opposite effect, activating signals. When PD-1 binds to either of its ligands (PD-L1 or PD-L2), which are primarily expressed within inflamed tissues and the tumor microenvironment, it results in inhibition of T-cell activity. APC, antigen-presenting cell (dendritic cell, macrophage or any cell that expresses antigen); TCR, T-cell receptor; MHC, major histocompatibility complex.
The CD28 family of cell surface receptors [CD28, cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), inducible costimulator (ICOS), program death-1 (PD-1), and B- and T-lymphocyte attenuator (BTLA)] plays a critical role in controlling the adaptive arm of the immune response and controlling T-cell activation. The counterpart (ligand) for CD28 is the “B7 family”, containing B7-1 (CD80) and B7-2 (CD86), which are usually present on APC. Although there is structural similarity between members of the CD28 family, functional heterogeneity is observed. For instance, ligation of CD28 and ICOS promotes T-cell activation, whereas engagement of CTLA-4, PD-1, and BTLA inhibits T-cell activation20. Other regulators of T-cell activation have recently been characterized and may have important roles. These include T-cell immunoglobulin and mucin domain-containing protein 3 (TIM3; also known as HAVCR2), lymphocyte activation gene-3 (LAG-3) and V-domain immunoglobulin suppressor of T-cell activation (VISTA)21-23.
CTLA-4 is expressed exclusively on T-cells and shares identical ligands (CD80 and CD86) with the T-cell co-stimulatory receptor CD28. When the T-cell receptor (TCR) is engaged by cognate antigen, CD28 induces T-cell activation. CTLA-4 has a much higher overall affinity for both ligands and inhibits the activation of T-cells by outcompeting CD28 in binding CD80 and CD86. At the same time, CTLA-4 activates the Src homology region 2 domain-containing phosphatase-2 (SHP2) and protein phosphatase 2A (PP2A) and counteracts kinase signals induced by TCR and CD28, sequestrates CD80 and CD86 from CD28 engagement, and actively removes CD80 and CD86 from the APC surface.
PD-1 signaling involves binding to several discrete ligands, including PD-L1 (also known as B7-H1 and CD274) and PD-L2 (also known as B7-DC and CD273), as well as to the co-stimulatory molecule CD80. The PD-1/PD-L1 interaction inhibits T-lymphocyte proliferation, survival and effector functions (cytotoxicity, cytokine release), induces apoptosis of tumor-specific T-cell and promotes differentiation of CD4 T-cells into Tregs and tumor cell resistance to cytotoxic T-lymphocytes (CTL) attack21. Because many tumors are highly infiltrated with Tregs that probably further suppress effector immune responses, blockade of the PD-1 pathway may also enhance antitumor immune responses by diminishing the number and/or suppressive activity of intratumoral Tregs. Chemnitz et al.24 revealed that the ability of PD-1 to block T-cell activation correlates with recruitment of SHP-1 and SHP-2. Indeed, PD-1 has a cytoplasmic immunoreceptor tyrosine based inhibitory motif (ITIM), as well as an immunoreceptor tyrosine-based switch motif (ITSM), and has been found to be capable of recruiting the phosphatases SHP-1 and SHP-2. Recruitment of SHP-1 and SHP-2 to ITIM within the PD-1 cytoplasmic tail inhibits positive signaling events downstream of the TCR, mainly PI3K/AKT activation25.
SHP-1 and SHP-2 are highly related tyrosine phosphatases that serve very distinct roles in signal transduction. SHP-1 expression is largely confined to hemopoietic cells and is thought to act as a negative regulator of STAT3 and other signaling pathways. SHP1 is encoded by the PTPN6 gene and the regulatory factor X-1 (RFX-1) is one transcription factor that can activate SHP-1 transcription26. SHP-2, in contrast, is widely expressed and generally acts in a positive manner to transduce signals from receptor protein tyrosine kinases. For instance, an established role of SHP-2 in EGFR or ALK signaling is to mediate ERK1/2 activation. However, SHP-2 also has been shown to inhibit the JAK-STAT signaling pathway27-29.
Immune response and cancer
Immunotherapies that boost the ability of endogenous T-cells to destroy cancer cells have demonstrated therapeutic efficacy in a variety of human malignancies. In 2010, the field was revitalized by a landmark randomized clinical trial that demonstrated that treatment with ipilimumab, an antibody targeting CTLA-4, improved overall survival (OS) of patients with metastatic melanoma30. Recent studies have demonstrated that T-cell–based immunotherapies are also effective in a range of other human malignancies. In particular, clinical trials of antibodies that interfere with PD-1 have shown clinical activity in tumor types as diverse as lung, bladder, stomach, renal cell, and head and neck cancer, as well as melanoma and Hodgkin’s lymphoma31.
T-cells in tumors—the so-called tumor infiltrating lymphocytes (TIL) have been studied intensively over the past years. The first evidence that T-cells could kill tumor cells was provided by L.R. Freedman and colleagues in 197232. Numerous studies suggest a positive prognostic impact of TIL but this still needs to be verified in large multi-center studies33. At present there is very limited knowledge as to why some tumors are heavily infiltrated by T-cells whereas others are not. Studies from the laboratory of Robert Schreiber have suggested the “Three Es of cancer immunoediting”34, or three phases of interaction between tumor and immune system: immune-Elimination of cancer cells, immune Equilibrium between cancer cells and cells of the immune system and immune Escape by cancer cells34. However, this notion is still unclear and TILs display a wide range of different phenotypes. Studies have shown that CD8 T-cells at the tumor site display markers of T-cell exhaustion to a higher extent than T-cells in the blood or from normal adjacent tissue35,36. In melanomas, CD8 and CD4 TILs display high expression of PD-1 and CTLA-4. Furthermore, the PD-1 positive fraction of the TILs displays impaired effector functions35.
Tumor and PD-L1 expression
Tumor cells can activate PD-L1 expression via multiple oncogenic signaling pathways involving IFN-γ/JAK2/IFN37, PI3K38, ALK/STAT339, MEK/ERK/STAT1, MYD88/TRAF640 or exposure to inflammatory cytokines such as IFN-γ41 produced by infiltrating immune cells. In breast cancer, PD-L1 expression is strongly associated with proliferative Ki-67 expression and cell cycle progression independent of host PD-142. In human glioma, loss of the tumor suppressor gene phosphatase and tensin homolog (PTEN) has been correlated with enhanced PD-L1 expression38. Similarly, in colorectal cancer, miR-20b, -21 and 130 inhibited PTEN expression, resulting in PD-L1 overexpression43. T-cell lymphoma cells carrying the oncogenic nucleophosmin (NPM)-ALK, involved in malignant transformation, induce high levels of PD-L1 expression via STAT3 and ERK activation39,44.
Abnormal expression of PD-L1 has been described in 19%-100% of NSCLCs and is associated with poor prognosis45-48. Reliable biomarkers associated with response to PD-1 blockade remain poorly understood49. Simultaneous activation of KRAS and inactivation of serine-threonine kinase 11 (also known as LKB1) induce lung squamous cell carcinoma formation50. Activation of the EGFR pathway might be involved in suppressing the immune response in murine melanoma models either through activating Tregs cells or reducing the levels of the T-cell chemoattractant49. Interestingly, Akbay et al.51 found that activation of the EGFR pathway induced PD-L1 expression to help NSCLC tumors to remodel tumor microenvironment to trigger immune escape and link tumor response to PD-1 inhibition. This role of EGFR signaling was independent of its effects on cell proliferation and survival, suggesting that the combination of PD-1 blockade with EGFR TKIs may be a promising therapeutic strategy to extend the duration of treatment response and delay development of resistance to EGFR inhibitors51. D’Incecco et al.52 found that PD-L1 positive NSCLC patients had higher sensitivity to EGFR-TKIs, longer time to progression and OS than PD-1 negative patients. They also reported that PD-L1 positive status was significantly associated with presence of EGFR mutations52. In the study of Azuma et al.53, inhibition of EGFR signaling by erlotinib down-regulated surface expression of PD-L1 in EGFR mutation-positive NSCLC cells, but not in the EGFR wild-type cells. In contrast, Mu et al.47 found no significant correlation between PD-L1 expression and EGFR/KRAS/BRAF/ALK expression in stage I NSCLC patients, similar to Zhang et al.54, who found no significant relationship between PD-L1 expression and EGFR/KRAS expression in lung adenocarcinoma. At the 2015 ASCO Annual Meeting, median progression free survival (PFS) and OS for EGFR TKIs were similar between PD-L1 positive and PD-L1 negative patients at baseline. Also, median PFS for ALK TKIs was similar in PD-L1 positive and PD-L1 negative patients at baseline, but median OS was shorter among PD-L1 positive patients. Expression was dynamic, with changes in PD-L1 expression and immune infiltrates observed over time and/or following treatment55.
Cancer immunotherapy in clinical practice
Three new immune checkpoint agents have now been approved by the U.S. Food and Drug Administration (FDA) for the treatment of melanoma31. The list of cancers that can be targeted with immunotherapy is growing and there are high expectations that immune checkpoint agents will also be approved for treatment of patients with lung, kidney, bladder and prostate cancer, as well as lymphoma and many other tumor types. Immune checkpoints inhibitors target molecules that regulate T cells rather than the T cells themselves in order to reverse the activation of inhibitory pathways and release antitumor T-cell responses.
Two phase III clinical trials with anti-CTLA-4 (ipilimumab) were conducted in patients with advanced melanoma and demonstrated improved OS with the drug30,56. Anti-CTLA-4, having more mature survival data than other agents, leads to durable clinical responses that can last a decade and more, but only in a fraction of patients. A recent analysis indicated survival of 10 years or more for a subset of patients57. Ipilimumab was approved in 2011.
Pembrolizumab and nivolumab, two antibodies against PD-1 were approved in September and December 2014, respectively, for treatment of metastatic melanoma31. A phase I clinical trial with pembrolizumab led to response rates of almost 38% in patients with advanced melanoma, and a subsequent study reported an overall response rate of 26% in patients who had progressive disease after prior ipilimumab treatment58,59. In a phase III trial, nivolumab improved OS of patients with metastatic melanoma in comparison with dacarbazine chemotharpy59. According to the results of the CheckMate 057 trial presented at the 2015 ASCO Annual Meeting, nivolumab is the first PD-1 inhibitor to significantly improve OS in comparison with docetaxel, in previously treated patients with advanced non-squamous NSCLC with 27% reduction in risk of death and significantly improved overall response rate. Tumor PD-L1 expression was found to be predictive of nivolumab benefit60. Nivolumab was FDA approved in March 2015 for patients with previously treated advanced or metastatic NSCLC based on a phase III clinical trial which reported an improvement in OS for patients treated with nivolumab as compared to patients treated with docetaxel chemotherapy31. In addition, nivolumab was recently found to be the first PD-1 inhibitor to demonstrate a survival benefit versus standard-of-care docetaxel in previously treated patients with advanced squamous NSCLC with 41% reduction in risk of death; benefit was independent of PD-1 expression61.
Biomarkers and response to immunotherapy; neoantigen load as a potential biomarker for cancer immunotherapy
There are ongoing studies to identify predictive biomarkers to select patients for treatment with a particular agent, but this is complicated by the complexity of the immune response. The expression of PD-L1 in cancer cells is an obvious candidate as it can directly turn off the immune response by inhibiting the activity of cytotoxic T-cells infiltrating the tumor. However, PD-L1 expression in tumor cells has little predictive power. Tumeh et al.62 established a set of conditions that correlates with good response of patients with melanoma to pembrolizumab therapy. These include the presence of cytotoxic T-cells in the tumor, the expression of PD-L1 and PD-1 in immune cells in the tumor margin, and less complexity (in terms of antigen receptors) in the tumor T-cell population62. Herbst et al.63 also observed that PD-L1 expression in immune cells is a good biomarker of response to immunotherapy.
Blockade of CTLA-4 and PD-1 has resulted in durable responses in many patients30,64. However it remains unclear why some have only transient or no response. A major hurdle in tumor immunotherapy is the fact that mechanisms of self-tolerance that prevent autoimmunity also impair T-cell responses against tumors. The nature of the antigens that allow the immune system to distinguish cancer cells from non-cancer cells has long remained obscure. Every tumor contains hundreds or thousands of somatic mutations and certain types of tumors display many more or less mutations. Melanomas and lung cancers are the outliers and contain approximately 200 nonsynchronous mutations per tumor, associated with environmental exposure to ultraviolet light and smoking65. It seems that response to immune-based drugs may be written in tumor DNA. Tumors with a high somatic mutation load are more likely to respond to immunotherapy as, in theory, they would have a higher diversity of neoantigens that can trigger an immune response when the CTLA-4/PD-1 inhibition is bypassed. In NSCLC patients treated with anti–PD-1, mutational load shows a strong correlation with clinical response66. Likewise, in melanoma patients treated with ipilimumab, long-term benefit is also associated with a higher mutational load, although the effect appears less profound in this setting67. In the study of Snyder and colleagues67, mutational burden was higher in patients with a sustained clinical benefit than in those without. While the data indeed show that high mutation load correlates with responsiveness to therapy in many cases, surprisingly some tumors with a high load of somatic mutations fail to respond to checkpoint blockade. Therefore, quality not quantity of mutations has the strongest predictive value. A number of tetrapeptide sequences common to patients with sustained clinical benefit, but completely absent in patients with a minimal or no benefit, were homologous to viral and bacterial antigens67. An interesting interpretation of these data is that the neoantigen-specific T-cell response is preferentially directed toward a subset of mutant sequences, something that could facilitate bioinformatic identification of neoantigens for therapeutic targeting68. However, other studies have not found the profound bias toward these tetrapeptide signatures that would be predicted if their role was central to the tumor-specific T-cell response, meaning that the identified tetrapeptide motifs may play a different role69.
Conclusion
Cancer immunotherapy relies on the ability of the immune system to identify and destroy tumor cells and elicit a long-lasting memory of this interaction. Various strategies are being developed to enhance anti-tumor immune responses, with a recent focus on antagonists of inhibitory signaling pathways to overcome immune checkpoints. Existing therapies are also being investigated for their ability to induce an anti-tumor immune response, something which could lead to administration of combination therapies providing a more efficacious and durable response. However, there are issues that remain to be understood. Soon many cancer immunotherapies will be made available, many combinations will be possible, and this choice will be quite challenging from a clinical, regulatory, and reimbursement perspective. Biomarkers and companion diagnostics may also play a big role in guiding the way, as will a deepening understanding of immunotherapy mechanisms and cancer response.
Footnotes
No potential conflicts of interest are disclosed.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014;64:9-29. [DOI] [PubMed] [Google Scholar]
- 2.Rosell R, Bivona TG, Karachaliou N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet 2013;382:720-731. [DOI] [PubMed] [Google Scholar]
- 3.Rosell R, Karachaliou N, Wolf J, Ou SH. ALK and ROS1 non-small-cell lung cancer: two molecular subgroups sensitive to targeted therapy. Lancet Respir Med 2014;2:966-968. [DOI] [PubMed] [Google Scholar]
- 4.Rosell R. Dynamic Evolution of ALK Positive Non-Small Cell Lung Cancers and Management of Associated Brain Metastases [podcast] J Clin Oncol 2015. [Epub ahead of print]. [Google Scholar]
- 5.Kilinc MO, Gu T, Harden JL, Virtuoso LP, Egilmez NK. Central role of tumor-associated CD8+ T effector/memory cells in restoring systemic antitumor immunity. J Immunol 2009;182:4217-4225. [DOI] [PubMed] [Google Scholar]
- 6.Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci. 2011;7:651-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luckheeram RV, Zhou R, Verma AD, Xia B. CD4+T cells: differentiation and functions. Clin Dev Immunol 2012;2012:925135. [DOI] [PMC free article] [PubMed]
- 8.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005;5:263-274. [DOI] [PubMed] [Google Scholar]
- 9.Kryczek I, Wei S, Gong W, Shu X, Szeliga W, Vatan L, et al. Cutting edge: IFN-gamma enables APC to promote memory Th17 and abate Th1 cell development. J Immunol 2008;181:5842-5846. [DOI] [PubMed] [Google Scholar]
- 10.Gershon RK, Kondo K. Infectious immunological tolerance. Immunology 1971;21:903-914. [PMC free article] [PubMed] [Google Scholar]
- 11.Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology 1970;18:723-737. [PMC free article] [PubMed] [Google Scholar]
- 12.Berendt MJ, North RJ. T-cell-mediated suppression of anti-tumor immunity. An explanation for progressive growth of an immunogenic tumor. J Exp Med 1980;151:69-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bursuker I, North RJ. Generation and decay of the immune response to a progressive fibrosarcoma. II. Failure to demonstrate postexcision immunity after the onset of T cell-mediated suppression of immunity. J Exp Med 1984;159:1312-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.North RJ, Bursuker I. Generation and decay of the immune response to a progressive fibrosarcoma. I. Ly-1+2- suppressor T cells down-regulate the generation of Ly-1-2+ effector T cells. J Exp Med 1984;159:1295-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995;155:1151-1164. [PubMed] [Google Scholar]
- 16.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003;299:1057-1061. [PubMed] [Google Scholar]
- 17.Heikkinen T, Järvinen A. The common cold. Lancet 2003;361:51-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol 2005;23:515-548. [DOI] [PubMed] [Google Scholar]
- 19.Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science 1993;259:368-370. [DOI] [PubMed] [Google Scholar]
- 20.Riley JL, June CH. The CD28 family: a T-cell rheostat for therapeutic control of T-cell activation. Blood 2005;105:13-21. [DOI] [PubMed] [Google Scholar]
- 21.Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res 2013;19:4917-4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res 2011;71:3540-3551. [DOI] [PubMed] [Google Scholar]
- 23.Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med 2011;208:577-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945-954. [DOI] [PubMed] [Google Scholar]
- 25.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 2012;209:1201-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su JC, Chiang HC, Tseng PH, Tai WT, Hsu CY, Li YS, et al. RFX-1-dependent activation of SHP-1 inhibits STAT3 signaling in hepatocellular carcinoma cells. Carcinogenesis 2014;35:2807-2814. [DOI] [PubMed] [Google Scholar]
- 27.Tang TL, Freeman RM, Jr, O’Reilly AM, Neel BG, Sokol SY. The SH2-containing protein-tyrosine phosphatase SH-PTP2 is required upstream of MAP kinase for early Xenopus development. Cell 1995;80:473-483. [DOI] [PubMed] [Google Scholar]
- 28.Bennett AM, Hausdorff SF, O’Reilly AM, Freeman RM, Neel BG. Multiple requirements for SHPTP2 in epidermal growth factor-mediated cell cycle progression. Mol Cell Biol 1996;16:1189-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehmann U, Schmitz J, Weissenbach M, Sobota RM, Hortner M, Friederichs K, et al. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J Biol Chem 2003;278:661-671. [DOI] [PubMed] [Google Scholar]
- 30.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56-61. [DOI] [PubMed] [Google Scholar]
- 32.Freedman LR, Cerottini JC, Brunner KT. In vivo studies of the role of cytotoxic T cells in tumor allograft immunity. J Immunol 1972;109:1371-1378. [PubMed] [Google Scholar]
- 33.Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012;12:298-306. [DOI] [PubMed] [Google Scholar]
- 34.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol 2004;22:329-360. [DOI] [PubMed] [Google Scholar]
- 35.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009;114:1537-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang QJ, Hanada K, Robbins PF, Li YF, Yang JC. Distinctive features of the differentiated phenotype and infiltration of tumor-reactive lymphocytes in clear cell renal cell carcinoma. Cancer Res 2012;72:6119-6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002;8:793-800. [DOI] [PubMed] [Google Scholar]
- 38.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 2007;13:84-88. [DOI] [PubMed] [Google Scholar]
- 39.Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A 2008;105:20852-20857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007;110:296-304. [DOI] [PubMed] [Google Scholar]
- 41.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012;4:127ra37. [DOI] [PMC free article] [PubMed]
- 42.Ghebeh H, Tulbah A, Mohammed S, Elkum N, Bin Amer SM, Al-Tweigeri T, et al. Expression of B7-H1 in breast cancer patients is strongly associated with high proliferative Ki-67-expressing tumor cells. Int J Cancer 2007;121:751-758. [DOI] [PubMed] [Google Scholar]
- 43.Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y, et al. MiR-20b, -21, and -130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol 2014;75:348-353. [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto R, Nishikori M, Tashima M, Sakai T, Ichinohe T, Takaori-Kondo A, et al. B7-H1 expression is regulated by MEK/ERK signaling pathway in anaplastic large cell lymphoma and Hodgkin lymphoma. Cancer Sci 2009;100:2093-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hirahara K, Ghoreschi K, Yang XP, Takahashi H, Laurence A, Vahedi G, et al. Interleukin-27 priming of T cells controls IL-17 production in trans via induction of the ligand PD-L1. Immunity 2012;36:1017-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konishi J, Yamazaki K, Azuma M, Kinoshita I, Dosaka-Akita H, Nishimura M. B7-H1 expression on non-small cell lung cancer cells and its relationship with tumor-infiltrating lymphocytes and their PD-1 expression. Clin Cancer Res 2004;10:5094-5100. [DOI] [PubMed] [Google Scholar]
- 47.Mu CY, Huang JA, Chen Y, Chen C, Zhang XG. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol 2011;28:682-688. [DOI] [PubMed] [Google Scholar]
- 48.Wölfle SJ, Strebovsky J, Bartz H, Sähr A, Arnold C, Kaiser C, et al. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol 2011;41:413-424. [DOI] [PubMed] [Google Scholar]
- 49.Pivarcsi A, Müller A, Hippe A, Rieker J, van Lierop A, Steinhoff M, et al. Tumor immune escape by the loss of homeostatic chemokine expression. Proc Natl Acad Sci U S A 2007;104:19055-19060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 2014;25:590-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 2013;3:1355-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D’Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L, et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br J Cancer 2015;112:95-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol 2014;25:1935-1940. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, Wang L, Li Y, Pan Y, Wang R, Hu H, et al. Protein expression of programmed death 1 ligand 1 and ligand 2 independently predict poor prognosis in surgically resected lung adenocarcinoma. Onco Targets Ther 2014;7:567-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gainor JF, Sequist LV, Shaw AT, Azzoli CG, Piotrowska Z, Huynh T, et al. Clinical correlation and frequency of programmed death ligand-1 (PD-L1) expression in EGFR-mutant and ALK-rearranged non-small cell lung cancer (NSCLC). J Clin Oncol 2015;33:abstr 8012.
- 56.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517-2526. [DOI] [PubMed] [Google Scholar]
- 57.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol 2015;33:1889-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320-330. [DOI] [PubMed] [Google Scholar]
- 60.Paz-Ares L, Horn L, Borghaei H, Spigel DR, Steins M, Ready N, et al. Phase III, randomized trial (CheckMate 057) of nivolumab (NIVO) versus docetaxel (DOC) in advanced non-squamous cell (non-SQ) non-small cell lung cancer (NSCLC). J Clin Oncol 2015;33:abstr LBA109.
- 61.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med 2015. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189-2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69-74. [DOI] [PubMed] [Google Scholar]
- 69.Schumacher TN, Kesmir C, van Buuren MM. Biomarkers in cancer immunotherapy. Cancer Cell 2015;27:12-14. [DOI] [PubMed] [Google Scholar]