

Abstract
Experimentally-determined or computationally-predicted protein phosphorylation sites for distinctive species are becoming increasingly common. In this paper, we compare the predictive performance of a novel classification algorithm with different encoding schemes to develop a rice-specific protein phosphorylation site predictor. Our results imply that the combination of Amino acid occurrence Frequency with Composition of K-Spaced Amino Acid Pairs (AF-CKSAAP) provides the best description of relevant sequence features that surround a phosphorylation site. A support vector machine (SVM) using AF-CKSAAP achieves the best performance in classifying rice protein phophorylation sites when compared to the other algorithms. We have used SVM with AF-CKSAAP to construct a rice-specific protein phosphorylation sites predictor, Rice_Phospho 1.0 (http://bioinformatics.fafu.edu.cn/rice_phospho1.0). We measure the Accuracy (ACC) and Matthews Correlation Coefficient (MCC) of Rice_Phospho 1.0 to be 82.0% and 0.64, significantly higher than those measures for other predictors such as Scansite, Musite, PlantPhos and PhosphoRice. Rice_Phospho 1.0 also successfully predicted the experimentally identified phosphorylation sites in LOC_Os03g51600.1, a protein sequence which did not appear in the training dataset. In summary, Rice_phospho 1.0 outputs reliable predictions of protein phosphorylation sites in rice, and will serve as a useful tool to the community.
Phosphorylation is one of the most important protein post-translation modification (PTMs) in eukaryotes. It plays essential roles in the majority of biological pathways, regulating cellular processes like metabolism, proliferation, differentiation and apoptosis1. More than 30% of all eukaryotic proteins are estimated to undergo reversible phosphorylation2. Biochemically, phosphorylation results in a transfer of a phosphate moiety from adenosine triphosphate (ATP) to the acceptor residue, thereby generating adenosine diphosphate (ADP) whilst inducing the residue to be phosphorylated. The process of protein phosphorylation usually involves distinct short peptide motifs, or patterns including phosphorylation of substrate sites, being recognized by different protein kinases which then leads to a phosphate moiety being typically attached to either Serine (Ser), Threonine (Thr) or Tyrosine (Tyr) residues.
Conventional experimental identifications and recent advances in high throughput Mass Spectrometry (MS) techniques have generated a large number of phosphorylated substrates with confirmed phosphorylation sites. In parallel, a series of algorithms have also been developed to predict phosphorylation sites from amino acid sequence. These range from simple motif or pattern searches to more complex machine learning methods like Artificial Neural Networks (ANN) and Support Vector Machines (SVM). Examples of such predictive algorithms include NetPhos3, NetPhosK4, KinasePhos5, DISPHOS6, Scansite7, PPSP8, GPS9, PredPhospho10 and Musite11.
Most computational phosphorylation site predictors are not organism-specific predictors. However, with the increases in experimentally verified protein phosphorylation sites for different organisms, an increasing goal is to develop organism-specific phosphorylation predictors, such has occurred for yeast12, Arabidopsis13 and rice1. The yeast-specific predictor, NetPhosYeast, outperforms existing generic predictors in the identification of phosphorylation sites in yeast12. PhosPhAt predicting phosphorylated-Serine sites for Arabidopsis is found to perform better with Arabidopsis sequences than other generic predictors13. Furthermore, a protein family specific phosphorylation site predictor, PhosTryp, was developed specifically for the trypanosomatidae family in parasitic protozoa14.
We have focused our efforts on Rice (Oryza sativa L.). Rice is considered a model plant species of the monocots group, it has a sequenced genome15 and serves as a cornerstone for the study of functional genomics in cereal plants16. Phosphorylation proteins have been identified in rice treated with various hormones17 and under different environmental conditions, including high salinity18, drought19 and high temperature20. Many phosphorylation sites in rice were identified by Nakagami et al.21 However, current predictors perform poorly when individually used to predict phosphorylation sites in rice phosphoproteins1. We have therefore established a meta-predictor for rice-specific phosphorylation sites1. However, this rice-specific predictor was not trained directly by the rice phosphorylation sites data, but was developed by integrating six newly predicting programs, including NetPhosK, NetPhos2.0, KinasePhos, PrePhospho 1.0, Scansite and DISPHOS. This paper augments this earlier work by building a Support Vector Machine (SVM) prediction model using experimentally identified rice phosphorylation sites directly.
Results
Performance of the 6 encoding schemes
The performance of the three sole encoding schemes was measured by using different sizes of datasets and with SVM used as the classifier. CKSAAP performed best among the three sole encoding schemes (Fig. 1). However, with the size of dataset increasing, the performance of SVM with CKSAAP decreased, SVM with AF kept fluctuating, while that of SVM with KNN increased (Table 1). The same changing trends (CKSSAP decreasing, AF fluctuating and KNN increasing) in performance of SVM with AF, KNN or CKSAAP was also true when the ratio of (+) sites to (−) sites increased (Table 1).
Figure 1. ROC curves of predicting performance of SVM with 3 different sole encoding schemes.
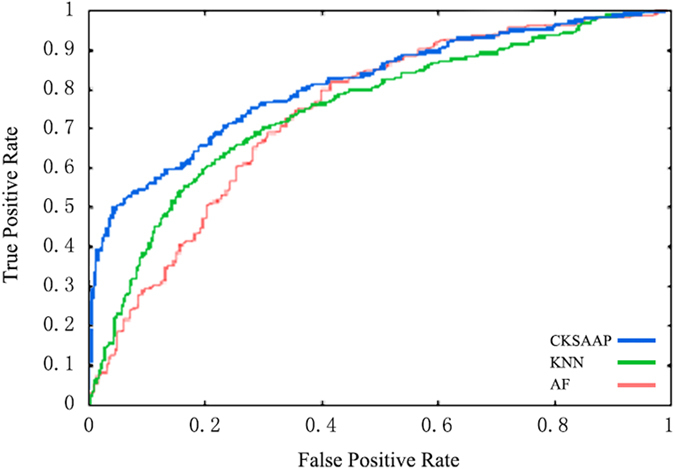
*In the diagrams, the increased area under the ROC indicates the improved classification performance. The same below.
Table 1. Performance of 3 sole encoding schemes on different size of dataset.
| Method | (+) sites | (−) sites | Ratio | Sn (%) | Sp (%) | ACC (%) | MCC |
|---|---|---|---|---|---|---|---|
| AF | 112 | 127 | 0.88:1 | 67.20 | 66.13 | 69.51 | 0.403 |
| 365 | 370 | 0.99:1 | 72.00 | 73.07 | 75.11 | 0.461 | |
| 853 | 937 | 0.91:1 | 68.31 | 69.43 | 72.30 | 0.408 | |
| 1530 | 1630 | 0.94:1 | 69.37 | 68.21 | 70.15 | 0.391 | |
| 2107 | 2018 | 1.04:1 | 75.33 | 76.86 | 74.94 | 0.477 | |
| KNN | 112 | 127 | 0.88:1 | 57.00 | 52.3 | 58.29 | 0.237 |
| 365 | 370 | 0.99:1 | 60.14 | 58.12 | 59.50 | 0.281 | |
| 853 | 937 | 0.91:1 | 68.22 | 63.10 | 67.20 | 0.306 | |
| 1530 | 1630 | 0.94:1 | 68.73 | 65.16 | 69.75 | 0.362 | |
| 2107 | 2018 | 1.04:1 | 75.35 | 71.03 | 72.39 | 0.407 | |
| CKSAAP | 112 | 127 | 0.88:1 | 82.77 | 80.30 | 83.37 | 0.633 |
| 365 | 370 | 0.99:1 | 82.27 | 80.63 | 82.84 | 0.617 | |
| 853 | 937 | 0.91:1 | 81.02 | 80.12 | 81.27 | 0.623 | |
| 1530 | 1630 | 0.94:1 | 77.84 | 79.70 | 80.56 | 0.612 | |
| 2107 | 2018 | 1.04:1 | 80.02 | 79.33 | 80.41 | 0.605 |
The performance of AF combined with CKAAP (AF-CKSAAP) was better than the sole encoding scheme, AF or CKSAAP (Fig. 2A). The same was true for AF combined with KNN (AF-KNN) (Fig. 2B). However, CKSAAP combined with KNN (CKSAAP-KNN) outperformed KNN, but did not outperform CKSAAP (Fig. 2C). In the preliminary experiment, we found that the combination of all the three encoding schemes did not significantly outperform CKSAAP (Data not shown) but increased feature dimensions. This result implies that AF, KNN and CKSAAP might be complementary to each other to some extent, especially AF and CKSAAP.
Figure 2. ROC curves of predicting performance of SVM with the combining encoding schemes.
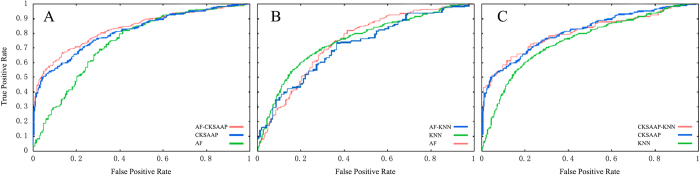
*A. ROC curves of SVM with AF-CKSAAP, AF and CKSAAP. B. ROC curves of SVM with AF-KNN, AF and KNN. C. ROC curves of SVM with CKSAAP-KNN, KNN and CKSAAP.
Performance of 4 different classifiers
The performance of the classifiers with the six different encoding schemes were firstly compared. The best results of a DT classifier were for CKSAAP, with an ACC of 71.14% and MCC of 0.314 (Fig. 3). The best results of a KNN classifier were for CKSAAP-KNN, with an ACC of 73.71% and MCC of 0.402 (Fig. 3). The best results of a RF classifier were for AF-KNN, with an ACC of 75.1% and MCC of 0.458 (Fig. 3). The best results of a SVM classifier were for AF-CKSAAP, with an ACC of 80.90% and MCC of 0.617 (Fig. 3).
Figure 3. MCC of predicting performance of different classification algorithms with different encoding schemes.
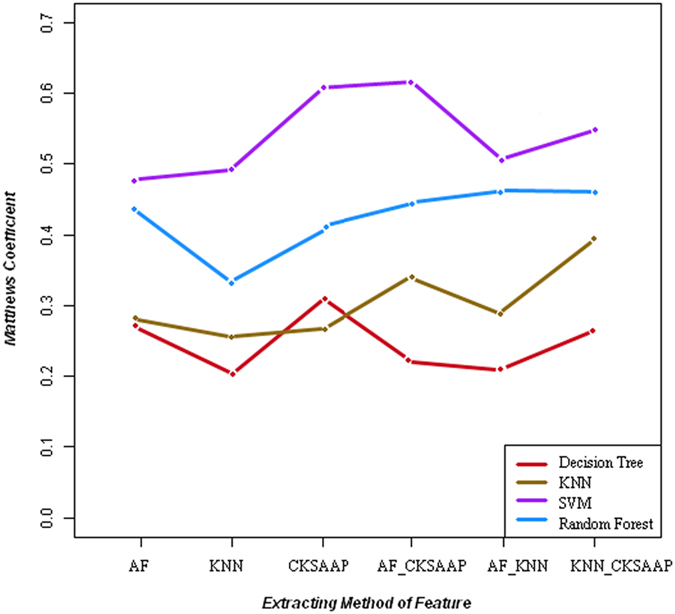
We then compared classifying ability of the 4 different classifiers on phosphorylation sites of proteins in rice. As shown in Fig. 3, SVM performed best on the phosphorylation sites among the 4 classifiers with any of the 6 encoding schemes. The SVM with AF-CKSAAP, CKSAAP and CKSAAP-KNN lay in the top 3 predicting models.
Performance of the top three predicting models on different phospho-amio acids
We used different phospho-amino acids datasets with different ratio of (+) sites to (−) sites to detect the performance of the top three predicting models (SVM with AF-CKSAAP, SVM with CKSAAP and SVM with CKSAAP-KNN). For phospho-serine, phospho-threonine or phospho-tyrosine sites, SVM with AF-CKSAAP out-performed SVM with CKSAAP or CKSAAP-KNN, even though the ratio of (+) sites to (−) sites in the dataset was unbalanced (Table 2).
Table 2. Predicting performance of SVM with 3 different encoding schemes on different phospho-amino acid sites.
| Methods | Phospho-amino acids | Ratio | Sn (%) | Sp (%) | ACC (%) | MCC |
|---|---|---|---|---|---|---|
| CKSAAP | Serine | 1:0.7 | 79.84 | 80.41 | 80.36 | 0.617 |
| 1:1 | 80.32 | 80.55 | 80.51 | 0.619 | ||
| 0.7:1 | 79.91 | 80.31 | 80.16 | 0.613 | ||
| Threonine | 1:0.34 | 79.89 | 80.11 | 79.95 | 0.597 | |
| 1:1 | 79.34 | 79.62 | 78.79 | 0.583 | ||
| 0.34:1 | 78.42 | 78.86 | 78.37 | 0.589 | ||
| Tyrosine | 1:0.17 | 84.17 | 83.36 | 84.00 | 0.638 | |
| 1:1 | 77.25 | 78.71 | 78.03 | 0.573 | ||
| 0.17:1 | 74.61 | 73.83 | 73.02 | 0.532 | ||
| AF- CKSAAP | Serine | 1:0.7 | 81.22 | 79.20 | 81.15 | 0.623 |
| 1:1 | 81.87 | 81.23 | 82.14 | 0.635 | ||
| 0.7:1 | 83.22 | 83.14 | 84.71 | 0.642 | ||
| Threonine | 1:0.34 | 78.57 | 78.27 | 79.52 | 0.601 | |
| 1:1 | 76.28 | 77.31 | 77.20 | 0.593 | ||
| 0.34:1 | 78.19 | 77.22 | 78.05 | 0.591 | ||
| Tyrosine | 1:0.17 | 80.19 | 79.75 | 80.72 | 0.623 | |
| 1:1 | 78.35 | 77.55 | 79.12 | 0.597 | ||
| 0.17:1 | 77.75 | 75.38 | 78.04 | 0.593 | ||
| CKSAAP -KNN | Serine | 1:0.7 | 77.14 | 75.43 | 76.31 | 0.542 |
| 1:1 | 76.18 | 74.83 | 75.12 | 0.526 | ||
| 0.7:1 | 80.86 | 80.17 | 81.14 | 0.624 | ||
| Threonine | 1:0.34 | 71.77 | 71.19 | 72.37 | 0.497 | |
| 1:1 | 72.46 | 72.81 | 73.34 | 0.509 | ||
| 0.34:1 | 75.53 | 73.11 | 74.27 | 0.520 | ||
| Tyrosine | 1:0.17 | 68.91 | 69.71 | 69.53 | 0.418 | |
| 1:1 | 69.92 | 70.24 | 70.11 | 0.433 | ||
| 0.17:1 | 72.86 | 73.42 | 70.34 | 0.465 |
Blom et al. (2004) suggested that a real non-phosphorylation site had to be solvent-inaccessible4. After discarding the predicting solvent-accessible non-phosphorylation sites from Table S2 and composing a new negative dataset (Table S3), we used the new training dataset which was extracted from Table S3 and its balancing positive dataset to re-train the top three predicting models. Table 3 also indicated that the overall performance of SVM with AF-CKSAAP was better than that of SVM with CKSAAP or CKSAAP-KNN.
Table 3. Predicting performance of the three top SVM models trained by the negative dataset in Table S3 and its balancing positive dataset.
| Models | Sn (%) | Sp (%) | ACC (%) | MCC | AUC |
|---|---|---|---|---|---|
| CKSAAP | 79.47 | 82.83 | 81.15 | 0.623 | 0.839 |
| AF-CKSAAP | 82.84 | 81.59 | 82.04 | 0.641 | 0.858 |
| CKSAAP-KNN | 77.46 | 79.86 | 78.63 | 0.579 | 0.820 |
AUC is the area under ROC curve.
Assessment of the predictor, Rice_Phospho 1.0, with the newly existing predictors
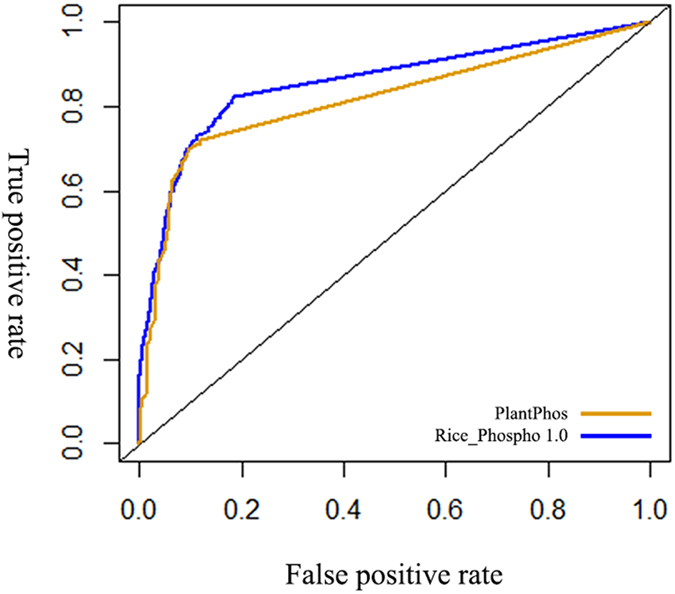
We used SVM with AF-CKSAAP to develop a new rice-specific predictor, Rice_Phospho 10. We applied the independent test dataset to compare the predicting performance of Rice_phospho 1.0 with the newly existing predictors, including Scansite, Musite, PlantPhos and PhosphoRice. The MCC of the prediction performance of Rice_phosphos 1.0 in comparison to Scansite, Musite and PhosphoRice were shown in Table 4. Rice_Phospho 1.0 had higher MCC value than the existing predictors, indicating that the performance of Rice_Phospho 1.0 was significantly better than that of Scansite, Musite and PhosphoRice. The Area Under ROC Curve (AUC) of Rice_Phospho1.0 was higher than that of PlantPhos (Fig. 4), implying that Rice_Phospho 1.0 also outperformed PlantPhos.
Table 4. Predicting performance of SVM models and newly developed predictors.
| Tools | Sn (%) | Sp (%) | ACC (%) | MCC |
|---|---|---|---|---|
| Musite | 55.18 | 81.91 | 73.32 | 0.368 |
| Scansite | 75.18 | 53.23 | 59.90 | 0.285 |
| PhosphoRice | 70.25 | 74.40 | 75.32 | 0.462 |
| Rice_Phospho 1.0 | 79.93 | 81.21 | 80.33 | 0.616 |
Figure 4. ROC curves of predicting performance of Rice_Phospho 1.0 and PlantPhos.
Construction online predictor, Rice_Phospho 1.0
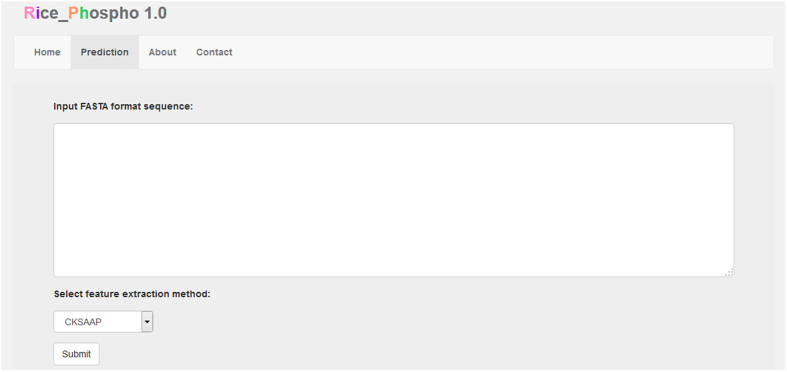
We constructed the online tool, Rice_Phospho 1.0, which was a specific SVM predictor on the protein phosphorylation sites in rice (Oryza sativa L.). The potential phosphorylation sites are retrieved after the user uploads a protein sequence in FASTA format into the text area and selects one of the encoding schemes (Fig. 5). Rice_Phospho 1.0 is accessible via http://bioinformatics.fafu.edu.cn/rice_phospho1.0.
Figure 5. Interface of the online predictor, Rice_Phospho 1.0, on rice protein phosphorylation sites.
Discussion
Our analysis indicates that CKSAAP encoding can extract the character around the phoshorylation sites more concisely than AF and KNN. Generally, AF and KNN methods select the position-specific feature of a sequence fragment, while the CKSAAP encoding pays attention to the co-location of amino acid pairs at different positions surrounding phosphorylation sites22. The sequence character extracted by CKSAAP can also reflect the composition of short linear motifs, which have been widely reported to be involved in many biological processes such as the communication of protein-protein interactions23. The results in this paper imply that short linear motifs maybe more important than position-specific patterns in recognizing protein phosphorylated substrates. CKSAAP encoding has been reported to predict the structural property of a sequence fragment24 and PTM sites, including ubiquitylated sites22 and mucin-type O-glycosylated sites25. Therefore, we might also expect a better performance of the CKSAAP encoding in the prediction of protein phosphorylated sites.
More importantly, the reasonably good performance of SVM with AF-CKSAAP reflected that the combining encoding schemes, AF and CKSAAP, can effectively capture the information of enriched/depleted residue pairs around phosphorylation sites in rice. The AF encoding scheme clearly characterizes amino acids in different positions surrounding a potential phosphorylated site, but it is weak in reflecting the coupling effect of amino acid pairs at different positions. On the other hand, the CKSAAP has the ability to detect the relationship between amino acid pairs at different positions, but it cannot capture the position specific amino acid information22. Therefore, the complementary capability of AF to CKSAAP results in a better performance for AF-CKSAAP in extracting the sequence character surrounding a potential phosphorylated site when compared with the individual encoding scheme. Meanwhile, in terms of different phospho-amino acid sites, SVM with AF-CKSAAP performed better than others, even though the ratio of (+) sites to (−) sites in the test dataset was unbalanced. This is because the accuracy of the predictors may be overestimated when the ratio of (+) sites to (−) sites in the training dataset was optimized1.
We used the model of SVM with AF-CKSAAP to develop an online tool, Rice_Phospho 1.0, which was a specific predictor on the phosphorylation sites in rice. To verify the performance of Rice_Phospho 1.0, one experimentally identified phosphorylated protein in rice which did not appear in the training dataset was used as a query sequence. A α-tubulin isoform (LOC_Os03g51600.1) was experimentally identified to be phosphorylated at Thr349 site by a comprehensive mutagenesis method26. Rice_Phospho 1.0 successfully predicted the experimentally identified pThr at the position 349. Moreover, three more sites, including Ser216, Tyr432 and Ser439, were predicted as novel phophorylated sites.
In summary, we have benchmarked the combination of several encoding schemes and classification routines to establish their relative effectiveness. This led to the choice of a SVM with AF-CKSAAP encoding scheme which we have incorporated into the development of an effective rice specific protein phosphorylation site predictor, Rice_phospho 1.0 (http://bioinformatics.fafu.edu.cn/rice_phospho1.0). Rice_Phospho 1.0 provides state of the art levels of reliability in predicting protein phosphorylation sites in rice and will be a useful tool to the community.
Methods
Preprocessing of dataset
We collected rice phosphorylation sites from the recent literature of Nakagami et al.21. We also used the feature table of Swiss-Prot database, from which records annotated as ‘predicted’ or ‘similarity’ were excluded. After removing the redundant phosphorylation sites, the number of serine (S), threonine (T) and tyrosine (Y) substrates were 4220, 605 and 141 respectively, and these phosphorylation sites were involved in 2162 proteins1.
The 25-mer sequences (−12 to +12) surrounding the phosphorylation sites were extracted from the protein sequences1. Because all of these phosphorylation sites were experimentally verified, they were regarded as (+) sites and compiled within a positive dataset (Table S1). The Ser, Thr and Tyr residues that were not annotated as phosphorylation sites within the dataset were regarded as (−) sites (i.e., non-phosphorylation sites), and the 25-mer sequences surrounding them were extracted and compiled within a negative dataset (Table S2). We extracted one-third of the data from each of these two datasets to compose an independent test dataset. We used the remainder of the data in the two datasets to construct a training dataset. The phosphorylation and non-phosphorylation sites were randomly chosen from the training dataset to compile a different ratio of (+) sites to (−) sites dataset during the cross-validation processes.
Because the residues buried in the core of a protein would not be accessible to any kinases4, the NetSurfP program27 was used to predict the surface accessibility of each non-phosphorylated site in Table S2. The solvent-inaccessible non-phosphorylation sites were compiled in Table S3. We randomly selected one third of the data to compose another independent test dataset, and re-trained a Support Vector Machine (SVM) by using the Composition of K-Spaced Amino Acid Pairs (CKSAAP), Amino acid occurrence frequency combined with CKSAAP (AF-CKSAAP), CKSAAP combined with K-Nearest Neighbor (CKSAAP-KNN) as feature selection methods.
A standard 10-fold cross validation was used to train the classifiers. We calculated the Sensitivity (Sn), Specificity (Sp), Accuracy (ACC) and the Matthew’s Correlation Coefficient (MCC) of each predictor1. The dataset was randomly partitioned into 10 subsets, including one testing subset and nine training subsets. Each predictor was trained by shifting the test subset stepwise so that all data is used for training and testing.
Encoding schemes and feature selection
K-Nearest Neighbor (KNN)
The KNN method is used to classify the samples based on their distances. For each sequence, the distances between all the sequences of positive datasets or negative datasets were measured using the formula introduced by Gao et al. (2009)28.
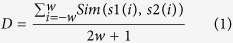 |
where two protein sequences s1 = {s1(−w), s1(−w + 1),…, s1(w)} and s2 = {s2(−w), s2(−w + 1),…, s2(w)}, w = 12, sim—amino acid similarity matrix—is derived from the normalized BLOSUM62.
The K nearest distances were selected, and the average distance among them was calculated. This process was repeated for different values of K (0.1%, 0.2%, 0.5%, 1%, 2%, 5% and 10% of the positive datasets or negative datasets). The ratios of the average distances between positive datasets and negative datasets were extracted as the feature values28.
Composition of K-Spaced Amino Acid Pairs (CKSAAP)
CKSAAP has been successfully used to represent sequence fragments22. A sequence fragment may contain 400 types (AxA, AxC, AxD, …, OxO) of K-spaced amino acid pairs (i.e. the pairs separated by K other amino acids). The flowchart and the calculation used for the CKSAAP feature selection approach are shown in Fig. S1. The value of NAA is the composition of the corresponding amino acid pairs in the sequence fragment, while Ntotal represent the total composition of amino acid pairs in the sequence fragment. For instance, if there are n AxA pairs in the sequence fragment, the value of corresponding component of NAA is n (Fig. S1).
When the value of K is increased, the prediction accuracy and sensitivity increases, but so does the computational complexity and the time required for training the models22,29. In this paper, we considered the CKSAAP encoding scheme with K = 0, 1, 2, 3, 4 and 5, meaning the total dimension of the 6-spaced feature vector is 2400.
Amino acid occurrence frequency (AF)
The frequency of one amino acid in each sequence fragment was calculated by the following equation:
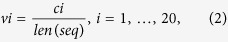 |
where ci and len (seq) denote the number of instances of amino acid i in the sequence fragment and the length of the sequence fragment, respectively. vi illustrates the frequency of the amino acids in the sequence.
Combined encoding schemes
We combined the three sole encoding schemes (KNN, CKSAAP and AF) to construct three bi-encoding schemes. Because of the high dimensionality of the CKSAAP, relief-F was used to decrease the total dimension for the combined methods. Relief-F is the extension to the original Relief algorithm, which is able to deal with noisy and multi-class problems rather than two-class problem30. Relif-F was run in Waikato Environment for Knowledge Analysis (WEKA) to decrease the dimension of the combining encoding schemes31, leading to total dimensions for AF-KNN, AF-CKSAAP and CKSAAP-KNN of 27, 952 and 939, respectively.
Classification Algorithms
Support Vector Machine (SVM)
A SVM is a supervised learning algorithm for two-group classification problems, whose goal is to find a rule that best maps each member of the training set to the correct classification25. Briefly, SVM constructs a hyperplane that separates two different groups of feature vectors in the training set with a maximum margin. The orientation of a test sample relative to the hyperplane gives the predicted score, and hence the predicted class can be derived32. Because of its solid mathematical foundation in statistics theory, and the ability of overcoming over-fitting, SVMs are popular and have been used to predict protein PTMs sites28, protein localization33 and protein-protein interaction34. In this paper, LibSVM in WEKA with Radial Basis Kernels (RBF) used with K (xi, yi) = exp(−γ||xi − yi||2)29.
Random Forest (RF)
The RF is an ensemble of unpruned decision trees35 which have already been used to predict protein-protein interactions36,37 and long disordered regions in proteins38. In RF, the number of trees in the forest is adjustable, and each tree is grown to full length using a subset of the training dataset. To classify an instance with an unknown class label, each tree casts a unit classification vote. The forest selects the classification having the most votes over all the trees in the forest. Therefore, there are two key parameters in RF. One is the number of the trees, M, the other is the number of features selected randomly, m. In this paper, we selected the optimal value of M = 100, and determined m based on the result of a preliminary evaluation.
Decision Tree (DT)
Decision trees are an attractive predictive modeling procedure because of their easy interpretation by non-statisticians. In DT algorithms, the classification tree analysis generates groups of individuals on the basis of a selected criterion, the Gini index, for splitting a group into two to maximize the probability of a single outcome, namely substantial renal deterioration39. The recursive process of partitioning data continues until the Gini index indicates that the tree fits, without overfitting, the information contained in the dataset. It can provide a practical model for dichotomous outcomes if the validity of the obtained model is proved sufficient. The missing values were replaced with values minimizing the impurity of the nodes, median values for continuous variables and most frequent categories for categorical variables, or distribution-based estimates. The decision trees and random forests were implemented using R platform with package e1071.
Performance Assessment
Predictors comparison
The current rice-specific phosphorylation sites predictor, PhosphoRice, two non-organism specific predictors, Musite and Scansite, and one plant-specific predictor, PlantPhos, were compared with the new predictor in this paper. In Musite online prediction, General phospho-serine/threonine and General phospho-tyrosine (Green plants) were selected and 25mer was input to the predictor with default settings. In Scansite, the setting of medium stringency level was selected and resulted in the production of Scansite_medium predictor. In PlantPhos, phosphorylation sites will be predicted with the score over −3.0 (HMMER bit score).
Evaluation
Sn, Sp, ACC and MCC were employed to evaluate the performance of the different predictors.
 |
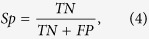 |
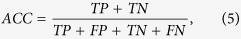 |
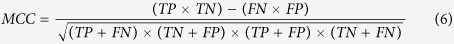 |
where TP, FP, FN, and TN denote true positives, false positives, false negatives, and true negatives. Sn and Sp illustrate the correct prediction ratios of positive and negative datasets, respectively. Because MCC is much less susceptible to the ratio of positive samples and negative samples in the dataset, it is the most widely used prediction measure for two-class prediction programs1.
Statistics
We used SPSS 16.0 to create receiver operating characteristic (ROC) curves to measure the performance of different predictors. For each possible threshold, the sensitivity and specificity were evaluated, and the ROC curves [sensitivity versus (1-specificity) curve] were used to compare the predictive performance of different classifiers with different encoding schemes.
Additional Information
How to cite this article: Lin, S. et al. Rice_Phospho 1.0: a new rice-specific SVM predictor for protein phosphorylation sites. Sci. Rep. 5, 11940; doi: 10.1038/srep11940 (2015).
Supplementary Material
Acknowledgments
This work was supported by the Natural Science Foundation of China and Fujian (No. 31270454, 61163047 and 2013J01077) and the Key Program of Ecology, Fujian, China (No. 0608507 and No. 6112C0600).
Footnotes
Author Contributions H.H. conceived of the study. Q.S. and W.W. designed the experiment, H.T. wrote the manuscript and A.H. revised it. S.L. and W.W. (Weifeng Wan) designed the encoding schemes, J.H. and C.X. developed the predictors. V.C. and S.Q. helped to test the predictors. J.K. maintained the online tool. All authors read and approved the final manuscript.
References
- Que S. et al. PhosphoRice: A meta-predictor of rice-specific phosphorylation sites. Plant Methods 8, 5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou K. C. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). J Theor Biol. 273, 236–247 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N., Gammeltoft S. & Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 294, 1351–1362 (1999). [DOI] [PubMed] [Google Scholar]
- Blom N., Sicheritz-Ponten T., Gupta R., Gammeltoft S. & Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–49 (2004). [DOI] [PubMed] [Google Scholar]
- Huang H. D., Lee T. Y., Tzeng S. W. & Horng J. T. KinasePhos: a web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 33, W226–229 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakoucheva L. M. et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer J. C., Cantley L. C. & Yaffe M. B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y., Li A., Wang L., Feng H. & Yao X. PPSP: prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinformatics 7, 163 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y. et al. GPS: a comprehensive www server for phosphorylation sites prediction. Nucleic Acids Res. 33, W184–187 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H., Lee J., Oh B., Kim K. & Koh I. Prediction of phosphorylation sites using SVMs. Bioinformatics 20, 3179–3184 (2004). [DOI] [PubMed] [Google Scholar]
- Gao J., Thelen J. J., Dunker A. K. & Xu D. Musite, a tool for global prediction of general and kinase specific phosphorylation sites. Mol Cell Proteomics 9, 2586–2600 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingrell C. R., Miller M. L., Jensen O. N. & Blom N. NetPhosYeast: prediction of protein phosphorylation sites in yeast. Bioinformatics 23, 895–897 (2007). [DOI] [PubMed] [Google Scholar]
- Heazlewood J. L. et al. PhosPhAt: a database of phosphorylation sites in Arabidopsis thaliana and a plant-specific phosphorylation site predictor. Nucleic Acids Res. 36, D1015–21 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmeri A. et al. PhosTryp: a phosphorylation site predictor specific for parasitic protozoa of the family trypanosomatidae. BMC Genomics 12, 614 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- IRGSP. The map-based sequence of the rice genome. Nature 436, 793–800 (2005). [DOI] [PubMed] [Google Scholar]
- Agrawal G. K. & Rakwal R. Rice proteomics: A Cornerstone for cereal food crop proteomics. Mass Spectrom Rev. 25, 1–53 (2006). [DOI] [PubMed] [Google Scholar]
- He H. & Li J. Proteomic analysis of phosphoproteins regulated by abscisic acid in rice leaves. Biochem Biophys Res Commun. 371, 883–888 (2008). [DOI] [PubMed] [Google Scholar]
- Chitteti B. R. & Peng Z. Proteome and phosphoproteome differential expression under salinity stress in rice (Oryza sativa L.) roots. J Proteome Res. 5, 1718–1727 (2007). [DOI] [PubMed] [Google Scholar]
- Ke Y., Han G., Chen X. & He H. Differential regulation of proteins and phosphoproteins in rice under drought stress. Biochem Biophys Res Commun. 379, 133–138 (2009). [DOI] [PubMed] [Google Scholar]
- Chen X. et al. Phosphosproteins regulated by heat stress in rice leaves. Proteome Sci. 9, 37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagami H., Sugiyama N., Mochida K. & Daudi A. Large-scale comparative phosphoproteomics identifies conserved phosphorylation sites in plants. Plant Physiol. 153, 1161–1674 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. et al. Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs. PLOS One 6, e22930 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neduva V. et al. Systematic discovery of new recognition peptides mediating protein interaction networks. PLOS Biol. 3, e405 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Kurgan L. A. & Ruan J. Prediction of flexible/rigid regions from protein sequences using k-spaced amino acid pairs. BMC Struct Biol. 7, 25 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. Z., Tang Y. R., Sheng Z. Y. & Zhang Z. Prediction of mucin-type O-glycosylation sites in mammalian proteins using the composition of k-spaced amino acid pairs. BMC Bioinformatics 9, 101 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban Y. et al. α-tubulin is rapidly phosphorylated in response to hyperosmotic stress in rice and Arabidopsis. Plant Cell Physio. 54, 848–58 (2013). [DOI] [PubMed] [Google Scholar]
- Petersen B., Petersen T. N., Andersen P., Nielsen M. & Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct Biol. 9, 51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J. et al. A new machine learning approach for protein phosphorylation site prediction in plants. Lect Notes Comput Sci. 5462, 18–29 (2009). [Google Scholar]
- Zhao X. W., Zhang W. Y., Xu X., Ma Z. Q. & Yin M. H. Prediction of protein phosphorylation sites by using the composition of k-spaced amino acid pairs. PLOS One 7, e46302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S. W., Abdul-Kareem S., Merican A. F. & Zain R. B. Oral cancer prognosis based on clinicopathologic and genomic markers using a hybrid of feature selection and machine learning methods. BMC Bioinformatics 14, 170 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. et al. The WEKA data mining software: an update. SIGKDD Exploration 11, 10–18 (2009). [Google Scholar]
- Noble W. S. Support Vector Machine applications in computational biology. In: Schollkopf B., Tsuda K., Vert J. P. Edited. Kernel Methods in Computational Biology. MIT Press, 61–95 (2004). [Google Scholar]
- Kumar R., Jain S., Kumari B. & Kumar M. Protein sub-nuclear localization prediction using SVM and Pfam domain information. PLOS One 9, e98345 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz M. M., Maleki M., Rueda L., Raza M. & Banerjee S. Prediction of biological protein-protein interactions using atom-type and amino acid properties. Proteomics 11, 3802–3810 (2011). [DOI] [PubMed] [Google Scholar]
- Breiman L. Random forests. Mach Learn. 45, 5–32 (2001). [Google Scholar]
- Chen X. W. & Liu M. Prediction of protein–protein interactions using random decision forest framework. Bioinformatics 21, 4394–4400 (2005). [DOI] [PubMed] [Google Scholar]
- Sikic M., Tomić S. & Vlahoviček K. Prediction of protein–protein interaction sites in sequences and 3D structures by random forests. PLOS Comput Biol. 5, e1000278 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P. et al. Large-scale prediction of long disordered regions in proteins using random forests. BMC Bioinformatics 10, 8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandomi A. H., Fridline M. M. & Roke D. A. Decision tree approach for soil liquefaction assessment. The Scientific World Journal 2013, 346285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.