

Abstract
A single double-strand break (DSB) induced by HO endonuclease triggers both repair by homologous recombination and activation of the Mec1-dependent DNA damage checkpoint in budding yeast1–6. Here we report that DNA damage checkpoint activation by a DSB requires the cyclin-dependent kinase CDK1 (Cdc28) in budding yeast. CDK1 is also required for DSB-induced homologous recombination at any cell cycle stage. Inhibition of homologous recombination by using an analogue-sensitive CDK1 protein7,8 results in a compensatory increase in non-homologous end joining. CDK1 is required for efficient 5′ to 3′ resection of DSB ends and for the recruitment of both the single-stranded DNA-binding complex, RPA, and the Rad51 recombination protein. In contrast, Mre11 protein, part of the MRX complex, accumulates at unresected DSB ends. CDK1 is not required when the DNA damage checkpoint is initiated by lesions that are processed by nucleotide excision repair. Maintenance of the DSB-induced checkpoint requires continuing CDK1 activity that ensures continuing end resection. CDK1 is also important for a later step in homologous recombination, after strand invasion and before the initiation of new DNA synthesis.
In budding yeast, a chromosomal DSB created by HO endonuclease has been used both to study the kinetics and efficiency of DSB repair and to analyse the induction of the DNA damage checkpoint dependent on Mec1 (an ATR homologue). In cells carrying HML or HMR mating-type switching donor sequences, a DSB at the MAT locus is efficiently repaired by gene conversion. In strains lacking donor sequences, induction of an unrepairable DSB causes arrest of cell cycle progression before anaphase1,2. In both instances, a key step is the 5′ to 3′ resection of DSB ends to produce single-stranded DNA (ssDNA), which is bound by the RPA complex. RPA binding is essential both for association of Mec1 checkpoint kinase9 and for loading of Rad51 recombination protein6.
Activation of the Mec1-dependent DNA damage checkpoint after a DSB is regulated by the cell cycle3, with no activation in G1-arrested cells. A DSB induced in cells that have been arrested in G1, and then released into S phase, results in hyperphosphorylation of the Mec1 target Rad53 after the completion of S phase, in G2 (Supplementary Fig. S1a). To test whether the checkpoint depends on the activity of cyclin-dependent kinases, we inactivated CDK1 in nocodazole-blocked G2 cells. We overexpressed the CDK1/Clb inhibitor, Sic1 (ref. 10), in G2 cells at the same time that an unrepairable DSB was induced at MAT. CDK1 inactivation is measured by the progressive dephosphorylation of the B subunit of DNA polymerase-α, a marker for Cdc28/Clb activity11 (Fig. 1). SIC1 overexpression prevents the accumulation of phosphorylated Rad53 and Chk1 (Fig. 1) and impairs hyperphosphorylation of the upstream checkpoint factors Ddc2 and Rad9 as well as Mre11 (Fig. 1). Because the phosphorylation of Ddc2 and Rad9 is directly mediated by Mec1 kinase, we conclude that CDK1 inactivation affects Mec1.
Figure 1.

CDK1 activity is required for DSB-induced phosphorylation of checkpoint proteins in G2 cells. The phosphorylation of checkpoint proteins in the presence of an HO-induced unrepaired DSB in G2/M cells arrested with nocodazole (N) is shown, comparing cells with active CDK1 or GAL::SIC1-inactivated CDK1.
To determine whether G1-arrested cells are able to perform homologous recombination (HR), we arrested MATa cells with α-factor and then induced a DSB with HO. Recombination between HMLα and MATa, monitored by Southern blots, was nearly absent in comparison with that in cells growing exponentially or in cells arrested in G2 with nocodazole, where replacement of MATa by MATα was efficient (Fig. 2a).
Figure 2.

CDK1 is required for homologous recombination. a, MAT switching is initiated by creating an HO-induced DSB at the MAT locus that is repaired by gene conversion from HML or HMR. MAT switching is shown in asynchronous cells or cells arrested in G1 (α-factor arrest) or G2/M (nocodazole arrest). b–d, MAT switching in cdc28-as1 (b), wild-type (c) and cdc7-as3 (d) strains with and without 1-NMPP1 inhibitor. e, Allelic recombination in a cdc28-as1 homozygous diploid strain. The position of two DNA probes is shown: MAT-distal (MD, verifies DSB induction) and MAT-proximal (MP, detects product). f, Efficiency of NHEJ in cdc28-as1 cells with either active or inactive CDK1 at different stages of the cell cycle. Error bars indicate s.d.
Inhibition of HR in G1-arrested cells was also seen in a diploid where a DSB at MAT could be repaired only by allelic recombination with an uncleavable MATa-inc locus on a homologous chromosome (Fig. 2e). We obtained similar inhibition of HO-induced recombination in G1 haploid cells when an HO-induced DSB was repaired by ectopic recombination between chromosomes III and V (ref. 5; data not shown).
To establish that the failure of HO-induced recombination in G1-arrested cells was attributable to a lack of Cdc28 activity, we examined recombination in cells expressing Cdc28-as1, a mutant sensitive to the ATP analogue inhibitor 1-NMPP1 (ref. 7). Galactose-induced HO cleavage was efficient, but cell-cycle-arrested cells failed almost completely to repair the DSB (Fig. 2b). 1-NMPP1 did not affect repair in wild-type cells (Fig. 2c).
CDK1 is also required for HR in the G2/M stage of cell cycle. We arrested cdc28-as1 cells in G2 with nocodazole and then induced the expression of HO. Whereas recombination was normal in G2-arrested cells, MAT switching was nearly abolished in Cdc28-inhibited cells (Fig. 2b).
Failure of both checkpoint activation and HR in G1-arrested cells and in both Sic1-inhibited and Cdc28-as1-inhibited G2 cells correlates with an absence of 5′ to 3′ resection of DSB ends. The effect of overexpressing GAL1::SIC1 in nocodazole-arrested G2 cells was shown by examining the rate of loss of the HO-cut MATa EcoRI restriction fragment in strains lacking HML or HMR, where the DSB is not repaired (Fig. 3a). Inhibiting CDK1 counteracted degradation of the HO-cut fragment as well as additional fragments a greater distance from the DSB. Resection was not significantly affected in G2-arrested cells deleted for Rad9, Rad17 or Mec1 checkpoint proteins12 (data not shown).
Figure 3.

CDK1 is needed for DSB resection. a, The 5′ to 3′ resection of DSB ends was analysed in the indicated mutants and at different points in the cell cycle by loss of the HO-cut EcoRI restriction fragment in a strain lacking HML or HMR. b, ChIP analysis of binding of RPA (left), Rad51 (centre) and Mre11 (right) to DSB ends in a donorless strain4,6, in asynchronous or G1 cells and in G2/M-arrested cells with or without Sic1 inhibition of CDK1. Where error bars are shown, results are means ± s.d. c, Lack of Rad53 phosphorylation in mre11Δ G2/M-arrested cells, compared with other conditions.
Similar defects in resection were seen in α-factor-arrested G1 cells and in all stages of the cell cycle when Cdc28-as1 was inhibited (Fig. 3a). Without resection, HO-cleaved MAT sequences fail to bind either RPA or Rad51 (Fig. 3b and Supplementary Fig. S1) as determined by chromatin immunoprecipitation (ChIP). A similar failure of RPA loading was seen when CDK1 was inhibited by Sic1 overexpression (Fig. 3b). In contrast, both resection and RPA and Rad51 binding are seen in nocodazole-arrested G2 cells, in which CDK1 is active (Fig. 3b). Without RPA and Rad51 binding, HR should not occur. The absence of RPA recruitment to DSB ends in CDK1-inhibited cells also accounts for the failure to activate the Mec1-dependent DNA damage checkpoint, because Mec1-Ddc2 recruitment depends on prior binding of RPA9.
The 5′ to 3′ resection of HO-induced DSB ends is reduced, but not eliminated, in cells deleted for MRE11, RAD50 or XRS2 (ref. 1). However, there are cell cycle differences in the control of resection. In G2-arrested cells, 5′ to 3′ resection depends almost completely on the MRX complex13, but in G1-arrested cells there is still residual resection when RAD50 is deleted (Fig. 3a). When Cdc28-as1 kinase was inhibited in wild-type G2-arrested cells, resection was as defective as in rad50Δ G2-arrested cells (Fig. 3a). We conclude that Cdc28 controls resection activity associated with the MRX complex. We note that Cdc2 in Schizosaccharomyces pombe has been suggested to control the resection of DSBs, but independently of Rad50 (ref. 14). Without resection, mre11Δ G2-arrested cells also fail to activate the DNA damage checkpoint15 (Fig. 3c).
Because Mre11 is required for DSB resection and checkpoint activation1,15,16, we asked whether defective resection caused by CDK1 inactivation reflects the inability to load Mre11 on broken chromosomes. We found Mre11 at DSBs in G2 cells (Fig. 3b and Supplementary Fig. S1), even when CDK1 is inactive. However, whereas in wild-type cells the amount of Mre11 crosslinked near the DSB declines by 2 h, in Sic1-overexpressing cells Mre11 remains associated with unresected ends. Transient association of Mre11 with DSB ends in wild-type cells might suggest that MRX proteins migrate down DNA as resection proceeds; however, we could not detect significant association of Mre11 with DNA 5 kilobases (kb) from the HO cut even after several hours (when resection has reached and passed this region6) (data not shown). However, it is possible that Mre11 becomes spread out and undetectable. CDK1 inactivation therefore does not prevent Mre11 loading onto DSBs.
Although G1-arrested cells fail to perform efficient DSB-induced HR, they exhibit an increased efficiency of non-homologous end joining (NHEJ). In strains lacking HML or HMR, repair of the DSB occurs only by Ku- and DNA ligase 4-dependent rejoining of the 4-base-pair (bp) 3′-overhanging complementary HO-cleaved ends17. When 1-NMPP1 was added 30 min before 1 h of HO induction in growing cells, NHEJ increased fivefold, from 12% to 65% (Fig. 2f). The same increase in NHEJ was observed when CDK1 inhibitor was added to nocodazole-arrested G2 cells (Fig. 2f). These results support and extend previous suggestions18,19 that the higher stability of DSB ends in G1-arrested cells stimulates NHEJ, which we propose might result from a lack of resection of these ends. In addition, NHEJ is probably aided by the retention of the end-joining protein Mre11 at DSB ends (Fig. 3b).
Lack of checkpoint activation in CDK1-inhibited cells proves to be specific for DSB damage and not for other conditions that induce Mec1-dependent checkpoint activation. For example, G1 cells exposed to the ultraviolet-mimetic agent 4-nitroquinoline 1-oxide (4NQO) exhibit Rad53 phosphorylation11 (Fig. 4a). CDK1 activity is therefore not required when the checkpoint is activated by presumably ssDNA intermediates of nucleotide excision repair. Furthermore, CDK1 inhibition does not affect checkpoint activation in response to S-phase arrest caused by hydroxyurea20, during which ssDNA intermediates arise at stalled replication forks21. We conclude that CDK1 influences Mec1-dependent checkpoint activation only when the DNA damage involves DSBs that must be resected to generate ssDNA.
Figure 4.
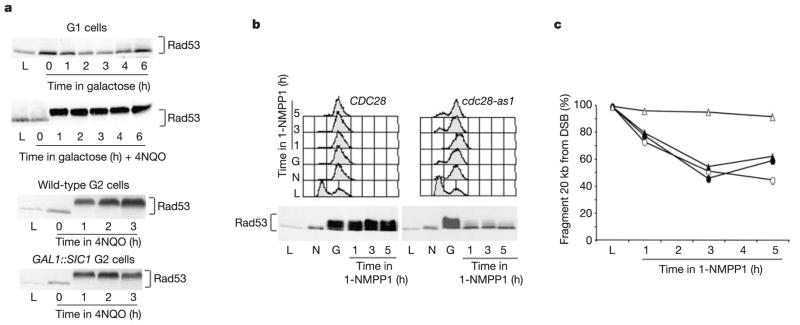
CDK1 is required to maintain Rad53 activation in response to a DSB but not after damage by 4NQO. a, In G1-arrested cells, Rad53 is not phosphorylated when an HO-mediated DSB is induced by galactose, but is triggered when 4NQO provokes nucleotide excision repair. In nocodazole-arrested G2/M cells, treatment with 4NQO causes Rad53 phosphorylation even when CDK1 is inhibited by Sic1. b, At 6 h after DSB induction, when the checkpoint is established and Rad53 is phosphorylated, 1-NMPP1 was added to either CDC28 or cdc28-as1 strains. c, Rad53 phosphorylation is lost when CDK1 is inactivated, and resection of a StyI fragment, 20 kb from the DSB, is inhibited. Open triangles, cdc28-as1 G2 + 1-NMPP1, filled triangles, cdc28-as1 G2, open circles, wild type G2 + 1-NMPP1.
We tested whether CDK1 activity was required for the maintenance of the active checkpoint state and continuing DSB resection. DSB formation was induced in wild-type and cdc28-as1 G2 cells. Both strains promote robust checkpoint activation, but when Cdc28-as1 was inactivated by adding 1-NMPP1, both the checkpoint and DSB resection were turned off (Fig. 4b, c). When inhibitor is added, a region 20 kb from the DSB fails to be degraded (Fig. 4c). Hence, CDK1 is required for ongoing resection and for maintaining the checkpoint. Indeed we believe that the signal for checkpoint maintenance is not simply the presence of ssDNA but rather some event that is linked to the continuing resection process.
Beyond affecting the formation of ssDNA, CDK1 seems to have a second function during HR. First, although in G2-arrested cells both the rad50Δ mutation and inhibition of Cdc28-as1 have the same effect on resection (Fig. 3a), recombination still occurs at about 20% of wild type in rad50Δ cells but is completely abolished when Cdc28-as1 is blocked (data not shown). This suggests that Cdc28 must affect other steps in DSB repair. To examine CDK1’s role more directly, we added inhibitor to cdc28-as1 cells at intervals after initiation of HO cleavage. When inhibitor was added 4–6 h after break induction, the extent of repair was unaffected, but at earlier times the addition of inhibitor reduced the completion of DSB repair (Fig. 5a). The most striking results were found when inhibitor was added 2 h after DSB induction. By this time, most DSB ends have been resected sufficiently to bind both Rad51 and RPA4,6. This is evident from ChIP analyses showing that, by 90 min, RPA and Rad51 had bound near the HO cut as efficiently as in wild-type cells. Moreover, Rad51-ssDNA filaments succeeded in strand invasion and synapsis with the donor, so that the HML locus (200 kb from MAT) could be immunoprecipitated by anti-Rad51 antibody4. As shown in Fig. 5b and Supplementary Fig. S1, Rad51-mediated synapsis between MAT and HML was comparable to that seen without inhibitor at each time up to 4 h, when MAT switching is complete without inhibitor. However, the next step in repair, the initiation of new DNA synthesis (primer extension) from the 3′ end of the invading strand, was severely impaired in cells to which inhibitor was added (Fig. 5c and Supplementary Fig. S1). We conclude that CDK1 is required after synapsis and before the initiation of new DNA synthesis, a process that requires the loading of the proliferating cell nuclear antigen (PCNA) clamp and the recruitment of DNA polymerase.
Figure 5.

Role of CDK1 in a late stage of MAT switching. a, Cdc28-as1 was inhibited in cells at different times before and after HO induction, and the amount of product formed at 6 h was determined. b, Strand invasion was tested by ChIP analysis for RPA and Rad51 in a cdc28-as1 strain. The diagram shows homologous sequences (dashed outlined boxes) and primers used to detect immunoprecipitation (IP) of MAT (p1, p2) or HML (p1, p3) with anti-Rad51 or Rfa1 antibodies. Symbols indicate the time at which inhibitor was added with respect to break induction: triangles, no inhibitor; squares, 0.5 h; circles, 1.5 h. c, Polymerase chain reaction detects the first new DNA synthesis primed by the 3 ′ invading end after strand invasion. Circles, no inhibitor; triangles, 1-NMPP1 added 1.5 h after HO induction. Where error bars are shown, results are means ± s.d.
The Cdc7 protein kinase is activated by CDK1 and is required for the initiation of DNA replication22. Using an analogue-sensitive allele of CDC7, cdc7-as3, we showed that HO-induced recombination is efficient in Cdc7-as3-inhibited cells (Fig. 2d). Thus, cells arrested in late G1, before the initiation of S phase but beyond the START point where CDK1 is activated, are capable of resecting DSB ends and completing HR.
Results presented here reveal a central role for budding yeast’s CDK1, Cdc28, in the regulation of DSB repair. First, when CDK1 is inactive, in G1 cells before passing START, HR is markedly reduced, as it is in G1-stage mammalian cells. This observation is supported by the finding that G1-arrested yeast cells fail to form DNA-damage-induced foci of Rad52 protein23. Moreover, the DNA damage checkpoint, which requires extensive resection of DSB ends, is not activated in G1-arrested cells3. We note that HR is not completely abolished in G1-arrested or Cdc28-inhibited cells; there is still residual 5′ to 3′ resection of DSB ends (apparently by a Cdc28-independent exonuclease) and about 5–10% of product is still formed. The CDK1-dependent regulation of HR and NHEJ might also help to explain the cell-cycle specificity of DSB repair in mammalian cells. Mammalian G1 cells predominantly repair ionizing radiation damage by NHEJ24. Similarly, V(D)J recombination by NHEJ during lymphoid development is restricted to G0/G1 phases of the cell cycle by the tight regulation of RAG2 gene expression and protein degradation. When RAG2 is expressed throughout the cell cycle it causes aberrant recombination products involving HR25. Once cells have passed START, HR is activated, allowing DSBs arising during replication to be repaired efficiently by HR.
We show that CDK1 is required for the establishment and maintenance of the Mec1/Rad53-mediated checkpoint response after DSB formation and for initiating and sustaining Mre11-dependent DSB resection. CDK1-mediated DSB resection influences checkpoint activation by allowing the formation of RPA-ssDNA filaments. Mre11 loading at DSB ends is not in itself sufficient to promote checkpoint activation, suggesting that Mec1-dependent checkpoint activation is instead influenced by the Mre11-mediated formation of RPA filaments. A tantalizing possibility is that in G2 CDK1 regulates the phosphorylation state of the MRX complex and/or other as yet unidentified factors implicated in DSB processing. Indeed several proteins implicated in DNA recombination are targeted by CDKs, including Xrs2, RPA and Srs2 (refs 8, 20). However, we note that neither mre11-T294V/S379A/S413A/T529V/S560A/T677V nor xrs2-T108V/S542A (in which the putative Cdc28-dependent phosphorylation sites have been mutagenized) exhibit hypersensitivity to DNA-damaging agents or appreciable defects in checkpoint activation after a DSB in G2 (data not shown). CDKs have been also implicated in the phosphorylation of checkpoint proteins (namely Rad9, Drc1/Sld2, Ddc1, Dun1 and Ptc3)8,26,27; however, it is not known whether these phosphorylation events have any role in checkpoint activation or rather in recovery from damage.
A hypothesis suggested by our data is that CDK1 has a key function in regulating alternative DSB repair pathways (NHEJ or HR) in G1 and G2. Hence, CDK1 inactivation in G2 seems to promote a G1-like repair response. Since Mre11 is required both for NHEJ and HR28, perhaps in CDK1-limiting G2 cells, Mre11 is ‘frozen’ in a NHEJ-proficient status, unable to resect DSB ends and therefore to generate checkpoint signals. This is also in accordance with observations in vitro showing that MRX complexes protect DNA ends from other exonucleases29.
Methods
Strains
Strains used to study DSB ends resection, DNA damage checkpoint activation and NHEJ were derivatives of JKM139 (hoΔ MATa hmlΔ::ADE1 hmrΔ::ADE1 ade1-100 leu2-3,112 lys5 trp1Δ::hisG ura3-52 ade3::GAL::HO) or JKM179 (same genotype except MATα), carrying GAL1::SIC1 (integrating pMHT plasmid with a stable version of the SIC1 gene under the GAL1 promoter11, kindly provided by J. Diffley); CHK1-13MYC; DDC-4HA; RAD9-13MYC; mre11Δ; mec1Δsml1Δ; rad50Δ; bar1Δ; cdc28-as1; cdc5-ad or the combination of these mutations. In MATa-inc strains the HO recognition site has a 1-bp substitution that prevents HO endonuclease cleavage. The diploid strain used to study DSB-induced checkpoint activation and resection was MATa/MATa-like, where part of the Yα sequence was replaced with KANMX. MAT switching was tested in MATa HMLα HMRa ade3::GAL::HO ade1-100 leu2-3,112 lys5 ura3-52 bar1Δ strains, some carrying cdc28-as1 or cdc7-as3. In the experiment shown in Fig. 2b, gene conversion from both HML and HMR yields cells that remain α-factor sensitive because both HMR and HML contain Ya information. In the strain used to study allelic recombination, the recipient chromosome has an insertion of URA3 at PHO87 located 5 kb to the left of MAT, whereas the donor chromosome has the Yα and Z1 sequences of MATα replaced by KANMX and is not cut by HO endonuclease.
Fluorescence-activated cell sorting analysis
Fluorescence-activated cell sorting was performed as described3.
CDK1 inactivation, α-factor arrest and nocodazole arrest
CDK1 and Cdc7 were inactivated in cdc28-as1 and cdc7-as3 strains by adding the ATP analogue inhibitor 1-NMPP1 at 5μM unless otherwise indicated. In cdc28-as1, cdc7-as3 and wild-type cells, inhibitor was added 1.25 doubling times before inducing HO unless otherwise indicated. Cells were arrested in G1 with 10μg ml−1 α-factor (or 0.5μg ml−1 in bar1Δ strains). Cells were arrested in G2/M with 20μg ml−1 nocodazole.
HO induction
Strains were grown in YP-lactate (1% Bacto yeast extract, 2% Bacto peptone, 3% lactic acid, pH 5.5) and HO induction was done as described previously4.
5′-3′ strand resection at a DSB
Resection was measured as a rate of HO-cut band disappearance. In some cases the resection of sequences distant from the break was also measured. Purified genomic DNA, digested with EcoRI (Fig. 3) or StyI (Fig. 4), was separated on a 1% agarose gel, transferred to Hybond N+ and probed with 32P-labelled DNA from MATa and from sequences 20 kb proximal to the DSB to establish the rate of resection. Hybridization to HIS4 or LEU2 (both more than 100 kb away) was used to normalize the amount of DNA at each time point. The rate of resection at each time point was plotted as the percentage of the density of the initial cut band. The density of the HO-cut band at t = 1 h was set to 100%.
NHEJ efficiency
NHEJ was examined in a donorless cdc28-as1 strain. 1-NMPP1 inhibitor was added 30 min before HO induction. Re-cutting of MATa by HO was prevented by filtering cells out of galactose-containing medium 30 min after DSB induction and diluting cells into YP-dextrose. (1% Bacto yeast extract, 2% Bacto peptone, 2% dextrose, pH 5.5). The efficiency of NHEJ was determined as the intensity of the MATa-containing restriction fragment 3 h after HO induction, normalized to the amount of DNA.
Analysis of homologous recombination
MAT switching and other homologous recombination events were analysed on Southern blots4. For MAT switching, genomic DNA was digested with StyI and BglII (Fig. 2a) or with StyI (Fig. 2b–d) and probed with a MAT-distal probe. For allelic recombination, DNA was digested with BglII and PvuI and probed with the MAT-distal probe to check the efficiency of HO induction and with a MAT-proximal probe to check the appearance of the product. Initial new DNA synthesis after strand invasion was determined by polymerase chain reaction as described4, normalized to the amount of ARG5,6 DNA.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as described previously4,6. Antibodies against Mre11, Rad51 and RPA were provided by J. H. J. Petrini, A. Shinohara and S. Brill, respectively.
Western blots
Protein extracts were performed as described3. Antibodies used for western blots were Rad53 polyclonal antibody JD47 (a gift from J. Diffley), Mre11 polyclonal antibody (produced by the IFOM antibody facility) and monoclonal antibodies 9E10 (anti-Myc epitope), 12CA5 (anti-HA (haemagglutinin) epitope) and 6D2 (ref. 11) (anti-B subunit).
Supplementary Material
Acknowledgments
The order of the co-first authors (G.I. and A.P.) and the order of the second two authors (A.B. and X.W.) were decided alphabetically. A.B. and X.W. made substantial and equal contributions to this work. We thank C. Zhang and K. Shokat (supported by NIH grant AI44009) for the cdc28-as1 allele and the 1-NMPP1 inhibitor. We note the ownership of the ASKA technology by Cellular Genomics Inc. and thank CGI for granting us the rights to perform the experiments discussed in the publication. We thank J. Petrini, J. Diffley, K. Labib, M. P. Longhese, S. Brill and the IFOM antibody facility for reagents. We are grateful to C. Lucca, E. Pandiani, M. B. Vaze, S. Francia, S. Lovett, M. Lichten and the members of our laboratories for discussions and technical support. N.M.H. was supported by the NIH. M.F. and A.P. were supported by grants from Associazione Italiana per la Ricerca sul Cancro and M.F. from Telethon-Italy and the European Community. G.I. was supported by Charles A. King Trust Postdoctoral Fellowship. G.I. is on leave from N. Copernicus University, Torun, Poland. J.E.H. was supported by the NIH and the DOE.
Footnotes
Supplementary Information accompanies the paper on www.nature.com/nature.
Competing interests statement The authors declare that they have no competing financial interests.
References
- 1.Lee SE, et al. Saccharomyces Ku70, Mre11/Rad50 and RPA proteins regulate adaptation to G2/M arrest DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 2.Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell. 1997;90:1097–1106. doi: 10.1016/s0092-8674(00)80375-x. [DOI] [PubMed] [Google Scholar]
- 3.Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from G2/M arrest. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- 4.Sugawara N, Wang X, Haber JE. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol Cell. 2003;12:209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- 5.Vaze MB, et al. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell. 2002;10:373–385. doi: 10.1016/s1097-2765(02)00593-2. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Haber JE. Role of Saccharomyces single-stranded DNA-binding protein RPA in the strand invasion step of double-strand break repair. PLoS Biol. 2004;2:104–111. doi: 10.1371/journal.pbio.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bishop AC, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 8.Ubersax JA, et al. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- 9.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 10.Nasmyth K. At the heart of the budding yeast cell cycle. Trends Genet. 1996;12:405–412. doi: 10.1016/0168-9525(96)10041-x. [DOI] [PubMed] [Google Scholar]
- 11.Pellicioli A, et al. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J. 1999;18:6561–6572. doi: 10.1093/emboj/18.22.6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naiki T, Wakayama T, Nakada D, Matsumoto K, Sugimoto K. Association of Rad9 with double-strand breaks through a Mec1-dependent mechanism. Mol Cell Biol. 2004;24:3277–3285. doi: 10.1128/MCB.24.8.3277-3285.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diede SJ, Gottschling DE. Exonuclease activity is required for sequence addition and Cdc13p loading at a de novo telomere. Curr Biol. 2001;11:1336–1340. doi: 10.1016/s0960-9822(01)00400-6. [DOI] [PubMed] [Google Scholar]
- 14.Caspari T, Murray JM, Carr AM. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 2002;16:1195–1208. doi: 10.1101/gad.221402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grenon M, Gilbert C, Lowndes NF. Checkpoint activation in response to double-strand breaks requires the Mre11/Rad50/Xrs2 complex. Nature Cell Biol. 2001;3:844–847. doi: 10.1038/ncb0901-844. [DOI] [PubMed] [Google Scholar]
- 16.D’Amours D, Jackson SP. The yeast Xrs2 complex functions in S phase checkpoint regulation. Genes Dev. 2001;15:2238–2249. doi: 10.1101/gad.208701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank-Vaillant M, Marcand S. Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell. 2002;10:1189–1199. doi: 10.1016/s1097-2765(02)00705-0. [DOI] [PubMed] [Google Scholar]
- 19.Karathanasis E, Wilson TE. Enhancement of Saccharomyces cerevisiae end-joining efficiency by cell growth stage but not by impairment of recombination. Genetics. 2002;161:1015–1027. doi: 10.1093/genetics/161.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liberi G, et al. Srs2 DNA helicase is involved in checkpoint response and its regulation requires a functional Mec1-dependent pathway and Cdk1 activity. EMBO J. 2000;19:5027–5038. doi: 10.1093/emboj/19.18.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 22.Foiani M, Liberi G, Lucchini G, Plevani P. Cell cycle-dependent phosphorylation and dephosphorylation of the yeast DNA polymerase α-primase B subunit. Mol Cell Biol. 1995;15:883–891. doi: 10.1128/mcb.15.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lisby M, Mortensen UH, Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nature Cell Biol. 2003;5:572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- 24.Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J, Desiderio S. Cyclin A/CDK2 regulates V(D)J recombination by coordinating RAG-2 accumulation and DNA repair. Immunity. 1999;11:771–781. doi: 10.1016/s1074-7613(00)80151-x. [DOI] [PubMed] [Google Scholar]
- 26.Toh GW, Lowndes NF. Role of the Saccharomyces cerevisiae Rad9 protein in sensing and responding to DNA damage. Biochem Soc Trans. 2003;31:242–246. doi: 10.1042/bst0310242. [DOI] [PubMed] [Google Scholar]
- 27.Masumoto H, Muramatsu S, Kamimura Y, Araki H. S-Cdk-dependent phosphorylation of Sld2 essential for chromosomal DNA replication in budding yeast. Nature. 2002;415:651–655. doi: 10.1038/nature713. [DOI] [PubMed] [Google Scholar]
- 28.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nature Rev Mol Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 29.Chen L, Trujillo K, Ramos W, Sung P, Tomkinson AE. Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell. 2001;8:1105–1115. doi: 10.1016/s1097-2765(01)00388-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.