

Abstract
Background
Schizophrenia is a neurodevelopmental disorder with genetic and environmental factors contributing to its pathogenesis, although the mechanism is unknown due to the difficulties in accessing diseased tissue during human neurodevelopment. The aim of this study was to find neuronal differentiation genes disrupted in schizophrenia and to evaluate those genes in post-mortem brain tissues from schizophrenia cases and controls.
Methods
We analyzed differentially expressed genes (DEG), copy number variation (CNV) and differential methylation in human induced pluripotent stem cells (hiPSC) derived from fibroblasts from one control and one schizophrenia patient and further differentiated into neuron (NPC). Expression of the DEG were analyzed with microarrays of post-mortem brain tissue (frontal cortex) cohort of 29 schizophrenia cases and 30 controls. A Weighted Gene Co-expression Network Analysis (WGCNA) using the DEG was used to detect clusters of co-expressed genes that werenon-conserved between adult cases and controls brain samples.
Results
We identified methylation alterations potentially involved with neuronal differentiation in schizophrenia, which displayed an over-representation of genes related to chromatin remodeling complex (adjP = 0.04). We found 228 DEG associated with neuronal differentiation. These genes were involved with metabolic processes, signal transduction, nervous system development, regulation of neurogenesis and neuronal differentiation. Between adult brain samples from cases and controls there were 233 DEG, with only four genes overlapping with the 228 DEG, probably because we compared single cell to tissue bulks and more importantly, the cells were at different stages of development. The comparison of the co-expressed network of the 228 genes in adult brain samples between cases and controls revealed a less conserved module enriched for genes associated with oxidative stress and negative regulation of cell differentiation.
Conclusion
This study supports the relevance of using cellular approaches to dissect molecular aspects of neurogenesis with impact in the schizophrenic brain. We showed that, although generated by different approaches, both sets of DEG associated to schizophrenia were involved with neocortical development. The results add to the hypothesis that critical metabolic changes may be occurring during early neurodevelopment influencing faulty development of the brain and potentially contributing to further vulnerability to the illness.
Electronic supplementary material
The online version of this article (doi:10.1186/s12920-015-0098-9) contains supplementary material, which is available to authorized users.
Keywords: Schizophrenia, Gene network, Neuronal differentiation, Module analyses, Oxidative stress
Background
Schizophrenia is a severe and debilitating neuropsychiatric disorder with a worldwide prevalence of approximately 1 % and an estimated heritability of 80–85 % [1]. Multiple factors contribute to its pathogenesis, including genetic and environmental factors [2–4], which are involved in the disturbance of the neuronal network formation and brain function [5].
Although the genetic component involvement is clear, the precise molecular and cellular defects that precedes illness onset and the sequence of mechanisms that initiate or attenuate disease progression, particularly in neurons, are still unknown. In addition to findings from animal models [6–8], post-mortem human brain studies in schizophrenia revealed significant alterations in genes with high expression in multiple cortical and hippocampal regions [9–13]. Data from prefrontal cortex obtained post-mortem revealed 28 modules of co-expressed genes associated to oxidative phosphorylation, energy production and metabolism, post-translation modifications, neurogenesis and neuronal differentiation. Additionally, these modules showed a robust effect of age suggesting that genes related to nervous system development process and neuronal differentiation are deregulated in schizophrenia [14]. A co-expression module analysis from patients with schizophrenia (SCZP) and controls of seven studies showed that the top-modules of each study displayed a significant enrichment of neuronal markers as well as genes related to the electron transport chain and oxidative phosphorylation biological processes [15]. Eventually, these alterations, through the analyses of genes and proteins, can be found in the peripheral blood suggesting that they are potential molecular markers for diagnosis or follow-up [16–19].
Promising approaches involve the study of tissue from the olfactory system by nose biopsies [20] and the development of human induced pluripotent cells (hiPSC) techniques. The later demonstrated that functional primarily glutamatergic neurons differentiated from hiPSC were able to fire action potentials after eight weeks in culture, showing the possibility of studying schizophrenia through this technique [21]. SCZP-hiPSC-derived neurons display diminished neuronal connectivity with reduced neuritis and changes in Wnt and cAMP signaling pathways when compared to controls [22]. Additionally, RNA sequencing analysis of hiPSCs-derived neurons highlighted the importance of long non-coding RNAs in neurogenesis [23].
In a previous study, we showed that neural progenitor cells (NPCs), derived from hiPSCs from skin fibroblasts of a schizophrenic patient had a 2-fold increase in extramitochondrial oxygen consumption compared to normal controls, and correspondingly elevated levels of reactive oxygen species (ROS), which was reduced to control levels by addition of valproic acid [24]. Mitochondrial dysfunction was also associated with impaired differentiation of neurons into both mature dopaminergic and glutamatergic phenotypes in schizophrenia-derived hiPSCs [25].
Although genes involved with ROS and other molecular mechanisms in hiPSC and NPC have been studied in patients with schizophrenia [26], the ones specifically disrupted in neuronal differentiation were not assessed directly in adult brain samples. The aim of this study was to disclose gene expression changes during neuronal differentiation disrupted in schizophrenia and to expand these findings to the brain tissue in adults. To achieve that, genes identified as involved with neuronal differentiation exclusively in schizophrenia were analyzed by co-expression network in post-mortem brain samples derived from adult patients and controls. Then, we identified the biological processes associated with a less conserved module between cases and controls brain samples.
Methods
Tissue collection
In short, biopsies of primary human fibroblasts from a 48 year-old woman with DSM-IV-TR schizophrenia defined as treatment-resistant and under clozapine treatment (SZCP) were collected in parallel with a control subject (CON) that negative for any major lifetime DSM-IV-TR diagnosis. Fibroblasts underwent primary culture procedures to generate hiPSCs and were subsequently differentiated into neurons (NPC). Details were previously described [24].
Total RNA was isolated from three plates of cell culture (with molecular confirmation for the cell type that was being studied [24]) using Trizol (TRI Reagent, Sigma-Aldrich, St Louis, MO, USA) according to the manufacturer’s instructions. RNA quantity and integrity were assessed by spectrophotometry (NanoDrop ND-2000 UV–Vis Spectrophotometer, NanoDrop Technologies) and microfluidics-based electrophoresis (Agilent 2100 Bioanalyzer; Agilent Technologies, Santa Clara, California), respectively, and obtained OD of ~2.0 and RIN > 9. Genomic DNA was extracted using standard proteinase K digestion followed by phenol/chloroform extraction. DNA quantity and integrity were assessed by spectrophotometry (NanoDrop ND-2000 UV Spectrophotometer, NanoDrop Technologies) and agarosis gel 0.8 %.
The study protocol was approved by the ethics committee of the Hospital de Clínicas de Porto Alegre, Porto Alegre (RS) with all subjects providing written informed consent co-signed by close relatives (Register 09-108). The study was performed in accordance with the Declaration of Helsinki.
Comparative genomic hybridization based on microarray (array-CGH)
We performed comparative genomic hybridization based on microarrays (array-CGH) in a commercial whole-genome 180 K platform containing 180,000 oligonucleotide probes (design 30864, Agilent Technologies, Palo Alto, California) using as reference DNA a commercially available human pool of healthy individuals (Promega). Purification, cohybridization of labelled test and reference DNA samples and washing were carried out according to the manufacturer’s instructions. Scanned images were processed using the Feature Extraction 10.7.3.1 software (Agilent Technologies, Palo Alto, California).
Methylation experiments
Bisulfite-conversion of 500 ng of DNA was performed according to the manufacturer’s recommendations (EZ DNA Methylation Gold Kit, Zymo Research, Irvine, California) for the Illumina Infinium Assay. The incubation profile was 16 cycles at 95 °C for 30 s, 50 °C for 60 min and a final holding step at 4 °C for 10 min. In total, 10 μl of bisulfite-converted DNA were used for hybridization on Infinium HumanMethylation 450 BeadChip with 485,557 probes, following the Illumina Infinium HD Methylation protocol (Illumina, Inc, San Diego, California). All arrays passed Illumina standard quality control metrics and were included in this study.
Microarray experiments
For mRNA expression, samples were prepared according to the Agilent mRNA Microarray System protocol using 250 ng of total RNA for amplification and labelled using the Agilent Low RNA Input Fluorescent Linear Amplification Kit. Samples were hybridized in the Agilent Whole Human Genome Microarray Kit, 4 × 44 K-G4112F in the Agilent SureHyb chambers (Agilent Technologies, Palo Alto, California) for 20 h at 55 °C, and the platforms were washed with the manufacturer’s washing buffers. Controls were used in all labelling reactions. Scanned images of the arrays were processed using Feature Extraction 10.7.3.1 software (Agilent Technologies, Palo Alto, California) with default parameters. Quality control of the probes (i.e. low intensity, saturation, controls, etc.) selected 30,635 probes corresponding to 22,186 transcripts for the analyses. From those, 14,491 probes correspond to unique genes and 7695 were not associates with an ENTREZID. Four genes had more than one ENTREZID with the same Official Symbol and were excluded from the analyses.
Frontal cortical samples from a previous study were used for further analyses, all presenting good quality total and amplified RNA [27]. Postmortem brain tissue was donated by The Stanley Medical Research Institute's brain collection. Details about ethics regarding these samples are available at the Neuropathology Consortium (http://www.stanleyresearch.org). Comparing patients with schizophrenia and controls, there were no significant differences concerning age, brain weight or PMI, with only pH showing a significant difference (p = 0.01). In short, 30 controls subjects (CTS) and 29 patients with schizophrenia (SZP) from the Stanley Foundation were hybridized in the Agilent 4x44K human oligonucleotide microarray (Agilent 4112 F, Agilent Technologies, Palo Alto, California) following manufacturer procedures. For analysis of replicate spots, the average intensity after background correction was calculated, followed by normalization using locally weighted linear regression (LOWESS) with α = 0.2 within slides using the R software version 2.11.1 (R Development Core Team, 2010).
Data were deposited in NCBI Gene Expression Omnibus and are accessible through GEO Series accession number GSE62191 and GSE62105.
Data analyses
DNA copy number variations (CNVs) were identified with Nexus software 7.0 (Biodiscovery, Hawthorne, CA, USA) using the FASST2 segmentation algorithm. To be considered a CNV, the criteria were: a minimum of 3 consecutive affected probes (effective resolution of ~50 Kb for CNV calling), a significance threshold set at 5.0e−8, and threshold log2 ratio Cy3/Cy5 of 0.4 for gain and −0.4 for loss. Data from sex chromosomes were excluded. Gene annotation was performed according to the GRCh37/hg19using the University of California Santa Cruz Genome Browser (http://genome.ucsc.edu/) and the Database of Genomic Variants (DGV; May 2014 update).
Due to the low number of samples, we simplified the methylation analyses. CpGs with β-values ≤0.2 were considered hypo-methylated and with β-values ≥0.8 were considered hyper-methylated, which were used to construct Venn-diagrams to compare the data in fibroblasts, hiPSC and NPC.
For gene expression analyses, it was applied HT-self [28]. Self–self experiments were performed by labelling the hIPSC from SZCP with either Cy3 or Cy5 dyes and hybridizing simultaneously on the same microarray slide to determine the intensity dependent cut-offs. These cut-offs were applied to the non-self–self experiment that was performed in triplicate (SCZP × CON). To identify differentially expressed genes, three criteria were applied: a. each gene must be represented by at least two probes; b. more than 50 % of the probes representing that gene presented reliable signal; c. there was at least 80 % of signal agreement among probes. To avoid technical variability, only genes that reached the three criteria in all replicates were selected.
To investigate if genes identified in this study could have a role during foetal development of the prefrontal cortex (dorsolateral prefrontal cortex, ventrolateral prefrontal cortex, anterior (rostral) cingulate (medial prefrontal) cortex), they were searched in the BrainSpan database (Allan Brain Atlas) [29]. Genes with at least 1 RPKM in more than 75 % of the samples or those who had an expression interquartile range >0.5 were considered for the analyses. Genes were assigned as foetal or non-foetal according to the development stage of the sample. It was considered differentially expressed those genes with fold-change >2.
The Wilcoxon Rank Sum Test was applied using TMEV software package [30] to identify differentially expressed genes between 29 SZP and 30 CTS considering those with p < 0.01 as significantly differentially expressed. As pH was significantly different in the brains between cases and controls, we accessed the DEG which expression was possibly being affected by pH. Hence, a multiple regression analysis was applied for all expressed genes in the microarray platform with a further comparison to 1000 permutated datasets. We observed that the difference of pH in the brain affects around 20 % of expressed genes (F, p < 0.05) of the whole array. These genes were compared to the DEGs between SZP and CTS.
The DEG sets found by both analyses were compared to the findings of a published study that defined 17 modules comprised of co-expressed genes associated with human cortical development. Briefly, to define human brain development modules, RNA-sequencing data from neocortical regions of the BrainSpan were analyzed through co-expression networks using the R package Weighted Gene Co-expression Network Analysis (WGCNA) [31]. The identified modules were characterized using GO-Elite [32] for controlling the network-wide false discovery rate. It was significant those pathways that had >10 genes at Z-summary >2 and FDR < 0.01 [31]. To estimate the enrichment of the DEG sets among the 17 modules, the Modular Single-set Enrichment Test (MSET) was applied with 10,000 permutations [33]. For that, we calculated the probability of the DEG being significantly represented in each module compared to 10,000 simulated sets of genes generated randomly from the microarray background. For each DEG and module, the p-value was calculated based on the number of simulated randomized sets that contains equal or higher number of genes belonging to the DEG in that module. Scatter smooth plots were construct only for the modules that had DEG over-representation (p-value ≤0.05) using the module eigengene values identified by the study [31].
In this study, the preservation module function from the WGCNA package in R were used to build unsigned co-expression networks to characterize features of co-expressed gene modules [34–36]. A pairwise correlation matrix was computed for all gene pairs across all samples, and an adjacency matrix was calculated by raising the correlation matrix to the power of 7. This threshold was chosen based on the scale-free topology criterion, which is the smallest threshold that resulted in a scale-free R2 fit about 0.8, and used for all networks: schizophrenia samples only and controls samples only. For each pair of genes, a robust measure of network interconnectedness (topological overlap measure) was calculated based on the adjacency matrix. The topological overlap based dissimilarity was then used as input for average linkage hierarchical clustering. Finally, modules were defined as branches of the resulting clustering tree. To cut the branches, we used the hybrid dynamic tree-cutting in order to have robustly defined modules. The minimum module size was set to 20 genes with the deepSplit parameter set to 2. Each module was summarized by the first principal component of the scaled (standardized) module expression profiles (ME). Hub genes were assigned as having high intramodular connectivity kME >0.75. To discriminate changes in modules comparing SZP and CTS, we performed a module Preservation analysis that identified which modules were preserved in both datasets. For that, a Z-summary was calculated to exclude the possibility of randomness in preservation based on 1000 permutations, and indicating if a module is strongly (Z-summary >10), moderately (2 < Zsummary <10) or not preserved (Z-summary <2). For visualization, the resulting modules were plotted using Cytoscape [37].
For DEG, functional and disease enrichment analyses were performed in WebGestalt [38] using the whole genome as background with a further analysis implemented in DAVID (database http://david.abcc.ncifcrf.gov/). It was considered those biological processes and pathways that comprised more than 10 genes and FDR < 0.05.
Results
Characterization of molecular alterations potentially interfering with gene expression
To refine the identification of genes regulated during neuronal differentiation, copy number alterations were characterized by array comparative genomic hybridization (CGH). Most of the detected copy number variations (CNVs) were present in all cell types (fibroblasts, hiPSCs and NPCs), which precludes their characterization as an acquired copy number alteration related to cell differentiation. There was no CNVs found exclusively in NPC from SCZP. Nevertheless, one large gain at 20q11.21 (1.2 MB) was acquired by the hiPSC and maintained in the NPC from the SCZP. The region contains 32 genes (Additional file 1: Figure S1). There were no CNVs found exclusively in NPC from SCZP.
To explore methylation alterations potentially involved in neuronal differentiation in schizophrenia, we compared NPCs, hiPSC and fibroblast from SCZP and CON, selecting CpG sites differentially methylated exclusively between hiPSCs and NPCs in SCZP and CON. A Venn-diagram identified 51 (33 genes) and 5 (3 genes) CpG sites as hypo- or hyperrmethylated, exclusively in SCZP, respectively (Additional file 1: Figure S2, Table S1). These genes were over-represented in specific locations of the genome, 1p36, 10q22, 12q22 and 14q24 (adjusted p-value, adjP < 0.05). We also found overrepresentation of chromatin remodeling complex (adjP = 0.04), represented by DPF3, SMARCA2 and RERE. DPF3b interacts with the histones H3 and H4 in an acetylation-sensitive manner to regulate transcription [39]. SMARCA2 is a member of the SWI/SNF family of proteins with helicase and ATPase activities and thought to activate transcription altering chromatin structure in specific sites [40]. RERE is a nuclear receptor co-repressor that binds to the histone methyltransferase G9a orchestrating molecular events that lead to a stable methylation of histone H3-lysine 9 [41].
Characterization of expression patterns in SCZP
Gene expression profiles from hiPSC (SCZP or CON) were compared to the corresponding groups of differentiated cells (i.e. NPCs) to identify differences during neuronal differentiation. We considered that the analyses in CON-derived cells identified DEG during neuronal differentiation; there were 1658 up-regulated (range from 0.50 to 6.76) and 1698 down-regulated (ranging from −0.43 to −8.49) genes. Comparison of the SCZP-derived cells revealed 752 up-regulated (range from 0.56 to 3.4) and 818 down-regulated (ranging from −0.54 to −4.97) genes. Subsequently, to exclude genes associated with normal neuronal differentiation, up- and down-regulated genes in CON and SCZP were compared to select those that were exclusive for SCZP. The analysis revealed 228 genes (151 up- and 77 down-regulated) altered only during neuronal-differentiation of the SCZP cells (Additional file 1: Table S2, Figure S3), from which ten were described by studies in schizophrenia. Most of them were related to nervous system development and/or plasticity: NDEL1, ATF4, AKT1, MDGA1, PDE4B, FEZ1, TF3, ADRBK2, CACNA1C, ADRA2A (Additional file 1: Table S3). In agreement with our data, 10 genes (ATF4, CACNA1C, CPE, CTSF, EPHA2, GPR155, HOXB9, MYBL1, NLGN1, and PMAIP1) were identified by an independent study that used a similar design (hiPSC x NPC in schizophrenia cases) [26].
Using expression data from the prefrontal cortex of Brain Span (Allen Brain Atlas), expression of the 228 gene set were compared between 16 foetal and 22 non-foetal samples. Nine genes were not present in Allen Atlas and 35 did not pass quality control (see material and methods). From the remaining 184 genes, 73 (40 %) were differentially expressed and therefore considered important for brain development.
To explore the functional significance of the genes from neurodevelopment associated with schizophrenia, we performed unbiased Gene Ontology term enrichment and pathway analyses of the 228 genes. The most noteworthy biological processes that were overrepresented were metabolic processes, signal transduction, nervous system development, regulation of neurogenesis and neuronal differentiation. Among the cellular pathways, we found over-representation of genes related to MAPK signaling pathway, pathways in cancer and metabolic pathways (Table 1). Finally, it was interesting to note that genes involved with cancer, infectious, metabolic and brain disorders (including stress, bipolar disorder and schizophrenia) were overrepresented diseases (Table 2).
Table 1.
Signaling cellular pathways over-represented among the 228 gene set
| Pathway | Statistical test | Genes |
|---|---|---|
| MAPK signalling pathway | C = 268;O = 13;E = 1.38;R = 9.42; p = 1.47e09;adjP = 1.50E-07 | PDGFA, ATF4, AKT1, CACNA1C, |
| DUSP6, CACNA1E, CACNA1H, NLK, | ||
| CHUK, MAP3K5, NTF3, FOS, PPM1V | ||
| Pathways in cancer | C = 326;O = 13;E = 1.68;R = 7.75; p = 1.80e08;adjP = 7.74E-07 | PDGFA, RARA, AKT1, ITGA2, E2F3, |
| FH, SMO, CHUK, FZD5, FOS, BID, | ||
| SPI1, HDAC2 | ||
| Metabolic pathways | C = 1130;O = 21;E = 5.82;R = 3.61; p = 4.84e07;adjP = 1.39E-05 | IMPA1, TRDMT1, HSD17B8, DPM1, |
| MGAT4B, CPOX, FH, SPHK1, DCT, | ||
| GAL3ST1, PNMT, PIK3C2A, ASNS, PC, MTHFS, PPAP2B, CDO1, HAAO, | ||
| ATP6V1C1, ALDH1A1, AGL |
Criteria: at least 10 genes represented in the pathway and adjpvalue ≤0.05 (hypergeometric t test with p value adjusted by multiple test adjustment, Benjamini and Hochberg [69])
C number of reference genes in the category number of genes in the gene set and O in the observed category, E expected number in the category, R ratio of enrichment, p p value from hypergeometric test, adjP value adjusted by the multiple test adjustment (adjP)
Table 2.
Diseases over-represented among the 228 gene set
| Disease | Statistical test | Genes |
|---|---|---|
| Cancer or viral infections | C = 951;O = 25;E = 4.90;R = 5.11; p = 3.35e-11;adjP = 8.71e-10 | RARA, WIF1, AKT1, LGALS3, RB1CC1, E2F3, FH, SPHK1, |
| DCT, FOS, BID, PDGFA, CD24, S100A7, EPHA2, PMAIP1, | ||
| SMO, RHOBTB2, CTNND1, SPI1, ETV1, ALDH1A1, CEACAM1, HDAC2, MYB | ||
| Neoplasms | C = 854;O = 22;E = 4.40;R = 5.00; p = 7.90e-10;adjP = 1.03e-08 | RARA, WIF1, AKT1, LGALS3, E2F3, FH, DCT, PDGFA, CD24, |
| S100A7, EPHA2, PMAIP1, FLT1, SMO, RHOBTB2, CTNND1, SSTR1, SPI1, ETV1, ALDH1A1, CEACAM1, MYB | ||
| Drug interaction with drug | C = 349;O = 13;E = 1.80;R = 7.24; p = 4.00e-08;adjP = 3.47e-07 | CEBPB, PPIB, KPNA4, RARA, AKT1, PMAIP1, ITGA2, ACTA1, PACS1, STX4, FOS, BID, HDAC2 |
| HIV | C = 755;O = 18;E = 3.89;R = 4.63; p = 9.30e-08;adjP = 6.04e-07 | ADARB1, PPIB, KHDRBS1, MGAT4B, FURIN, PSMD7, PACS1, |
| FOS, EED, PIK3C2A, CEBPB, PSMB10, KPNA4, MANEA, DDX6, ACTA1, GTF2H1, HLA-DOA | ||
| Bipolar disorder | C = 344;O = 12;E = 1.77;R = 6.78; p = 2.71e-07;adjP = 1.41e-06 | IMPA1, ATF4, DUSP6, CACNA1C, MDGA1, CTSF, CIT, PDE4B, FEZ1, NLGN1, NTF3, ADRBK2 |
| Infection | C = 516;O = 14;E = 2.66;R = 5.27; p = 5.72e-07;adjP = 2.48e-06 | MIF, PSMB10, PPIB, KPNA4, TBK1, MICB, FURIN, PACS1, NKRF, IL13RA1, TICAM1, HLA-DOA, CEACAM1, PIK3C2A |
| Metabolic diseases | C = 612;O = 15;E = 3.15;R = 4.76; p = 8.02e-07;adjP = 2.61e-06 | PC, PEX6, JAZF1, CST3, CPOX, TOP3A, APOA2, NEFH, ADIPOR2, PYGL, LIPA, AGL, APOA1, BCL11A, ZMPSTE24 |
| Leukemia | C = 452;O = 13;E = 2.33;R = 5.59; p = 7.65e-07;adjP = 2.61e-06 | RCBTB1, RARA, ASNS, AKT1, HUWE1, PMAIP1, PRDM16, DOK1, CASC5, BID, SPI1, BCL11A, MYB |
| Stress | C = 464;O = 13;E = 2.39;R = 5.44; p = 1.02e-06;adjP = 2.95e-06 | ASNS, ATF4, MTF1, AKT1, PMAIP1, MICB, ACTA1, SGK1, CHUK, MAP3K5, FOS, PRDX4, RPL11 |
| Mental disorders | C = 564;O = 14;E = 2.90;R = 4.82; p = 1.62e-06;adjP = 3.79e-06 | NDEL1, CACNA1C, MDGA1, ASPM, BACE1, CST3, PDE4B, NEFH, FEZ1, NLGN1, NTF3, ADRBK2, CDK5R1, ADRA2A |
| Adhesion | C = 647;O = 15;E = 3.33;R = 4.50; p = 1.59e-06;adjP = 3.79e-06 | TEK, CD24, AKT1, LGALS3, EPHA2, RB1CC1, MDGA1, ITGA2, |
| ACTA1, CADM3, PTPN12, NLGN1, CTNND1, CEACAM1, CLDN11 | ||
| Neoplastic processes | C = 411;O = 12;E = 2.12;R = 5.67; p = 1.75e-06;adjP = 3.79e-06 | WASF3, CD24, WIF1, AKT1, LGALS3, EPHA2, FLT1, SPHK1, CTNND1, ETV1, ALDH1A1, CEACAM1 |
| Death | C = 343;O = 11;E = 1.77;R = 6.23; p = 1.94e-06;adjP = 3.88e-06 | RBM20, ATF4, AKT1, PMAIP1, SPHK1, HRK, BCLAF1, CHUK, MAP3K5, BID, CDK5R1 |
| Leukemia, myeloid | C = 279;O = 10;E = 1.44;R = 6.96; p = 2.14e-06;adjP = 3.97e-06 | RARA, HUWE1, PMAIP1, PRDM16, DOK1, CUX1, HRK, BID, SPI1, MYB |
| Neoplasm | C = 298;O = 10;E = 1.53;R = 6.52; p = 3.84e-06;adjP = 6.24e-06 | WASF3, CD24, AKT1, LGALS3, EPHA2, FURIN, CUX1, CTNND1, ETV1, CEACAM1 |
| Invasiveness | ||
| Nervous system diseases | C = 694;O = 15;E = 3.57;R = 4.20; p = 3.73e-06;adjP = 6.24e-06 | ATXN7, USH1G, PC, PEX6, ASPM, BACE1, DUX4, CST3, SOX3, NEFH, CACNA1H, FOXG1, GIGYF2, XK, CDK5R1 |
| Breast | C = 377;O = 11;E = 1.94;R = 5.67; p = 4.79e-06;adjP = 7.33e-06 | S100A7, CD24, AKT1, LGALS3, EPHA2, RB1CC1, FXYD3, RHOBTB2, ALDH1A1, HDAC2, MYB |
| Neoplasms | ||
| Brain diseases | C = 411;O = 11;E = 2.12;R = 5.20; p = 1.08e-05;adjP = 1.56e-05 | ATXN7, PC, PEX6, BACE1, CST3, NEFH, AQP1, CACNA1H, FOXG1, GIGYF2, CDK5R1 |
| Schizophrenia | C = 360;O = 10;E = 1.85;R = 5.40; p = 1.98e-05;adjP = 2.57e-05 | NDEL1, ATF4, AKT1, CACNA1C, MDGA1, PDE4B, FEZ1, NTF3, ADRBK2, ADRA2A |
| Central nervous system diseases | C = 438;O = 11;E = 2.25;R = 4.88; p = 1.94e-05;adjP = 2.57e-05 | ATXN7, PC, PEX6, BACE1, CST3, NEFH, CACNA1H, FOXG1, GIGYF2, XK, CDK5R1 |
| Virus diseases | C = 488;O = 11;E = 2.51;R = 4.38; p = 5.16e-05;adjP = 6.39e-05 | CXCL5, PSMB10, KPNA4, TBK1, MICB, ACTA1, PSMD7, TICAM1, HLA-DOA, FOS, PIK3C2A |
| Carcinoma | C = 522;O = 11;E = 2.69;R = 4.09; p = 9.37e-05;adjP = 0.0001 | S100A7, CD24, WIF1, AKT1, LGALS3, EPHA2, SMO, FH, CTNND1, ALDH1A1, CEACAM1 |
| Pathologic processes | C = 561;O = 10;E = 2.89;R = 3.46; p = 0.0007;adjP = 0.0008 | MIF, CXCL5, CD24, HTRA1, PRDM16, JAZF1, ITGA2, FLT1, ZMIZ1, LOXL1 |
| Syndrome | C = 654;O = 10;E = 3.37;R = 2.97; p = 0.0022;adjP = 0.0024 | WASF3, USH1G, PEX6, WASL, TOP3A, FH, FOXG1, LOXL1, XK, ZMPSTE24 |
| Genetic | C = 808;O = 10;E = 4.16;R = 2.40; p = 0.0095;adjP = 0.0099 | MIF, CACNA1C, HTRA1, JAZF1, ITGA2, PDE4B, ZMIZ1, LOXL1, GIGYF2, ADRA2A |
| Predisposition to | ||
| Disease |
Criteria: at least 10 genes represented in the pathway and adjpvalue ≤0.05 (hyper-geometric t test with p value adjusted by multiple test adjustment, Benjamini and Hochberg [69])
C number of reference genes in the category number of genes in the gene set and O in the observed category, E expected number in the category, R ratio of enrichment, p p value from hypergeometric test,. adjP value adjusted by the multiple test adjustment (adjP)
There were no overlap between the 228 DEG and genes from the 20q11.1 gain observed in the schizophrenic case. BACE1 was the only gene that was also identified by the methylation analysis. This gene seems to regulate proteins with important roles in developmental process such as NGR1 [42], which overexpression results in a schizophrenia-like behavior in a mouse model [43].
Comparison of deregulated genes from individual cells and brain bulk samples
Comparison of gene expression profiling from 29 SZP and 30 CTS (brain bulks) revealed 233 differentially expressed transcripts (p < 0.01), of which 69 are unknown transcripts. We observed 127 (75 genes) up- and 106 (89 genes) down-regulated transcripts, none of them surviving the multiple correction. Furthermore, difference in pH of the brain between SZP and CTS is affecting 78 (33 %) of the differentially expressed transcripts (Additional file 1: Table S4).
To gain further information of the 228 and 233 DEG sets, they were mapped to the gene co-expression modules from human cortical development [31]. The enrichment analysis point out that each set of DEGs had over-represented genes but in different modules (M). While the set of 228 genes was enriched in M8, the set of 233 genes was enriched in M1, M5 and M15 (Additional file 1: Table S5). M8 and M15 were representative of genes preferentially expressed in “early” and “late” foetal period in human brain development, respectively (Additional file 1: Figure S4). This finding suggests that both datasets are related to neurodevelopment genes, but to different stages of the brain development.
To expand our findings from a cellular model of neuronal differentiation to postmortem samples from human brains, the 228 gene set, found exclusively in SCZP were compared to the 233 differentially expressed genes from adult brain samples. There were 194 genes out of the 228 represented in both platforms, from which four genes were common to both analyses: AQP1, DUSP6, FOS and TRIM24. Only DUSP6 and FOS have been described in schizophrenia [44, 45]. None of the them were affected by the pH difference.
Further, to identify functional modules in patients and controls, a co-expression analyses of the 194 genes was performed in WGCNA. Modules for CTS and SZP were generated using the Scale-free Topology Criterion with power 7. Four modules were identified plus the grey module (Fig. 1a). Genes and biological processes overrepresented in each module are displayed in Tables 3 and 4, respectively. The best preserved module in SZP compared to CTS is the blue (blue, Z-summary >10, Fig. 1B) which had an overrepresentation of genes related to cell cycle.
Fig. 1.
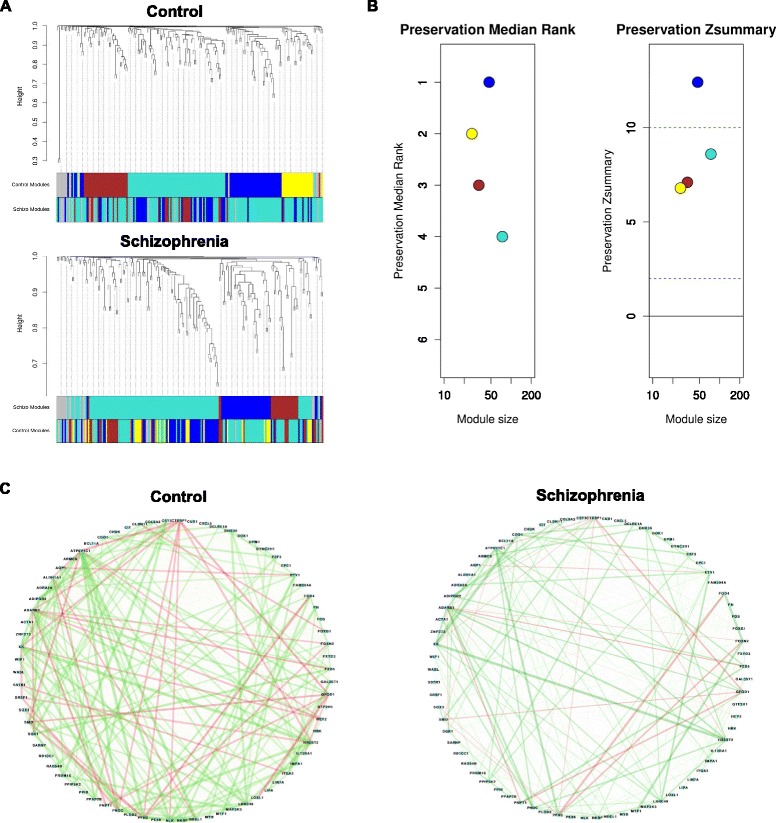
Module preservation analysis. a Dendrograms produced by average hierarchical clustering using topological overlapping matrix dissimilarity. Colours represent different modules. Upper panel shows modules in control subjects (CTS; blue, brown, yellow and turquoise) compared to patients with schizophrenia (SZP). Lower panel shows modules in patients with schizophrenia (blue, brown and turquoise) compared to modules in control subjects. b Left panel shows the median preservation rank (y-axis) in relation to module size (x-axis). Each circle represents a module labelled in different colours (blue, turquoise, yellow and brown). Right panel shows the Zsummary (y-axis) in function of module size. Dashed lines represent thresholds 2 and 10: ≥10: high preservation; 2 < Zsummary <10: moderate preservation; <2: low preservation. The panels show that the blue module is more preserved in control and patients whilst the turquoise module is less preserved. c Connectivity patterns (correlation network adjacencies) between genes from the turquoise module in controls (control) and patients (schizophrenia) showing a large loss of connectivity among genes in patient’s module compared to control modules. Line thickness represents the connectivity pattern and line colour reflects the absolute correlation: −1 (negative) to 1 (positive). This module is enriched in genes related to response to negative regulation of cell differentiation and oxidative stress
Table 3.
Genes preserved in the modules of the respective samples
| Module | Genes |
|---|---|
| Blue | AGL, ANAPC4, APOA1, ARMCX6, ASPM, ATF4, BCLAF1, BID, CAPRIN1, CENPJ, CLIP4, CPOX, CTNND1, |
| EDNRA, EIF3A, EIF5, EPHA2, FAM133B, GMFB, GRB10, HLA-DOA, HSD17B8, ING5, KHDRBS1, KPNA4, | |
| LYPD6, LZTS1, METTL16, MOCS3, NLGN1, NPR1, NRL, ORMDL1, PC, PSMB10, PSTK, RANBP2, RINT1, SMG5, SPHK1, STX4, USH1G, WASF3, WDR44, ZMIZ1, ZNF426, ZNF99 | |
| Brown | CACNA1C, CACNA1E, CCAR1, CEACAM1, EED, EIF3J, FURIN, GJD3, HDAC2, HUWE1, ING3, LGALS3, |
| LOC389634, MANEA, METAP2, MRPL19, MTHFS, NEK6, NOC3L, NUP107, PDK2, PMAIP1, PPM1B, | |
| PRDX4, PTPN12, PYGL, RCBTB1, SCAF8, SNTB1, TEK, TRIM24, USP1, ZMPSTE24 | |
| Grey | CCDC149, CEBPB, DDX6, FLT1, GIGYF2, KCNK15, KDELC2, RARA, RARS, SLC38A4, TBK1, TMEM208, TRDMT1, ZNF614 |
| Turquoise | ACTA1, ADARB1, ADIPOR2, ADRA2A, ALDH1A1, AQP1, ARMC8, ATP6V1C1, BCL11A, CDO1, CHUK, CIT, |
| CLDN11, COL9A3, CST3, CTDSP1, CUX1, CXCL5, DCLRE1A, DHX36, DOK1, DPM1, DYNC2H1, E2F3, | |
| EPC1, ETV1, FAM204A, FGD4, FH, FOS, FOXG1, FOXN2, FXYD3, FZD5, GAL3ST1, GFOD1, GTF2H1, | |
| HEY2, HRK, HS6ST2, IL13RA1, IMPA1, ITGA2, LIN7A, LIPA, LOXL1, LRRC49, MAP3K5, MTF1, MYB, | |
| NDEL1, NKRF, NLK, PEX6, PFN2, PLOD2, PNOC, PNPT1, PPAP2B, PPIB, PPIP5K2, PRDM16, RAD54B, | |
| RB1CC1, SARNP, SGK1, SMO, SOX3, SRSF1, SSTR1, WASL, WIF1, XK, ZNF273 | |
| Yellow | ADRBK2, AKT1, ASIC2, ASNS, CACNA1H, CADM3, CD24, CDK5R1, DUSP6, FBRS, GPR155, HNRNPH1, MDGA1, MGAT4B, MICB, MYBL1, PACS1, PDE4B, PDGFA, PNMT, PPP1R13B, RANGRF, RHOBTB2, RIPK4, SPRY4, TICAM1 |
Table 4.
Biological processes overrepresented in the modules
| Module | GO ID | Ontology name | Ontology definition |
|---|---|---|---|
| Blue | GO:0010564 | regulation of cell cycle process | Any process that modulates a cellular process that is involved in the progression of biochemical and morphological phases and events that occur in a cell during successive cell replication or nuclear replication events. |
| GO:0000086 | G2/M transition of mitotic cell cycle | Progression from G2 phase to M phase of the mitotic cell cycle. The molecular event responsible for this transition is the activation of the major cell cycle cyclin-dependent kinase (e.g. Cdc2 in S. pombe, CDC28 in S. cerevisiae, Cdk1 in human). | |
| GO:0051325 | interphase | A cell cycle process comprising the steps by which a cell progresses through interphase, the stage of cell cycle between successive rounds of chromosome segregation. Canonically, interphase is the stage of the cell cycle during which the biochemical and physiologic functions of the cell are performed and replication of chromatin occurs. | |
| GO:0051329 | interphase of mitotic cell cycle | Interphase occurring as part of the mitotic cell cycle. Canonically, interphase is the stage of the cell cycle during which the biochemical and physiologic functions of the cell are performed and replication of chromatin occurs. A mitotic cell cycle is one which canonically comprises four successive phases called G1, S, G2, and M and includes replication of the genome and the subsequent segregation of chromosomes into daughter cells. | |
| GO:0007346 | regulation of mitotic cell cycle | Any process that modulates the rate or extent of progress through the mitotic cell cycle. | |
| GO:0000278 | mitotic cell cycle | Progression through the phases of the mitotic cell cycle, the most common eukaryotic cell cycle, which canonically comprises four successive phases called G1, S, G2, and M and includes replication of the genome and the subsequent segregation of chromosomes into daughter cells. In some variant cell cycles nuclear replication or nuclear division may not be followed by cell division, or G1 and G2 phases may be absent. | |
| GO:0045184 | establishment of protein localization | The directed movement of a protein to a specific location. | |
| GO:0016482 | cytoplasmic transport | The directed movement of substances (such as macromolecules, small molecules, ions) into, out of, or within the cytoplasm of a cell. | |
| Brown | GO:0006508 | proteolysis | The hydrolysis of proteins into smaller polypeptides and/or amino acids by cleavage of their peptide bonds. |
| GO:0030163 | protein catabolic process | The chemical reactions and pathways resulting in the breakdown of a protein by the destruction of the native, active configuration, with or without the hydrolysis of peptide bonds. | |
| GO:0044257 | cellular protein catabolic process | The chemical reactions and pathways resulting in the breakdown of a protein by individual cells. | |
| GO:0044267 | cellular protein metabolic process | The chemical reactions and pathways involving a specific protein, rather than of proteins in general, occurring at the level of an individual cell. Includes cellular protein modification. | |
| GO:0042593 | glucose homeostasis | Any process involved in the maintenance of an internal steady state of glucose within an organism or cell. | |
| GO:0019538 | protein metabolic process | The chemical reactions and pathways involving a specific protein, rather than of proteins in general. Includes protein modification. | |
| GO:0006325 | chromatin organization | Any process that results in the specification, formation or maintenance of the physical structure of eukaryotic chromatin. | |
| GO:0016568 | chromatin modification | The alteration of DNA, protein, or sometimes RNA, in chromatin, which may result in changing the chromatin structure. | |
| Grey | GO:0017148 | negative regulation of translation | Any process that stops, prevents, or reduces the frequency, rate or extent of the chemical reactions and pathways resulting in the formation of proteins by the translation of mRNA. |
| GO:0035264 | multicellular organism growth | The increase in size or mass of an entire multicellular organism, as opposed to cell growth. | |
| GO:0042035 | regulation of cytokine biosynthetic process | Any process that modulates the frequency, rate or extent of the chemical reactions and pathways resulting in the formation of cytokines. | |
| GO:0042089 | cytokine biosynthetic process | The chemical reactions and pathways resulting in the formation of cytokines, any of a group of proteins that function to control the survival, growth and differentiation of tissues and cells, and which have autocrine and paracrine activity. | |
| GO:0042107 | cytokine metabolic process | The chemical reactions and pathways involving cytokines, any of a group of proteins or glycoproteins that function to control the survival, growth and differentiation of tissues and cells, and which have autocrine and paracrine activity. | |
| GO:0006399 | tRNA metabolic process | The chemical reactions and pathways involving tRNA, transfer RNA, a class of relatively small RNA molecules responsible for mediating the insertion of amino acids into the sequence of nascent polypeptide chains during protein synthesis. Transfer RNA is characterized by the presence of many unusual minor bases, the function of which has not been completely established. | |
| GO:0014065 | phosphatidylinositol 3-kinase cascade | A series of reactions, mediated by the intracellular phosphatidylinositol 3-kinase (PI3K). PI3K cascades lie downstream of many cell surface receptor linked signaling pathways and regulate numerous cellular functions. | |
| GO:0014066 | regulation of phosphatidylinositol 3-kinase cascade | Any process that modulates the frequency, rate or extent of signal transduction mediated by the phosphatidylinositol 3-kinase cascade. | |
| GO:0014068 | positive regulation of phosphatidylinositol 3-kinase cascade | Any process that activates or increases the frequency, rate or extent of signal transduction mediated by the phosphatidylinositol 3-kinase cascade. | |
| Turquoise | GO:0006366 | transcription from RNA polymerase II promoter | The synthesis of RNA from a DNA template by RNA polymerase II, originating at an RNA polymerase II promoter. Includes transcription of messenger RNA (mRNA) and certain small nuclear RNAs (snRNAs). |
| GO:0000003 | reproduction | The production by an organism of new individuals that contain some portion of their genetic material inherited from that organism. | |
| GO:0045596 | negative regulation of cell differentiation | Any process that stops, prevents, or reduces the frequency, rate or extent of cell differentiation. | |
| GO:0006357 | regulation of transcription from RNA polymerase II promoter | Any process that modulates the frequency, rate or extent of transcription from an RNA polymerase II promoter. | |
| GO:0022414 | reproductive process | A biological process that directly contributes to the process of producing new individuals by one or two organisms. The new individuals inherit some proportion of their genetic material from the parent or parents. | |
| GO:0006979 | response to oxidative stress | Any process that results in a change in state or activity of a cell or an organism (in terms of movement, secretion, enzyme production, gene expression, etc.) as a result of oxidative stress, a state often resulting from exposure to high levels of reactive oxygen species, e.g. superoxide anions, hydrogen peroxide (H2O2), and hydroxyl radicals. | |
| GO:0032990 | cell part morphogenesis | The process in which the anatomical structures of a cell part are generated and organized. | |
| GO:0006351 | transcription, DNA dependent | The cellular synthesis of RNA on a template of DNA. | |
| Yellow | GO:0045859 | regulation of protein kinase activity | Any process that modulates the frequency, rate or extent of protein kinase activity. |
| GO:0043549 | regulation of kinase activity | Any process that modulates the frequency, rate or extent of kinase activity, the catalysis of the transfer of a phosphate group, usually from ATP, to a substrate molecule. | |
| GO:0043409 | negative regulation of MAPK cascade | Any process that stops, prevents, or reduces the frequency, rate or extent of signal transduction mediated by the MAPKKK cascade. | |
| GO:0002764 | immune response-regulating signaling pathway | The cascade of processes by which a signal interacts with a receptor, causing a change in the level or activity of a second messenger or other downstream target, and ultimately leading to the activation, perpetuation, or inhibition of an immune response. | |
| GO:0050865 | regulation of cell activation | Any process that modulates the frequency, rate or extent of cell activation, the change in the morphology or behavior of a cell resulting from exposure to an activating factor such as a cellular or soluble ligand. | |
| GO:0002682 | regulation of immune system process | Any process that modulates the frequency, rate, or extent of an immune system process. | |
| GO:0002684 | positive regulation of immune system process | Any process that activates or increases the frequency, rate, or extent of an immune system process. | |
| GO:0010741 | negative regulation of intracellular protein kinase cascade | Any process that decreases the rate, frequency or extent of a series of reactions, mediated by protein kinases, which occurs as a result of a single trigger reaction or compound. | |
| GO:0001932 | regulation of protein phosphorylation | Any process that modulates the frequency, rate or extent of addition of phosphate groups into an amino acid in a protein. | |
| GO:0010627 | regulation of intracellular protein kinase cascade | Any process that modulates the rate, frequency or extent of a series of reactions, mediated by protein kinases, which occurs as a result of a single trigger reaction or compound. |
Although showing a moderate Z-summary, using the median rank value based on the observed values (less dependent on modules sizes), the turquoise module was only marginally preserved (2 < Z-summary <10). Since this module includes negative regulation of cell differentiation and response to oxidative stress processes, identified deregulated in these same cells [24], Pearson correlation of paired genes were plotted (Fig. 1C). We can observe a change of correlations in SZP compared to CTS (change of color and thickness of lines linking two genes). Whilst co-expressed genes related to regulation of cell cycle processes, protein phosphatase binding and nonsense mRNA decay were extremely conserved between SZP and CTS, genes related to response to negative regulation of cell differentiation and oxidative stress were not.
Next, we defined the hub genes of the modules as those genes with high module connectivity (kME >0.75). This resulted in 13 hubs (CAPRIN1, CLIP4, CTNND1, GRB10, KHDRBS1, KPNA4, LYPD6, NRL, PSTK, RANBP2, RINT1, WASF3 and ZNF99) in the highly preserved blue module of CTS, all conserved in SZP (which gained additional four hubs: ZMIZ1, ORMDL1, EIF5 and FAM133B). It is interesting to note that in the yellow and brown modules, the number of hubs in SZP is smaller than in CTS, but all hubs identified in SZP were conserved from CTS, with only one exception in the yellow module (AKT) and one in the brown module (ZMPSTE24).
The marginally preserved turquoise module had 13 and 16 hub genes in CTS and SZP, respectively, with an overlap of only eight hubs, from which three were previously associated with schizophrenia: ADARB1 [46], PNOC [47] and XK [48]. Furthermore, three out of seven hubs found only in SZP were previously associated with schizophrenia. ADRA2A is required for normal pre-synaptic control of transmitter release in noradrenergic neurons and has polymorphisms associated to the increased risk of developing the disease [49]. CIT is a serine/threonine protein kinase involved in central nervous system development and cancer cells proliferation. Polymorphisms in CIT also seems to have an impact in the risk of developing schizophrenia [50]. LIN7A encodes a protein involved with the synaptic function and neuron migration in cerebral cortex development [51] and was associated to autism and attention deficit hyperactivity disorder [52, 53]. Even though the other four hubs (ARMC8, BCL11A, CDO1 and FOXG1) have not been linked to schizophrenia, they are candidates for further studies.
Discussion
In the present study, we characterized molecular alterations during the differentiation of hiPSCs to NPCs from a patient with schizophrenia and health control and analyzed the identified genes in the adult brain tissue from schizophrenia cases and controls to study biological processes that could be disrupted in the disease. As we faced the limitation of a direct comparison due to the different nature of samples (single cell type–neuron–versus all cells types–frontal cortex), we decided to make the analyses using gene co-expression network complemented with the exploration of biological processes. This allowed us to explore genes expressed during neuronal differentiation and important for the development of schizophrenia in the context of adult brain bulk.
It is known that cultured cells are more likely to have genomic instability, genetic and epigenetic mutations [54, 55]. However, we only found one alteration by aCGH, a 1.2 MB gain in SCZP-hiPSC that was maintained in the NPC, suggesting that genes in this region may not be essential for the initial stages of neuron differentiation. Even though none of the 228 DEG was located in this region, it might contains regulatory variants with some role in the disease, although they were not explored in this study. The differentially methylated sites found during neuronal differentiation were preferentially associated with chromatin remodeling complex biological function, an interesting finding considering the previous study where the oxidative stress present in SCZP-NPC was reverted to NPC control levels after treatment of valproic acid (VPA), a histone deacetylase (HDAC) inhibitor [24].
Despite the fact that total RNA was extracted from three independent cell cultures (hiPSC and NPC), we are aware of the limitation of comparing one case with one control, which is the reason why we used the HT-self model for gene expression analyses. This consists in a mathematical model developed to analyze low numbers of samples [28]. Moreover, we used very stringent criteria in all analyses to identify the DEG and overrepresented ontologies (e.g. to be considered overrepresented, each class should contain more than 10 genes and a significant p-value after correction for multiple comparisons). Yet, data interpretations present certain limitations.
Concerning the genes involved with neuronal differentiation (hiPSC versus NPC), our comparisons revealed that cells derived from health control displayed twice more DEG than cells derived from the patient with schizophrenia. This finding may be involved with the disruption of cell differentiation that has been described by other studies in schizophrenia. The 228 gene set associated with neuronal differentiation in a patient with schizophrenia had over-representation of pathways classically related to cancer, such as MAPK and metabolic cellular pathways. Whilst new categories may appear with the advance of research in the field, many aspects of normal development and cancer have association to neuronal disorders [56]. There are no implications for tumorigenesis risk, although patients with schizophrenia are at a reduced risk of developing cancer [57]. In our previous study with these cells, we demonstrated that SZCP-NPC had altered metabolism compared to CON-NPC [24]. Likewise, the 228 set had over-representation of genes related to energy, lipid and carbohydrate metabolism, which are altered in cancer [58] and stem cells [59] adding evidence for the implication of tumor-suppressor and oncogenes to neurodevelopment and associated disorders.
Additionally, gene expression profiling from post-mortem brain tissue of patients with schizophrenia and controls were studied. To begin with, although there were no significant differences in age or PMI, the pH was slightly lower in the brain of patients with schizophrenia, a characteristic that has been described although not considered for the analyses [15]. The pH may have an effect on the expression of some genes or these genes might be regulating the pH change. We found that 78 out of 233 DEG identified by the comparison between brain bulks from patients with schizophrenia and health controls had their expression also associated to differences in pH, an issue that needs to be deeper investigated.
Likewise, to explore the relationship between genes involved in neuronal differentiation and fully differentiated brains in patients with schizophrenia, we have to consider some additional known limitations: (1) Use of medication, hypoxia and/or injury are known confounding factors in the samples derived from the patients; (2) Comparison between data generated by single cell types (hiPSC and NPC) with data representing a mixed cell population at the end stage of the disease (a “whole brain tissue” that includes the glia, immunological and vascular cells); 3) Different methods were used to identify differentially expressed genes; (4) Genes from neurodevelopment are not being expressed or are being expressed at very low levels once the brain has ended its maturation. Supporting this later hypothesis, the comparison of the 228 and 233 DEG with modules derived from developing brains [31], showed that even though both sets have genes associated with neurodevelopment, they are representative of different stages. Thus, the overlap of only four genes was not a surprise.
Consequently, we decided to study the 228 gene set using co-expression modules in the expression data from the cohort of post-mortem cases of schizophrenia and controls identifying a well preserved module (blue) and, although not completely, a less conserved module (turquoise). The blue module was associated with regulation of cell cycle progress, protein phosphatase binding and nonsense mRNA decay. Genes from this module have function related to genome maintenance, RNA integrity, cell proliferation and/or differentiation, such as ANPC4, SPHK1 and SPP [60–62].
The turquoise module displays a set of genes involved in negative regulation of cell differentiation and oxidative stress response, more specifically reactive oxygen species (ROS) production, mitochondria function and neuron differentiation, which are closely related to the functional study of these cells that exhibited deregulation of mitochondria function in SZCP-NPC [24]. Among the genes represented in the module, three genes have a described function associated with ROS generation. ASK1 is a member of the mitogen-activated protein kinase family activated by cellular stress, activating c-jun N-terminal kinase (JNK) and p38 in response to oxidative stress, endoplasmic reticulum stress, infection and calcium influx [63]. PNPT1 is predominantly localized in the mitochondrial inter-membrane space and is implicated in RNA targeting to human mitochondria, small noncoding nuclear RNAs (5S rRNA, MRP RNA, some tRNAs, and miRNAs) [64]. cAMP-induced cFos is a transcriptional repressor of CD44 that triggers ROS generation [65]. Additionally, some hub genes found exclusively in SZP have been associated to neuronal plasticity and different cancers, most of them associated to cell proliferation. Likewise, some of the hub genes have already been associated to neurodevelopment disorders.
Mitochondria activity and energy metabolism undergo dramatic remodeling during embryonic development. Early embryos are initially dependent upon oxidative metabolism due to inheritance of maternal mitochondria which are subsequently segregated among daughter cells. A predominantly glycolytic metabolism provides sufficient energy to support stem cells in basal state, but a robust metabolic network is required to adequate the increasingly energetic demands [66]. The established initial mechanism support flux through the tricarboxylic acid cycle and electron transport chain whilst mitochondrial oxidization of pyruvate and glutamine to CO2 is optimized to extract maximal energetic supply. It has been suggested that a concomitant rise in mitochondrial reactive oxygen species may prime stem cells for lineage differentiation [66, 67]. The large number of observations identifying stem cell properties affected by energy-responsive molecules and signaling pathways raises questions about the fate of stem cells under conditions when metabolic homeostasis is perturbed. The literature suggests that abnormal endocrine signaling in organisms in extreme metabolic states has a substantial impact on proliferation and differentiation of multiple stem cell populations throughout the body [68]. It is also possible that variations in metabolism during gestation contribute to changes in offspring through their effects on stem cells. For example, low energy levels and the associated hormonal signals that occur in the pregnant mother could be directly transmitted to the offspring through the placenta, resulting in either transient or permanent changes in embryonic stem cells. Therefore, it seems that risk factors for schizophrenia such as famine, hipoxia and low birth weight could be implicated in permanent changes to stem cells leading to altered brain neural-development, as well as some alterations that are still presented in adulthoods, like disturbed neural plasticity and increased ROS production.
Conclusions
In this study, data integration provided by different sample types (single cell versus tissue bulk) and methodologies were used to contribute to a better understating the schizophrenia. This model allowed us to examine the influence of the neuronal cells to the schizophrenic brain through the involvement of genes related to neuronal differentiation and cell metabolism. We identified a non-conserved module based on a co-expression network in schizophrenia compared to health controls that enabled us to predict that metabolic changes involving oxidative stress and negative regulation of cell differentiation may be occurring during early neurodevelopment and prompting to the development if schizophrenia. Several genes from this module were associated to schizophrenia by other studies and we revealed new candidates (ARMC8, BCL11A, CDO1 and FOXG1) to be disrupted during neuronal differentiation in patients with schizophrenia.
Additional file
This is a PDF document containing tables (1 to 5), images (1 to 4) with respective legends that exhibit selected findings from the research that were deemed less important to the overall message than the figure and tables included in the main body of this manuscript.
Acknowledgements
We thank the patients, doctors and nurses involved with sample collection and the Stanley Medical Research Institute. This research was supported by either Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq #17/2008) and Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ). MM (CNPq 304429/2014-7), ACT (FAPESP 2014/00041-1), LL (CAPES 10682/13-9) HV (CAPES) and BP (PPSUS 137270) were supported by their fellowships.
Abbreviations
- hiPSC
Human induced pluripotent cells
- SCZP
Schizophrenia
- CON
Control individual
- NPCs
Neural progenitor cells
- ROS
Reactive oxygen species
- CNVs
Copy number variations
- CGH
Comparative genomic hybridization
- CTS
Controls subjects
- SZP
Patients with schizophrenia
- ROS
Reactive oxygen species
- SAM
Significant Analysis of Microarray
- WGCNA
Weighted Gene Co-expression Network Analysis
- TOM
Topological overlap measure
- adjP
Adjusted p-value
- FDR
False discovery rate
- PC
Pearson Correlation
Footnotes
Mariana Maschietto and Ana C Tahira contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BSP performed cell transformation and differentiation, SR supervised experiments. MM extracted DNA and RNA, carried out library preparation for bisulfite sequencing, performed array experiments and analyzed expression arrays. DCM supervised expression array experiments. ACVK and HV analyzed copy number and methylation arrays, respectively. RP performed self-self analyses. ACT, LL and DM made the network and module analyses. PB-A provided sample’s collection and coordinated clinical study. HB and JAP conceived the study. MM and HB wrote the manuscript with input from all authors. All authors read and approved the final manuscript.
Contributor Information
Mariana Maschietto, Email: marianamasc@gmail.com.
Ana C Tahira, Email: tahira.ana@gmail.com.
Renato Puga, Email: renatopuga@gmail.com.
Leandro Lima, Email: lelimaufc@gmail.com.
Daniel Mariani, Email: danielbmariani@gmail.com.
Bruna da Silveira Paulsen, Email: bruna.paulsen@lance-ufrj.org.
Paulo Belmonte-de-Abreu, Email: 00006525@ufrgs.br.
Henrique Vieira, Email: enrikevieira@gmail.com.
Ana CV Krepischi, Email: ana.krepischi@gmail.com.
Dirce M Carraro, Email: dirce.carraro@cipe.accamargo.org.br.
Joana A Palha, Email: japalha@ecsaude.uminho.pt.
Stevens Rehen, Email: srehen@gmail.com.
Helena Brentani, Email: helena.brentani@gmail.com.
References
- 1.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60(12):11871192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 2.Tsuang MT, Stone WS, Faraone SV. Genes, environment and schizophrenia. Br J Psychiatry Suppl. 2001;40:s18–24. doi: 10.1192/bjp.178.40.s18. [DOI] [PubMed] [Google Scholar]
- 3.Palha JA, Goodman AB. Thyroid hormones and retinoids: a possible link between genes and environment in schizophrenia. Brain Res Rev. 2006;51(1):61–71. doi: 10.1016/j.brainresrev.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Sun J, Jia P, Fanous AH, van den Oord E, Chen X, Riley BP, et al. Schizophrenia gene networks and pathways and their applications for novel candidate gene selection. PLoS One. 2010;5(6):e11351. doi: 10.1371/journal.pone.0011351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christian K, Song H, Ming GL. Adult neurogenesis as a cellular model to study schizophrenia. Cell Cycle. 2010;9(4):636–7. doi: 10.4161/cc.9.4.10932. [DOI] [PubMed] [Google Scholar]
- 6.Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54(3):387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 7.Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci U S A. 2007;104(36):14501–6. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brennand KJ, Gage FH. Modeling psychiatric disorders through reprogramming. Dis Model Mech. 2012;5(1):26–32. doi: 10.1242/dmm.008268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao L, Vawter MP. Shared gene expression alterations in schizophrenia and bipolar disorder. Biol Psychiatry. 2008;64(2):89–97. doi: 10.1016/j.biopsych.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhan L, Kerr JR, Lafuente MJ, Maclean A, Chibalina MV, Liu B, et al. Altered expression and coregulation of dopamine signalling genes in schizophrenia and bipolar disorder. Neuropathol Appl Neurobiol. 2011;37(2):206219. doi: 10.1111/j.1365-2990.2010.01128.x. [DOI] [PubMed] [Google Scholar]
- 11.Iwamoto K, Kato T. Gene expression profiling in schizophrenia and related mental disorders. Neuroscientist. 2006;12(4):349–61. doi: 10.1177/1073858406287536. [DOI] [PubMed] [Google Scholar]
- 12.Arion D, Unger T, Lewis DA, Levitt P, Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2007;62(7):711–21. doi: 10.1016/j.biopsych.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haroutunian V, Katsel P, Dracheva S, Stewart DG, Davis KL. Variations in oligodendrocyte-related gene expression across multiple cortical regions: implications for the pathophysiology of schizophrenia. Int J Neuropsychopharmacol. 2007;10(4):565–73. doi: 10.1017/S1461145706007310. [DOI] [PubMed] [Google Scholar]
- 14.Torkamani A, Dean B, Schork NJ, Thomas EA. Coexpression network analysis of neural tissue reveals perturbations in developmental processes in schizophrenia. Genome Res. 2010;20(4):403–12. doi: 10.1101/gr.101956.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mistry M, Gillis J, Pavlidis P. Meta-analysis of gene coexpression networks in the post-mortem prefrontal cortex of patients with schizophrenia and unaffected controls. BMC Neurosci. 2013;14:105. doi: 10.1186/1471-2202-14-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Middleton FA, Pato CN, Gentile KL, McGann L, Brown AM, Trauzzi M, et al. Gene expression analysis of peripheral blood leukocytes from discordant sib-pairs with schizophrenia and bipolar disorder reveals points of convergence between genetic and functional genomic approaches. Am J Med Genet B Neuropsychiatr Genet. 2005;136B(1):12–25. doi: 10.1002/ajmg.b.30171. [DOI] [PubMed] [Google Scholar]
- 17.Domenici E, Willé DR, Tozzi F, Prokopenko I, Miller S, McKeown A, et al. Plasma protein biomarkers for depression and schizophrenia by multi analyte profiling of case-control collections. PLoS One. 2010;5(2):e9166. doi: 10.1371/journal.pone.0009166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowden NA, Weidenhofer J, Scott RJ, Schall U, Todd J, Michie PT, et al. Preliminary investigation of gene expression profiles in peripheral blood lymphocytes in schizophrenia. Schizophr Res. 2006;82(2–3):175–83. doi: 10.1016/j.schres.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 19.Maschietto M, Silva AR, Puga RD, Lima L, Pereira CB, Nakano EY, et al. Gene expression of peripheral blood lymphocytes may discriminate patients with schizophrenia from controls. Psychiatry Res. 2012;200(2–3):1018–21. doi: 10.1016/j.psychres.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 20.Benitez-King G, Riquelme A, Ortiz-Lopez L, Berlanga C, Rodriguez-Verdugo MS, Romo F, et al. A noninvasive method to isolate the neuronal linage from the nasal epithelium from schizophrenic and bipolar diseases. J Neurosci Methods. 2011;201(1):35–45. doi: 10.1016/j.jneumeth.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Pedrosa E, Sandler V, Shah A, Carroll R, Chang C, Rockowitz S, et al. Development of patient-specific neurons in schizophrenia using induced pluripotent stem cells. J Neurogenet. 2011;25(3):88–103. doi: 10.3109/01677063.2011.597908. [DOI] [PubMed] [Google Scholar]
- 22.Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473(7346):221–5. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Lin M, Foxe JJ, Pedrosa E, Hrabovsky A, Carroll R, et al. Transcriptome comparison of human neurons generated using induced pluripotent stem cells derived from dental pulp and skin fibroblasts. PLoS One. 2013;8(10):e75682. doi: 10.1371/journal.pone.0075682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paulsen BD, Maciel RD, Galina A, da Silveira MS, Souza CD, Drummond H, et al. Altered oxygen metabolism associated to neurogenesis of induced pluripotent stem cells derived from a schizophrenic patient. Cell Transplant. 2011;21(7):1547–59. doi: 10.3727/096368911X600957. [DOI] [PubMed] [Google Scholar]
- 25.Robicsek O, Karry R, Petit I, Salman-Kesner N, Muller FJ, Klein E, et al. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol Psychiatry. 2013;18(10):1067–76. doi: 10.1038/mp.2013.67. [DOI] [PubMed] [Google Scholar]
- 26.Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry. 2014;20(3):361–8. doi: 10.1038/mp.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Baumont A, Maschietto M, Lima L, Carraro DM, Olivieri EH, Fiorini A, et al. Innate immune response is differentially dysregulated between bipolar disease and schizophrenia. Schizophr Res. 2015;161(2–3):215–21. doi: 10.1016/j.schres.2014.10.055. [DOI] [PubMed] [Google Scholar]
- 28.Vêncio RZ, Koide T. HTself: self-self based statistical test for low replication microarray studies. DNA Res. 2005;12(3):211–4. doi: 10.1093/dnares/dsi007. [DOI] [PubMed] [Google Scholar]
- 29.Sunkin SM, Ng L, Lau C, Dolbeare T, Gilbert TL, Thompson CL, et al. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 2013;41(Database issue):D996–1008. doi: 10.1093/nar/gks1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, et al. TM4: a free, open-source system for microarray data management and analysis. BioTechniques. 2003;34(2):374–8. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 31.Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155(5):1008–21. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zambon AC, Gaj S, Ho I, Hanspers K, Vranizan K, Evelo CT, et al. GO-Elite: a flexible solution for pathway and ontology overrepresentation. Bioinformatics. 2012;28(16):2209–10. doi: 10.1093/bioinformatics/bts366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisinger BE, Saul MC, Driessen TM, Gammie SC. Development of a versatile enrichment analysis tool reveals associations between the maternal brain and mental health disorders, including autism. BMC Neurosci. 2013;14:147. doi: 10.1186/1471-2202-14-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008;24(5):719–20. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 36.Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7(1):e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33(Web Server issue):W741–8. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeng L, Zhang Q, Li S, Plotnikov AN, Walsh MJ, Zhou MM. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466(7303):258–62. doi: 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, et al. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26(2):151–62. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Charroux B, Kerridge S, Tsai CC. Atrophin recruits HDAC1/2 and G9a to modify histone H3K9 and to determine cell fates. EMBO Rep. 2008;9(6):555562. doi: 10.1038/embor.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fleck D, Garratt AN, Haass C, Willem M. BACE1 dependent neuregulin processing: review. Curr Alzheimer Res. 2012;9(2):178–83. doi: 10.2174/156720512799361637. [DOI] [PubMed] [Google Scholar]
- 43.Luo X, He W, Hu X, Yan R. Reversible overexpression of bace1-cleaved neuregulin-1 N-terminal fragment induces schizophrenia-like phenotypes in mice. Biol Psychiatry. 2014;76(2):120–7. doi: 10.1016/j.biopsych.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee KY, Ahn YM, Joo EJ, Chang JS, Kim YS. The association of DUSP6 gene with schizophrenia and bipolar disorder: its possible role in the development of bipolar disorder. Mol Psychiatry. 2006;11(5):425–6. doi: 10.1038/sj.mp.4001807. [DOI] [PubMed] [Google Scholar]
- 45.Grant P, Gabriel F, Kuepper Y, Wielpuetz C, Hennig J. Psychosis-proneness correlates with expression levels of dopaminergic genes. Eur Psychiatry. 2014;29(5):304–6. doi: 10.1016/j.eurpsy.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 46.Kubota-Sakashita M, Iwamoto K, Bundo M, Kato T. A role of ADAR2 and RNA editing of glutamate receptors in mood disorders and schizophrenia. Mol Brain. 2014;7:5. doi: 10.1186/1756-6606-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blaveri E, Kalsi G, Lawrence J, Quested D, Moorey H, Lamb G, et al. Genetic association studies of schizophrenia using the 8p21-22 genes: prepronociceptin (PNOC), neuronal nicotinic cholinergic receptor alpha polypeptide 2 (CHRNA2) and arylamine Nacetyltransferase 1 (NAT1) Eur J Hum Genet. 2001;9(6):469–72. doi: 10.1038/sj.ejhg.5200646. [DOI] [PubMed] [Google Scholar]
- 48.Shimo H, Nakamura M, Tomiyasu A, Ichiba M, Ueno S, Sano A. Comprehensive analysis of the genes responsible for neuroacanthocytosis in mood disorder and schizophrenia. Neurosci Res. 2011;69(3):196–202. doi: 10.1016/j.neures.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 49.Lochman J, Balcar VJ, Stastny F, Sery O. Preliminary evidence for association between schizophrenia and polymorphisms in the regulatory Regions of the ADRA2A, DRD3 and SNAP-25 Genes. Psychiatry Res. 2013;205(1–2):7–12. doi: 10.1016/j.psychres.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 50.Nicodemus KK, Callicott JH, Higier RG, Luna A, Nixon DC, Lipska BK, et al. Evidence of statistical epistasis between DISC1, CIT and NDEL1 impacting risk for schizophrenia: biological validation with functional neuroimaging. Hum Genet. 2010;127(4):441–52. doi: 10.1007/s00439-009-0782-y. [DOI] [PubMed] [Google Scholar]
- 51.Matsumoto A, Mizuno M, Hamada N, Nozaki Y, Jimbo EF, Momoi MY, et al. LIN7A depletion disrupts cerebral cortex development, contributing to intellectual disability in 12q21-deletion syndrome. PLoS One. 2014;9(3):e92695. doi: 10.1371/journal.pone.0092695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mizuno M, Matsumoto A, Hamada N, Ito H, Miyauchi A, Jimbo EF, et al. Role of an adaptor protein Lin-7B in brain development: possible involvement in autism spectrum disorders. J Neurochem. 2015;132(1):61–9. doi: 10.1111/jnc.12943. [DOI] [PubMed] [Google Scholar]
- 53.Lanktree M, Squassina A, Krinsky M, Strauss J, Jain U, Macciardi F, et al. Association study of brain-derived neurotrophic factor (BDNF) and LIN-7 homolog (LIN-7) genes with adult attention-deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(6):945–51. doi: 10.1002/ajmg.b.30723. [DOI] [PubMed] [Google Scholar]
- 54.Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT, et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7(4):521–31. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 55.Laurent LC, Ulitsky I, Slavin I, Tran H, Schork A, Morey R, et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8(1):106–18. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vestin A, Mills AA. The tumor suppressor Chd5 is induced during neuronal differentiation in the developing mouse brain. Gene Expression Patterns: GEP. 2013;13(8):482–9. doi: 10.1016/j.gep.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bushe CJ, Hodgson R. Schizophrenia and cancer: in 2010 do we understand the connection? Can J Psychiatry. 2010;55(12):761–7. doi: 10.1177/070674371005501203. [DOI] [PubMed] [Google Scholar]
- 58.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rafalski VA, Mancini E, Brunet A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J Cell Sci. 2012;125(Pt 23):5597–608. doi: 10.1242/jcs.114827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wasch R, Robbins JA, Cross FR. The emerging role of APC/CCdh1 in controlling differentiation, genomic stability and tumor suppression. Oncogene. 2010;29(1):1–10. doi: 10.1038/onc.2009.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spiegel S, Milstien S. Sphingosine-1-phosphate: signaling inside and out. FEBS Lett. 2000;476(1–2):55–7. doi: 10.1016/S0014-5793(00)01670-7. [DOI] [PubMed] [Google Scholar]
- 62.Lou CH, Shao A, Shum EY, Espinoza JL, Huang L, Karam R, et al. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Reports. 2014;6(4):748–64. doi: 10.1016/j.celrep.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shiizaki S, Naguro I, Ichijo H. Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. Advances in Biological Regulation. 2013;53(1):135–44. doi: 10.1016/j.jbior.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 64.Vedrenne V, Gowher A, De Lonlay P, Nitschke P, Serre V, Boddaert N, et al. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am J Hum Genet. 2012;91(5):912–8. doi: 10.1016/j.ajhg.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carrier JL, Javadi P, Bourrier E, Camus C, Segal-Bendirdjian E, Karniguian A. cFos mediates cAMP-dependent generation of ROS and rescue of maturation program in retinoid-resistant acute promyelocytic leukemia cell line NB4-LR1. PLoS One. 2012;7(11):e50408. doi: 10.1371/journal.pone.0050408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11(5):596–606. doi: 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Mitochondria in control of cell fate. Circ Res. 2012;110(4):526–9. doi: 10.1161/RES.0b013e31824ae5c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ochocki JD, Simon MC. Nutrient-sensing pathways and metabolic regulation in stem cells. J Cell Biol. 2013;203(1):23–33. doi: 10.1083/jcb.201303110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a pratical and powerful approach to multiple testing. J R Statist Soc B. 1995;57(1):289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This is a PDF document containing tables (1 to 5), images (1 to 4) with respective legends that exhibit selected findings from the research that were deemed less important to the overall message than the figure and tables included in the main body of this manuscript.