

Preface
Lysine methyltransferase 2 family (KMT2) proteins methylate lysine 4 on the histone H3 tail at important regulatory regions in the genome and thus impart critical functions through modulating chromatin structures and DNA accessibility. While the human KMT2 family was initially named the mixed lineage leukemia (MLL) family, due to the role of the founding member KMT2A (also called MLL, MLL1, ALL-1, HRX) in this disease, recent exome-sequencing studies revealed KMT2 genes to be among the most frequently mutated genes in many types of human cancers. Efforts to integrate the molecular mechanisms of KMT2 with its roles in tumorigenesis have led to the development of first-generation inhibitors of KMT2 function, which could become novel cancer therapies.
Introduction
Epigenetic regulation lies at the heart of cellular identity in a multicellular organism, as each cell carries the same DNA while expressing a highly choreographed set of genes.1,2 Each cell type has a unique epigenome, characterized by specific modifications of histone proteins and DNA methylation patterns that divide the genome into active, poised, and silent domains. The lysine methyltransferase 2 (KMT2, also known as mixed lineage leukemia (MLL)) family methylates histone H3 lysine 4 (H3K4) to promote genome accessibility and transcription. The KMT2 family is highly conserved, having evolved from an ancient lineage of proteins present in unicellular eukaryotes.3,4 In cancer, transcriptional dysregulation subverts cellular identity by aberrantly linking proliferation and migration programs present during embryonic development to an otherwise differentiated, often quiescent, phenotype required for postnatal homeostasis.5,6 Dysregulation or mutation of the KMT2-family in human cancer exemplifies how this can occur: in hematopoietic cells, KMT2A (also known as MLL1) translocations result in oncogenic fusion-proteins that recruit the H3K79 methyltransferase DOT1L. This changes the epigenetic identity of the cells and drives a subset of infantile and adult leukemia.7,8
Driven in part by the historic discoveries of KMT2A rearrangements as causes of MLL 7,8 and KMT2A H3K4 methyltransferase activity,9,10 intense efforts have been made to interrogate KMT2 function and regulation. Here, we review the key advances that have defined how KMT2 proteins function within multimeric complexes whose subunits modulate KMT2 activity on chromatin. Recent exome-sequencing studies show that KMT2 dysregulation extends well beyond a rare cancer like KMT2A-rearranged MLL.11,12 Instead, KMT2-family mutations are among the most frequent alterations in human cancer 13 and are associated with some of the most common and deadly solid tumors, such as lung 11 and colon 12 carcinomas. Finally, we discuss the development of inhibitors that target co-factors in KMT2-containing complexes and highlight the promise of these agents against a variety of human cancers. Although much of what is known for KMT2 enzymes was initially characterized in yeast,3 due to limited space, this review will focus on KMT2 family of proteins in higher eukaryotes.
Structural-functional insights on the regulation of KMT2 activities
The KMT2 family is highly conserved throughout eukaryotes3,4. Three subgroups of KMT2 are present in Drosophila: trx, trr and dSET1.4 Chromosomal duplication during mammalian evolution resulted in two paralogs in each KMT2 subgroup.14 They are trx-related KMT2A and KMT2B (also known as MLL2, WBP7, sometimes called MLL4), trr-related KMT2C (also known as MLL3, HALR) and KMT2D (also known as MLL4, ALR, sometimes called MLL2), as well as dSET1 related KMT2F (also known as hSET1A, SETD1A) and KMT2G (also known as hSET1B, SETD1B) genes (Figure 1A). KMT2E (also known as MLL5) is more homologous to the SET3 family and has no intrinsic methyltransferase activity.15 Therefore KMT2E will not be included in the current review.
Figure 1. Metazoan KMT2 family histone methyltransferases.

a. Schematic representation of domain structures for each KMT2. KMT2A and KMT2B have the consensus D/GVDD sites (indicated by arrow) that are cleaved by Taspase 1, at two conserved sites for KMT2A and at one conserved site for KMT2B. This cleavage unique feature for this KMT2 subgroup. The resulting large N-terminal fragment and a smaller C-terminal fragment subsequently associate through FYRN and FYRC domains to generate a functional, noncovalent heterodimeric complex. KMT2C and KMT2D contain two clusters of PHD domains as well as the juxtaposed FYRN, FYRC and the SET domains on the C-terminus. One HMG-I binding motif and multiple nuclear receptor interacting motifs (LXXLL) are present in the protein sequences. These motifs are frequently found in transcription factors and cofactors. The PHD4–6 domains of KMT2C and KMT2D are able to bind unmethylated and asymmetrically di-methylated H4 arginine 3 (H4R3me0 and H4R3me2a), supporting a coordinated function with protein arginine methyltransferases.49 KMT2F and KMT2G are the smallest KMT2 subgroup and contain an N-terminal RNA recognition motif (RRM) and a C-terminal N-SET domain, which interact with WDR82 and ubiquitylated histone H2B respectively.
b. Schematic representation of interactions among KMT2A core subunits as well as their interactions with chromatin. KMT2A core complex is stabilized by pair-wise interactions between KMT2A (WIN)-WDR5, WDR5-RbBP5 (VDV), RbBP5-ASH2L and ASH2L-DPY30. KMT2A, pink; WDR5, blue; RbBP5, green; ASH2L, purple; DPY30, red. Furthermore, KMT2A is stabilized on chromatin through multivalent interactions, which include interactions of the AT hook and CxxC domain with AT-rich sequence 189 and non-methylated CpG dinucleotides 70, respectively; as we as interactions between PHD and Bromo-domains with H3K4me3 and acetylated lysine residues, respectively.72 The coupling of the enzyme with binding domains for its methylation product(s) is important for feed-forward maintenance of H3K4me in cells. Transient interactions between SET and histone H3 tail as well as ASH2L PHD and ubiquitin are also reported.
KMT2 proteins reside in large, multi-subunit complexes composed of unique sets of interacting proteins (Table 1).10,16–20 Despite the diversity, four subunits (i.e. WD repeat protein 5 (WDR5), retinoblastoma binding protein 5 (RbBP5), ASH2L and DPY30) are commonly found in all KMT2 complexes. Biochemical reconstitution shows that WDR5, RbBP5 and ASH2L form a stoichiometric core entity (Figure 1B), 21 which stably interact with the KMT2 enzymes and enhances their otherwise weak activities (~50–500 fold). 21,22 Dimeric DPY30 further contributes to overall KMT2 activity on the H3 peptide by ~2 fold.22 A three-dimensional view of the KMT2A core complex shows that the catalytic SET (Su(var)3–9, Enhancer of zester and Trithorax) domain is positioned in the center while RbBP5–WDR5 and ASH2L–DPY30 pairs are on opposite ends of the SET domain.23 Although the atomic-resolution structure of the KMT2 core complex is not yet reported, X-ray crystal structures of most individual components and key interaction interfaces among them have been solved. 24–31 They show that the KMT2A SET domain adopts a conformation that is inefficient in catalysis,32 which necessitates a conformational change by interacting with the WDR5-RbBP5-ASH2L core entity.
Table 1.
Subunit composition of the metazoan KMT2 complexes
| KMT2A or KMT2B complex | KMT2C or KMT2D complex | KMT2F or KMT2G complex | |
|---|---|---|---|
| Common subunits |
|
|
|
| Unique subunits |
|
|
|
Abbreviations: CFP1, CXXC finger protein 1; HCF1, host cell factor 1; KMT, histone–lysine N-methyltransferase; NCOA6, nuclear receptor coactivator 6; PA1, PTIP associated 1; PTIP, PAX transactivation- domain interacting protein; RBBP5, retinoblastoma-binding protein 5; UTX, ubiquitously transcribed tetratricopeptide repeat, X chromosome; WDR, WD repeat-containing protein.
In vitro biochemical studies show that ASH2L and RbBP5 form a stable heterodimer,21 which is required for the activities of all KMT2 complexes.21,33 It is likely that the RbBP5/ASH2L heterodimer facilitates and stabilizes the interaction between the SET domain and KMT2 substrates.34 While transient interactions between RbBP5/ASH2L and the KMT2A SET domain are documented 34,35, WDR5 stably bridges RbBP5 and KMT2A via direct binding to a conserved WDR5 interacting (WIN) motif in KMT2A 24,30,36 and a valine-aspartate-valine (VDV) motif in RbBP5 37. Disruption of WDR5-KMT2A binding by the WIN peptides or their derivatives leads to disintegration of the KMT2A complex and inhibition of KMT2A catalytic activity in vitro 25,38,39. Interestingly, despite conservation of the core entity, WDR5 is not necessary in regulating other KMT2 complexes 40. The unique regulation of KMT2A activity by WDR5 40 has recently been exploited to develop specific KMT2A methyltransferase inhibitors (discussed below) 40–42.
In addition to regulation by core components, activities of KMT2A and KMT2B are also regulated by Taspase1, a threonine aspartase. 43 KMT2A contains two consensus sites while KMT2B has one for Taspase1 recognition (Figure 1). Following Taspase1-mediated proteolysis, the resulting large N-terminal fragment and a smaller C-terminal fragment associate through FY-rich, N-terminal (FYRN) and FY-rich, C-terminal (FYRC) domains to generate a functional, noncovalent heterodimeric complex. 43,44 While it is still being debated whether the cleavage per se is important,45 the cleavage-facilitated intra-molecular interactions are able to regulate KMT2A activity and expression of target genes in cells.43
Substrate specificity of the KMT2 enzymes
The requirement of WDR5 for overall enzymatic activity is only one of many distinguishing biochemical properties among KMT2 enzymes. The KMT2 core complexes also diverge in their in vitro substrate specificity. Mass spectrometry studies show that the recombinant KMT2F and KMT2G core complexes are capable of mono-, di- and tri-methylation on nucleosomal H3 K4, similar to the yeast ScSet1.3 Recombinant KMT2A and KMT2B core complexes are able to mono- and di-methylate H3 38,46 and have low tri-methylation activity. By comparison, the recombinant KMT2C and KMT2D core complexes are predominantly mono-methyltransferases (Lee and Dou, unpublished observation). The basis for distinct substrate specificity remains poorly understood and will benefit from future structural studies of respective KMT2 SET domains. Notably, the substrate specificities of the bacterially expressed core complexes are not always consistent with those of purified holo-complexes, 16,17,47–49 or the core complex reconstituted in insect cells 21. It is likely that additional factors may contribute to the KMT2 regulation in the holo-complex or KMT2s are subject to additional regulation by protein post-translational modifications (PTMs). For example, it has been reported that KMT2A and KMT2F, but not KMT2C, are regulated by histone H2B ubiquitination (H2Bub)-mediated trans-activation.46 Furthermore, SUMOylation of RbBP5 substantially inhibits KMT2A activity in cells by disrupting its association with ASH2L.50 Given the extensive PTMs of KMT2 core subunits revealed by recent proteomic studies, it will be of interest to determine whether PTMs modulate the substrate specificity of the KMT2 complexes.
Subunit compositions of the KMT2 enzymes
Label-free quantitative mass spectrometry shows that subunit composition of KMT2 complexes has a high degree of heterogeneity in cells.51 In fact, one distinguishing feature among the three KMT2 subgroups is their unique set of associating transcription factors and transcription cofactors (Table 1). Specifically, MENIN encoded by multiple endocrine neoplasia type 1 (MEN1)17 and LEDGF (lens epithelium-derived growth factor) 52,53 are a unique subunits for KMT2A and KMT2B, and mediate their recruitment to target genes via interactions with transcription factors such as estrogen receptor-α (ER-α).54 PTIP (Paired box (Pax) transactivation-domain interacting protein) and NCOA6 (nuclear receptor coactivator 6, also called ASC2) are unique subunits in the KMT2C and KMT2D complexes.16,18,55 They interact with the PAX family transcription factors 55 as well as nuclear receptors such as PPAR-γ (peroxisome proliferator-activated receptor γ) and LXR (liver X receptor).16 Unique components of KMT2F and KMT2G complexes include WDR82 (WD40 repeat protein 82) and CFP1 (CxxC finger protein 1). WDR82 interacts directly with RNA polymerase II (RNAPII) that is phosphorylated at serine 5 within the heptad repeats of the C-terminal domain (CTD) and CFP1 binds to non-methylated CpG dinucleotides.19 Unique subunits within each of the KMT2 subgroups probably, to some extent, dictate functional specificity since they are directly or indirectly involved in recruiting KMT2s to distinct regions in the genome. In addition to stable complex subunits, the KMT2 core complexes also dynamically interact with sequence-specific transcription factors including E2Fs,56–58 p53,57,59,60 MEF2D (myocyte enhancer factor 2D),61 PAX7,62 NFE2 (nuclear factor, erythroid 2),63 Ap2δ (activating protein),64 NF-Y,65 USF1 (upstream transcription factor 1) 66 and OCT4 (octamer-binding transcription factor 4, also called POU5F1). 67 These interactions likely contribute to context-dependent functions of the KMT2 complexes.
The recruitment and chromatin binding of KMT2 complexes
Distinct sets of interacting proteins underlie the largely variable distributions of KMT2 proteins throughout the genome (Figure 2). Chromatin-immunoprecipitation-sequencing ((ChIP-seq) studies show that KMT2F and KMT2G generally bind at transcription start sites (TSSs) 66 while KMT2C and KMT2D are highly enriched at enhancers.48,68 Genome-wide binding sites for KMT2A and KMT2B are less clear, as recent studies indicate their binding at both gene promoters and enhancers.68,69
Figure 2. Distinct distributions of KMT2 enzymes at transcription regulatory regions.
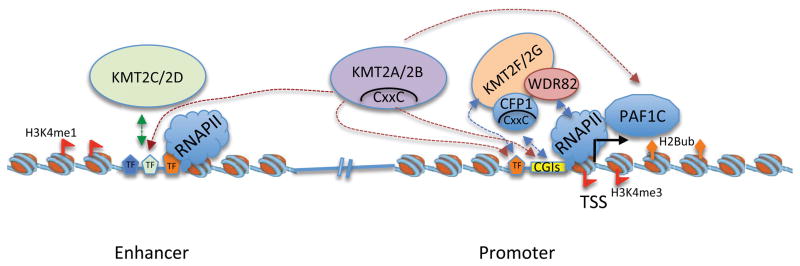
Based on KMT2 functional domains, their respective interaction with chromatin and substrate specificity, it is envisioned that KMT2C and KMT2D are the predominant KMT2s at distal enhancers while KMT2F and KMT2G are the major enzymes at gene promoters. KMT2A and KMT2B could function at both regulatory regions. The distribution of KMT2 enzymes in the genome is consistent with distribution of histone H3 K4 methylation, i.e. H3K4me1 is a predominant histone mark at cell-type specific distal regulatory enhancers and H3K4me3 is mainly found at actively transcribed gene promoters. Recruitment of KMT2 to different genomic regions is also facilitated by their interactions with sequence specific transcription factors or RNA polymerase II (RNAPII), which leads to non-coding and coding transcription at enhancers and promoters, respectively.
The patterns of genome-wide distribution of KMT2 proteins are consistent with that of H3K4me states (i.e. mono-, di- or tri-methylation), as expected for the substrate specificity of each KMT2 protein (Figure 2). They also underscore the intrinsic affinity of KMT2 proteins for different chromatin regions. For example, KMT2A and KMT2B contain the CxxC domains that bind non-methylated CpG dinucleotides 70, a common feature for a large subset of metazoan gene promoters.71 The binding of KMT2A and KMT2B to gene promoters can be further stabilized by the PHD3 (plant homeotic domain 3) and Bromo-domains, which bind to H3K4me3 and acetylated lysine residues, respectively, that are highly enriched around TSS.72 In contrast, KMT2C and KMT2D contain binding motifs (e.g. high mobility group (HMG-I) and Leu-Xaa-Xaa-Leu-Leu (LxxLL) motifs) that are commonly found in transcription factors and cofactors.73,74 They also bind to unmethylated and asymmetrically di-methylated H4 arginine 3 (H4R3me0 and H4R3me2a) via PHD4, PHD5 and PHD6.49 These modifications are not necessarily enriched at gene promoters, consistent with the preferred binding of KMT2C and KMT2D at intergenic regions. Recruitment of KMT2 proteins to specific chromatin regions (e.g. the homeobox A (HOXA) cluster) by long noncoding RNAs has also been reported 75,76.
Despite these progresses, many unanswered questions regarding KMT2 recruitment and chromatin binding remain. For example, confocal microscopy has revealed that two paralogs in each KMT2 subgroup (i.e. KMT2F vs. KMT2G, KMT2A vs. KMT2B) exhibit largely non-overlapping subnuclear distribution despite identical domain structure and interacting proteins 20,77. In addition, highly localized KMT2 proteins are often found in small areas within much broader H3K4me domains, especially at intergenic regions.48 It is not clear how H3K4me is spread in these regions without concurrent spreading of the KMT2 enzymes. Furthermore, it is not clear whether KMT2 complexes are recruited as a whole to chromatin or assembled at target genes following step-wise recruitment. The prevalence of overlapping binding sites in the genome for only a subset of core subunits (e.g. WDR5, ASH2L or KMT2) seems to support the latter. 63 Alternatively, KMT2 subunits can bind to distinct regions near the target loci (e.g. β-globin ) and methylate H3 through a scanning model.63 These remaining questions warrant further studies.
Physiological functions of KMT2 family histone methyltransferases
KMT2s in transcription regulation
KMT2 play important roles in transcription regulation. KMT2 enzymes are enriched at multiple transcription regulatory regions including gene promoters and enhancers.3,4 Despite being the major enzymes for H3K4me, individual Kmt2 deletion in mouse embryonic fibroblasts (MEFs) leads to surprisingly few changes in gene expression.78 However, KMT2C and KMT2D are crucial to induce transcriptome changes during adipogenesis and trans-differentiation of pre-adipocytes into myocytes.48 Large scale KMT2-dependent transcriptome changes are also described during embryonic stem cell (ESC) differentiation 67, leukemic transformation 40 or cellular response to external stimuli (e.g. lipopolysaccharide79, retinoic acid 80). These results support a dynamic and/or context-dependent requirement of KMT2s for proper gene expression. They also highlight the epigenetic nature of H3K4me, as deletions of KMT2s in differentiated cells show fewer changes in gene expression81 when compared to deletions in mitotically active, progenitor cell types. In other words, H3K4me may have inherent stability once set.
Direct and causal functions of several KMT2s in transcription initiation have been elegantly demonstrated in vitro 57,59,82, and are further supported by KMT2-dependent RNAPII binding at promoters and enhancers in various cells.48,68 The function of H3K4me3 in transcription has been extensively discussed.83,84 It generally involves the recruitment of a large repertoire of ‘reader’ proteins 85,86, which communicate the epigenetic changes to the basic transcription machinery leading to either productive transcription or gene repression.83,84 Despite the high level of H3K4me3 at active gene promoters, H3K4me3 alone does not necessarily dictate transcription outcome given that ‘reader’ proteins can be either coactivators or corepressors.85,86 Instead, a combinatorial readout that involves H3K4me3 and other histone modifications is more likely to be predictive.85 H3K4me also contributes to transcription regulation by preventing ectopic silencing of euchromatin,87 facilitating pre-mRNA processing 88 and regulation of antisense transcripts.89
ChIP-seq studies for genome-wide histone modifications show that H3K4me1 is perhaps the most important histone modification that marks the distal regulatory enhancers, which can be further divided into ‘active’ and ‘poised’ enhancers depending on H3K27 modifications.90,91 In fact, H3K4me1 enrichment is a widely used criteria to catalog lineage-specific and developmental stage-specific enhancers due to its remarkable fidelity.92,93 Recent studies show that de novo establishment of H3K4me1-enriched lineage specific enhancers foreshadows cell type specific transcription programs.92 Dysregulation of enhancer activity leads to disruption of the precise spatiotemporal regulation of gene expression, and therefore, is linked to human cancers.94,95 Characterizing the functions of KMT2 enzymes at enhancers is an emerging field. It is recently shown that KMT2C and KMT2D are the major histone methyltransferases responsible for H3K4me1 at adipocyte- and myocyte-specific enhancers.45 They function to prime these enhancers in coordination with lineage specific transcription factors by maintaining open chromatin structures and facilitating RNAPII access.48 H3K4me1 at macrophage-specific enhancers is deposited by KMT2A and KMT2D and is required for active non-coding transcription at enhancers.68 Deciphering KMT2-dependent enhancer regulation is likely to further shed light on the roles of these enzymes in development and human diseases.
KMT2 and DNA methylation
Stable binding of KMT2A to mitotic chromatin has been reported,81 raising the question on whether H3K4me distribution represents a bona fide, epigenetic mark, similar to DNA methylation. Several lines of evidence suggest extensive interplay between H3K4 methylation and DNA methylation. H3K4me3 and DNA methylation show almost mutually exclusive distribution patterns due to incompatible binding of H3K4me3 by the DNA methyltransferase complexes 96,97 as well as preferred binding by the CxxC domains in KMT2 complexes to unmethylated CpG islands (CGIs).98 CGIs, which mark a major class of mammalian promoters that lack the core promoter elements,99 can serve as a signaling module that locally influences chromatin structures through recruiting the CxxC domain-containing KMT2 complexes (i.e. those with KMT2A, KMT2B, KMT2F or KMT2G) for de novo accumulation of H3K4me3. Reciprocally, high levels of H3K4me3 and concurrent on-going transcription protect non-methylated CGIs in the early embryo from de novo DNA methylation.99 In leukemia cells, binding to CGIs by KMT2A protects CpG clusters within HOXA9 from methylation.100–102 Loss-of function mutation in the KMT2A CxxC domain leads to increased DNA methylation at CGI, silencing of HOXA9 expression and subsequent inhibition of leukemia cell growth.102 Furthermore, unmethylated CGIs also ensure that KMT2F and KMT2G localize to the correct promoters, and safeguard against ectopic KMT2F and KMT2G binding at enhancers and the aberrant transcriptional activity that would result from such binding.103 Recently, interplay between KMT2 and the DNA methylcytosine dioxygenases TET2 (ten-eleven translocation) and TET3 has been reported.104 TET2 and TET3 promote GlcNAcylation of HCF1 (host cell factor) by the O-GlcNAc transferase (OGT), which is important for KMT2F-mediated H3K4me3.104 Functional coupling of TET2 and TET3 that are responsible for DNA 5-hydroxymethylation to KMT2 mediated H3K4 methylation is intriguing and clearly warrants further investigation.
Regulation of KMT2 during cell cycle and development
In vivo functions of KMT2 family enzymes are context-dependent and dynamic (see Box 1). This is, at least in part, due to the regulatory proteolysis of KMT2s. It is reported that KMT2A and its Drosophila homolog Trx remain associated with DNA during DNA replication and mitosis to ensure perpetuation of transcriptional states.81,105 Despite S- and M-phase bookmarking, KMT2A is degraded at G1/S and M/G1 transitions during cell cycle by E3 ubiquitin ligases, SKP1–cullin 1–F-box protein (SCF)SKP2 and anaphase promoting complex (APC)CDC20, respectively. 106–108 Furthermore, the KMT2A level is tightly regulated by ankyrin repeat and suppressor of cytokine signaling (SOCS) box-containing protein 2 (ASB2) mediated degradation during hematopoietic differentiation.107 The PHD2 domains of KMT2A and KMT2B also possess intrinsic E3 ubiquitin ligase activity.106 Importantly, the regulatory proteolysis of KMT2A is disrupted in leukemogenic KMT2A fusion proteins, indicating the importance of KMT2A turnover in a physiological context.109 Proteolytic turnover for other KMT2s has yet been studied. A recent study shows that stability of yeast ScSET1 could be regulated by the level of cellular H3K4me.110 Whether similar feedback control exists for KMT2 in higher eukaryotes remains to be determined. Multi-level regulation of KMT2 activity ensures tight spatiotemporal control of H3K4me, a necessity in higher eukaryotes.
Box 1. KMT2s in development.
Humans and transgenic mice carrying mutant KMT2 alleles have defined a developmental role for the KMT2 family. Interestingly, homozygous deletion of each murine KMT2 results in a severe but distinct phenotype, indicating unique functional specification. Kmt2f is required at earliest stage of development, as its deletion leads to gastrulation failure and embryonic lethality at E5.5. In vitro studies show that Kmt2f is essential for establishment and/or maintenance of embryonic stem cells (ESC), epiblast stem cells and neural stem cells.161 In contrast, homozygous deletion of all other KMT2s leads to lethality at late stage of development. Widespread developmental defects have been reported for constitutive homozygous deletion of Kmt2b and Kmt2g, which die around E10.5 and E11.5, respectively. Most organs and structures arise normally and no major morphological defects are reported except growth retardation.123–125 Deletion of Kmt2d leads to embryonic lethality around E9.5. Although the developmental defects of Kmt2d are not yet defined, conditional deletion of Kmt2d in somitic precursor cells leads to reduced brown adipose tissue and muscle mass.48 Germline homozygous deletion of Kmt2a and Kmt2c results in specific defects. The abnormalities are mostly in axial skeleton patterning and definitive hematopoiesis for Kmt2a,162–164 and reduced white adipose tissue (WAT) for Kmt2c.165 Most of the heterozygous KMT2 alleles do not have reported gross defects in mice. The essential functions of KMT2 in development are probably not due to loss of SET domain activity since mice expressing Kmt2a lacking the SET domain are viable with only minor defects 166 and mice expressing Kmt2c lacking the SET domain have only partial embryonic lethality.165 Combinatorial deletion of the closely related SET domains of each KMT2 subgroup has not been performed to rule out the essential, yet redundant functions of H3K4me activities within each KMT2 subgroup. One interesting observation is that the developmental requirement of KMT2 genes can be highly specific and transient. For example, Kmt2b deletion before E10.5 and after E11.5 has different consequences, as only the latter results in diabetic live-born mice.167 Kmt2b deletion in adults results in defects such as male and female sterility.125 Similarly, conditional Kmt2a deletion in adult hematopoietic system has relatively weaker effects as compared to general deletion in utero.164,168 Transition of the major enzyme for global H3K4me3 from KMT2B to KMT2F is also reported during inner cell mass formation.161
KMT2 mutations span a diverse set of human cancers
Exome-sequencing of human tumors has revealed a surprising variety of disease-specific KMT2 mutations.13 Although previous analyses had focused mainly on KMT2A somatic rearrangements in MLL, newly identified KMT2 mutations greatly expand the potential role of KMT2s in cancer.13 An analysis of the nearly 2200 mutations in KMT2 familial cancers in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (cancer.sanger.ac.uk/) (see Table 2), together with recent in vivo modeling of KMT2 in disease, provide tantalizing clues to the pathogenesis of KMT2-driven cancers, and to the development of novel cancer therapeutics.
Table 2.
| KMT2 | Disease (Top 3 or 5) | No. | Mutation (Top 3) | Domain Affected |
|---|---|---|---|---|
| KMT2A | Large Intestine Lung Bladder Endometrium Breast |
305 | Nonsense Frameshift Missense |
SET – 22.9% PHD – 18.0% |
| KMT2B | Endometrium Large Intestine Lung Brain (Glioma) Liver |
236 | Missense Nonsense Frameshift |
SET – 26.2% PHD – 13.1% |
| KMT2C | Lung Large Intestine Breast Endometrium Bladder |
845 | Nonsense Missense Frameshift |
SET – 28.3% PHD – 17.1% |
| KMT2D | Blood/Lymph Node Large Intestine Lung Endometrium Brain (Medulloblastoma) |
627 | Nonsense Frameshift Missense |
SET – 37.0% PHD – 12.9% |
| KMT2F | Large Intestine Lung Endometrium Skin (Melanoma) Liver |
141 | Missense Frameshift Nonsense |
SET – 15.6% RRM – 3.5% |
| KMT2G | Endometrium Large Intestine Kidney |
32 | Frameshift Missense Nonsense |
SET – 21.9% RRM – 9.4% |
This information was derived from authors’ analysis of COSMIC (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/). All information was accessed on August 25, 2014, and search terms for genes included “MLL” (KMT2A), “MLL4” (KMT2B), “MLL3” (KMT2C), “KMT2D,” “SETD1A” (KMT2F), “SETD1B” (KMT2G). All non-coding and coding synonymous mutations were excluded. The histologic origin of tumors was broad and distribution was gene-specific. COSMIC data also included zygosity, and many other characteristics that detailed each sample deposited in the database. The amino acid change for each KMT2 was then mapped to the Universal Protein Resource (www.uniprot.org) for the corresponding KMT2 gene (search terms “KMT2A,” “KMT2B,” “KMT2C,” etc.). Any mutation falling in a functional domain (e.g. SET, PHD, etc.) was annotated; therefore a single mutation (e.g. frameshift or nonsense) may affect more than one domain. The COSMIC data do not specify whether the mutation is a driver or passenger mutation.
KMT2A
KMT2A rearrangements, tandem duplications and copy number amplifications have been extensively studied and thoroughly discussed in the context of MLL (see Box 2).7,111 In comparison, mutations in the KMT2A region are under-represented in hematopoietic and lymphoid malignancies, accounting for only 2.3% of 305 non-synonymous mutations in the KMT2A region in the COSMIC database (Table 2). Yet, recent profiling studies show that mutations in the KMT2A region are prevalent in a large spectrum of solid tumors (e.g. colon, lung, bladder, endometrial, and breast cancers).11,112–114 Most KMT2A mutations are nonsense (NS) and frame shift (FS) mutations that lead to early termination of translation, with about 40% of these resulting in truncated proteins that have no methyltransferase activity. Truncations that impact PHD and CxxC domains account for 18% and 9% of mutations, respectively. Missense mutations or in-frame deletions in the SET, WIN, PHDs and CxxC domains of KMT2A are also reported, albeit at much lower frequencies. It remains unclear how the missense mutations in cancers affect the functions of the specific domains and/or on the overall stability of the protein. While the loss of heterozygosity remains unknown for 60% of total reported KMT2A mutations, heterozygous mutations account for nearly all the known mutations for which zygosity has been determined. Thus, similar to KMT2A rearranged MLL and KMT2A germline mutations (see Box 2), it seems that at least one wild-type KMT2A allele remains intact in vast majority of cancers.
BOX 2. Pathogenesis of MLL.
KMT2A abnormalities on chromosome 11q23 identify a group of biphenotypic leukemia, where leukemic blasts express both lymphoid and myeloid surface antigens, leading to the name mixed lineage leukemia (MLL). KMT2A rearrangements include translocations, tandem duplications and amplifications. Together, they account for approximately 10% of human leukemias and are associated with poor prognosis.7,8,111
Most KMT2A rearrangements are balanced translocations on one allele and lead to production of in-frame gain-of-function oncogenic fusion proteins. The remaining KMT2A wild-type allele and its methyltransferase activity are essential for MLL at onset or after leukemic transformation. To date, over 70 fusion proteins have been identified, suggesting heterogeneity in pathogenesis.7,8,111 The most frequent rearrangements in MLL involve fusion partners in the ENL (Eleven Nineteen Leukemia)-associated protein (EAP) complex169, which account for 90% and 70% of KMT2A-rearranged acute lymphblastic leukemia and acute myeloid leukemia, respectively. Since EAP complex components interact with transcriptional cofactors such as PAF1C (RNA polymerase II associated factor 1 complex) 170,171, DOT1L (Dot1-like) 172,173, P-TEFb (positive transcription elongation factor b)–BRD4 (bromo-domain protein 4) 145,146 and CBX8 (chromobox 8)–TIP60 (Tat-interacting protein 60) 174, it is postulated that MLL fusion proteins function to maintain a high level of expression of genes that are composed of a leukemic signature by regulating transcription elongation. Recently, it is reported that the reciprocal fusion protein (i.e. AF4-MLL) is also able to induce acute lymphoblastic leukemia by maintaining the ability to interact with DOT1L, P-TEFb as well as KMT2A core subunits and to influence the transcription elongation process.175,176
Mechanistic studies show that MLL fusion proteins are able to induce leukemic transformation from hematopoietic stem cells in a dosage dependent manner.177 They confer stem cell like properties such as self-renewal and high expression of anti-apoptotic genes and genes for drug efflux pumps.7,8 This function is likely mediated by a limited number of KMT2A direct targets such as HOXA9, HOXA10, and MEIS1, which in turn regulate LSC specific transcription programs.177,178 Interestingly, the mouse models that either constitutively express fusion proteins or produce fusion proteins by inter-chromosomal translocations from the endogenous KMT2A locus only develop leukemia and not other tumor types,179,180 suggesting a hematopoietic-specific tumorigenic capability.
Genetic studies in mice have demonstrated that deletion of one Kmt2a allele is not sufficient to induce leukemic transformation or spontaneous tumorigenesis in other tissues, consistent with the fact that patients carrying haploinsufficient germline KMT2A mutations do not have a reported predisposition to cancer.115 Instead, in MLL, malignant transformation requires the gain-of-function KMT2A rearrangements that lead to production of MLL fusion proteins.
Therefore, additional KMT2A-independent oncogenic events are likely needed in cancers harboring KMT2A loss-of-function mutations. This raises the question of whether KMT2A mutations initiate transformation or simply provide a permissive environment for tumorigenesis. Future exome sequencing of KMT2A in cancer patients at various time points (early vs. late, primary vs. metastasis, and pre- vs. post-treatment) will be important to show when KMT2A mutations arise in the disease processes, and their relationship to metastasis and relapse.
Accumulating evidence seems to suggest that KMT2A plays distinctive roles after cancer onset. While loss of one wild-type KMT2A allele is recurrent, in vitro studies show that a variety of malignant (cervical, colon, lung, choriocarcinoma) cell lines are more sensitive to KMT2A knockdown than normal cell lines.116 Following establishment of HeLa xenografts in athymic nude mice, antisense-mediated KMT2A reduced tumor size and vascularity as compared to controls.116 Furthermore, KMT2A, together with hypoxia-inducible factor 1α (HIF1α), is overexpressed in hypoxic regions of the tumor, and its knockdown reduced expression of HIF1α and VEGF (vascular endothelial growth factor) in a cervical cancer xenograft.7,116 KMT2A has also been shown to be a key downstream effector of the hepatocyte growth factor (HGF)–MET signaling pathway and facilitates metastasis in a hepatocellular carcinoma model.117 The essential functions of wild type KMT2A have also been demonstrated in KMT2A-rearranged MLL (see Box 2).118 Specifically, knockdown of KMT2A by shRNA, or genetic deletion of wild type KMT2A leads to decreased growth of MLL-AF9 transformed cells and the mice receiving KMT2A depleted cells do not succumb to leukemia.118 Mechanistically, KMT2A promotes MYC transcription via binding to MYC super-enhancers.119 KMT2A also positively regulates cell cycle regulators, including CDKN1B (cyclin-dependent kinase inhibitor 1B), CDKN2C, and CDKN2A.120,121 Deregulation of cell cycle is one of common mechanisms for malignant transformation.122 The potentially different functions of KMT2A in normal vs. malignant cells suggest that targeting KMT2A could specifically inhibit cancer cells.
KMT2B
Analysis of currently available data reveals that there are fewer KMT2B non-synonymous mutations in human cancers. Endometrial, large intestine, lung, glioma, and liver carcinomas contain 72% of identified mutations in the KMT2B coding region (Table 2). Similar to KMT2A, mutations affecting the SET domain are most common, and the majority of these are due to FS and NS alterations, while missense mutations in the SET domain are responsible for 5% of total KMT2B mutations. Of the KMT2B mutations for which zygosity is known, over 90% are heterozygous while the remaining are homozygous. Studies of germline or conditional Kmt2b haploinsufficiency in mice have failed to show an oncogenic propensity.123–126 Indeed, KMT2B has been described generally as a positive regulator of cell growth. Homozygous inactivation of Kmt2b in ESCs and in germline knockout mouse models resulted in proliferation defects and embryonic lethality due to increased apoptosis.123,127 Like KMT2A, KMT2B is recruited to the MYC enhancer through a β-catenin-dependent process to promote MYC transcription via H3K4me3 methylation.119
KMT2C and KMT2D
To date, hundreds of KMT2C and KMT2D mutations have been identified, making them among the most frequently mutated genes in human cancer.13 For KMT2C, mutations are the most prevalent in lung128, large intestine,129 breast,129 endometrial and bladder carcinomas,113 which together account for 60% of the total KMT2C mutations identified (Table 2). Non-Hodgkin lymphoma130 and medulloblastoma or other primitive neuroectodermal tumors 131 constitute nearly a quarter of KMT2D mutant cancers. KMT2D mutations in acute myeloid leukemia, lung, large intestine and endometrial carcinomas are also widely reported.13
Most cancer-associated KMT2C and KMT2D mutations are FS and NS alterations, which probably lead to protein truncations (Table 2). Mutations that impact the SET and PHD domains in both proteins account for 28% and 25% of total KMT2C mutations and for 37% and 60% of total KMT2D mutations, respectively. In comparison, missense mutations in the SET domain of KMT2C and KMT2D are exceedingly rare, accounting for less than 1% of total KMT2C and KMT2D mutations. Interestingly, disproportionally high percentage of missense mutations in the PHD domains are found in KMT2C (16%) and KMT2D (6%), which are among the highest of all types of missense mutations in the KMT2 gene family. These findings suggest that the PHD domains of KMT2C and KMT2D are probably important for tumor suppression, and thus functional characterization of these missense mutations may be a fertile area of future investigation.
Zygosity of most KMT2C and KMT2D cancer mutations is unknown. In cases where zygosity is determined, the majority are heterozygous, with ~15% (53/322) of KMT2C and KMT2D homozygous mutations. Interestingly, half of the homozygous KMT2D mutations are seen in infrequent tumors such as medulloblastoma, lymphoma (diffuse large B cell lymphoma (DLBCL) and follicular lymphoma), kidney carcinoma, and malignant melanoma. Homozygous mutations were never reported in Kabuki Syndrome (Box 2), indicating that complete loss of KMT2D is not tolerated in the germ line, but can uncommonly occur somatically in a particular set of sporadic human cancers.
Compared with KMT2A and KMT2B, KMT2C and KMT2D have different functions in cancer. KMT2C and KMT2D have known negative effects on cell growth.13,60,132–135 Several lines of evidence suggest that KMT2C is a tumor suppressor. In Drosophila mosaic eye imaginal discs, clones containing loss-of-function mutant trr (an ortholog to mammalian KMT2C and KMT2D) alleles have a growth advantage over wild-type clones.132 Nearly half of mice carrying a loss-of-function, homozygous SET deletion Kmt2c allele develop ureter epithelial tumors.60 Full penetrance is achieved when Kmt2c mutant alleles are introduced into a Trp53 heterozygous background.60 However, this mouse model does not recapitulate the KMT2C haploinsufficient state observed in human bladder carcinomas, which represent about 5% of KMT2C-mutated malignancies in the COSMIC database. Interestingly, a recent report showed that adoptive transfer of hematopoietic stem cells with knockdown of Kmt2c and neurofibromin1 (Nf1) into Trp53 null mice resulted in acute myeloid leukemia.133 This model mimics the haploinsufficient state seen in human diseases, in that KMT2C protein levels were ~50% of control.133 For KMT2D, the picture is more complicated. On one hand, KMT2D interacts directly with p53 to promote expression of p53 target genes.60 Kmt2d deletion is able to abrogate MLL-AF9-induced leukemogenesis through a CDKN1A-dependent process.134 On the other hand, high levels of KMT2D portend poor prognosis in patients with breast cancer, and KMT2D knockdown impairs proliferation and invasion in human breast cancer xenografts in mice, as well as in human colorectal and medulloblastoma cell lines.136–138 These studies show that the effect of KMT2D mutations might be cell-type or context dependent.
What would be the potential epigenetic mechanism by which KMT2C and KMT2D exert their effects in tumorigenesis? Since KMT2C and KMT2D are crucial for H3K4me1 at cell type specific distal enhancers,48,68 it is possible that loss-of-function KMT2C and KMT2D mutations would promote tumorigenesis by dysregulation of enhancer activity and thus, by disruption of normal developmental programs, subversion of cell identities and/or alteration of gene networks that are regulated by tumor suppressors (e.g. p53) and oncogenes (e.g. MYC). Consistent with this view, variant enhancers defined by H3K4me1 comprise a signature in colon cancers that is robustly predictive of the cancer transcriptome and is associated with cancer predisposition.94,139 Closer examination of the KMT2C and KMT2D dependent enhancer landscape in normal and cancer cells should yield important mechanistic insights.
KMT2F and KMT2G
KMT2F and KMT2G catalyze the bulk of H3K4me3 in most cells. Large intestine, lung, endometrial carcinomas, and malignant melanoma together account for 62% (87/141) of cancers with KMT2F mutations in COSMIC, and endometrial, large intestine, and kidney carcinomas comprise 63% (20/32) of KMT2G-mutant cancers. The NS, FS and missense mutations that impact the SET domain contribute to 16% (22/141) of KMT2F mutations in cancer. In contrast, no missense mutations have been identified that involve the KMT2G SET domain. KMT2F mutations that affect the CFP1-interacting N-SET domain including missense mutations account for ~10% of total KMT2F mutations. Missense mutations in the RRM domain of both KMT2F and KMT2G are reported, though at much lower frequency. Consistent with their importance in development, among KMT2F mutations with known zygosity, only 13% were homozygous. No homozygous cancer mutations were identified for KMT2G. Although heterozygous loss-of-function KMT2F mutations may be linked to the elevated risk of colon cancer in patients with schizophrenia (see box 3),140,141 these mutations (and other KMT2F and KMT2G mutations recorded in COSMIC) have not yet been modeled in vivo to determine whether the human mutations induce cancers in transgenic animals.
BOX 3. Germline KMT2 mutations in neuropsychiatric disorders.
Germline mutations in KMT2 family genes are widely reported in persons exhibiting intellectual disability (ID) or other neuropsychiatric features, and some may have a cancer disposition. All reported germline mutations are haploinsufficient, consistent with the essential functions of KMT2 genes during development.
To date, several hundred KMT2D mutations have been associated with 55–80% of cases of Kabuki syndrome (KS), characterized by distinctive facial features, ID, among other features.181 KS has been modeled in mice carrying a heterozygous allele that lacked the Kmt2d SET domain.182 Kmt2d+/− mice demonstrated loss of global H3K4me3, and similar to KS patients, displayed ID.183 Many KS patients show predisposition to tumors, such as pilomatricomas, synovial sarcoma, fibromyxoid sarcoma, acute lymphoblastic leukemia, Burkitt lymphoma, neuroblastoma, and hepatoblastoma.184 Nearly three-quarters of KMT2D KS mutations share an identical pattern of domain loss as the somatic cancer mutations. Missense KS mutations in the SET domain have been shown to affect KMT2 complex assembly.185
Germline mutations of other KMT2 genes are also reported. De novo KMT2A germline mutations have been linked to Weidemann-Steiner Syndrome (WSS), which features “hairy elbows,” ID, distinctive facial appearance,115 and correlates with growth retardation observed in Kmt2a+/− mice.162 NS mutations for KMT2C have been reported in Kleefstra spectrum syndrome (KSS) 186 and autism spectrum disorder (ASD) 187, both of which features ID and reduced social communication. De novo heterozygous KMT2F mutations have been linked to the common neuropsychiatric disease schizophrenia.141 Cancer predisposition has not been described in WSS, KSS and ASD. This is despite the fact that schizophrenia patients have a 190% higher risk for developing large intestine carcinoma, the most common KMT2F-mutant malignancy.140,188
Mechanism-based therapeutic targeting of KMT2 enzymes
Extensive mechanistic studies show transcriptional deregulation in mixed lineage leukemia (MLL) and have unraveled many key intermediates in disease progression (Box 2). Several promising therapeutic targets have recently emerged as the result. Here we summarize recent progress and insights gained from development of novel pharmacological inhibitors for KMT2A-rearranged MLL, which provides insights targeting KMT2 in other human cancers.
Targeting MLL fusion protein dependent pathways
Targeting the gain-of-function KMT2A rearrangements in the MLL fusion protein pathway represents a major translational effort. Currently, several small molecular inhibitors against MLL fusion-interacting proteins are yielding promising results in MLL (Figure 3). Interestingly, these inhibitors substantially alter the gene signatures of leukemia stem cells (LSCs) and induce prominent myeloid differentiation of LSCs in murine models. While the current DOT1L inhibitors have only modest effects on MLL in murine models, substantial changes in global H3K79me are detected 1–2 days after treatment, which are followed by phenotypic alterations that occur at ~10–14 days of treatment.142,143 Targeting DOT1L also benefit leukemia that does not have KMT2A rearrangement.144 Recruitment of P-TEFb is also considered a critical step in transcriptional deregulation leading to leukemia.145,146 P-TEFb is a cyclin dependent kinase that contains the catalytic subunit, cyclin-dependent kinase 9 (CDK9), and a regulatory subunit, cyclin T. P-TEFb interacts with both MLL fusion proteins.145,146 and bromodomain protein 4 (BRD4) 147,148, which is essential for MYC expression in leukemia cells.149,150 Inhibitors of CDK9 (e.g. flavopiridol 151) and BRD4 (e.g. JQ1143 and iBET152) have shown promising results in treating MLL in mice and are in clinical trials for a variety of hematologic and solid tumors (e.g. NCT02141828, NCT01587703 and NCT00012181). Interestingly, the KMT2A fusion protein MLL-AF4 and P-TEFb also promote cytotoxicity induced by bortezomib (a proteasome inhibitor) via PAX5-dependent activation of CDKN1B in subset of human leukemia,153 suggesting that proteasome inhibitors might be an option for specific MLL. Targeting recruitment of MLL fusion proteins by blocking MENIN-MLL interaction is also an effective therapeutic strategy.52,154 MENIN inhibitors have shown great cellular potency in MLL by blocking expression of HOXA9 and other leukemic genes.155 Interestingly, since MENIN is also a sub-stoichiometric component of the wild type KMT2A and KMT2B complex,51,156 blocking MENIN-MLL interaction leads to reduction of both H3K4me and H3K79me at the HOXA9 locus.155
Figure 3. Schematic for the pathogenesis of MLL.
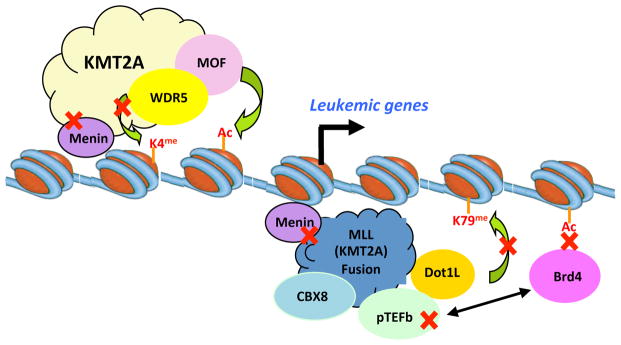
As detailed in Box 2, both wild type KMT2A and MLL fusion proteins contribute to the regulation of leukemia signature genes. While KMT2A functions in transcription initiation through its H3K4 methyltransferase activity, MLL fusion proteins function to recruit transcription cofactors such as MENIN, DOT1L, P-TEFb, LEDGF, and CBX8–TIP60 to promote transcription elongation. Small molecules targeting each of these steps have shown efficacies in MLL cell lines and represent potential therapeutic strategy. References see text.
Targeting KMT2A methyltransferase activity
Patients with MLL usually harbor at least one wild-type KMT2A allele. Using mouse models, it has been shown that wild-type Kmt2a is required for survival of MLL-AF9 leukemia cells.118 The essential function of wild-type KMT2A in MLL is mainly due to its histone methyltransferase activity since specific blocking of KMT2A activity by the peptidomimetic MM401, which disrupts KMT2A and WDR5 interaction,40 phenocopies KMT2A gene deletion in this context.40 While MM401 has no toxicity on normal bone marrow cells, it induces analogous morphological changes as DOT1L or MENIN inhibitors after ~4–7 days.40 Targeting wild-type KMT2A activity may have broad applications outside MLL, including myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) that harbor KMT2A amplification or tandem duplication. Despite the importance of KMT2A activity in leukemia, the KMT2A SET domain is in fact dispensable for leukemia initiation.157 This paradox is reminiscent that of HOXA9, which is a critical downstream target of KMT2A. Although HOXA9 is essential for MLL and is invariably up regulated in MLL patients, Hoxa9-null bone marrow cells can still be efficiently transformed by MLL fusion proteins.158,159 It is likely that KMT2A SET activity is sufficient, but not necessary, for leukemic transformation by MLL fusion proteins. We speculate that fusion proteins, by initially enhancing expression of HOXA9, gradually drive leukemia cells towards HOXA9 dependence/addiction that in turn requires wild type KMT2A to maintain. In absence of KMT2A activity, MLL fusion proteins could induce leukemia through alternative pathways, consistent with rare forms of MLL that carry null KMT2A alleles.160
Perspectives on KMT2s in cancers
Recent studies on KMT2 mutations in human patients show that these genes are frequently altered in a broad spectrum of cancers in addition to previously characterized hematological malignancies. Most KMT2 mutations in non-hematological cancers are heterozygous nonsense mutations that lead to protein truncations. Despite being found as only a small subset, KMT2 missense mutations impact most conserved protein domains including SET and PHD. Mouse genetic studies show that heterozygous deletion of KMT2 genes does not lead to spontaneous tumorigenesis, suggesting that haploinsufficient KMT2 mutations, by themselves, are probably not the ‘drivers’ of human cancers. Furthermore, homozygous deletion of KMT2s in murine cancer models often show distinct phenotypes, suggesting that despite the shared substrate, KMT2 family genes probably have diverse and non-redundant functions in cancer. Future characterizing and modeling of KMT2 mutations in cancer are important to delineate whether they are drivers or passengers, gain- or loss- of function, dominant or recessive mutations. It is also important to differentiate the functions of KMT2 in malignant vs. pre-malignant cells.
Mechanistic studies on KMT2s show that these intrinsically different enzymes are tightly regulated by their interacting proteins, by proteolysis and by distinct chromatin recruitment mechanisms. Advances in understanding of how KMT2A and its oncogenic fusion proteins are regulated and their mode of actions have already led to identification of several novel therapeutic targets, which are now actively pursued for novel cancer therapies for MLL. We envision similar efforts to study other KMT2 genes in cancers will lead to novel discoveries that bear clinical implications.
Acknowledgments
We would like to thank Drs. Gregory Dresslor and Kai Ge for critical reading of the manuscript. We recognize that some important aspects of KMT2 biology, especially discoveries carried out in model organisms as well as KMT2A aberration in MLL, that we were unable to cover in this Review. We apologize to those that we have not been able to reference due to space constraint. The funding is provided by the US National Institutes of Health (GM082856 to Y.D. and EY022299 to R.C.R.), the Leukemia and Lymphoma Society (LLS), American Cancer Society (ACS) and Stand Up to Cancer to Y.D; Research to Prevent Blindness, and Knights Templar Eye Foundation to R.C.R.
GLOSSARY TERM
- H3K79me
Histone H3 lysine 79 methylation is deposited by histone methyltransferase DOT1L. H3K79me is commonly found in the transcribed gene coding regions and considered a histone mark for transcription elongation
- CpG dinucleotides
CpG dinucleotides (CpGs) refer to cytosine and guanine separated by one phosphate. The frequency of CpG dinucleotides in human genomes is extremely low (~1%) and most CpGs are methylated to form 5methyl-cytosine. Relatively high level of non-methylated CpGs is usually associated with promoters of actively transcribed genes
- AT-rich sequence
Higher frequency of adenosine and thymidine can be found in specific genomic regions such as transcription start sites (TATA box) or replication initiation sites in bacteria. It can be specifically bound by several protein motifs such as AT-hook, HMG motif as well as B-box zinc finger
References
- 1.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 2.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 3.Shilatifard A. Chromatin Modifications by Methylation and Ubiquitination: Implications in the Regulation of Gene Expression. Annu Rev Biochem. 2006 doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 4.Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol. 2011;12:799–814. doi: 10.1038/nrm3230. [DOI] [PubMed] [Google Scholar]
- 5.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 7.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 8.Liedtke M, Cleary ML. Therapeutic targeting of MLL. Blood. 2009;113:6061–6068. doi: 10.1182/blood-2008-12-197061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milne TA, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura T, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 11.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.FitzGerald KT, Diaz MO. MLL2: A new mammalian member of the trx/MLL family of genes. Genomics. 1999;59:187–192. doi: 10.1006/geno.1999.5860. [DOI] [PubMed] [Google Scholar]
- 15.Pijnappel WW, et al. The S. cerevisiae SET3 complex includes two histone deacetylases, Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev. 2001;15:2991–3004. doi: 10.1101/gad.207401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goo YH, et al. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol Cell Biol. 2003;23:140–149. doi: 10.1128/MCB.23.1.140-149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes CM, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 18.Cho YW, et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem. 2007;282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JH, Skalnik DG. CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem. 2005;280:41725–41731. doi: 10.1074/jbc.M508312200. [DOI] [PubMed] [Google Scholar]
- 20.Lee JH, Tate CM, You JS, Skalnik DG. Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J Biol Chem. 2007;282:13419–13428. doi: 10.1074/jbc.M609809200. [DOI] [PubMed] [Google Scholar]
- 21.Dou Y, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 22.Patel A, Dharmarajan V, Vought VE, Cosgrove MS. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J Biol Chem. 2009;284:24242–24256. doi: 10.1074/jbc.M109.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi YH, et al. Structural analysis of the core COMPASS family of histone H3K4 methylases from yeast to human. Proc Natl Acad Sci U S A. 2011;108:20526–20531. doi: 10.1073/pnas.1109360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel A, Dharmarajan V, Cosgrove MS. Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J Biol Chem. 2008;283:32158–32161. doi: 10.1074/jbc.C800164200. [DOI] [PubMed] [Google Scholar]
- 25.Dharmarajan V, Lee JH, Patel A, Skalnik DG, Cosgrove MS. Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferases. J Biol Chem. 2012;287:27275–27289. doi: 10.1074/jbc.M112.364125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Cao F, Wan B, Dou Y, Lei M. Structure of the SPRY domain of human Ash2L and its interactions with RbBP5 and DPY30. Cell Res. 2012;22:598–602. doi: 10.1038/cr.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, et al. Crystal structure of the N-terminal region of human Ash2L shows a winged-helix motif involved in DNA binding. EMBO Rep. 2011;12:797–803. doi: 10.1038/embor.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuetz A, et al. Structural basis for molecular recognition and presentation of histone H3 by WDR5. Embo J. 2006;25:4245–4252. doi: 10.1038/sj.emboj.7601316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Couture JF, Collazo E, Trievel RC. Molecular recognition of histone H3 by the WD40 protein WDR5. Nat Struct Mol Biol. 2006;13:698–703. doi: 10.1038/nsmb1116. [DOI] [PubMed] [Google Scholar]
- 30.Zhang P, Lee H, Brunzelle JS, Couture JF. The plasticity of WDR5 peptide-binding cleft enables the binding of the SET1 family of histone methyltransferases. Nucleic Acids Res. 2012;40:4237–4246. doi: 10.1093/nar/gkr1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarvan S, et al. Crystal structure of the trithorax group protein ASH2L reveals a forkhead-like DNA binding domain. Nat Struct Mol Biol. 2011;18:857–859. doi: 10.1038/nsmb.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol Cell. 2009;33:181–191. doi: 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 33.Steward MM, et al. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol. 2006;13:852–854. doi: 10.1038/nsmb1131. [DOI] [PubMed] [Google Scholar]
- 34.Cao F, et al. An Ash2L/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 SET domain. PLoS One. 2010;5:e14102. doi: 10.1371/journal.pone.0014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patel A, et al. Automethylation activities within the mixed lineage leukemia-1 (MLL1) core complex reveal evidence supporting a “two-active site” model for multiple histone H3 lysine 4 methylation. J Biol Chem. 2014;289:868–884. doi: 10.1074/jbc.M113.501064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song JJ, Kingston RE. WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J Biol Chem. 2008;283:35258–35264. doi: 10.1074/jbc.M806900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Avdic V, et al. Structural and biochemical insights into MLL1 core complex assembly. Structure. 2011;19:101–108. doi: 10.1016/j.str.2010.09.022. [DOI] [PubMed] [Google Scholar]
- 38.Patel A, Vought VE, Dharmarajan V, Cosgrove MS. A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J Biol Chem. 2008;283:32162–32175. doi: 10.1074/jbc.M806317200. [DOI] [PubMed] [Google Scholar]
- 39.Karatas H, Townsend EC, Bernard D, Dou Y, Wang S. Analysis of the binding of mixed lineage leukemia 1 (MLL1) and histone 3 peptides to WD repeat domain 5 (WDR5) for the design of inhibitors of the MLL1-WDR5 interaction. J Med Chem. 2010;53:5179–5185. doi: 10.1021/jm100139b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao F, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 2014;53:247–261. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karatas H, et al. High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J Am Chem Soc. 2013;135:669–682. doi: 10.1021/ja306028q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Senisterra G, et al. Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem J. 2013;449:151–159. doi: 10.1042/BJ20121280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsieh JJ, Cheng EH, Korsmeyer SJ. Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell. 2003;115:293–303. doi: 10.1016/s0092-8674(03)00816-x. S009286740300816X [pii] [DOI] [PubMed] [Google Scholar]
- 44.Yokoyama A, Kitabayashi I, Ayton PM, Cleary ML, Ohki M. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood. 2002;100:3710–3718. doi: 10.1182/blood-2002-04-1015. 2002-04-1015 [pii] [DOI] [PubMed] [Google Scholar]
- 45.Yokoyama A, et al. MLL becomes functional through intra-molecular interaction not by proteolytic processing. PLoS One. 2013;8:e73649. doi: 10.1371/journal.pone.0073649. PONE-D-13-22945 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu L, et al. ASH2L regulates ubiquitylation signaling to MLL: trans-regulation of H3 K4 methylation in higher eukaryotes. Mol Cell. 2013;49:1108–1120. doi: 10.1016/j.molcel.2013.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sebastian S, et al. MLL5, a trithorax homolog, indirectly regulates H3K4 methylation, represses cyclin A2 expression, and promotes myogenic differentiation. Proc Natl Acad Sci U S A. 2009;106:4719–4724. doi: 10.1073/pnas.0807136106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JE, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dhar SS, et al. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26:2749–2762. doi: 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nayak A, Viale-Bouroncle S, Morsczeck C, Muller S. The SUMO-specific isopeptidase SENP3 regulates MLL1/MLL2 methyltransferase complexes and controls osteogenic differentiation. Mol Cell. 2014;55:47–58. doi: 10.1016/j.molcel.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 51.van Nuland R, et al. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol Cell Biol. 2013;33:2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. S1535-6108(08)00158-X [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murai MJ, et al. The same site on the integrase-binding domain of lens epithelium-derived growth factor is a therapeutic target for MLL leukemia and HIV. Blood. 2014;124:3730–3737. doi: 10.1182/blood-2014-01-550079. blood-2014-01-550079 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mo R, Rao SM, Zhu YJ. Identification of the MLL2 complex as a coactivator for estrogen receptor alpha. J Biol Chem. 2006;281:15714–15720. doi: 10.1074/jbc.M513245200. [DOI] [PubMed] [Google Scholar]
- 55.Patel SR, Kim D, Levitan I, Dressler GR. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell. 2007;13:580–592. doi: 10.1016/j.devcel.2007.09.004. S1534-5807(07)00345-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tyagi S, Chabes AL, Wysocka J, Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell. 2007;27:107–119. doi: 10.1016/j.molcel.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 57.Dou Y, et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 58.Takeda S, et al. Proteolysis of MLL family proteins is essential for taspase1-orchestrated cell cycle progression. Genes Dev. 2006;20:2397–2409. doi: 10.1101/gad.1449406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang Z, et al. SET1 and p300 act synergistically, through coupled histone modifications, in transcriptional activation by p53. Cell. 2013;154:297–310. doi: 10.1016/j.cell.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee J, et al. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc Natl Acad Sci U S A. 2009;106:8513–8518. doi: 10.1073/pnas.0902873106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aziz A, Liu QC, Dilworth FJ. Regulating a master regulator: establishing tissue-specific gene expression in skeletal muscle. Epigenetics. 2010;5:691–695. doi: 10.4161/epi.5.8.13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawabe Y, Wang YX, McKinnell IW, Bedford MT, Rudnicki MA. Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell. 2012;11:333–345. doi: 10.1016/j.stem.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Demers C, et al. Activator-mediated recruitment of the MLL2 methyltransferase complex to the beta-globin locus. Mol Cell. 2007;27:573–584. doi: 10.1016/j.molcel.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tan CC, et al. Transcription factor Ap2delta associates with Ash2l and ALR, a trithorax family histone methyltransferase, to activate Hoxc8 transcription. Proc Natl Acad Sci U S A. 2008;105:7472–7477. doi: 10.1073/pnas.0711896105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fossati A, Dolfini D, Donati G, Mantovani R. NF-Y recruits Ash2L to impart H3K4 trimethylation on CCAAT promoters. PLoS One. 2011;6:e17220. doi: 10.1371/journal.pone.0017220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deng C, et al. USF1 and hSET1A mediated epigenetic modifications regulate lineage differentiation and HoxB4 transcription. PLoS Genet. 2013;9:e1003524. doi: 10.1371/journal.pgen.1003524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ang YS, et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell. 2011;145:183–197. doi: 10.1016/j.cell.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaikkonen MU, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guenther MG, et al. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A. 2005;102:8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Allen MD, et al. Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. Embo J. 2006;25:4503–4512. doi: 10.1038/sj.emboj.7601340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Z, et al. Pro isomerization in MLL1 PHD3-bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression. Cell. 2010;141:1183–1194. doi: 10.1016/j.cell.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Plevin MJ, Mills MM, Ikura M. The LxxLL motif: a multifunctional binding sequence in transcriptional regulation. Trends Biochem Sci. 2005;30:66–69. doi: 10.1016/j.tibs.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 74.Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol. 1999;19:5237–5246. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang YW, et al. Essential role of lncRNA binding for WDR5 maintenance of active chromatin and embryonic stem cell pluripotency. Elife. 2014;3:e02046. doi: 10.7554/eLife.02046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang KC, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bach C, Mueller D, Buhl S, Garcia-Cuellar MP, Slany RK. Alterations of the CxxC domain preclude oncogenic activation of mixed-lineage leukemia 2. Oncogene. 2009;28:815–823. doi: 10.1038/onc.2008.443. [DOI] [PubMed] [Google Scholar]
- 78.Wang P, et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Austenaa L, et al. The histone methyltransferase Wbp7 controls macrophage function through GPI glycolipid anchor synthesis. Immunity. 2012;36:572–585. doi: 10.1016/j.immuni.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 80.Guo C, et al. Global identification of MLL2-targeted loci reveals MLL2’s role in diverse signaling pathways. Proc Natl Acad Sci U S A. 2012;109:17603–17608. doi: 10.1073/pnas.1208807109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Blobel GA, et al. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol Cell. 2009;36:970–983. doi: 10.1016/j.molcel.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang H, et al. Regulation of transcription by the MLL2 complex and MLL complex-associated AKAP95. Nat Struct Mol Biol. 2013;20:1156–1163. doi: 10.1038/nsmb.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 84.Vermeulen M, Timmers HT. Grasping trimethylation of histone H3 at lysine 4. Epigenomics. 2010;2:395–406. doi: 10.2217/epi.10.11. [DOI] [PubMed] [Google Scholar]
- 85.Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leung A, et al. Histone H2B ubiquitylation and H3 lysine 4 methylation prevent ectopic silencing of euchromatic loci important for the cellular response to heat. Mol Biol Cell. 2011;22:2741–2753. doi: 10.1091/mbc.E11-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Auriemma LB, Shah S, Linden LM, Henriksen MA. Knockdown of menin affects pre-mRNA processing and promoter fidelity at the interferon-gamma inducible IRF1 gene. Epigenetics Chromatin. 2012;5:2. doi: 10.1186/1756-8935-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Dijk EL, et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 2011;475:114–117. doi: 10.1038/nature10118. [DOI] [PubMed] [Google Scholar]
- 90.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Buecker C, Wysocka J. Enhancers as information integration hubs in development: lessons from genomics. Trends Genet. 2012;28:276–284. doi: 10.1016/j.tig.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lara-Astiaso D, et al. Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014;345:943–949. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Akhtar-Zaidi B, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336:736–739. doi: 10.1126/science.1217277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Herz HM, Hu D, Shilatifard A. Enhancer malfunction in cancer. Mol Cell. 2014;53:859–866. doi: 10.1016/j.molcel.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ooi SK, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang Y, et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010;38:4246–4253. doi: 10.1093/nar/gkq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thomson JP, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–1086. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.203751125/10/1010. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Erfurth FE, et al. MLL protects CpG clusters from methylation within the Hoxa9 gene, maintaining transcript expression. Proc Natl Acad Sci U S A. 2008;105:7517–7522. doi: 10.1073/pnas.0800090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Risner LE, et al. Functional specificity of CpG DNA-binding CXXC domains in mixed lineage leukemia. J Biol Chem. 2013;288:29901–29910. doi: 10.1074/jbc.M113.474858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cierpicki T, et al. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat Struct Mol Biol. 2010;17:62–68. doi: 10.1038/nsmb.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Clouaire T, et al. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26:1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Deplus R, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. Embo J. 2013;32:645–655. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Petruk S, et al. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell. 2012;150:922–933. doi: 10.1016/j.cell.2012.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang J, Muntean AG, Wu L, Hess JL. A subset of mixed lineage leukemia proteins has plant homeodomain (PHD)-mediated E3 ligase activity. J Biol Chem. 2012;287:43410–43416. doi: 10.1074/jbc.M112.423855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang J, Muntean AG, Hess JL. ECSASB2 mediates MLL degradation during hematopoietic differentiation. Blood. 2012;119:1151–1161. doi: 10.1182/blood-2011-06-362079. blood-2011-06-362079 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu H, Cheng EH, Hsieh JJ. Bimodal degradation of MLL by SCFSkp2 and APCCdc20 assures cell cycle execution: a critical regulatory circuit lost in leukemogenic MLL fusions. Genes Dev. 2007;21:2385–2398. doi: 10.1101/gad.1574507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu H, et al. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature. 2010;467:343–346. doi: 10.1038/nature09350. nature09350 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Soares LM, Radman-Livaja M, Lin SG, Rando OJ, Buratowski S. Feedback control of Set1 protein levels is important for proper H3K4 methylation patterns. Cell Rep. 2014;6:961–972. doi: 10.1016/j.celrep.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ayton PM, Cleary ML. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene. 2001;20:5695–5707. doi: 10.1038/sj.onc.1204639. [DOI] [PubMed] [Google Scholar]
- 112.Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 113.Gui Y, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kandoth C, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jones WD, et al. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am J Hum Genet. 2012;91:358–364. doi: 10.1016/j.ajhg.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ansari KI, Kasiri S, Mandal SS. Histone methylase MLL1 has critical roles in tumor growth and angiogenesis and its knockdown suppresses tumor growth in vivo. Oncogene. 2013;32:3359–3370. doi: 10.1038/onc.2012.352. onc2012352 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Takeda S, et al. HGF-MET signals via the MLL-ETS2 complex in hepatocellular carcinoma. J Clin Invest. 2013;123:3154–3165. doi: 10.1172/JCI65566. 65566 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Thiel AT, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer cell. 2010;17:148–159. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sierra J, Yoshida T, Joazeiro CA, Jones KA. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 2006;20:586–600. doi: 10.1101/gad.1385806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Milne TA, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Xia ZB, et al. The MLL fusion gene, MLL-AF4, regulates cyclin-dependent kinase inhibitor CDKN1B (p27kip1) expression. Proc Natl Acad Sci U S A. 2005;102:14028–14033. doi: 10.1073/pnas.0506464102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. S0092-8674(11)00127-9 [pii] [DOI] [PubMed] [Google Scholar]
- 123.Glaser S, et al. Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development. 2006;133:1423–1432. doi: 10.1242/dev.02302. [DOI] [PubMed] [Google Scholar]
- 124.Glaser S, et al. The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenetics Chromatin. 2009;2:5. doi: 10.1186/1756-8935-2-5. 1756-8935-2-5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Andreu-Vieyra CV, et al. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000453. e1000453 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kerimoglu C, et al. Histone-methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J Neurosci. 2013;33:3452–3464. doi: 10.1523/JNEUROSCI.3356-12.2013. 33/8/3452 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lubitz S, Glaser S, Schaft J, Stewart AF, Anastassiadis K. Increased apoptosis and skewed differentiation in mouse embryonic stem cells lacking the histone methyltransferase Mll2. Mol Biol Cell. 2007;18:2356–2366. doi: 10.1091/mbc.E06-11-1060. E06-11-1060 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Peifer M, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 130.Morin RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Parsons DW, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kanda H, Nguyen A, Chen L, Okano H, Hariharan IK. The Drosophila ortholog of MLL3 and MLL4, trithorax related, functions as a negative regulator of tissue growth. Mol Cell Biol. 2013;33:1702–1710. doi: 10.1128/MCB.01585-12. MCB.01585-12 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen C, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014;25:652–665. doi: 10.1016/j.ccr.2014.03.016. S1535-6108(14)00124-X [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Santos MA, et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature. 2014;514:107–111. doi: 10.1038/nature13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. nature12912 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ansari KI, Hussain I, Kasiri S, Mandal SS. HOXC10 is overexpressed in breast cancer and transcriptionally regulated by estrogen via involvement of histone methylases MLL3 and MLL4. J Mol Endocrinol. 2012;48:61–75. doi: 10.1530/JME-11-0078. JME-11-0078 [pii] [DOI] [PubMed] [Google Scholar]
- 137.Kim JH, et al. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014;74:1705–1717. doi: 10.1158/0008-5472.CAN-13-1896. 0008-5472.CAN-13-1896 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Guo C, et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget. 2013;4:2144–2153. doi: 10.18632/oncotarget.1555. 1555 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Corradin O, et al. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 2014;24:1–13. doi: 10.1101/gr.164079.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hippisley-Cox J, Vinogradova Y, Coupland C, Parker C. Risk of malignancy in patients with schizophrenia or bipolar disorder: nested case-control study. Arch Gen Psychiatry. 2007;64:1368–1376. doi: 10.1001/archpsyc.64.12.1368. 64/12/1368 [pii] [DOI] [PubMed] [Google Scholar]
- 141.Takata A, et al. Loss-of-function variants in schizophrenia risk and SETD1A as a candidate susceptibility gene. Neuron. 2014;82:773–780. doi: 10.1016/j.neuron.2014.04.043. S0896-6273(14)00358-4 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bernt KM, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. S1535-6108(11)00228-5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Daigle SR, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. S1535-6108(11)00227-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Deshpande AJ, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell. 2014;26:896–908. doi: 10.1016/j.ccell.2014.10.009. S1535-6108(14)00413-9 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lin C, et al. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell. 2010;17:198–212. doi: 10.1016/j.ccr.2009.12.040. S1535-6108(10)00003-6 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang Z, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 148.Jang MK, et al. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 149.Zuber J, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mueller D, et al. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS biology. 2009;7:e1000249. doi: 10.1371/journal.pbio.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dawson MA, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liu H, et al. Proteasome inhibitors evoke latent tumor suppression programs in pro-B MLL leukemias through MLL-AF4. Cancer Cell. 2014;25:530–542. doi: 10.1016/j.ccr.2014.03.008. S1535-6108(14)00117-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yokoyama A, et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 155.Grembecka J, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8:277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Scacheri PC, et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet. 2006;2:e51. doi: 10.1371/journal.pgen.0020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mishra BP, et al. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep. 2014;7:1239–1247. doi: 10.1016/j.celrep.2014.04.015. S2211-1247(14)00302-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.So CW, Karsunky H, Wong P, Weissman IL, Cleary ML. Leukemic transformation of hematopoietic progenitors by MLL-GAS7 in the absence of Hoxa7 or Hoxa9. Blood. 2004;103:3192–3199. doi: 10.1182/blood-2003-10-3722. 2003-10-3722 [pii] [DOI] [PubMed] [Google Scholar]
- 159.Kumar AR, et al. Hoxa9 influences the phenotype but not the incidence of Mll-AF9 fusion gene leukemia. Blood. 2004;103:1823–1828. doi: 10.1182/blood-2003-07-2582. [DOI] [PubMed] [Google Scholar]
- 160.Ohyashiki K, Kocova M, Ryan DH, Rowe JM, Sandberg AA. Secondary acute myeloblastic leukemia with a Ph translocation in a treated Wegener’s granulomatosis. Cancer Genet Cytogenet. 1986;19:331–333. doi: 10.1016/0165-4608(86)90062-2. [DOI] [PubMed] [Google Scholar]
- 161.Bledau AS, et al. The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development. 2014;141:1022–1035. doi: 10.1242/dev.098152. [DOI] [PubMed] [Google Scholar]
- 162.Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 163.Hess JL, Yu BD, Li B, Hanson R, Korsmeyer SJ. Defects in yolk sac hematopoiesis in Mll-null embryos. Blood. 1997;90:1799–1806. [PubMed] [Google Scholar]
- 164.McMahon KA, et al. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell. 2007;1:338–345. doi: 10.1016/j.stem.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 165.Lee J, et al. Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc Natl Acad Sci U S A. 2008;105:19229–19234. doi: 10.1073/pnas.0810100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Terranova R, Agherbi H, Boned A, Meresse S, Djabali M. Histone and DNA methylation defects at Hox genes in mice expressing a SET domain-truncated form of Mll. Proc Natl Acad Sci U S A. 2006;103:6629–6634. doi: 10.1073/pnas.0507425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Goldsworthy M, et al. Mutations in Mll2, an H3K4 methyltransferase, result in insulin resistance and impaired glucose tolerance in mice. PLoS One. 2013;8:e61870. doi: 10.1371/journal.pone.0061870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jude CD, et al. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1:324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mueller D, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110:4445–4454. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Milne TA, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell. 2010;38:853–863. doi: 10.1016/j.molcel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Muntean AG, et al. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer cell. 2010;17:609–621. doi: 10.1016/j.ccr.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Krivtsov AV, et al. Global increase in H3K79 dimethylation in murine and human MLL-AF4 lymphoblastic leukemias. Blood. 2007;110:108A–108A. [Google Scholar]
- 173.Jo SY, Granowicz EM, Maillard I, Thomas D, Hess JL. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood. 2011;117:4759–4768. doi: 10.1182/blood-2010-12-327668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tan J, et al. CBX8, a polycomb group protein, is essential for MLL-AF9-induced leukemogenesis. Cancer cell. 2011;20:563–575. doi: 10.1016/j.ccr.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bursen A, et al. The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood. 2010;115:3570–3579. doi: 10.1182/blood-2009-06-229542. [DOI] [PubMed] [Google Scholar]
- 176.Benedikt A, et al. The leukemogenic AF4-MLL fusion protein causes P-TEFb kinase activation and altered epigenetic signatures. Leukemia. 2011;25:135–144. doi: 10.1038/leu.2010.249. [DOI] [PubMed] [Google Scholar]
- 177.Chen W, et al. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer cell. 2008;13:432–440. doi: 10.1016/j.ccr.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Krivtsov AV, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 179.Corral J, et al. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell. 1996;85:853–861. doi: 10.1016/s0092-8674(00)81269-6. [DOI] [PubMed] [Google Scholar]
- 180.Collins EC, Pannell R, Simpson EM, Forster A, Rabbitts TH. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-loxP in mouse development. EMBO Rep. 2000;1:127–132. doi: 10.1038/sj.embor.embor616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ng SB, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. ng.646 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bjornsson HT, et al. Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci Transl Med. 2014;6:256ra135. doi: 10.1126/scitranslmed.3009278. 6/256/256ra135 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Huang PH, et al. Histone deacetylase inhibitors stimulate histone H3 lysine 4 methylation in part via transcriptional repression of histone H3 lysine 4 demethylases. Mol Pharmacol. 2011;79:197–206. doi: 10.1124/mol.110.067702. mol.110.067702 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Berdasco M, Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet. 2013;132:359–383. doi: 10.1007/s00439-013-1271-x. [DOI] [PubMed] [Google Scholar]
- 185.Shinsky SA, et al. A non-active-site SET domain surface crucial for the interaction of MLL1 and the RbBP5/Ash2L heterodimer within MLL family core complexes. J Mol Biol. 2014;426:2283–2299. doi: 10.1016/j.jmb.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kleefstra T, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet. 2012;91:73–82. doi: 10.1016/j.ajhg.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.De Rubeis S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014 doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. S0140-6736(09)60995-8 [pii] [DOI] [PubMed] [Google Scholar]
- 189.Zeleznik-Le NJ, Harden AM, Rowley JD. 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc Natl Acad Sci U S A. 1994;91:10610–10614. doi: 10.1073/pnas.91.22.10610. [DOI] [PMC free article] [PubMed] [Google Scholar]