

Abstract
Recent studies have shown that nuclear transcription factor cyclic adenosine monophosphate response element binding protein (CREB) is overexpressed in many different types of cancers. Therefore, CREB has been pursued as a novel cancer therapeutic target. Naphthol AS-E and its closely related derivatives have been shown to inhibit CREB-mediated gene transcription and cancer cell growth. Previously, we identified naphthamide 3a as a different chemotype to inhibit CREB’s transcription activity. In a continuing effort to discover more potent CREB inhibitors, a series of structural congeners of 3a was designed and synthesized. Biological evaluations of these compounds uncovered compound 3i (666-15) as a potent and selective inhibitor of CREB-mediated gene transcription (IC50 = 0.081 ± 0.04 μM). 666-15 also potently inhibited cancer cell growth without harming normal cells. In an in vivo MDA-MB-468 xenograft model, 666-15 completely suppressed the tumor growth without overt toxicity. These results further support the potential of CREB as a valuable cancer drug target.
Introduction
The cAMP-response element binding protein (CREB) is a nuclear transcription factor that can be activated to initiate gene transcription in response to hormones, growth factors, and neuronal activity.1,2 These stimuli activate intracellular protein serine/threonine kinases such as mitogen-activated protein kinase (MAPK), protein kinase A (PKA), protein kinase B (PKB/Akt), and p90 ribosomal S6 kinase (p90RSK).3 All these kinases have been shown to be able to phosphorylate Ser133 in CREB.1,3 Phosphorylation at Ser133 is crucial in CREB’s binding with histone acetyl transferase and mammalian transcription coactivator CREB-binding protein (CBP) and its paralog p300 to initiate CREB-dependent gene transcription. The binding interaction between CREB and CBP/p300 is mediated by the activation domain in CREB called kinase-inducible domain (KID) and KID-interacting (KIX) domain in CBP/p300.4 Three protein phosphatases, protein phosphatase 1 (PP1),5 protein phosphatase 2A (PP2A),6 and phosphatase and tensin homolog (PTEN),7 have been shown to dephosphorylate Ser133 in phosphorylated CREB to turn off CREB-dependent gene transcription.
The protein kinases leading to CREB activation are frequently overactivated, while the three phosphatases to dephosphorylate CREB are often inactivated in various cancer cells. Therefore, it was predicted that CREB would be overactivated in cancer cells. Consistent with this prediction, CREB and phosphorylated CREB have been consistently shown to be overexpressed in cancer tissues from brain,8,9 breast,10,11 lung,12 prostate,13 and bone marrow.14 Because of its aberrant activation in cancer cells, CREB has been pursued as a novel cancer therapeutic target.3 We recently identified naphthol AS-E (1, Figure 1) as a cell-permeable inhibitor of CREB-mediated gene transcription through inhibiting KID-KIX interaction,15 the essential protein–protein interaction to activate CREB-dependent gene transcription.4 Consistent with the important roles of CREB in the maintenance of cancer cells, we found that 1 and its close related derivatives selectively inhibited proliferation of a large panel of cancer cell lines from different organs in the low micromolar concentration range without harming normal cells in vitro.16
Figure 1.

Chemical structures of previously reported CREB inhibitors: naphthol AS-E (1) and compounds 2 and 3a. Compound 2 is rapidly transformed into 3a through an O,N-acyl transfer reaction at pH 7.4.
During our course of studies to improve the aqueous solubility and biological activity of 1, we designed and synthesized compound 2 (Figure 1). Compound 2 presented significantly improved antiproliferative activity against a panel of different cancer cells.17 Unexpectedly, we found that 2 was rapidly converted into 3a under physiological conditions and was considered as a prodrug of 3a, where a long-range O,N-acyl transfer reaction was involved (Figure 1).17 While 2 displayed in vivo antibreast cancer activity, its CREB inhibition potency remained modest.17 In this report, we detail our optimization of 3a and identification of 3i (666-15) as a potent CREB inhibitor with highly efficacious in vivo antibreast cancer activity.
Results and Discussion
Analog Design Rationale
A series of structural congeners of 3a shown in Figure 2 was designed to improve its biological activities and physicochemical properties. Compound 3a contains a phenolic hydroxyl group that is a potential site for glucuronidation, which would limit its metabolic stability and bioavailability.18−20 To test if this potential metabolic liability can be removed without compromising bioactivity, compound 3b was designed to interrogate the role of the phenol group in 3a in contributing to its bioactivity. Compound 3a also has relatively high polar surface area (PSA, 123.2 Å2) and high cLogP (5.30) (Table 1). To improve these two physicochemical parameters, compounds 3c,d were designed by removing one of the conjugated planar naphthyl rings. Truncating one of the naphthyl ring systems into a benzene system decreases the PSA to ∼98 Å2 and cLogP to ∼4.9 (Table 1). Compounds 3e–g were designed to probe the role of the primary amino group in 3a. If this primary amino group tolerates structural changes, additional functional groups may be attached to the primary amino group. Analogs 3h–j were designed by varying the lengths of the linker and side chain to understand their roles in biological activities. As presented in Table 1, compounds 3e–j show decreased PSA and 3g–i also present decreased cLogP compared to 3a.
Figure 2.
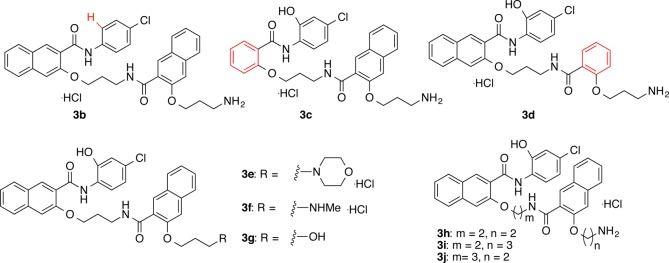
Chemical structures of newly designed structural congeners (3b–j) of 3a. The key structural difference between 3b–d and 3a is highlighted in red.
Table 1. Physiochemical Properties and CREB Inhibition Activity of 3a–j.
| compd | PSAa (Å2) | cLogPa | CREB inhibition IC50 (μM)b |
|---|---|---|---|
| 3a | 123.2 | 5.30 | 2.22 ± 0.38 |
| 3b | 86.9 | 6.41 | 4.69 ± 1.28 |
| 3c | 98.0 | 4.91 | 10.05 ± 2.29 |
| 3d | 97.9 | 4.92 | 5.30 ± 1.41 |
| 3e | 96.1 | 5.42 | >50 |
| 3f | 92.45 | 5.87 | 18.53 ± 8.68 |
| 3g | 105.8 | 5.26 | 7.30 ± 1.66 |
| 3h | 100.2 | 4.43 | 0.30 ± 0.12 |
| 3i | 100.2 | 4.83 | 0.081 ± 0.04 |
| 3j | 100.0 | 5.39 | 5.23 ± 0.36 |
The polar surface area (PSA) and calculated log P (cLogP) values were computed from their global energy minima using QikProp.
CREB inhibition refers to inhibition of CREB-mediated gene transcription in HEK 293T cells using a CREB reporter assay. The IC50 was presented as the mean ± SD of at least two independent experiments in triplicate or >50 in the cases where the IC50 was not reached at the highest tested concentration (50 μM).
Chemistry
The synthesis of compounds 3b–j is presented in Schemes 1–7 and is overall similar to the synthesis of 3a as described before.17 All the final products were prepared in good to excellent yields. This synthesis of 3b is shown in Scheme 1. Mitsunobu coupling (Ph3P/DEAD)21 between 1 and Boc-protected 3-amino-1-propanol (A1) gave 6b, whose Boc protecting group was removed under acidic condition to generate free base 7b after neutralization with NaHCO3. Amide formation between amine 7b and previously reported acid 5a(17) under the BOP/DIPEA coupling condition yielded amide 8b. Deprotection of Boc in 8b with 2 N HCl delivered product 3b. Compound 3c was prepared in a similar fashion with the exception of a need for 7c as the key intermediate (Scheme 2). The commercially available starting materials methyl salicylate (1c) and A1 were coupled together under Mitsunobu reaction condition. Saponification of methyl ester 4c generated acid 5c, which was then coupled with aniline 9 to yield 6c with MsCl as the activating reagent.22 The activating reagent MsCl was found to be superior to BOP in achieving high selectivity for forming desired amide versus the alternative undesired ester.17 Removal of the Boc group from 6c provided amine 7c, which was further coupled with acid 5a followed by acidic deprotection of Boc to give desired product 3c. The activating reagent MsCl was again found to be superior to BOP in achieving high selectivity for forming desired amide versus the alternative ester and was selected for all the subsequent amide formation reactions. Compound 3d was prepared in two steps by coupling between amine 7a(17) and acid 5c followed by Boc deprotection (Scheme 3).
Scheme 1. Synthesis of Compound 3b.

Scheme 7. Synthesis of Compound 3j.

Scheme 2. Synthesis of Compound 3c.

Scheme 3. Synthesis of Compound 3d.

The preparation of morpholine substituted compound 3e is shown in Scheme 4. The morpholine side chain was incorporated into 1a(23) by Mitsunobu reaction with A2. Saponification of ester 4e provided acid 5e, which was then coupled with amine 7a by MsCl/Et3N to afford amide 8e. Treatment of 8e with HCl yielded corresponding hydrochloride salt 3e. A similar sequence of reactions was employed to prepare 3f and 3g. The side chain in 4g was installed by O-alkylation of naphthol 1a, while the N-methyl group in 4f was introduced by methylation of carbamate 4a by NaH/MeI.24
Scheme 4. Synthesis of Compounds 3e–g.

Compounds 3h–j having different linker and side chain lengths were synthesized as shown in Schemes 5–7. Intermediate 7h was prepared essentially the same as that for 7a(17) with the use of A4 as the Mitsunobu coupling partner followed by saponification, amide formation, and Boc deprotection. Amide coupling between the amine 7h and acid 5h generated amide 8h, whose Boc was removed under acidic condition to provide 3h. Intermidiates 8i and 8j were prepared by assembling building blocks 5a and 7h, 5h and 7a, respectively (Schemes 6 and 7). Final deprotection of Boc in 8i and 8j delivered desired compounds 3i and 3j uneventfully.
Scheme 5. Synthesis of Compound 3h.

Scheme 6. Synthesis of Compound 3i.

Inhibition of CREB-Mediated Gene Transcription by 3b–j
The newly synthesized final compounds 3b–j were evaluated for their activity in inhibiting CREB-mediated gene transcription in HEK 293T cells using a CREB Renilla luciferase (RLuc) reporter assay.15 In this assay, HEK 293T cells were transfected with a RLuc reporter under the control of a synthetic CREB promoter containing three copies of cAMP-response elements (CRE). The transfected cells were then treated with different concentrations of compounds for 30 min before the induction of RLuc synthesis by forskolin (10 μM), an activator of adenylate cyclase to activate CREB’s transcription activity.25 The results from this CREB reporter assay are summarized in Table 1, where the concentrations required to inhibit 50% of CREB’s transcription activity (IC50) are shown. For comparison purpose, the potency of previously reported compound 3a (IC50 = 2.22 μM) was also included in Table 1.17
In comparison to 3a, compound 3b without the phenol group showed about 2-fold decrease of activity in CREB inhibition (IC50 = 4.69 μM), indicating that the phenol group in 3a has a beneficial effect on CREB inhibition. Therefore, the rest of the compounds were designed to retain this crucial phenol group. Compounds 3c and 3d, with one of the naphthyl rings being trimmed down to a benzene ring, displayed approximately 2- to 5-fold less potent CREB inhibition activity than 3a, suggesting that the two naphthyl rings could not be simplified to phenyl rings without compromising CREB inhibitory activity. Replacement of the primary amino group in 3a with morpholine (3e), N-methylamino (3f), or hydroxyl (3g) group resulted in a total loss of CREB inhibition activity for 3e (IC50 > 50 μM) and significant decrease in CREB inhibition activity for 3f (IC50 = 18.53 μM) and 3g (IC50 = 7.30 μM). These data indicated that the primary amino group at the side chain of 3a is critical for maintaining CREB inhibition activity and not suitable for even minor modifications like methylation.
We then focused on the modification of the lengths of the linker between the two naphthyl rings and the side chain in compound 3a to interrogate their roles in CREB inhibition activity. Specifically, compounds 3h–j with two- or three-carbon chains were designed and synthesized. Gratifyingly, compound 3h with a two-carbon linker and a two-carbon side chain showed significantly increased CREB inhibition activity (IC50 = 0.30 μM) compared to 3a. Furthermore, compound 3i with a two-carbon linker and a three-carbon side chain exhibited even more potent CREB inhibition activity (IC50 = 81 nM), which is approximately 28-fold improvement over 3a. The enhancement of CREB inhibition activity seen for 3i is very structure-specific because its structural isomer 3j, which has a three-carbon linker and two-carbon side chain, was a much weaker CREB inhibitor with an IC50 of 5.23 μM. The CREB inhibitory potency difference among 3a and 3h–j demonstrated that the length of the linker between the two naphthyl rings can dramatically affect the activity and the length of the side chain also has a critical role on the CREB inhibition activity.
Inhibition of Cancer Cell Proliferation by 3b–j
We also evaluated the antiproliferative activity of compounds 3b–j in four different cancer cell lines: A549 (non-small-cell lung cancer), MCF-7 (breast cancer), MDA-MB-231 (breast cancer), and MDA-MB-468 (breast cancer) using the colorimetric MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay.16,26 The concentrations required to inhibit 50% of the cancer cell growth (GI50) are presented in Table 2. As reported before,17 compound 3a was a submicromolar inhibitor of proliferation of all the four cancer cell lines tested. The analogs 3b–e all presented less potent antiproliferative activity than 3a, in agreement with their reduced CREB inhibition potency. Although 3e was inactive in the CREB reporter assay, it showed weak growth inhibition activity in MDA-MB-468 cells (GI50 = 13.74 μM) but no activity in the other three cancer cell lines. It is unlikely that the weak activity in MDA-MB-468 cells was a result of inhibition of CREB’s transcription activity. Similar discrepancy was also observed for 3f, which exhibited weak CREB inhibition activity while displaying robust antiproliferative activity in these four cancer cell lines. The GI50 for 3f is 0.26, 1.65, 0.26, and 0.20 μM in A549, MCF-7, MDA-MB-231, and MDA-MB-468 cells, respectively. Compound 3g displayed modest CREB inhibition activity, but it was a rather weak inhibitor of proliferation in all four cancer cell lines tested with GI50 values ranging from 10.19 to 82.67 μM. Finally, in the series of compounds 3h–j with different lengths of the linker and side chain, we observed that potent CREB inhibitor 3i also potently inhibited cancer cell growth. In MDA-MB-231 and MDA-MB-468 cells, the GI50 for 3i was 73 and 46 nM, respectively. In A549 and MCF-7 cells, it exhibited robust activity as well with GI50 of 0.47 and 0.31 μM. Compared to 3i, 3h retained reasonable CREB inhibition activity and inhibition of cancer cell growth while 3j was much less potent. Therefore, compound 3i represents the most potent CREB inhibitor bearing potent anticancer activity reported to date.15−17,27,28
Table 2. Antiproliferative Activities of 3a–ja.
| GI50 (μM) |
||||
|---|---|---|---|---|
| compd | A549 | MCF-7 | MDA-MB-231 | MDA-MB-468 |
| 3a | 0.29 ± 0.01 | 0.14 ± 0.03 | 0.37 ± 0.13 | 0.22 ± 0.07 |
| 3b | 0.95 ± 0.77 | 1.72 | 1.00 ± 0.58 | 1.62 ± 0.48 |
| 3c | 2.44 ± 0.40 | 2.30 ± 0.67 | 4.61 ± 2.20 | 1.82 ± 0.20 |
| 3d | 2.32 ± 0.35 | 2.66 ± 1.30 | 5.82 ± 4.61 | 2.32 ± 0.39 |
| 3e | >100 | >100 | >100 | 13.74 ± 3.98 |
| 3f | 0.26 ± 0.06 | 1.65 ± 0.44 | 0.26 ± 0.04 | 0.20 ± 0.02 |
| 3g | 39.15 ± 32.09 | 28.99 ± 10.27 | 82.67 ± 24.52 | 10.19 ± 1.47 |
| 3h | 1.16 ± 0.05 | 0.81 ± 0.78 | 1.21 ± 0.15 | 0.25 ± 0.01 |
| 3i | 0.47 ± 0.06 | 0.31 ± 0.10 | 0.073 ± 0.04 | 0.046 ± 0.04 |
| 3j | 2.17 ± 0.11 | 1.89 ± 0.60 | 2.71 ± 0.32 | 1.85 ± 0.23 |
GI50 is the concentration required to inhibit the cancer cell growth by 50% as evaluated by the MTT assay. The compounds were incubated with cells for 72 h. The GI50 was presented as mean ± SD of at least two independent experiments in duplicates or >100 in the cases where GI50 was not reached at the highest tested concentration (100 μM). When SD was not presented, only one experiment was performed in duplicate.
Previously, it was shown that 3a only weakly inhibited CREB-CBP interaction (IC50 = 19.72 ± 1.78 μM) as assayed by a split RLuc complementation assay.17 We also investigated if the more potent CREB inhibitor 3i could inhibit CREB-CBP interaction using the same assay. It was also found to be a rather weak inhibitor of CREB-CBP interaction with IC50 = 18.27 ± 2.81 μM. We conclude that 3i inhibits CREB’s transcription activity in living cells independent of direct CREB or CBP binding interaction. Further studies are needed to understand if 3i will modulate the upstream components of CREB activation.3 Or alternatively, an unbiased chemoproteomics approach29 may be utilized to identify the direct target of 3i to understand its mechanism of inhibiting CREB-mediated gene transcription.
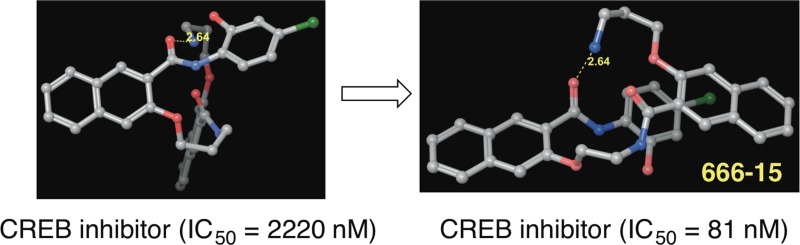
The results presented above showed that the bioactivities of 3a are very sensitive to structural modifications to either increase or decrease its activity. The physicochemical property parameters like PSA30 and cLogP31 that are associated with cell membrane permeability do not seem to be the major determinants. For example, compounds 3h–j bear similar PSA, but their bioactivities do not correlate with their cLogP (Table 1). To identify the structural basis for the observed bioactivity differences among 3a and 3h–j, we performed conformational searches to identify their global conformational minima using MacroModel. The conformational ensemble was generated by systematically rotating all the rotatable bonds in 3a and 3h–j. The identified global conformational minima are shown in Figure 3. All the four compounds form an intramolecular hydrogen bond between the protonated ammonium nitrogen and amide carbonyl oxygen. However, the more potent CREB inhibitors 3h and 3i adopt a more compact conformation by forming π–π stacking interaction between one of the naphthyl rings and chlorophenyl ring (Figure 3). On the other hand, the same naphthyl ring in the less potent compounds 3a and 3j do not form π–π stacking interaction with the chlorophenyl ring by assuming a more extended conformation at their global minima. These differences suggest that the unique conformation associated with 3h and 3i may contribute to their potent CREB inhibitory activity and antiproliferative activity.
Figure 3.

Conformation of the global energy minimum of 3a (A), 3j (B), 3h (C), and 3i (D). The distance between the amide oxygen and ammonium nitrogen was labeled in each conformation to indicate formation of an intramolecular hydrogen bond.
Compound 3i Selectively Inhibited CREB-Mediated Gene Transcription
In the CREB RLuc reporter assay with transfected HEK 293T cells, compound 3i was very potent in inhibiting CREB’s transcription activity. In order to investigate if 3i also inhibited endogenous CREB target gene expression, the transcript level of nuclear receptor related 1 protein (Nurr1/NR4A2), a well-defined CREB target gene in HEK 293T cells, was evaluated.17,32 The cells were treated with 3i followed by stimulation with forskolin (10 μM). Then the relative mRNA of Nurr1/NR4A2 was determined by quantitative reverse transcription polymerase chain reaction (qRT-PCR). As shown in Figure 4, forskolin robustly stimulated Nurr1/NR4A2 level to ∼31-fold. 3i dose-dependently inhibited transcription of Nurr1/NR4A2. Significant inhibition was observed even at 50 nM of 3i. In contrast, the weaker CREB inhibitor 3a only started to show significant inhibition at 1000 nM (Figure S1 in Supporting Information). These results are consistent with those from the CREB reporter assay.
Figure 4.

Compound 3i decreased endogenous CREB target gene expression. HEK 293T cells were treated with different concentrations of 3i followed by treatment with forskolin. Then the relative mRNA level of Nurr1/NR4A2 was determined by qRT-PCR analysis.
To investigate 3i’s selectivity on different transcription activators, we employed RLuc reporter assays to monitor individual transcription factor activity in HEK 293T cells. VP16-CREB is a fusion protein by fusing the potent activation domain VP16 to the full-length CREB. It requires CREB-CRE interaction for transcriptional activation, but it is a constitutively active transcription factor independent of phosphorylation as opposed to wild type CREB.33 As shown in Table 1 and Figure 5, 3i potently (IC50 = 81 nM) and efficaciously inhibited CREB’s transcription activity in HEK 293T cells. On the other hand, it showed much less efficacious inhibition of VP16-CREB and p53-mediated gene transcription. And even this weak inhibition only occurred at high concentrations (>1 μM). In a separate transcription reporter assay with NF-κB, much higher concentrations of 3i were required to inhibit NF-κB-mediated gene transcription (IC50 = 5290 nM, Figure S2), which is distinct from 1 and its phosphate.32 Collectively, these results indicate that 3i selectively inhibited CREB-mediated gene transcription.
Figure 5.

Compound 3i selectively inhibited CREB-mediated gene transcription. HEK 293T cells were transfected with indicated combinations of plasmids. Then the cells were treated with different concentrations of 3i before RLuc activity measurement. Forskolin (Fsk, 10 μM) was added to CRE-Rluc only transfected cells at 30 min after drug treatment to stimulate CREB’s activity. The RLuc activity was normalized to the protein concentration and presented as relative luciferase unit (RLU)/μg protein.
Compound 3i Selectively Inhibited the Growth of Cancer Cells but Not Normal Cells
With a potent and specific CREB inhibitor 3i in hand, we tested if it was toxic to normal cells. Previous genetic studies have shown that normal cells tolerate well with reduced levels of CREB.34,35 As shown in Table 2 and Figure 6A,B, 3i potently inhibited growth of MDA-MB-231 and MDA-MB-468 cells with GI50 in the midnanomolar concentration range. On the other hand, no significant inhibition of growth was observed in two different normal cell lines, human mammary epithelial cells (HMEC) and human foreskin fibroblasts (HFF), up to 1 μM concentration, which is more than 10-fold higher than its GI50 in MDA-MB-231 and MDA-MB-468 breast cancer cells. This selective toxicity is in strong contrast to conventional chemotherapeutics like doxorubicin, which did not show differential toxicity between cancer and normal cells under the same assay conditions.36 Therefore, pharmacological inhibition of CREB’s transcription activity is well tolerated in normal cells, which is consistent with the idea of cancer cells’ addiction to CREB.3,37,38
Figure 6.

Compound 3i selectively inhibited tumor cell growth. Shown are antiproliferative dose–response curves of 3i in breast cancer MDA-MB-468 (A) and MDA-MB-231 (B) cells as well as normal HMEC (C) and HFF (D) cells. The cells were incubated with 3i for 72 h, and then the remaining live cells were quantified by the MTT assay.
Compound 3i Completely Suppressed the Tumor Growth in Vivo
The selective in vitro toxicity of 3i against cancer cells versus normal cells prompted us to investigate its in vivo antitumor activity. Preliminary toxicity studies showed that intraperitoneal (ip) injection of 10 mg/kg of 3i is well tolerated in mice (Figure S3). This dose was chosen for in vivo antitumor efficacy studies in the MDA-MB-468 xenografts. The MDA-MB-468 tumor was allowed to grow to an average size of 100 mm3 in nude mice. Then the mice were randomized to receive either vehicle or 3i at 10 mg/kg once a day, 5 days per week for 5 weeks by ip injection. The tumor volumes and body weights were measured 2–3 times/week. The data in Figure 7A showed the tumor growth in the mice treated with 3i was efficaciously inhibited with complete tumor stasis. During the same period, the tumor volume in the vehicle-treated group was more than tripled (Figure 7A). The body weights of 3i-treated animals and vehicle-treated ones were indistinguishable from each other during the entire treatment period (Figure 7B), indicating no overt toxicity with this compound treatment. These results are consistent with in vitro studies with compound 3i (Figure 6C,D) where normal cells could tolerate much higher concentrations of 3i than cancer cells. These data further support the notion that pharmacologically targeting CREB is a promising strategy for development of novel cancer therapeutics.
Figure 7.
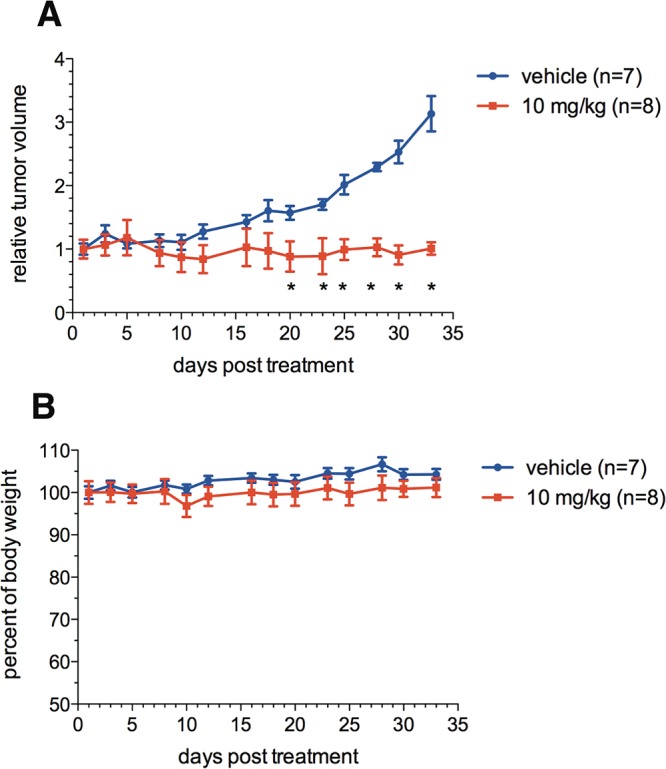
Compound 3i suppressed MDA-MB-468 tumor growth in vivo. MDA-MB-468 tumor-bearing mice were treated either with vehicle or with 3i at 10 mg/kg once a day for 5 days a week. The duration of treatment was 5 weeks. The relative tumor volume (A) and body weight (B) of the treated mice are shown: (∗) P < 0.05 by Student’s t test.
Conclusion
In an important extension of previous work,17 we have prepared a series of naphthamide derivatives based on the structure of 3a. Overall, the observed antiproliferative activities of these naphthamides correlated well with their CREB inhibition activity. Structure–activity relationships observed for members of this series revealed that many structural elements present in 3a are crucial for maintaining CREB inhibition and cancer cell growth inhibition activity. The phenol in the chlorophenyl ring, the primary amino group in the side chain, and two naphthyl rings in 3a are all important for maintaining 3a’s bioactivity. Importantly, the carbon chain length of the linker between the two naphthyl rings, and the carbon chain length of the side chain are absolutely critical for optimal activities. We identified compound 3i, which we named as 666-15, as a potent and efficacious inhibitor toward CREB-mediated gene transcription. 666-15 also displayed potent and efficacious growth inhibition activity against cancer cells in vitro and in vivo. 666-15 should give us a new tool to further investigate CREB signaling.39
Experiment Section
Chemistry. General
Glass Contout solvent purification system was used to purify all the anhydrous solvents to be used for reactions. Melting points were determined in capillary tubes using Mel-Temp and are uncorrected. All 1H and 13C NMR spectra were obtained in a Bruker Avance 400 MHz spectrometer using CDCl3 or DMSO-d6 as the solvent, and the chemical shifts of the residual CHCl3 (δ 7.24) or DMSO (δ 2.50) were taken as references. Chemical shifts (δ) are reported in parts per million (ppm), and the signals are described as brs (broad singlet), d (doublet), dd (doublet of doublet), td (triplet of doublet), m (multiplet), q (quartet), s (singlet), and t (triplet). Coupling constants (J values) are given in Hz. Silica gel flash chromatography was performed using 230–400 mesh silica gel (EMD). All reactions were monitored using thin-layer chromatography (TLC) on silica gel plates (EMD). Yields were of purified compounds. All final compounds for biological evaluations were confirmed to be of >95% purity based on reverse phase HPLC (Waters, Milford, MA) analysis using an XBridge C18 column (4.6 mm × 150 mm) and detected at 254 nm. The mobile phases for HPLC are water and acetonitrile, both of which contained 0.1% TFA. The mass spectra were obtained from a Thermo Electron LTQ-Orbitrap Discovery high resolution mass spectrometer (Thermo Scientific) with electrospray operated in either positive or negative mode.
General Procedure A: Mitsunobu Reaction
To a solution of phenol (1 equiv), alcohol (1.2–1.5 equiv), and PPh3 (1.2–1.5 equiv) in THF (1.5–2 mL/mmol) was added DEAD (1.2–1.5 equiv) in THF (0.2–0.3 mL/mmol) dropwise at 0 °C. The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure and the residue was purified by silica gel flash column chromatography to give the corresponding product.
General Procedure B: Saponification of the Methyl Esters with LiOH
To a solution of methyl ester (1 equiv) in MeOH–THF–water (1:1:1, 9 mL/mmol) was added LiOH·H2O (5 equiv) at room temperature. The resulting mixture was stirred at room temperature overnight. The organic solvents were removed under reduced pressure, and the residue was acidified with 2 N HCl at 0 °C to pH ∼2 (pH ∼7 for 5e). The reaction mixture was extracted with ethyl acetate or THF for 5e. The organic layer was separated and washed with brine and dried over Na2SO4. The solution was filtered and the solvent was evaporated to give the corresponding acid.
General Procedure C: Amide Formation by MsCl and TEA
To a stirred solution of 5 (1 equiv) and TEA (1.0 equiv) in THF (3 mL/mmol) was added MsCl (1 equiv) dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 30 min, when the corresponding ammonium salt 7 or aniline 9 (1 equiv) was added. The reaction mixture was stirred at room temperature overnight. Another portion of TEA (1.0 equiv) was added if the salt 7 was used. The reaction mixture was diluted with 5% NaHCO3 and extracted with ethyl acetate. The organic layer was separated, washed with brine, and dried over Na2SO4. The solution was filtered and the solvent was removed to give a residue, which was purified by silica gel flash column chromatography to yield the corresponding amide.
General Procedure D: Removal of the Boc and MOM with 2 N HCl
An HCl solution in Et2O (2 M, 2–10 equiv) was added to a stirred solution of 6 or 8 (1.0 equiv) in CHCl3–MeOH (1:1, 6–10 mL/mmol). The resulting mixture was stirred at room temperature overnight. The solvents were removed under reduced pressure, and the solid was treated with acetone or ethyl ether. The solid was collected by filtration to give the corresponding product.
3-(3-Aminopropoxy)-N-(3-((3-((4-chlorophenyl)carbamoyl)naphthalen-2-yl)oxy)propyl)-2-naphthamide Hydrochloride (3b)
Compound 3b (33 mg, 83%) was obtained as a white solid from 8b following general procedure D: mp 247–248 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1 H), 8.59 (t, J = 5.7 Hz, 1 H), 8.20 (s, 1 H), 8.05 (s, 1 H), 8.00 (brs, 3 H), 7.96 (d, J = 8.1 Hz, 1 H), 7.89 (d, J = 7.9 Hz, 1 H), 7.87 (d, J = 8.4 Hz, 1 H), 7.85 (d, J = 8.3 Hz, 1 H), 7.81 (d, J = 8.9 Hz, 2 H), 7.58–7.50 (m, 3 H), 7.45–7.36 (m, 5 H), 4.31 (t, J = 5.9 Hz, 2 H), 4.26 (t, J = 5.8 Hz, 2 H), 3.52 (q, J = 6.1 Hz, 2 H), 3.00 (q, J = 5.5 Hz, 2 H), 2.10 (quintet, J = 5.8 Hz, 4 H); 13C NMR (100 MHz, DMSO-d6) δ 167.01, 165.23, 153.75, 153.57, 138.58, 135.46, 135.12, 130.46, 129.93, 129.17, 128.78, 128.62, 128.24, 128.05, 127.95, 127.56, 127.45, 127.15, 126.99, 126.90, 124.83, 124.81, 121.54, 107.81, 107.71, 66.36, 66.32, 37.20, 36.52, 29.14, 26.79. HRESI-MS for C34H32ClN3O4 + H, calcd 582.21541, found 582.21557.
3-(3-Aminopropoxy)-N-(3-(2-((4-chloro-2-hydroxyphenyl)carbamoyl)phenoxy)propyl)-2-naphthamide Hydrochloride (3c)
Compound 3c (90 mg, 71%) was obtained as a white solid from 8c following general procedure D: mp 179–180 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.05 (s, 1 H,), 10.44 (s, 1 H), 8.61 (t, J = 5.8 Hz, 1 H,), 8.43 (d, J = 9.0 Hz, 1 H), 8.11 (dd, J = 7.8, 1.9 Hz, 1 H), 8.06 (s, 1 H), 8.01 (brs, 3 H,), 7.89 (d, J = 8.1 Hz, 1 H), 7.84 (d, J = 8.2 Hz, 1 H), 7.59 (td, J = 7.9, 1.7 Hz, 1 H), 7.52 (t, J = 7.1 Hz, 1 H), 7.43 (s, 1 H), 7.40 (t, J = 7.8 Hz, 1 H), 7.30 (d, J = 8.5 Hz, 1 H), 7.14 (t, J = 7.6 Hz, 1 H), 7.05 (d, J = 2.2 Hz, 1 H), 6.87 (dd, J = 8.8, 2.3 Hz, 1 H), 4.37 (t, J = 6.1 Hz, 2 H), 4.25 (t, J = 5.9 Hz, 2 H), 3.55 (q, J = 6.1 Hz, 2 H), 3.01 (q, J = 5.8 Hz, 2 H), 2.24 (quintet, J = 6.3 Hz, 2 H), 2.08 (quintet, J = 6.3 Hz, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 166.59, 162.27, 156.72, 153.09, 147.55, 134.61, 133.69, 131.55, 129.42, 128.14, 127.55, 127.44, 126.71, 126.68, 126.42, 126.27, 124.30, 121.05, 120.92, 120.63, 118.66, 114.29, 113.29, 107.11, 67.23, 65.92, 36.85, 36.21, 28.66, 26.26. HRESI-MS for C30H30ClN3O5 + H, calcd 548.19468, found 548.19474.
3-(3-(2-(3-Aminopropoxy)benzamido)propoxy)-N-(4-chloro-2-hydroxyphenyl)-2-naphthamide Hydrochloride (3d)
Compound 3d (12 mg, 53%) was obtained as a white solid from 8d following general procedure D: mp 157–158 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.94 (s, 1 H), 10.50 (s, 1 H), 8.72 (s, 1 H), 8.46 (d, J = 8.7 Hz, 1 H), 8.38 (t, J = 6.0 Hz, 1 H), 8.04 (d, J = 8.3 Hz, 1 H), 7.90 (d, J = 7.8 Hz, 1 H), 7.88 (brs, 3 H), 7.62 (s, 1 H), 7.59 (td, J = 7.4, 1.0 Hz, 1 H), 7.54 (dd, J = 7.6, 1.8 Hz, 1 H), 7.45 (td, J = 7.8, 1.0 Hz, 1 H), 7.43 (t, J = 8.0, 1.2 Hz, 1 H), 7.09 (d, J = 8.3 Hz, 1 H), 7.02 (d, J = 2.6 Hz, 1 H), 7.00 (t, J = 7.4 Hz, 1 H), 6.92 (dd, J = 8.8, 2.5 Hz, 1 H), 4.42 (t, J = 6.1 Hz, 2 H), 4.14 (t, J = 5.8 Hz, 2 H), 3.54 (q, J = 6.1 Hz, 2 H), 2.95 (q, J = 6.1 Hz, 2 H), 2.26 (quintet, J = 6.2 Hz, 2 H), 2.01 (quintet, J = 6.0 Hz, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 167.04, 162.70, 156.01, 154.08, 148.16, 136.12, 133.56, 132.13, 130.03, 129.41, 129.03, 128.06, 127.51, 126.85, 126.69, 125.22, 125.18, 123.00, 121.53, 120.99, 119.34, 114.84, 113.20, 108.55, 67.62, 66.41, 37.33, 36.62, 29.04, 26.81. HRESI-MS for C30H30ClN3O5 + H, calcd 548.19468, found 548.19467.
N-(4-Chloro-2-hydroxyphenyl)-3-(3-(3-(3-morpholinopropoxy)-2-naphthamido)propoxy)-2-naphthamide Hydrochloride (3e)
Compound 3e (50 mg, 85%) was obtained as a white solid from 8e following general procedure D: mp 144–145 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1 H), 10.55 (s, 1 H), 10.36 (brs, 1 H), 8.73 (s, 1 H), 8.53 (t, J = 5.6 Hz, 1 H), 8.44 (d, J = 8.8 Hz, 1 H), 8.09 (s, 1 H), 8.05 (d, J = 8.1 Hz, 1 H), 7.91 (d, J = 8.4 Hz, 1 H), 7.88 (d, J = 8.5 Hz, 1 H), 7.83 (d, J = 8.2 Hz, 1 H), 7.66 (s, 1 H), 7.59 (t, J = 7.5 Hz, 1 H), 7.53 (t, J = 7.5 Hz, 1 H), 7.45 (t, J = 7.5 Hz, 1 H), 7.44 (s, 1 H), 7.39 (t, J = 7.6 Hz, 1 H), 7.02 (d, J = 2.1 Hz, 1 H), 6.88 (dd, J = 8.8, 2.4 Hz, 1 H), 4.47 (t, J = 6.0 Hz, 2 H), 4.23 (t, J = 5.8 Hz, 2 H), 3.82–3.71 (m, 4 H), 3.62 (q, J = 5.7 Hz, 2 H), 3.40–3.33 (m, 2 H), 3.23 (q, J = 6.3 Hz, 2 H), 2.95 (q, J = 10.2 Hz, 2 H), 2.31 (quintet, J = 6.3 Hz, 2 H), 2.19 (quintet, J = 6.4 Hz, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 166.14, 162.12, 153.63, 153.03, 147.68, 135.66, 134.62, 133.13, 129.72, 128.94, 128.55, 127.55, 127.53, 126.55, 126.41, 126.38, 126.19, 124.69, 124.34, 122.41, 120.92, 118.76, 114.32, 108.10, 107.45, 67.30, 65.81, 62.94, 53.84, 51.06, 36.36, 28.64, 22.47. HRESI-MS for C38H38ClN3O6 + H, calcd 668.25219, found 668.25229.
N-(4-Chloro-2-hydroxyphenyl)-3-(3-(3-(3-(methylamino)propoxy)-2-naphthamido)propoxy)-2-naphthamide Hydrochloride (3f)
Compound 3f (75 mg, 92%) was obtained as a white solid from 8f following general procedure D: mp 139–140 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.06 (s, 1 H), 10.55 (s, 1 H), 8.73 (s, 1 H), 8.71 (t, J = 5.7 Hz, 1 H), 8.66 (brs, 2 H), 8.46 (d, J = 8.4 Hz, 1 H), 8.08 (s, 1 H), 8.05 (d, J = 7.7 Hz, 1 H), 7.91 (d, J = 8.3 Hz, 1 H), 7.88 (d, J = 7.6 Hz, 1 H), 7.83 (d, J = 8.1 Hz, 1 H), 7.66 (s, 1 H), 7.59 (t, J = 7.5 Hz, 1 H), 7.53 (t, J = 7.5 Hz, 1 H), 7.44 (t, J = 8.4 Hz, 1 H), 7.43 (s, 1 H), 7.40 (t, J = 7.2 Hz, 1 H), 7.05 (d, J = 2.2 Hz, 1 H), 6.90 (dd, J = 8.6, 2.1 Hz, 1 H), 4.47 (t, J = 5.5 Hz, 2 H), 4.24 (t, J = 5.2 Hz, 2 H), 3.60 (q, J = 5.6 Hz, 2 H), 3.06 (t, J = 6.2 Hz, 2 H), 2.49 (s, 3 H), 2.31 (quintet, J = 5.9 Hz, 2 H), 2.10 (quintet, J = 6.0 Hz, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 166.88, 162.13, 153.62, 153.02, 147.71, 135.67, 134.63, 133.12, 129.47, 128.93, 128.54, 128.15, 127.62, 127.55, 127.43, 126.95, 126.44, 126.39, 126.20, 124.68, 124.37, 122.40, 120.89, 118.74, 114.35, 108.13, 107.15, 67.22, 66.25, 46.53, 36.29, 32.50, 28.55, 24.79. HRESI-MS for C35H34ClN3O5 + H, calcd 612.22598, found 612.22608.
N-(4-Chloro-2-hydroxyphenyl)-3-(3-(3-(3-hydroxypropoxy)-2-naphthamido)propoxy)-2-naphthamide (3g)
Compound 3g (80 mg, 95%) was obtained as a white solid from 8g following general procedure D: mp 139–140 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1 H), 10.53 (s, 1 H), 8.73 (s, 1 H), 8.53 (t, J = 5.8 Hz, 1 H), 8.45 (d, J = 8.8 Hz, 1 H), 8.20 (s, 1 H), 8.05 (d, J = 8.9 Hz, 1 H), 7.92–7.87 (m, 2 H), 7.83 (d, J = 8.2 Hz, 1 H), 7.63 (s, 1 H), 7.59 (t, J = 7.6 Hz, 1 H), 7.51 (t, J = 7.5 Hz, 1 H), 7.44 (t, J = 7.0 Hz, 1 H), 7.43 (s, 1 H), 7.37 (t, J = 7.6 Hz, 1 H), 6.98 (d, J = 2.3 Hz, 1 H), 6.89 (dd, J = 8.7, 2.3 Hz, 1 H), 4.45 (t, J = 6.2 Hz, 2 H), 4.21 (t, J = 6.0 Hz, 2 H), 3.59 (q, J = 6.0 Hz, 2 H), 3.52 (t, J = 5.8 Hz, 2 H), 2.30 (quintet, J = 5.9 Hz, 2 H), 1.90 (quintet, J = 5.7 Hz, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 165.85, 162.64, 154.16, 148.18, 136.15, 135.39, 133.61, 130.92, 129.40, 128.99, 128.78, 128.06, 127.90, 127.48, 126.84, 126.71, 126.14, 125.14, 124.62, 122.93, 121.50, 119.27, 114.81, 108.51, 107.52, 67.74, 66.65, 58.40, 36.65, 32.10, 29.16. HRESI-MS for C34H31ClN2O6 + Na, calcd 621.17628, found 621.17501.
3-(2-Aminoethoxy)-N-(2-((3-((4-chloro-2-hydroxyphenyl)carbamoyl)naphthalen-2-yl)oxy)ethyl)-2-naphthamide Hydrochloride (3h)
Compound 3h (48 mg, 89%) was obtained as a white solid from 8h following general procedure D: mp 265–266 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1 H), 10.58 (s, 1 H), 8.77–8.73 (m, 2 H), 8.45 (d, J = 8.8 Hz, 1 H), 8.25 (brs, 3 H), 8.13 (s, 1 H), 8.06 (d, J = 8.1 Hz, 1 H), 7.93 (d, J = 8.4 Hz, 1 H), 7.86 (d, J = 8.0 Hz, 1 H), 7.79 (d, J = 8.0 Hz, 1 H), 7.75 (s, 1 H), 7.61 (td, J = 7.7, 1.0 Hz, 1 H), 7.55 (s, 1 H), 7.55 (td, J = 7.5, 1.0 Hz, 1 H), 7.46 (td, J = 7.4, 1.0 Hz, 1 H), 7.41 (td, J = 7.6, 1.1 Hz, 1 H), 7.01 (d, J = 2.2 Hz, 1 H), 6.90 (dd, J = 8.1, 2.4 Hz, 1 H), 4.54 (t, J = 6.3 Hz, 2 H), 4.41 (t, J = 4.9 Hz, 2 H), 3.98 (q, J = 6.2 Hz, 2 H), 3.29–3.32 (m, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 166.21, 162.09, 153.11, 152.64, 147.78, 135.66, 134.74, 133.15, 130.66, 128.92, 128.53, 128.24, 127.86, 127.74, 127.70, 127.05, 126.50, 126.20, 125.98, 124.79, 124.69, 122.47, 120.99, 118.79, 114.38, 108.67, 108.38, 67.19, 65.66, 38.41, 38.21. HRESI-MS for C32H28ClN3O5 + H, calcd 570.17903, found 570.17881.
3-(3-Aminopropoxy)-N-(2-((3-((4-chloro-2-hydroxyphenyl)carbamoyl)naphthalen-2-yl)oxy)ethyl)-2-naphthamide Hydrochloride (3i or 666-15)
Compound 3i (35 mg, 65%) was obtained as a white solid from 8i following general procedure D: mp 209–210 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.05 (s, 1 H), 10.59 (s, 1 H), 8.78 (t, J = 5.7 Hz, 1 H), 8.75 (s, 1 H), 8.47 (d, J = 9.1 Hz, 1 H), 8.07 (d, J = 8.7 Hz, 1 H), 8.05 (s, 1 H), 7.93 (d, J = 8.3 Hz, 1 H), 7.90 (brs, 3 H), 7.83 (d, J = 8.3 Hz, 1 H), 7.74 (s, 1 H),7.72 (d, J = 7.7 Hz, 1 H), 7.61 (td, J = 7.6, 1.1 Hz, 1 H), 7.53 (td, J = 7.6, 1.2 Hz, 1 H), 7.46 (td, J = 7.5, 1.0 Hz, 1 H), 7.43 (s, 1 H), 7.38 (td, J = 7.6, 1.2 Hz, 1 H), 7.02 (d, J = 2.2 Hz, 1 H), 6.92 (dd, J = 8.7, 2.4 Hz, 1 H), 4.53 (t, J = 6.2 Hz, 2 H), 4.24 (t, J = 5.7 Hz, 2 H), 3.96 (q, J = 5.9 Hz, 2 H), 3.01–2.93 (m, 2 H), 2.06 (quintet, J = 6.3 Hz, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 166.56, 162.10, 153.12, 153.05, 147.76, 135.65, 134.77, 133.19, 130.00, 128.93, 128.56, 128.15, 127.78, 127.70, 127.36, 127.04, 126.50, 126.41, 125.69, 124.81, 124.39, 122.44, 120.98, 118.83, 114.41, 108.36, 107.32, 67.46, 65.99, 38.41, 36.84, 26.14. HRESI-MS for C33H30ClN3O5 + H, calcd 584.19468, found 584.19456.
3-(2-Aminoethoxy)-N-(3-((3-((4-chloro-2-hydroxyphenyl)carbamoyl)naphthalen-2-yl)oxy)propyl)-2-naphthamide Hydrochloride (3j)
Compound 3j (44 mg, 90%) was obtained as a white solid from 8j following general procedure D: mp 200–201 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.00 (s, 1 H), 10.56 (s, 1 H), 8.73 (s, 1 H), 8.64 (t, J = 5.9 Hz, 1 H), 8.47 (d, J = 9.0 Hz, 1 H), 8.17 (brs, 3 H), 8.14 (s, 1 H), 8.05 (d, J = 8.0 Hz, 1 H), 7.91 (d, J = 8.4 Hz, 2 H), 7.86 (d, J = 8.6 Hz, 1 H), 7.66 (s, 1 H), 7.59 (t, J = 7.7 Hz, 1 H), 7.56 (s, 1 H),7.55 (t, J = 7.0 Hz, 1 H), 7.45 (t, J = 7.8 Hz, 1 H), 7.42 (td, J = 7.1 Hz, 1 H), 7.03 (d, J = 2.6 Hz, 1 H), 6.90 (dd, J = 8.8, 2.4 Hz, 1 H), 4.47 (t, J = 5.9 Hz, 2 H), 4.41 (t, J = 4.7 Hz, 2 H), 3.61 (q, J = 5.7 Hz, 2 H), 3.22–3.29 (m, 2 H), 2.32 (quintet, J = 6.1 Hz, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 166.26, 162.15, 153.63, 152.66, 147.71, 135.68, 134.61, 133.14, 130.26, 128.93, 128.53, 128.23, 127.83, 127.72, 127.56, 126.97, 126.75, 126.51, 126.41, 126.22, 124.68, 122.39, 120.92, 118.77, 114.34, 108.83, 108.14, 67.34, 65.70, 38.29, 36.46, 28.55. HRESI-MS for C33H30ClN3O5 + H, calcd 584.19468, found 584.19433.
tert-Butyl (3-((3-((4-Chlorophenyl)carbamoyl)naphthalen-2-yl)oxy)propyl)carbamate (6b)
Compound 6b (700 mg, 77%) was prepared as a white solid from 1 (596 mg, 2 mmol) and A1 (420 mg, 2.4 mmol) following general procedure A. The product was eluted from the column with hexanes–ethyl acetate (4:1 to 2:1): mp 134–135 °C. 1H NMR (400 MHz, CDCl3) δ 9.98 (s, 1 H), 8.63 (s, 1 H), 7.83 (d, J = 8.0 Hz, 1 H), 7.67 (d, J = 8.4 Hz, 2 H), 7.63 (d, J = 8.8 Hz, 1 H), 7.45 (t, J = 8.4 Hz, 1 H), 7.35 (t, J = 7.6 Hz, 1 H), 7.27 (d, J = 8.4 Hz, 2 H), 7.10 (s, 1 H), 4.87 (brs, 1 H), 4.21 (t, J = 5.2 Hz, 2 H), 3.37 (q, J = 6.0 Hz, 2 H), 2.06 (quintet, J = 5.6 Hz, 2 H), 1.36 (s, 9 H). HRESI-MS for C25H27ClN2O4 + Na, calcd 477.15516, found 477.15411.
tert-Butyl (3-(2-((4-Chloro-2-hydroxyphenyl)carbamoyl)phenoxy)propyl)carbamate (6c)
Compound 6c (0.89 g, 59%) was prepared as a white solid from 5c (1.06 g, 3.6 mmol) and 9 (0.515 g, 3.6 mmol) following general procedure C. The product was eluted from the column with dichloromethane–ethyl acetate (20:1): mp 147–148 °C. 1H NMR (400 MHz, CDCl3) δ 10.48 (s, 1 H), 9.81 (brs, 1 H), 8.26 (dd, J = 7.9, 1.9 Hz, 1 H), 7.91 (d, J = 7.1 Hz, 1 H), 7.48 (td, J = 7.7, 2.1 Hz, 1 H), 7.12 (td, J = 7.6, 1.2 Hz, 1 H), 7.00 (d, J = 8.5 Hz, 1 H), 6.96 (d, J = 2.2 Hz, 1 H), 6.89 (dd, J = 8.6, 2.2 Hz, 1 H), 4.86 (brs, 1 H), 4.22 (t, J = 5.4 Hz, 2 H), 3.40 (q, J = 6.1 Hz, 2 H), 2.16 (quintet, J = 5.1 Hz, 2 H), 1.45 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 163.37, 156.45, 156.32, 147.33, 133.03, 132.00, 128.95, 126.13, 121.32, 121.03, 120.70, 119.93, 116.87, 111.56, 79.95, 66.68, 38.15, 29.68, 27.86. HRESI-MS for C21H25ClN2O5 + H, calcd 421.15248, found 421.15243.
tert-Butyl (2-((3-((4-Chloro-2-hydroxyphenyl)carbamoyl)naphthalen-2-yl)oxy)ethyl)carbamate (6h)
Compound 6h (1.63 g, 60%) was prepared as a white solid from 5h (1.987 g, 6 mmol) and 9 (0.86 g, 6 mmol) following general procedure C. The product was eluted from the column with dichloromethane–ethyl acetate (10:1): mp 185–186 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.87 (s, 1 H), 10.46 (s, 1 H), 8.72 (s, 1 H), 8.44 (d, J = 8.5 Hz, 1 H), 8.04 (d, J = 7.7 Hz, 1 H), 7.90 (d, J = 8.4 Hz, 1 H), 7.66 (s, 1 H), 7. 60 (t, J = 7.5 Hz, 1 H), 7.45 (t, J = 7.5 Hz, 1 H), 7.07 (t, J = 5.7 Hz, 1 H), 6.96 (d, J = 2.3 Hz, 1 H), 6.91 (dd, J = 8.4, 2.2 Hz, 1 H), 4.31 (t, J = 6.5 Hz, 2 H), 3.59 (q, J = 6.2 Hz, 2 H), 1.35 (s, 9 H); 13C NMR (100 MHz, DMSO-d6) δ 162.01, 155.66, 153.18, 147.74, 135.64, 133.15, 128.89, 128.49, 127.64, 126.99, 126.43, 126.23, 124.73, 122.34, 121.08, 118.79, 114.31, 108.19, 77.96, 67.94, 38.98, 28.13. HRESI-MS for C24H25ClN2O5 + Na, calcd 479.13442, found 479.13402.
3-(3-Aminopropoxy)-N-(4-chlorophenyl)-2-naphthamide (7b)
The hydrochloride salt (500 mg, 83%) of 7b was obtained from 6b following general procedure D. The free amine 7b was obtained by neutralizing the hydrochloride salt with 5% NaHCO3 solution and was extracted with CHCl3. The CHCl3 solution was dried over Na2SO4. The solvent was removed to give the free amine 7b in a quantitative yield: mp 234–235 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1 H), 8.20 (s, 1 H), 7.96 (d, J = 8.0 Hz, 1 H), 7.87 (d, J = 8.4 Hz, 1 H), 7.79 (d, J = 8.0 Hz, 2 H), 7.54 (t, J = 7.4 Hz, 1 H), 7.51 (s, 1 H), 7.42 (d, J = 8.8 Hz, 2 H), 7.39 (t, J = 6.8 Hz, 1 H), 4.26 (t, J = 6.0 Hz, 2 H), 2.72 (t, J = 6.4 Hz, 2 H), 1.88 (quintet, J = 6.0 Hz, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 165.59, 153.43, 138.53, 135.30, 130.02, 129.18, 128.73, 128.29, 127.96, 127.69, 127.58, 126.99, 124.96, 121.66, 107.75, 66.04, 37.17, 27.14. HRESI-MS for C20H19ClN2O2 + H, calcd 355.12078, found 355.12104.
2-(3-Aminopropoxy)-N-(4-chloro-2-hydroxyphenyl)benzamide Hydrochloride (7c)
Compound 7c (570 mg, 80%) was obtained as an off-white solid from 6c following general procedure D: mp 301–302 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.05 (s, 1 H), 10.32 (s, 1 H), 8.41 (d, J = 8.7 Hz, 1 H), 8.09 (dd, J = 7.8, 1.7 Hz, 1 H), 8.01 (brs, 3 H), 7.59 (td, J = 5.8, 1.8 Hz, 1 H), 7.26 (d, J = 8.4 Hz, 1 H), 7.16 (t, J = 7.5 Hz, 1 H), 7.05 (d, J = 1.4 Hz, 1 H), 6.89 (dd, J = 8.6, 2.2 Hz, 1 H), 4.36 (t, J = 5.7 Hz, 2 H), 3.04 (q, J = 6.2 Hz, 2 H), 2.23 (quintet, J = 6.5 Hz, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 162.22, 156.37, 147.32, 133.62, 131.51, 126.75, 126.23, 121.22, 121.03, 120.58, 118.83, 114.27, 113.21, 66.26, 36.49, 26.59. HRESI-MS for C16H17ClN2O3 + H, calcd 321.10005, found 321.10001.
3-(2-Aminoethoxy)-N-(4-chloro-2-hydroxyphenyl)-2-naphthamide Hydrochloride (7h)
Compound 7h (1.39 g, 97%) was obtained as a white solid from 6h following general procedure D: mp 305–306 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.05 (s, 1 H), 10.29 (s, 1 H), 8.68 (s, 1 H), 8.41 (d, J = 8.6 Hz, 1 H), 8.28 (brs, 3 H), 8.06 (d, J = 8.0 Hz, 1 H), 7.94 (d, J = 8.2 Hz, 1 H), 7.66 (s, 1 H), 7.61 (td, J = 7.6, 1.3 Hz, 1 H), 7.47 (td, J = 7.6, 1.0 Hz, 1 H), 7.03 (d, J = 2.7 Hz, 1 H), 6.93 (dd, J = 8.5, 2.5 Hz, 1 H), 4.56 (t, J = 4.9 Hz, 1 H), 3.51–3.46 (m, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 162.16, 152.71, 147.80, 135.41, 132.98, 128.86, 128.55, 127.79, 127.01, 126.49, 126.37, 124.93, 123.07, 121.17, 118.88, 114.63, 108.18, 65.88, 38.23. HRESI-MS for C19H17ClN2O3 + H, calcd 357.10005, found 357.10020.
tert-Butyl (3-((3-((3-((3-((4-Chlorophenyl)carbamoyl)naphthalen-2-yl)oxy)propyl)carbamoyl)naphthalen-2-yl)oxy)propyl)carbamate (8b)
DIPEA (26 μL, 0.15 mmol) and BOP (67 mg, 0.15 mmol) were sequentially added to a solution of 5a (52 mg, 0.15 mmol) in dichloromethane (4 mL) at room temperature. The resulting mixture was stirred at room temperature for 5 min. Then an additional portion of DIPEA (35 μL, 0.19 mmol) and 7b (53 mg, 0.15 mmol) were added. The resulting mixture was stirred at room temperature overnight. The solid precipitated from the reaction mixture was collected by filtration and washed with dichloromethane (20 mL) to give product 8b (68 mg, 67%) as a white solid: mp 160–161 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1 H), 8.41 (t, J = 5.2 Hz, 1 H), 8.22 (s, 1 H), 8.13 (s, 1 H), 7.96 (d, J = 8.4 Hz, 1 H), 7.87 (d, J = 8.2 Hz, 1 H), 7.86 (d, J = 7.5 Hz, 1 H), 7.83–7.78 (m, 3 H), 7.56–7.47 (m, 3 H), 7.44–7.34 (m, 5 H), 6.95 (t, J = 5.8 Hz, 1 H), 4.30 (t, J = 5.7 Hz, 2 H), 4.14 (t, J = 5.7 Hz, 2 H), 3.56 (q, J = 6.2 Hz, 2 H), 3.12 (q, J = 5.5 Hz, 2 H), 2.11 (quintet, J = 5.6 Hz, 2 H), 1.89 (quintet, J = 6.2 Hz, 2 H), 1.33 (s, 9 H); 13C NMR (100 MHz, DMSO-d6) δ 166.23, 165.06, 156.27, 153.91, 153.75, 138.53, 135.47, 135.28, 130.69, 130.60, 129.15, 128.82, 128.72, 128.28, 128.03, 127.97, 127.92, 127.55, 127.27, 126.92, 126.82, 126.61, 124.84, 124.66, 121.52, 107.68, 107.59, 78.07, 66.31, 66.22, 37.26, 36.45, 29.35, 29.24, 28.65. HRESI-MS for C39H40ClN3O6 + H, calcd 682.26784, found 682.26830.
tert-Butyl (3-((3-((3-(2-((4-Chloro-2-hydroxyphenyl)carbamoyl)phenoxy)propyl)carbamoyl)naphthalen-2-yl)oxy)propyl)carbamate (8c)
Compound 8c (170 mg, 66%) was prepared as a white foam solid from 5a (138 mg, 0.4 mmol) and 7c (143 mg, 0.4 mmol) following general procedure C. The product was eluted from the column with dichloromethane–ethyl acetate (10:1): mp 170–171 °C. 1H NMR (400 MHz, CDCl3) δ 10.66 (s, 1 H), 8.67 (brs, 1 H), 8.67 (s, 1 H), 8.28 (dd, J = 7.9, 2.0 Hz, 1 H), 8.18 (d, J = 8.8 Hz, 1 H), 7.91 (d, J = 8.1 Hz, 1 H), 7.70 (d, J = 8.5 Hz, 1 H), 7.52 (t, J = 7.3 Hz, 1 H), 7.46 (t, J = 7.7 Hz, 1 H), 7.41 (t, J = 7.3 Hz, 1 H), 7.17 (s, 1 H), 7.11 (t, J = 7.4 Hz, 1 H), 7.06 (d, J = 2.1 Hz, 1 H), 6.99 (d, J = 8.2 Hz, 1 H), 6.85 (dd, J = 8.8, 2.2 Hz, 1 H), 4.78 (t, J = 7.4 Hz, 1 H), 4.29 (t, J = 5.5 Hz, 2 H), 4.23 (t, J = 5.8 Hz, 2 H), 3.83 (q, J = 6.7 Hz, 2 H), 3.35 (q, J = 6.2 Hz, 2 H), 2.37 (quintet, J = 6.0 Hz, 2 H), 2.02 (quintet, J = 6.1 Hz, 2 H), 1.40 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 165.90, 162.88, 156.40, 156.01, 153.02, 146.95, 135.18, 132.86, 131.87, 128.48, 128.06, 127.86, 127.49, 126.50, 125.83, 124.13, 121.68, 120.93, 120.84, 120.72, 119.57, 115.85, 111.52, 106.64, 79.07, 67.38, 65.05, 38.13, 36.74, 29.12, 29.05, 27.87. HRESI-MS for C35H38ClN3O7 + Na, calcd 670.22905, found 670.22723.
tert-Butyl (3-(2-((3-((3-((4-Chloro-2-hydroxyphenyl)carbamoyl)naphthalen-2-yl}oxy)propyl)carbamoyl)phenoxy)propyl)carbamate (8d)
Compound 8d (60 mg, 40%) was prepared as a white solid from 5c (59 mg, 0.2 mmol) and 7a (82 mg, 0.2 mmol) following general procedure C. The product was eluted from the column with hexanes–ethyl acetate (1:1): mp 179–180 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1 H), 10.50 (s, 1 H), 8.72 (s, 1 H), 8.45 (d, J = 8.5 Hz, 1 H), 8.27 (t, J = 5.1 Hz, 1 H), 8.04 (d, J = 8.1 Hz, 1 H), 7.87 (d, J = 7.9 Hz, 1 H), 7.65 (d, J = 7.6 Hz, 1 H), 7.58 (t, J = 7.1 Hz, 1 H), 7.57 (s, 1 H), 7.46–7.39 (m, 2 H), 7.07 (d, J = 8.4 Hz, 1 H), 6.98 (t, J = 7.6 Hz, 1 H), 6.95 (d, J = 2.3 Hz, 1 H), 6.92–6.86 (m, 2 H), 4.42 (t, J = 6.3 Hz, 2 H), 4.03 (t, J = 6.1 Hz, 2 H), 3.58 (q, J = 6.6 Hz, 2 H), 3.04 (q, J = 6.4 Hz, 2 H), 2.28 (quintet, J = 5.5 Hz, 2 H), 1.80 (quintet, J = 6.2 Hz, 2 H), 1.29 (s, 9 H); 13C NMR (100 MHz, DMSO-d6) δ 165.97, 162.60, 156.46, 156.24, 154.11, 148.25, 136.12, 133.62, 132.33, 130.70, 129.40, 128.96, 128.05, 127.49, 126.82, 126.73, 125.13, 124.22, 122.92, 121.51, 120.82, 119.23, 114.78, 113.06, 108.43, 78.04, 67.69, 66.09, 37.03, 36.58, 29.39, 29.12, 28.60. HRESI-MS for C35H38ClN3O7 + Na, calcd 670.22905, found 670.22757.
N-(4-Chloro-2-hydroxyphenyl)-3-(3-(3-(3-morpholinopropoxy)-2-naphthamido)propoxy)-2-naphthamide (8e)
Compound 8e (90 mg, 34%) was prepared as a white solid from 5e (126 mg, 0.40 mmol) and 7a (163 mg, 0.40 mmol) following general procedure C. When the reaction mixture was diluted with 5% NaHCO3, the crude product was precipitated and collected, which was then purified by flash column chromatography, eluting with dichloromethane–methanol (20:1) to give the desired product: mp 247–248 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1 H), 10.52 (s, 1 H), 8.74 (s, 1 H), 8.44 (d, J = 9.4 Hz, 1 H), 8.37 (t, J = 5.9 Hz, 1 H), 8.14 (s, 1 H), 8.05 (d, J = 8.2 Hz, 1 H), 7.88 (d, J = 8.4 Hz, 2 H), 7.83 (d, J = 8.4 Hz, 1 H), 7.62 (s, 1 H), 7.59 (t, J = 7.6 Hz, 1 H), 7.51 (t, J = 7.7 Hz, 1 H), 7.45 (t, J = 7.5 Hz, 1 H), 7.40 (s, 1 H), 7.37 (t, J = 7.5 Hz, 1 H), 6.94 (d, J = 1.6 Hz, 1 H), 6.86 (dd, J = 8.5, 2.1 Hz, 1 H), 4.46 (t, J = 6.4 Hz, 2 H), 4.11 (t, J = 6.2 Hz, 2 H), 3.60 (q, J = 5.7 Hz, 2 H), 3.41–3.36 (m, 4 H), 2.31 (quintet, J = 5.9 Hz, 2 H), 2.25 (t, J = 7.1 Hz, 2 H), 2.16–2.08 (m, 4 H), 1.83 (quintet, J = 6.3 Hz, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 165.51, 162.06, 153.60, 153.47, 147.58, 135.65, 134.80, 133.22, 130.09, 128.96, 128.57, 128.23, 127.60, 127.53, 127.44, 126.95, 126.38, 126.35, 126.19, 126.03, 124.70, 124.17, 122.33, 120.92, 118.79, 114.19, 107.97, 107.10, 67.30, 66.62, 65.97, 54.68, 53.17, 36.13, 28.65, 25.49.
tert-Butyl (3-((3-((3-((3-((4-Chloro-2-hydroxyphenyl)carbamoyl)naphthalen-2-yl)oxy)propyl)carbamoyl)naphthalen-2-yl)oxy]propyl)(methyl)carbamate (8f)
Compound 8f (110 mg, 34%) was prepared as a white solid from 5f (160 mg, 0.45 mmol) and 7a (183 mg, 0.45 mmol) following general procedure C. The product was eluted from the column with dichloromethane–methanol (20:1): mp 219–220 °C. 1H NMR (400 MHz, CDCl3) δ 10.86 (s, 1 H), 10.52 (s, 1 H), 8.88 (t, J = 5.6 Hz, 1 H), 8.85 (s, 1 H), 8.65 (s, 1 H), 8.27 (d, J = 8.9 Hz, 1 H), 7.92 (d, J = 8.5 Hz, 1 H), 7.89 (d, J = 8.2 Hz, 1 H), 7.72 (d, J = 8.1 Hz, 1 H), 7.70 (d, J = 8.0 Hz, 1 H), 7.55–7.48 (m, 2 H), 77.43–7.37 (m, 2 H), 7.24 (s, 1 H), 7.17 (s, 1 H), 7.08 (d, J = 2.2 Hz, 1 H), 6.90 (dd, J = 8.7, 2.2 Hz, 1 H), 4.40 (t, J = 5.2 Hz, 2 H), 4.18 (t, J = 5.2 Hz, 2 H), 3.89 (q, J = 5.6 Hz, 2 H), 3.48 (t, J = 5.9 Hz, 2 H), 2.88 (s, 3 H), 2.44 (quintet, J = 5.9 Hz, 2 H), 2.06 (quintet, J = 6.1 Hz, 2 H), 1.40 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 165.98, 162.75, 155.71, 153.62, 153.04, 147.15, 135.39, 135.17, 133.86, 133.20, 128.77, 128.66, 128.42, 127.98, 127.83, 127.76, 127.64, 126.79, 125.77, 125.71, 124.20, 124.12, 122.31, 122.06, 120.97, 119.88, 116.52, 106.79, 106.34, 79.47, 67.51, 64.35, 44.34, 38.59, 33.58, 27.86, 26.10, 25.14. HRESI-MS for C40H42ClN3O7 + Na, calcd 734.26035, found 734.25755.
N-(4-Chloro-2-hydroxyphenyl)-3-(3-(3-(3-(methoxymethoxy)propoxy)-2-naphthamido)propoxy)-2-naphthamide (8g)
Compound 8g (189 mg, 56%) was prepared as a white solid from 5g (153 mg, 0.52 mmol) and 7a (215 mg, 0.52 mmol) following general procedure C. The product was eluted from the column with dichloromethane–ethyl acetate (20:1 to 10:1): mp 186–187 °C. 1H NMR (400 MHz, CDCl3) δ 10.72 (s, 1 H), 8.84 (s, 1 H), 8.75 (s, 1 H), 8.70 (t, J = 5.8 Hz, 1 H), 8.18 (d, J = 8.7 Hz, 1 H), 7.94–7.90 (m, 2 H), 7.73 (d, J = 7.8 Hz, 1 H), 7.71 (d, J = 7.8 Hz, 1 H), 7.55–7.50 (m, 2 H), 7.43–7.38 (m, 2 H), 7.25 (s, 1 H), 7.19 (s, 1 H), 7.10 (d, J = 2.3 Hz, 1 H), 6.89 (dd, J = 8.5, 2.2 Hz, 1 H), 4.62 (s, 2 H), 4.38 (t, J = 5.3 Hz, 2 H), 4.31 (t, J = 5.9 Hz, 2 H), 3.81 (q, J = 6.8 Hz, 2 H), 3.74 (t, J = 5.6 Hz, 2 H), 3.30 (s, 3 H), 2.40 (quintet, J = 5.8 Hz, 2 H), 2.16 (quintet, J = 5.8 Hz, 2 H); 13C NMR (100 MHz, CDCl3) δ 166.26, 163.19, 153.86, 153.79, 147.65, 135.85, 135.83, 134.30, 133.86, 129.15, 128.96, 128.51, 128.22, 128.08, 127.04, 126.30, 126.25, 124.76, 124 72, 122.37, 121.47, 120.31, 116.84, 107.37, 107.26, 96.56, 67.62, 66.94, 65.10, 55.32, 38.71, 29.73, 29.32. HRESI-MS for C36H35ClN2O7 + Na, calcd 665.20250, found 665.20026.
tert-Butyl (2-((3-((2-((3-((4-Chloro-2-hydroxyphenyl)carbamoyl)naphthalen-2-yl)oxy)ethyl)carbamoyl)naphthalen-2-yl)oxy)ethyl)carbamate (8h)
Compound 8h (85 mg, 42%) was prepared as a white solid from 5h (100 mg, 0.3 mmol) and 7h (118 mg, 0.3 mmol) following general procedure C. The product was eluted from the column with dichloromethane–ethyl acetate (10:1): mp 220–221 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1 H), 10.60 (s, 1 H), 8.74 (s, 1 H), 8.67 (t, J = 5.8 Hz, 1 H), 8.43 (d, J = 9.4 Hz, 1 H), 8.40 (s, 1 H), 8.05 (d, J = 8.2 Hz, 1 H), 7.90 (d, J = 7.8 Hz, 1 H), 7.89 (d, J = 7.9 Hz, 1 H), 7.83 (d, J = 8.2 Hz, 1 H), 7.74 (s, 1 H), 7.58 (t, J = 7.4 Hz, 1 H), 7.54 (t, J = 7.6 Hz, 1 H), 7.46 (s, 1 H), 7.44 (t, J = 7.1 Hz, 1 H), 7.40 (t, J = 7.4 Hz, 1 H), 7.13 (t, J = 5.6 Hz, 1 H), 6.89–6.84 (m, 2 H), 4.54 (t, J = 6.4 Hz, 2 H), 4.09 (t, J = 5.0 Hz, 2 H), 4.04 (q, J = 5.7 Hz, 2 H), 1.25 (s, 9 H), (one CH2 is hidden in the water peak at 3.35 ppm); 13C NMR (100 MHz, DMSO-d6) δ 165.05, 161.98, 155.97, 153.54, 153.11, 147.68, 135.67, 135.15, 133.21, 131.71, 128.92, 128.51, 127.94, 127.71, 127.49, 126.99, 126.43, 126.38, 126.17, 124.76, 124.41, 123.77, 122.36, 120.93, 118.75, 114.20, 108.42, 107.63, 77.91, 68.25, 67.34, 38.31, 28.02. HRESI-MS for C37H36ClN3O7 + Na, calcd 692.21340, found 692.21189.
tert-Butyl (3-((3-((2-((3-((4-Chloro-2-hydroxyphenyl)carbamoyl)naphthalen-2-yl)oxy)ethyl)carbamoyl)naphthalen-2-yl)oxy)propyl)carbamate (8i)
Compound 8i (82 mg, 40%) was prepared as a white solid from 5a (104 mg, 0.3 mmol) and 7h (118 mg, 0.3 mmol) following general procedure C. The product was eluted from the column with dichloromethane–ethyl acetate (10:1): mp 139–140 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1 H), 10.59 (s, 1 H), 8.75 (s, 1 H), 8.65 (t, J = 4.9 Hz, 1 H), 8.47 (d, J = 8.5 Hz, 1 H), 8.29 (s, 1 H), 8.05 (d, J = 8.5 Hz, 1 H), 7.89 (d, J = 8.1 Hz, 1 H), 7.87 (d, J = 8.3 Hz, 1 H), 7.81 (d, J = 8.2 Hz, 1 H), 7.70 (s, 1 H), 7.59 (t, J = 7.2 Hz, 1 H), 7.52 (t, J = 7.6 Hz, 1 H), 7.45 (t, J = 7.1 Hz, 1 H), 7.41 (s, 1 H), 7.38 (t, J = 7.6 Hz, 1 H), 6.93–6.83 (m, 3 H), 4.52 (t, J = 5.8 Hz, 2 H), 4.10 (t, J = 5.6 Hz, 2 H), 4.05 (q, J = 5.2 Hz, 2 H), 3.01 (t, J = 6.3 Hz, 2 H), 1.81 (quintet, J = 6.2 Hz, 2 H), 1.23 (s, 9 H); 13C NMR (100 MHz, DMSO-d6) δ 165.44, 161.97, 155.69, 153.48, 153.19, 147.69, 135.67, 135.04, 133.26, 131.08, 128.93, 128.52, 128.40, 127.82, 127.69, 127.38, 127.00, 126.46, 126.32, 126.20, 124.77, 124.64, 124.28, 122.23, 120.92, 118.80, 114.22, 108.27, 107.30, 77.52, 67.78, 65.76, 38.41, 36.54, 28.67, 28.03. HRESI-MS for C38H38ClN3O7 + Na, calcd 706.22905, found 706.22719.
tert-Butyl (2-((3-((3-((3-((4-Chloro-2-hydroxyphenyl)carbamoyl)naphthalen-2-yl)oxy)propyl)carbamoyl)naphthalen-2-yl)oxy)ethyl)carbamate (8j)
Compound 8j (74 mg, 54%) was prepared as a white solid from 5h (66 mg, 0.2 mmol) and 7a (81 mg, 0.2 mmol) following general procedure C. The product was eluted from the column with dichloromethane–ethyl acetate (10:1): mp 228–229 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1 H), 10.56 (s, 1 H), 8.73 (s, 1 H), 8.51 (t, J = 5.9 Hz, 1 H), 8.43 (d, J = 9.1 Hz, 1 H), 8.30 (s, 1 H), 8.04 (d, J = 8.3 Hz, 1 H), 7.90 (d, J = 7.4 Hz, 1 H), 7.88 (d, J = 7.9 Hz, 1 H), 7.83 (d, J = 8.5 Hz, 1 H),7.62 (s, 1 H), 7.57 (t, J = 7.6 Hz, 1 H), 7.52 (t, J = 7.6 Hz, 1 H), 7.46 (s, 1 H), 7.44 (t, J = 7.2 Hz, 1 H), 7.38 (t, J = 7.5 Hz, 1 H), 7.13 (t, J = 5.8 Hz, 1 H), 6.94 (d, J = 2.3 Hz, 1 H), 6.85 (dd, J = 8.5, 1.9 Hz, 1 H), 4.47 (t, J = 5.6 Hz, 2 H), 4.11 (t, J = 5.1 Hz, 2 H), 3.65 (q, J = 6.1 Hz, 2 H), 2.32 (quintet, J = 5.6 Hz, 2 H), 1.29 (s, 9 H) (one CH2 is hidden in the water peak at 3.35 ppm); 13C NMR (100 MHz, DMSO-d6) δ 164.87, 162.08, 155.95, 153.68, 153.48, 147.63, 135.68, 134.97, 133.16, 131.26, 128.92, 128.48, 128.41, 127.75, 127.53, 126.91, 126.35, 126.23, 124.73, 124.62, 124.32, 122.35, 120.95, 118.75, 114.19, 107.92, 107.40, 77.88, 68.19, 67.25, 36.36, 28.60, 28.06. HRESI-MS for C38H38ClN3O7 + Na, calcd 706.22905, found 706.22788.
Cell Culture
HEK 293T, HFF and A549 cell lines were obtained from American Tissue Culture Collection (ATCC, Manassas, VA). MDA-MB-231, MDA-MB-468, and MCF-7 cell lines were obtained from Developmental Therapeutics Program at the National Cancer Institute (NCI). The cells were maintained in DMEM (Life Technologies, Grand Island, NY) with nonessential amino acids (Life Technologies) and 10% (v/v) HyClone fetal bovine serum (FBS, GE Healthcare Life Science, Logan, UT) at 37 °C with 5% CO2. HMEC cells were obtained from Lonza (Walkersville, MA) and were cultured in MEGM complete media (Lonza) supplemented with 10 μg/mL penicillin and 10 μg/mL streptomycin (Life Technologies) at 37 °C under 5% CO2.
Inhibition of CREB-Mediated Gene Transcription
HEK 293T cells in a 10 cm plate were transfected with pCRE-RLuc (6 μg) with Lipofectamine2000 (Life Technologies) following the manufacturer’s instructions. Three hours after transfection, the cells were collected and replated into 96-well plates at ∼10 000 cells/well. The cells were allowed to attach to the bottom of the plates overnight. The cells were then treated with different concentrations of different compounds for 30 min, when forskolin (10 μM) was added to each well. The cells were incubated for further 5 h before cell lysis using 1× 30 μL Renilla luciferase lysis buffer (Promega, Madison, WI). An amount of 5 μL of the lysate was combined with 30 μL of benzyl-coelenterazine (Nanolight, Pinetop, AZ) solution in PBS (pH 7.4, 10 μg/mL). The protein concentration in each well was determined by Dye Reagent Concentrate (Bio-Rad, Hercules, CA). The Renilla luciferase activity was normalized to protein content in each well and expressed as relative luciferase unit/μg protein (RLU/μg protein). The IC50 was derived from nonlinear regression analysis of the RLU/μg protein–concentration curve in Prism 5.0 (La Jolla, CA).
Cell Growth Inhibition Assay
The growth inhibition of different cell types was assessed by MTT assay using MTT reagent (Sigma, St. Louis, MO). Briefly, the cells were plated into 96-well plates and the cells were allowed to attach to the bottom of the plates overnight. Then the cells were treated with different concentrations of different drugs for 72 h. The media were removed, and MTT reagent in complete tissue culture media (0.5 mg/mL) was added to each well and incubated at 37 °C for 3 h. The incubation media were removed and 100 μL of DMSO was added to each well. The absorbance of the formed purple formazan solution was read at 570 nm using Packard Fusion plate reader. The percent of growth is defined as 100 × (Atreated – Ainitial)/(Acontrol – Ainitial), where Atreated represents absorbance in wells treated with a compound, Ainitial represents the absorbance at time 0, and Acontrol denotes media-treated cells. The GI50 was derived from nonlinear regression analysis of the percent of growth–concentration curve in Prism 5.0.
qRT-PCR
The qRT-PCR assay was carried out essentially in the same way as described before.17 Briefly, HEK 293T cells were treated with different compounds for 1 h followed by treatment with DMSO or forskolin (10 μM) for 45 min. Then total RNA was isolated and treated with DNase I using NucleoSpin RNA kit (Clontech). The first-strand cDNA was synthesized using SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). Quantitative real-time PCR was performed on QuantStudio 7 Flex using SYBR Advantage qPCR Premix (Clontech). The 2–ΔΔCT method was used to analyze the relative changes in gene expression16,40 with hypoxanthine phosphoribosyltransferase 1 (HPRT) as the reference gene. The primers used were the same as before.17
In Vivo Xenograft Study
All the procedures for animal handling, care, and the treatment in this study were performed according to the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of Oregon Health & Science University following the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Each 6- to 8-week old BALB/c nude mouse (Charles River Laboratories) was inoculated subcutaneously at the right flank with MDA-MB-468 cells (5 × 106) in 0.1 mL of HBSS with Matrigel (1:1) for tumor development. When the tumor volume reached approximately 100 mm3, the mice were randomized to be treated with either vehicle or 3i at 10 mg/kg. 3i was dissolved in 1% N-methylpyrrolidone (NMP), 5% Tween-80 in H2O. The dosing solution was prepared weekly. The mice were treated once a day for 5 consecutive days a week, and the treatment lasted for 5 weeks. During the treatment, the tumor size and body weight were measured 2–3 times a week. The tumors were measured in two dimensions using a digital caliper, and the volume was expressed in mm3 using the formula V = 0.5ab2, where a and b represent the long and short diameters of the tumor, respectively. The tumor volume was normalized to the initial tumor volume at the time of the first treatment. Student t-test was used for statistical analysis.
Molecular Modeling
All the molecular modeling studies were conducted in Schrodinger Small Molecule Drug Discovery Suite (Portland, OR). All the structures were first optimized using MacroModel with MMFFs force field in the absence of any solvent. The charges were from the force field. Powell–Reeves conjugate gradient (PRCG) minimization algorithm was used, and all minimizations were converged to 0.05 kJ mol–1 Å–1. To identify the global conformational minimum for each compound, the minimized structure was subjected to a conformational search by rotating all the rotatable bonds in each structure. MMFFs force field was used. The global energy minimum was taken for cLogP and PSA calculation using QikProp module.
Acknowledgments
This work was financially supported by NIH RO1GM087305 (X.X.), RO1CA100855 (R.C.S.), Susan G. Komen KG100458 (X.X.) and DOD BC103625 (R.C.S.). We thank Andrea DeBarber for expert mass spectroscopic analyses. X.X. dedicates this paper to Professor Donglu Bai on the occasion of his 80th birthday.
Glossary
Abbreviations Used
- BOP
(benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
- cAMP
cyclic adenosine monophosphate
- cLogP
calculated log P
- CRE
cAMP response element
- CREB
cAMP response element binding protein
- CBP
CREB binding protein
- DEAD
diethyl azodicarboxylate
- DIPEA
diisopropylethylamine
- HFF
human foreskin fibroblast
- HMEC
human mammary epithelial cell
- KID
kinase-inducible domain
- KIX
KID-interacting domain
- MAPK
mitogen-activated protein kinase
- MMFF
Merck molecular force field
- Nurr1
nuclear receptor related 1 protein
- PKA
protein kinase A
- PKB
protein kinase B
- PP1
protein phosphatase 1
- PP2A
protein phosphatase 2A
- PSA
polar surface area
- PTEN
phosphatase and tensin homolog
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- RLuc
renilla luciferase
- SD
standard deviation
- TEA
triethylamine
Supporting Information Available
Additional supplemental figures, experimental procedures, NMR spectra of 3b–3j, and a csv file of molecular formula strings. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00468.
Author Contributions
∥ F.X. and B.X.L. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Shaywitz A. J.; Greenberg M. E. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 1999, 68, 821–861. [DOI] [PubMed] [Google Scholar]
- Mayr B.; Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [DOI] [PubMed] [Google Scholar]
- Xiao X.; Li B. X.; Mitton B.; Ikeda A.; Sakamoto K. M. Targeting CREB for cancer therapy: friend or foe. Curr. Cancer Drug Targets 2010, 10, 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan I.; Perez-Alvarado G. C.; Parker D.; Dyson H. J.; Montminy M. R.; Wright P. E. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:coactivator interactions. Cell 1997, 91, 741–752. [DOI] [PubMed] [Google Scholar]
- Hagiwara M.; Alberts A.; Brindle P.; Meinkoth J.; Feramisco J.; Deng T.; Karin M.; Shenolikar S.; Montminy M. Transcriptional attenuation following cAMP induction requires PP-1-mediated dephosphorylation of CREB. Cell 1992, 70, 105–113. [DOI] [PubMed] [Google Scholar]
- Wadzinski B. E.; Wheat W. H.; Jaspers S.; Peruski L. F.; Lickteig R. L.; Johnson G. L.; Klemm D. J. Nuclear-protein phosphatase-2A dephosphorylates protein kinase A phosphorylated CREB and regulates CREB transcriptional stimulation. Mol. Cell. Biol. 1993, 13, 2822–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu T.; Zhang Z.; Wang J.; Guo J.; Shen W. H.; Yin Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 2011, 71, 2821–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X.; Wang S.; Zhu L.; Wu C.; Yin B.; Zhao J.; Yuan J.; Qiang B.; Peng X. cAMP response element-binding protein promotes gliomagenesis by modulating the expression of oncogenic microRNA-23a. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 15805–15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodon L.; Gonzalez-Junca A.; Inda M. D.; Sala-Hojman A.; Martinez-Saez E.; Seoane J. Active CREB1 promotes a malignant TGF-beta2 autocrine loop in glioblastoma. Cancer Discovery 2014, 4, 1230–1241. [DOI] [PubMed] [Google Scholar]
- Chhabra A.; Fernando H.; Watkins G.; Mansel R. E.; Jiang W. G. Expression of transcription factor CREB1 in human breast cancer and its correlation with prognosis. Oncol. Rep. 2007, 18, 953–958. [PubMed] [Google Scholar]
- Zhang S.; Chen L.; Cui B.; Chuang H.-Y.; Yu J.; Wang-Rodriguez J.; Tang L.; Chen G.; Basak G. W.; Kipps T. J. ROR1 is expressed in human breast cancer and associated with enhanced tumor-cell growth. PLoS One 2012, 7, e31127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H. S.; Liu D. D.; Bekele B. N.; Kim M. K.; Pisters K.; Lippman S. M.; Wistuba I. I.; Koo J. S. Cyclic AMP response element-binding protein overexpression: A feature associated with negative prognosis in never smokers with non-small cell lung cancer. Cancer Res. 2008, 68, 6065–6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.; Zhau H. E.; Huang W. C.; Iqbal S.; Habib F. K.; Sartor O.; Cvitanovic L.; Marshall F. F.; Xu Z.; Chung L. W. K. cAMP-responsive element-binding protein regulates vascular endothelial growth factor expression: implication in human prostate cancer bone metastasis. Oncogene 2007, 26, 5070–5077. [DOI] [PubMed] [Google Scholar]
- Crans-Vargas H. N.; Landaw E. M.; Bhatia S.; Sandusky G.; Moore T. B.; Sakamoto K. M. Expression of cyclic adenosine monophosphate response-element binding protein in acute leukemia. Blood 2002, 99, 2617–2619. [DOI] [PubMed] [Google Scholar]
- Li B. X.; Xiao X. Discovery of a small-molecule inhibitor of the KIX-KID interaction. ChemBioChem 2009, 10, 2721–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. X.; Yamanaka K.; Xiao X. Structure-activity relationship studies of naphthol AS-E and its derivatives as anticancer agents by inhibiting CREB-mediated gene transcription. Bioorg. Med. Chem. 2012, 20, 6811–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. X.; Xie F.; Fan Q.; Barnhart K. M.; Moore C. E.; Rheingold A. L.; Xiao X. Novel type of prodrug activation through a long-range O,N-acyl transfer: a case of water-soluble CREB inhibitor. ACS Med. Chem. Lett. 2014, 5, 1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Basu S.; Meng S.; Wang X.; Hu M. Regioselective sulfation and glucuronidation of phenolics: insights into the structural basis. Curr. Drug Metab. 2011, 12, 900–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Kulkarni K.; Basu S.; Zhang S.; Hu M. First-pass metabolism via UDP-glucuronosyltransferase: a barrier to oral bioavailability of phenolics. J. Pharm. Sci. 2011, 100, 3655–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson M.; Johnson D. S.; Subramanyam C.; Bales K. R.; am Ende C. W.; Fish B. A.; Green M. E.; Kauffman G. W.; Mullins P. B.; Navaratnam T.; Sakya S. M.; Stiff C. M.; Tran T. P.; Xie L.; Zhang L.; Pustilnik L. R.; Vetelino B. C.; Wood K. M.; Pozdnyakov N.; Verhoest P. R.; O’Donnell C. J. Design, synthesis, and pharmacological evaluation of a novel series of pyridopyrazine-1,6-dione gamma-secretase modulators. J. Med. Chem. 2014, 57, 1046–1062. [DOI] [PubMed] [Google Scholar]
- Mitsunobu O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981, 1–28. [Google Scholar]
- Kim S. J.; Baek H. S.; Rho H. S.; Kim D. H.; Chang I. S.; Lee O. S.; Shin H. J.. Hydroxybenzamide derivatives, the method for preparing thereof and the cosmetic composition containing the same. WO2007021067A1, 2007
- Jiang M.; Li B. X.; Xie F.; Delaney F.; Xiao X. Design, synthesis, and biological evaluation of conformationally constrained analogues of naphthol AS-E as inhibitors of CREB-mediated gene transcription. J. Med. Chem. 2012, 55, 4020–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X.; Antony S.; Kohlhagen G.; Pommier Y.; Cushman M. Design, synthesis, and biological evaluation of cytotoxic 11-aminoalkenylindenoisoquinoline and 11-diaminoalkenylindenoisoquinoline topoisomerase I inhibitors. Bioorg. Med. Chem. 2004, 12, 5147–5160. [DOI] [PubMed] [Google Scholar]
- Seamon K. B.; Padgett W.; Daly J. W. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 3363–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival—application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [DOI] [PubMed] [Google Scholar]
- Xie F.; Li B. X.; Broussard C.; Xiao X. Identification, synthesis and evaluation of substituted benzofurazans as inhibitors of CREB-mediated gene transcription. Bioorg. Med. Chem. Lett. 2013, 23, 5371–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge J. M.; Rettenmaier T. J.; Wells J. A.; Pomerantz W. C.; Mapp A. K. FP tethering: a screening technique to rapidly identify compounds that disrupt protein–protein interactions. MedChemComm 2014, 5, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moellering R. E.; Cravatt B. F. How chemoproteomics can enable drug discovery and development. Chem. Biol. 2012, 19, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson M. P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [DOI] [PubMed] [Google Scholar]
- Yazdanian M.; Glynn S. L.; Wright J. L.; Hawi A. Correlating partitioning and caco-2 cell permeability of structurally diverse small molecular weight compounds. Pharm. Res. 1998, 15, 1490–1494. [DOI] [PubMed] [Google Scholar]
- Best J. L.; Amezcua C. A.; Mayr B.; Flechner L.; Murawsky C. M.; Emerson B.; Zor T.; Gardner K. H.; Montminy M. Identification of small-molecule antagonists that inhibit an activator: coactivator interaction. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 17622–17627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X.; Finkbeiner S.; Arnold D. B.; Shaywitz A. J.; Greenberg M. E. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 1998, 20, 709–726. [DOI] [PubMed] [Google Scholar]
- Hummler E.; Cole T. J.; Blendy J. A.; Ganss R.; Aguzzi A.; Schmid W.; Beermann F.; Schutz G. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 5647–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y. G.; Nesterova M.; Agrawal S.; Cho-Chung Y. S. Dual blockade of cyclic AMP response element- (CRE) and AP-1-directed transcription by CRE-transcription factor decoy oligonucleotide. Gene-specific inhibition of tumor growth. J. Biol. Chem. 1999, 274, 1573–1580. [DOI] [PubMed] [Google Scholar]
- Chen J.; Kassenbrock A.; Li B. X.; Xiao X. Discovery of a potent anti-tumor agent through regioselective mono-N-acylation of 7H-pyrrolo[3,2-f]quinazoline-1,3-diamine. MedChemComm 2013, 4, 1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K. M.; Frank D. A. CREB in the pathophysiology of cancer: implications for targeting transcription factors for cancer therapy. Clin. Cancer Res. 2009, 15, 2583–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkright M. D.; Montminy M. CREB: the unindicted cancer co-conspirator. Trends Cell Biol. 2005, 15, 457–459. [DOI] [PubMed] [Google Scholar]
- Kang X.; Lu Z.; Cui C.; Deng M.; Fan Y.; Dong B.; Han X.; Xie F.; Tyner J. W.; Coligan J. E.; Collins R. H.; Xiao X.; You M. J.; Zhang C. C. The ITIM-containing receptor LAIR1 is essential for acute myeloid leukaemia development. Nat. Cell Biol. 2015, 17, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.