

Abstract
Skeletal muscle redox homoeostasis is transcriptionally regulated by nuclear erythroid-2-p45-related factor-2 (Nrf2). We recently demonstrated that age-associated stress impairs Nrf2-ARE (antioxidant response element) transcriptional signaling. Here, we hypothesize that age-dependent decline or genetic ablation of Nrf2 leads to accelerated apoptosis and skeletal muscle degeneration. Under basal-physiological conditions, disruption of Nrf2 significantly down regulates antioxidants and causes oxidative stress. Surprisingly, Nrf2-null mice had enhanced antioxidant capacity identical to wild-type (WT) upon acute endurance exercise stress (AEES), suggesting activation of Nrf2-independent mechanisms (i.e. PGC1α) against oxidative stress. Analysis of pro-survival pathways under the basal state reveals decreased Akt levels, while pp53, a repressor of Akt, was increased in Nrf2-null versus WT mice. Upon AEES, Akt and p-Akt levels were significantly (p<0.001) increased (>10 fold) along with profound down regulation of pp53 (p<0.01) in Nrf2-null versus WT skeletal muscle, indicating the onset of pro-survival mechanisms to compensate the loss of Nrf2 signaling. However, we found a decreased stem cell population (Pax7) and MyoD expression (differentiation) along with profound activation of ubiquitin and apoptotic pathways in Nrf2- null versus WT mice upon AEES, suggesting that compensatory pro-survival mechanisms failed to overcome the programed cell death and degeneration in skeletal muscle. Further, the impaired regeneration was sustained in Nrf2-null vs. WT mice after 1 week of post-AEES recovery. In an age-associated oxidative stress condition, ablation of Nrf2 results in induction of apoptosis and impaired muscle regeneration.
Introduction
About 40% of mammalian body mass is composed of skeletal muscle. With aging, healthy individuals exhibit 20-30% reduction in muscle mass (1). A decline in mass and function of skeletal muscle due to aging contributes to age-related musculo-skeletal diseases such as sarcopenia (muscle fiber atrophy), impaired regeneration of satellite cells, pathologic remodeling of synaptic neuronal structures, decreased protein synthesis, oxidative stress, mitochondrial dysfunction and impaired energy production (2). Skeletal muscle myocytes are dynamic cells that respond to a variety of stresses by inducing compensatory mechanisms (2-7). One of the primary stresses of skeletal muscle is oxidative stress; the response to this stress, including an antioxidant defense response, is critically regulated by nuclear erythroid-2-p45-related factor-2 (Nrf2) (8, 9).
Regulation of Nrf2 signaling is believed to preserve redox homeostasis and protect the structure and function of skeletal muscle. Recently we demonstrated that while disruption of Nrf2 has a minimal role on the mouse skeletal muscle antioxidant defense system at a young (2 months) age, Nrf2 disruption promotes oxidative stress and impairs antioxidant mechanisms upon aging (8, 9). In general, Nrf2 signaling is dependent on its activation in response to reactive oxygen species (ROS) generation, and toxic and electrophilic stresses (10-14). Excessive production of reactive oxygen and nitrogen species (ROS/RNS) cause oxidative or nitrosative stress, which are key signals for the onset of several musculoskeletal diseases (15, 16). Upon aging, the magnitude of generation and accumulation of ROS is significantly higher in aged Nrf2-null mice compared to WT controls. This increased oxidative stress is accompanied by a striking decline of several antioxidants due to low levels and poor activation of cellular defense mechanisms (8, 9). However, it is unknown whether Nrf2-independent pathways are activated as compensatory measure in response to aging, endurance exercise and oxidative stress.
It has been previously shown that apoptotic events are directly linked with the cellular aging of post mitotic tissues such as brain, heart and skeletal muscle (17-30). A decline in the number of viable, fat-free myocytes results in skeletal muscle dysfunction. We have shown that induction of pro-apoptotic (BAD, BAX) and apoptotic (ASK1, AIF caspase-3 and 9) signals in aged Nrf2-null mice are due to increased generation/accumulation of ROS/RNS and glutathione (GSH) depletion (9). Further, in Nrf2-null mice age-associated oxidative stress and apoptotic events are accompanied by increased ubiquitination of a wide range of proteins and lipid peroxidation (4-hydroxy nonenol, 4-HNE) (9). Increased ROS/RNS generation and prolonged oxidative/nitrosative stress contribute to damage and dysfunction of organelles and cells in several age-associated diseases in humans (4, 15, 31-36). However, it is currently unknown whether Nrf2 is required for proliferation and differentiation of myoblasts undergoing severe oxidative stress.
Typically, in response to chronic oxidative stress and skeletal muscle injury, it is expected that factors for regeneration (satellite cells) and differentiation (MyoD lineage) to be activated to overcome the loss of myoblasts. The role of Nrf2 in such conditions has not been defined. Skeletal muscle progenitor stem cells express cell context-specific genes involved in tissue regeneration (37). Embryonic skeletal muscle requires paired-box transcriptional factors (Pax3 and Pax7) and canonical myogenic regulatory factors (MRFs; Myf-5, MyoD, myogenin and Mrf4) for its development (38). During regeneration of skeletal muscle, Pax7 and MyoD expressed in quiescent and activated satellite stem cells specifically determines and regulates cell lineage during skeletal muscle regeneration (39-42). However, it is currently unknown if the satellite cell pool is affected by aging and/or loss of Nrf2. Thus, using a genetic approach, we explored whether Nrf2 plays a critical role in skeletal muscle damage or repair in response to endurance stress on aging. We demonstrate that Nrf2 is an important regulator of redox state and determines stem cell lineage during the regeneration of aging skeletal muscle.
2. Materials and Methods
2.1 Animals
Nrf2-deficient (Nrf2- null) mice with a hybrid background were originally generated by Itoh and colleagues (43) and generously provided by Dr. Li Wang (University of Utah, Salt Lake City, UT). They were backcrossed with the C57/BL6 mice to the fifth generation. Nrf2-null mice and their littermate controls, Nrf2+/+ (wild-type) used in this study were obtained from intercrossing Nrf2+/- mice. Genotyping of mice was performed by PCR amplification of genomic DNA using the following primers: Nrf2 forward: 5-GCCTGAGAGCTGTAGGCCC-3, Nrf2 reverse: 5-GGAATGGAAAATAGCTCCTGCC-3 and Nrf2 mutant: 5-GGGTTTTCCCAGTCACGAC-3. The mice were housed under controlled temperature, humidity and a 12-hour light/dark cycle, and fed with a standard rodent diet and water ad libitum. Experimental protocols conducted on the mice were approved by IACUC at the University of Utah in accordance with the standards established by the U.S. Animal Welfare Act.
2.2 Antibodies and Reagents
The following antibodies and reagents were used: NQO-1 (ab34173), GAPDH (ab9485), lamin-B1 (ab16048), catalase (219010, Calbiochem, Merck GaA, Germany), CA, USA), SOD-1 (Enzo-CAD-SOD-100), SOD-2 (ab13534), GPX1 (ab22604), GSR (ab16801), PGC1α (ab54481), I-kBα (ab32518), I-kBβ (ab32518), Nrf1 (Kap-Tf125, Stressgen), AKT (CS#9272), pGSK3β (CS#9322), pp53 (CS#9282), BAX (CS#2772), BAD (CS#9292), ASK1 (CS#3762), PAX7 (ab34360), MyoD (ab64159), caspase-3 (CS#9662), caspase-9 (CS#9504), cleaved PARP (CS#9544s) and ubiquitin-ab (Enzo-BML-PW0150). Secondary antibodies conjugated with horseradish peroxidase IgG (Rabbit and Mouse/PI-1000 & PI-2000, Vector labs, Burlingame, USA) were used. A Bio-Rad Protein Assay (500-0006, Bio-Rad, Hercules, CA) was used to determine protein levels in skeletal muscle tissue extracts. Fluorescent probes – DCFDA, DHE and MitoSox Red to detect ROS (Molecular Probes/Invitrogen Corp). All reagents and primers for RNA extraction and real-time RT-PCR quantification were purchased from Qiagen Inc., Valencia, CA.
2.3 Acute endurance exercise stress (AEES) and preparation of tissues/samples for analysis
Wild type (WT, C57/Bl6/SJ) and Nrf2-null mice >23 months of age (n = 6/group) were subjected to acute endurance exercise on a treadmill for 2 weeks [∼90-120 minutes per day; 20-30m/min; 12° angle]. The duration of exercise was optimized based on the endurance of 23 months old mice. Mice performed run on a treadmill (Columbus Instruments, Columbus, Ohio) calibrated for angle and speed. Endurance capacity was determined in a group of non-trained mice (n≥10/group) to identify a maximal running endurance capacity. Mice were started at a speed of 20m/min for first 10 minutes and every 2 min the treadmill speed was increased by 1.0 m/min and at an angle of 12° (not plain). In general, a soft brush is attached to the treadmill via the back door in order to encourage the animals to run when they stop. Mice were considered exhausted when they could no longer move forward from the back of the lane despite the manual prompting using the soft brush prodding. Mice that were reluctant to run on the treadmill, despite receiving stimuli, a tickle on the hind feet using brush were not included in the study. This protocol was followed to induce severe oxidative stress as opposed to moderate training [treadmill running at 10-15m/min speed; 0° angle (plain) for 45 minutes per day] that activates compensatory cytoprotective mechanisms such as Nrf2-antioxidant signaling (8). After exercise, mice were sacrificed and the skeletal muscles (gastrocnemius) were processed/frozen (−80°C) for subsequent analysis (44, 45). For histology, 10 mg portions of muscle were immediately washed and processed in EPR buffer and analyzed. A portion (10-20 mg) of the skeletal muscle tissue was placed into RNA-later solution and stored in −80°C for RNA extraction.
2.4 Detection of ROS using fluorescent probes (DCFDA, DHE and MitoSox Red)
Superoxide and peroxides in the skeletal muscle of old (>23 months) WT and Nrf2-null mice were determined immediately after last exercise (on day 14 of AEES) using DHE/H2DCFDA fluorescence (D11347/C6827, Invitrogen Corp. USA) by following our previous reported protocols (46). Cell-permeable non-fluorescent DHE is oxidized to fluorescent 2-hydroxyethidium, which can be measured by appropriate excitation/emission filters for rhodamine. In the presence of superoxide (O2−.), oxidized ethidium intercalates with DNA, staining the nucleus a bright red fluorescence. H2DCFDA oxidized to a fluorescent DCF by hydrogen peroxide is detected by fluorescence microscopy using appropriate excitation/emission filters as reported previously (47). Briefly, 5-μm-thick transverse sections of cryo-fixed skeletal muscle tissues on clean glass slides were covered with the probe solution containing 10 μM of DHE/H2DCFDA and incubated in a light-protected chamber at 37°C for 30 min. MitoSox Red was used to determine the ROS generation in mitochondria. After washing with 1× PBS thrice, sections were fixed and mounted using DAPI containing Vector-Flouromount-G. Fluorescent images of the DCF and DHE were obtained using a FV-1000 laser scanning confocal microscope (Olympus Inc.). Fluorescence intensity was quantified by automated image analysis using Simple PCI 6 Imaging software (Hamamatsu Corporation, Sewickley, PA).
2.5 Gene expression analysis by quantitative real time PCR
Harvested skeletal muscle tissues were washed with RNase free PBS and stored in RNAlater reagent. About 20 mg of skeletal muscle tissue from 24 month old WT and Nrf2-null mice (N=4-6) was used to extract RNA using Qiagen RNA isolation kits, following the manufacturer's instructions. The quality and quantity of the RNA was analyzed using a Nano drop-analyzer. To synthesize cDNA, the reverse transcription reaction was performed on 2.0 μg of RNA using a Qiagen Reverse Transcription Kit (205311) as per manufacturer's instructions. For qPCR analysis, 100 ng of cDNA template, 10 μL of SYBR Green Master Mix (Qiagen-204054) and respective Qiagen primer sets for NQO-1 (QT00094367), catalase (QT01058106), G6PD (QT00120750), GCLC (QT QT00130543), Gclm (QT00174300), Gpx1 (QT01195936), Sod-1 (QT01762719), Sod-2 (QT00161707), Gsr (QT01758232), TRXN1 (QT01060297), Gstα (QT0098987), Gst-mu (QT00121191), Pax7 (QT00147728) and MyoD1 (QT00101983) were used and analyzed in a Roche-Light Cycler. Copy numbers of cDNA targets were quantified using Ct values. Respective gene expression levels were calculated by normalizing to the level of the housekeeping gene Arbp1 (QT00249375) or GAPDH (QT01658692).
2.6 Skeletal muscle tissue homogenization and immunoblotting
Skeletal muscle cytosolic proteins from WT and Nrf2-null mice under sedentary or AEES conditions were prepared by homogenizing the tissues using cytosolic buffer (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 0.5 mM MgCl2, with freshly prepared 1 mM dithiothreitol, 0.1 mM phenyl methylsulfonyl fluoride (PMSF) and 1% Triton-X100, pH 7.9), followed by centrifugation at 5000 rpm for 5-6 minutes. Cytosolic contaminants in the nuclear fraction were removed by washing with 4 volumes of cytosol buffer. Nuclear fractions were dissolved in nuclear extraction buffer (NEB; 20 mM HEPES, 420 mM NaCl, 0.1 mM EDTA, 1.5 mM MgCl2, 25% glycerol and 1 mM dithiothreitol, 0.5 mM PMSF, pH 7.9), incubated on ice with mild shaking and centrifuged at 8200rpm for 10 minutes. Then the proteins were quantified using Bio-Rad Bradford reagent. Protein samples for immunoblots were prepared in 4× Laemmli buffer with 5% freshly added β-mercaptoethanol and boiled for 5 minutes. About 30-40 μg of cytosolic proteins were separated on 10 or 12% SDS-PAGE and transferred to PVDF membranes. The membranes were treated with antibodies to catalase, SOD-1, GSR, G6PD, NQO-1, ubiquitin, γ-GCS, caspase-3/9, PAX7, MYOD1, GAPDH and lamin-B1. Horseradish peroxidase IgG (rabbit and mouse) conjugated secondary antibodies were used for chemiluminescence detection of specific proteins.
2.7 Immunoprecipitation and in-gel AKT kinase assay
AKT kinase activity in skeletal muscle was determined using an Akt/PKB protein kinase kit (Cat#9840, Cell-Signaling). Proteins (∼100-200 μg) extracted from WT and Nrf2-null skeletal muscle were immunoprecipated by incubating with agarose beads containing anti-Akt-antibody overnight (12 – 14 hrs) at 4°C and gentle mixing. Next, antigen-antibody complexes bound to the immunosorbant beads was purified by repeated washing with lysis buffer (1× TBS) containing 0.1% Tween 20 (TBS/T), and the pellet was used for kinase assay. Precipitated AKT from WT and Nrf2-null skeletal muscles was treated with 10 μl of 10× kinase buffer (#9802), 2 μl of 10 mM ATP (#9804), 1 μl of Akt/PKB protein kinase (10 units/μl) and sterile water to make up to 100 μl. This reaction mixture was incubated for 30 or 60 minutes at 30°C and terminated by adding 25 μL 3× SDS sample buffer, vortex and centrifuged for 30 seconds at 14,000 × G. Then the phosphorylated GSK3α/β (Ser21/9) was detected by immunoblotting and quantified using Image-J.
2.8 Immunofluorescence analysis (IF) of PAX7 expression
Frozen skeletal muscle in OCT medium were sectioned at 5 μm thickness from WT and Nrf2-null mice > 23 months old and immunostained with PAX7 primary antibodies as previously described (46, 48). Briefly, skeletal muscle tissue sections were fixed in 4.0% paraformaldehyde for 10 minutes, permeabilized with 0.25% Triton X-100 in PBS containing 0.01% Tween-20 for 5 min, and blocked with 5% goat serum in 0.01% PBST for 60 min. Primary antibody diluted in 0.01% PBST containing 1% BSA was added to the tissue sections and incubated overnight at 4°C. After 3 × 5 minutes washes with PBST, the sections were incubated with secondary anti-mouse Alexa-Fluor 488-conjugated or anti-Rabbit Alexa-Fluor 647-conjugated antibodies (1:1000 dilution) for 1 hour at room temperature. After three washes (5 minutes each) with 1× PBST, the sections were mounted with DAPI containing mounting medium (Vector Shield) and imaged using a FV-1000 Confocal (Olympus Inc.) microscope at a magnification of 60× for visualization and specific localization of PAX7 (1:200). Intensity of the PAX fluorescence was quantified by automated image analysis using the Simple PCI 6 Imaging Software (Hamamatsu Corporation, Sewickley, PA).
2.9 Statistical analysis
Data are expressed as mean ± standard deviation. All data were statistically analyzed using a Student's t-test, and P-values ≤0.05 were considered statistically significant. For each parameter, the WT-sedentary group was compared to corresponding Nrf2-null mice. Additional comparisons also were made between the sedentary vs. AEES groups.
Results
Abrogation of Nrf2 exacerbates oxidative stress induced by acute endurance exercise stress (AEES) in WT and Nrf2-null mice
Wild type (WT) and Nrf2-null mice >23 months old were subjected to AEES for 2 weeks and the skeletal muscles (gastrocnemius) were used to evaluate the role of Nrf2 and AEES on oxidative stress. First, we analyzed cytoplasmic and mitochondrial-dependent reactive oxygen species (ROS) using DCFHDA, DHE and MitoSox Red fluorescent probes. In the cytoplasm, the ROS levels were similar among the skeletal muscle of WT and Nrf2-null mice (Fig. 1A). Further, AEES induced excess ROS generation in WT and Nrf2-null mice skeletal muscle indicating oxidative stress. Importantly, a much greater increase in ROS was observed in Nrf2-null when compared to WT following AEES. Co-localization of DHE and MitoSOX Red was noted in mice that underwent AEES in relation to sedentary group, indicating that mitochondria might be a major source of ROS in response to AEES (Fig. 1B).
Figure 1. Aging and lack of Nrf2 increases oxidative stress in skeletal muscle.
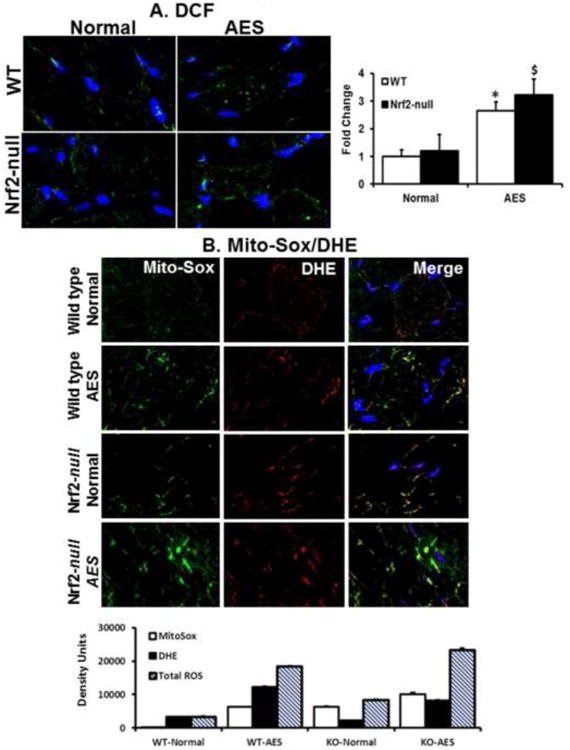
A. Determination of ROS in the skeletal muscle tissue sections using fluorescent (dihydro difluro diacetate (H2DCFDA)/dihydroethidium (DHE) probes and microscopy. Frozen tissue sections were incubated with 10μM of H2DCFDA for 30 min at 37° C. Sections were then washed with 1XPBS, mounted and analyzed by Zeiss 510 Meta confocal microscopy. Significantly increased ROS levels were seen in the WT and Nrf2-null mice in response to AEES compared with SED at 23 months (p<0.01), but these values were comparable between WT and Nrf2-null under sedentary condition.
B. Determination of the source of ROS generation using mitosox green and DHE fluorescent probes. Frozen sections of SM were stained with 10μM mitosox green and dihydroethidium (DHE) for 30 minutes and imaged using confocal microscope. Images from green and red fluorescence were merged (purple) to locate the mitochondrial source of ROS generation. The purple color is indicating ROS within the mitochondria. Both AEES and abrogation of Nrf2 showing increased ROS generation in the mitochondria of SM. Histograms showing density of the fluorescent signals from WT and Nrf2-null mice under sedentary and AEES conditions. The data are from n=3 mice/group and *p<0.05.
Nrf2 knockout significantly alters the transcriptional bio-signature of antioxidants in response to AEES
Nrf2 is a master transcriptional regulator of several antioxidant and cytoprotective genes (13, 14, 43, 46, 49-52). To evaluate the molecular pathways involved in AEES-mediated oxidative stress in skeletal muscle myocytes, we analyzed transcript levels of Nrf2-dependent antioxidant genes. Real-time qPCR analysis using Qiagen primer sets for the targets of Nrf2 showed significant differences between the WT and Nrf2-null mice (Fig. 2). Under a basal state, most of the antioxidant genes (Nqo1, G6pd, catalase, Gpx1, Gclc, Gsr, Txn1 Gst-α, and Gst-μ) were significantly down regulated in Nrf2-null skeletal muscle when compared to that of WT at >23 months of age. Our previous findings indicated that the levels of antioxidant enzymes were comparable between WT and Nrf2-null mice at younger (2 months) age (9). Previously, we demonstrated that acute exercise stress for 2 consecutive days resulted in significant induction of Nrf2-dependent antioxidant genes in WT while Nrf2-null mouse hearts exhibited down regulation of these genes (8). Thus, we assessed the effect of AEES on transcriptional regulation of the antioxidants in skeletal muscle. Interestingly, in WT some of the antioxidant (Nqo1, catalase, Gsr, G6pd and Gst-μ) genes were upregulated significantly (p<0.05) after AEES in comparison to sedentary controls. In contrast, Nrf2-null showed either decrease (Nqo1, Gclm, Gst-α and Gst-μ) or blunted (Gclc and Txn1) transcription of most antioxidants in response to AEES. However, a few of the antioxidant genes (G6pd, catalase and Gpx1) were significantly upregulated in response to AEES when compared to sedentary Nrf2-null cohorts, suggesting Nrf2-independent mechanisms.
Figure 2. Abrogation of Nrf2 down regulates the bio-signature of antioxidants upon AEES.
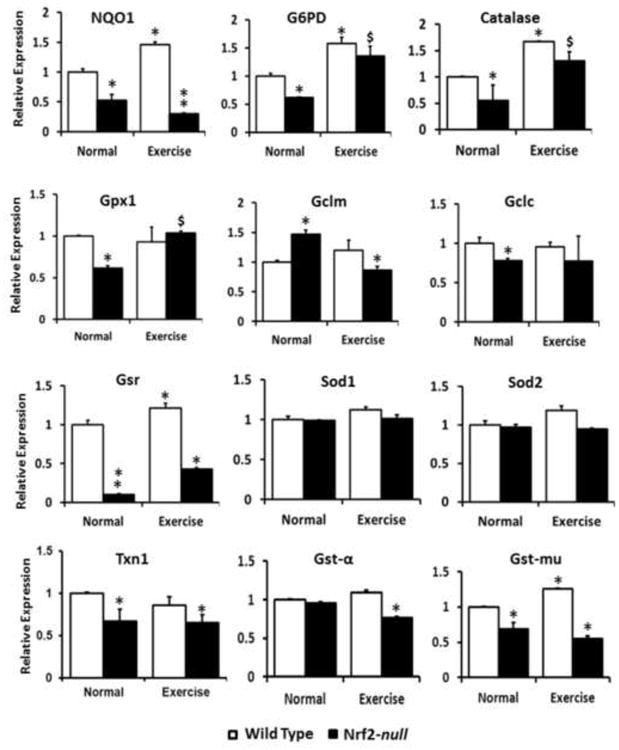
Real-time RT-PCR determinations of Nrf2 target genes in WT and Nrf2-null mice under sedentary and AEES conditions at >23 months were performed using Qiagen-mouse primer sets (n=4/gp.). Data were first normalized to Arbp1 expression and then to the corresponding gene expression in the WT group (Delta-delta-CT method). While there is an increasing trend in the messenger RNA (mRNA) expression for major targets of Nrf2 (i.e. catalase, G6pd, Nqo1 and Gst-mu) were significantly upregulated in WT upon AEES, most of these genes were down regulated in Nrf2-null mice under sedentary or AEES conditions. The data are from n=3-4 mice/group. (*p<0.05 vs. WT-SED).
Ablation of Nrf2 and impaired transcriptional regulation of antioxidant genes results in diminished defense mechanisms
To verify whether abrogation of Nrf2 and AEES-mediated oxidative stress evokes an antioxidant protein response in skeletal muscle, we performed immunoblotting analyses for major antioxidant enzymes in WT and Nrf2-null sedentary or AEES mice. Under basal state, protein expressions of NQO1, catalase, G6PD, SOD1 and GPX1 were significantly decreased in Nrf2-null in relation to WT (Fig. 3A-B). Upon AEES, while protein levels for catalase increased, G6PD, GPX1, NQO1 and SOD1 were comparatively decreased in WT mice suggesting differential translational regulation of the antioxidant enzymes. Interestingly, most of the antioxidants (catalase, G6PD, SOD1 and GPX1) were significantly increased in Nrf2-null after AEES when compared to Nrf2-null sedentary mice. Thus, it appears that AEES induces the antioxidants through activating the antioxidant transcription machinery in skeletal muscle, as we have previously reported in the heart (12). However, this acute response is not sufficient to overcome the excess ROS generation and chronic oxidative stress (Fig. 1).
Figure 3. Abrogation of Nrf2 impairs the protein levels of antioxidants.
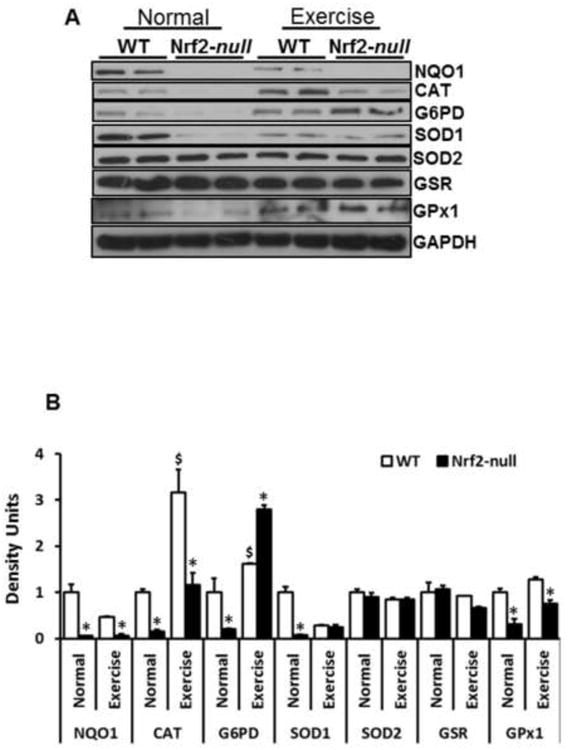
A) Representative Immunoblotting (IB) experiments of cytosol from WT and Nrf2-null mice under sedentary and AEES conditions at >23 months were performed using appropriate antibodies (n=4/group.). Protein blots were probed with anti-Nqo1, catalase, glucose-6-phosphate dehydrogenase, super oxide dismutase-1/2 (Sod-1/2), glutathione reductase, glutathione peroxidase and GAPDH. Each lane indicates an individual mouse.
B) Densitometry analysis was performed as relative intensity values calculated as mean arbitrary units obtained from the IB shown in (A). Significant decreases in NQO1, CAT, G6PD, SOD1 and GPX1 in Nrf2-null when compared to WT were evident under sedentary state. AEES upregulates most of the antioxidant proteins, but this effect was moderate in Nrf2-null mice. (*p<0.05, **p< 0.01, vs. WT-SED).
PGC1α is activated in response to acute endurance exercise stress in skeletal muscle
Next, we assessed whether alternative pathways to Nrf2 were activated in response to AEES. Since PGC1α and NF-kB have been proposed to mediate stress response induction of antioxidants (53-55), we determined their expression by immunoblotting. PGC1α is a master regulator of mitochondrial biogenesis (56-58). Under basal condition, Nrf2-null skeletal muscle had lower PGC1α protein in comparison that of WT mice. Interestingly, PGC1α protein levels were significantly increased in WT and Nrf2-null skeletal muscle in response to AEES (Fig. 4A). These results suggest that activation of PGC1α might be a primary response in skeletal muscle during AEES. Further, as shown in Fig 4B, significant activation of IkBα and IkBβ, negative regulators of NF-kB, in WT and Nrf2-null was evident in response to AEES. These observations indicate that NF-kB is not activated by either AEES or Nrf2 abrogation.
Figure 4. Proliferator–activated receptor co-activator-1-alpha (PGC1α) is activated in response to acute exercise stress in skeletal muscle.
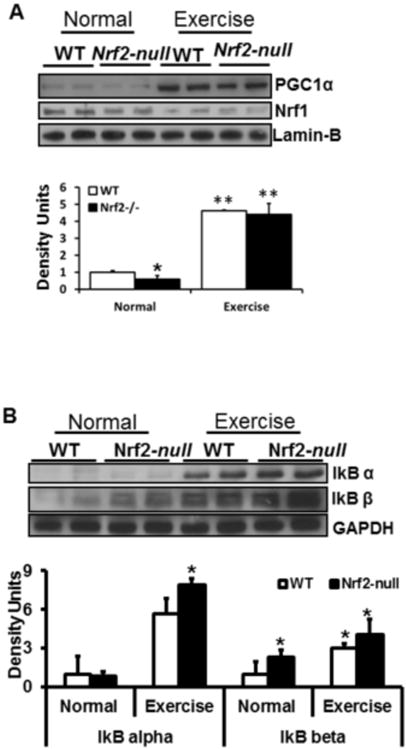
A-B) Analysis of nuclear levels of PGC1α and Nrf1 in WT and Nrf2-null mice under sedentary and AEES conditions at >23 months of age. Representative Immunoblotting (IB) experiments from nuclear extracts were performed using anti-PGC1α and anti-Nrf1 and lmin-B (A), and anti-IkBα, IkBβ, and GAPDH antibodies (n=4/group.). In sedentary mice, PGC1α levels were decreased in Nrf2-null versus WT mice. AEES augmented the PGC1α and IkBα/β levels in both genotypes. Each lane indicates an individual mouse. Protein blots/values represent n=4 or more mice from each group. *p<0.05; **p<0.01 between WT-SED vs. other experimental groups.
AKT pro-survival and p53 pathways are activated in response to AEES
Next, we analyzed whether, in response to PGC1α activation, pro-survival mechanisms protect myocytes from oxidative stress caused by AEES and/or abrogation of Nrf2. Here, we probed the skeletal muscle proteins from WT and Nrf2-null sedentary and AEES mice for AKT, pGSK3 and pp53 (Fig. 5B). Under basal conditions, both transcript and protein levels for AKT were significantly lower in Nrf2-null in comparison to WT skeletal muscle (Fig. 5A-B). Upon AEES, WT and Nrf2-null skeletal muscle significantly increased protein levels for AKT. In response to AEES, increased AKT was associated with profound upregulation of phosphorylated GSK3β, a direct downstream substrate for AKT. In-gel kinase assays for AKT function indicated activation of AKT in response to AEES (Fig. 5C). Surprisingly, after AEES the AKT activity was markedly higher in Nrf2-null compared to WT mice. These results indicate excessive activation of pro-survival pathways under Nrf2 abrogation. In agreement with the decreased AKT levels in Nrf2-null skeletal muscle at rest, profound activation of pp53 was observed. Upon AEES, pp53 protein levels were significantly increased in WT when compared to sedentary mice. However, after AEES pp53 levels were drastically decreased in Nrf2-null mice suggesting possible diminution of cell cycle mechanisms.
Figure 5. Acute exercise stress augments AKT pro-survival and P53 signaling in skeletal muscle.
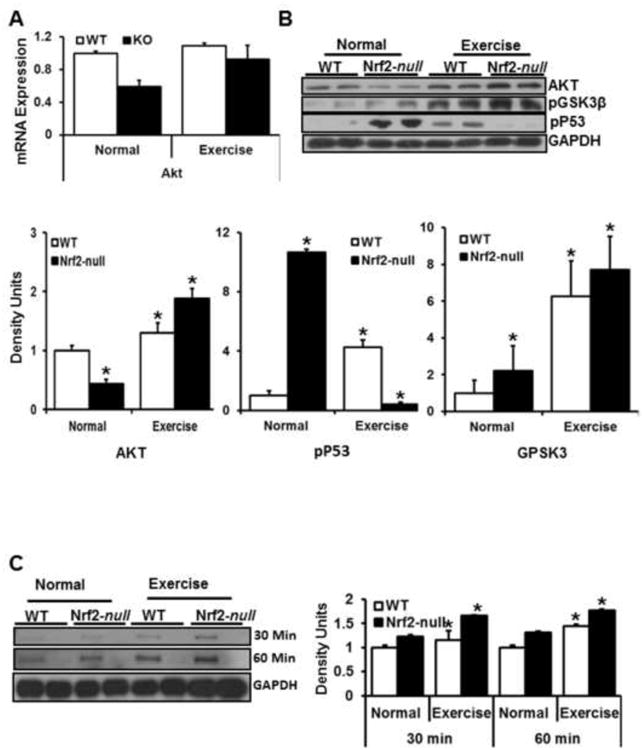
A. QPCR analysis using Qiagen primers showing down regulation of AKT in Nrf2-null versus WT mice under sedentary condition. AEES upregulates AKT mRNA in Nrf2-null, but has no effect on WT mice. N=3-4 mice/group; *p<0.05 vs. WT-SED.
B. Representative immunoblots (IB) and corresponding densitometry analyses for AKT, phosphor-GSK3β and phospho-P53 in WT and Nrf2-null mice under sedentary and AEES conditions. Upon AEES, pro-survival (AKT and GSK3β) mechanisms were activated along with decreased cell cycle (phospho-P53) mechanisms in Nrf2-null mice.
C. AKT kinase (in-gel) assay and Immunoblotting. Representative IB from WT and Nrf2-null mice indicating time dependent activation of AKT on phosphorylation of GSK3β. Increased AKT kinase function is evident in Nrf2-null versus WT mice after AEES. N=3 mice/group; *p<0.05 vs. WT-SED.
Apoptotic pathways override the AKT/pro-survival mechanisms in Nrf2-null mice
Activation of AKT/pro-survival mechanisms is known to protect cells from apoptosis (59-62). Since pp53 levels were also dramatically decreased in Nrf2-null skeletal muscle following AEES, we next analyzed whether apoptotic pathways were activated under Nrf2 abrogation due to AEES-induced oxidative stress. Immunoblot analyses for BAX, BAD, ASK1, caspase-3/9 and cleaved-PARP were performed in skeletal muscle of WT and Nrf2-null mice before or after AEES (Fig. 6A-D). Under basal conditions, apoptotic markers (BAD, BAX, ASK1, caspse3/9 and cleaved-PARP) were significantly increased in Nrf2-null compared to WT mice. Upon AEES, most of the apoptotic markers were also substantially increased in Nrf2-null compared to WT mice. Although a moderate increase of apoptotic markers was observed in WT in response to AEES, these parameters were lower when compared to Nrf2-null mice. These observations indicate that activation of pro-survival mechanisms in response to AEES in Nrf2-null mice is inadequate to suppress the induction of apoptotic pathways.
Figure 6. Apoptosis revokes AKT/pro-survival mechanisms in Nrf2-null mice.
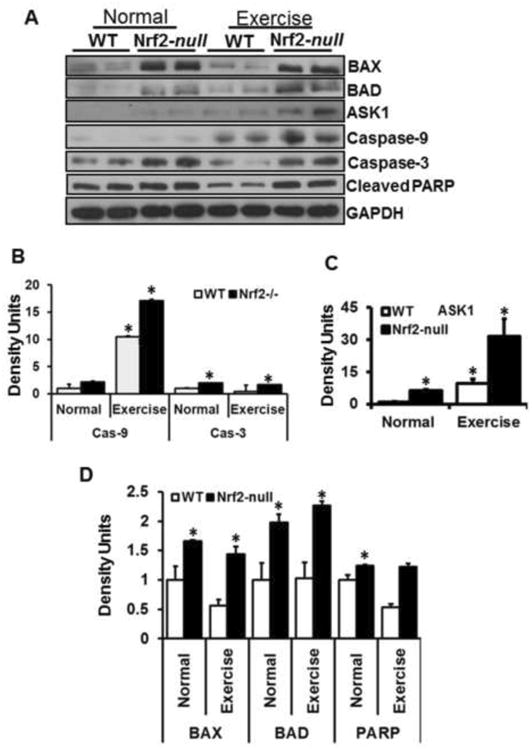
A. Representative immunoblots of cytosolic protein extracts from skeletal muscles of WT and Nrf2-null mice under sedentary and AEES conditions. Protein blots were probed with respective antibodies as indicated. Individual lanes represent separate animals (n=4/group). Analyses of apoptotic pathway revealed significant changes (*p<0.05 vs. WT-SED) in various markers including BAX, BAD, ASK1, caspase-3, caspase-9 and cleaved PARP between WT vs. Nrf2-null and SED vs. AEES.
B – D. Densitometry analyses of representative protein signals were performed using image-J and expressed relative to mean values of the WT-SED group. Overall apoptotic signals were significantly (*p<0.05 – 0.01) increased in Nrf2-null vs. WT mice.
AEES induced oxidative stress is associated with increased oxidation and ubiquitination of proteins
Chronic oxidative stress promotes ubiquitination of proteins (35, 63, 64). Degradation and subsequent removal of oxidatively modified or misfolded proteins are facilitated through the ubiquitin-proteasome systems (UPS). We examined whether Nrf2 abrogation or age-dependent increases in ROS results in oxidative modification and/or ubiquitination of skeletal muscle proteins. We determined the level of oxidation of skeletal muscle by measuring 4-hydroxynonenol (4-HNE) content in aged WT and Nrf2-null mice under sedentary or AEES conditions. There was a significant increase in 4-HNE positive proteins observed in Nrf2-null relative to WT mice under basal conditions. Robust induction of 4-HNE signals for a wide range of proteins (from 15 to 250 kDa) in old WT mice occurred in response to AEES. Upon AEES, Nrf2-null mice exhibited additional increase in 4-HNE positive proteins when compared to sedentary-Nrf2-null mice (Fig. 7A). Under resting state, ubiquitin-protein conjugates were significantly higher in Nrf2-null compared to WT skeletal muscle. Upon AEES, both WT and Nrf2-null mice had increased levels of ubiquitinated skeletal muscle proteins and the magnitude of ubiquitination was much greater in Nrf2-null compared to WT skeletal muscle (Fig. 7B). These results suggest that abrogation of Nrf2, coupled with induction of oxidative stress, could accelerate the age-associated augmentation of protein degradation pathways that contribute to skeletal muscle loss.
Figure 7. AEES induces oxidative stress and augments ubiquitination of proteins in Nrf2-null mice.
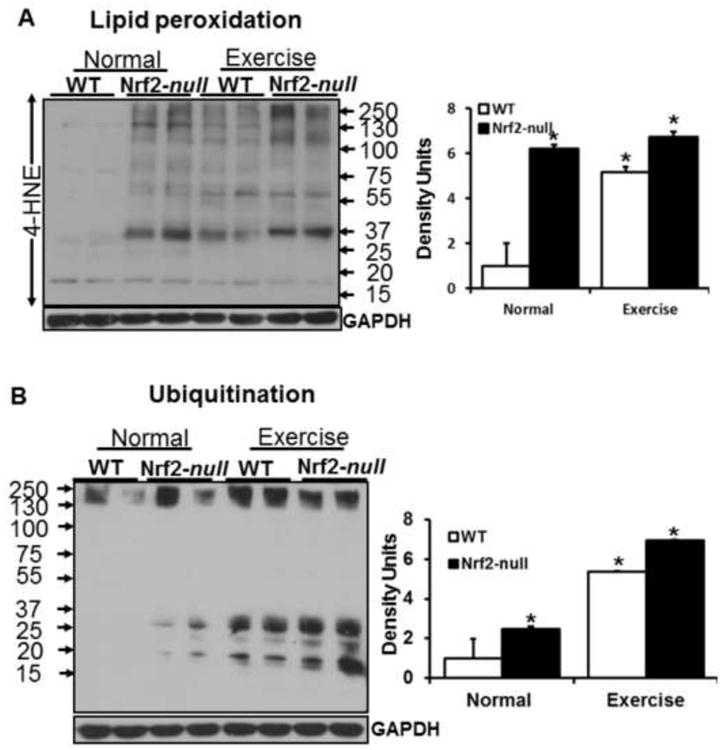
Oxidative stress induced lipid peroxidation and ubiquitination in the skeletal muscles of Nrf2−/− mice. (A) Immunoblots probed for anti-HNE-ab showing ∼6 fold increased signals in skeletal muscles of Nrf2-null versus WT at 23 months of age. After AEES, significant increases of HNE-modified proteins are evident in the SM, but these were comparable among WT and Nrf2-null mice (n=4/group; *p<0.05; **p<0.01 vs. WT-SED).
(B) Immunoblots probed for mono-/poly-ubiquitination-ab revealing increased ubiquitination of SM proteins in Nrf2-null versus WT mice. Densitometry analysis showing ∼2 fold increase in poly-ubiquitinated proteins in aged Nrf2-null when compared to age matched WT Mice, but AEES resulted in increased ubiquitination in both genotypes with a higher efficiency in Nrf2-null mice.
Nrf2 abrogation and AEES-induced oxidative stress mediated apoptosis impairs muscle regeneration and stem cell lineage
Regenerative capacity of skeletal muscle in response to oxidative stress or aging might require activation of quiescent muscle precursor cells (65-67). Activation of satellite cells is essential for regeneration, maintenance and repair of adult skeletal muscle damage due to aging and/or chronic stress states. Therefore, we analyzed the protein expression for PAX7 (stem cell marker) and MyoD (muscle differentiation factor) by immunoblotting. Under basal conditions, both the PAX7 and MyoD1 protein levels were significantly higher in Nrf2-null compared to WT skeletal muscle. However, after AEES, WT mice had increased PAX7 and MyoD1, but these levels were dramatically reduced in Nrf2-null mice (Fig. 8A). Next, we assessed the PAX7 population of skeletal muscle cells using immunofluorescence followed by microscopic examination (Fig. 8B). Under basal conditions, the number of Pax7 cells was higher in Nrf2-null than WT mice. In response to AEES, the Pax7 positive cell number significantly increased in WT, but decreased in Nrf2-null mice (Fig. 8B-C). Moreover, regenerative capacity was significantly decreased in Nrf2-null compared to WT mice. Although both the WT and Nrf2-null mice exhibited impaired regenerative capacity after AEES, the magnitude of decrease was significantly lower in Nrf2-null relative to WT mice (Fig. 8B-C). These results indicate that AEES abrogation of Nrf2 might be a confounding factor contributing to age-associated degeneration of myocytes. These results suggest that WT and Nrf2-null mice respond differently to AEES, and this could be related to the differences in the degree of oxidative stress and compensatory defense mechanisms in these genotypes.
Figure 8. AEES and loss of Nrf2 diminishes stem cell lineage and muscle regeneration in Nrf2-null mice.
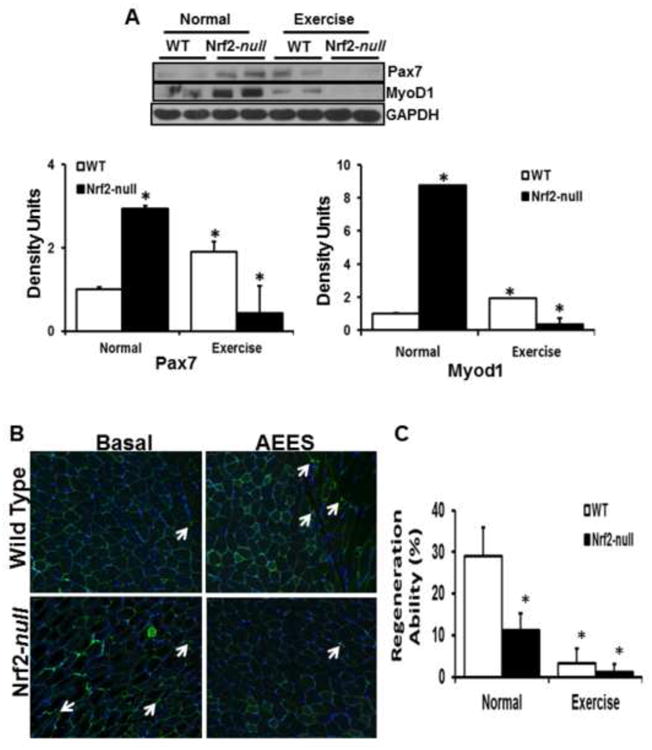
Abrogation of Nrf2 and AEES modulates the expression of stem cell and muscle regeneration factors (PAX7 and MYOD). (A) Representative immunoblots showing significantly diminished PAX7 and MYOD proteins in SM of Nrf2-null versus WT mice in response to AEES (n=4/group; *p<0.05). (B) Immunofluorescence analyses of PAX7 positive myocytes from WT and Nrf2-null mice (n=3 mice/group; *p<0.05 vs. WT-SED). C. Histograms of regeneration ability for WT and Nrf2-null mice were determined by calculating the percent of myocytes with active nuclei. Decreased satellite cell population is evident in Nrf2-null in response to AEES. Green – PAX7 positive; red – laminin and blue – DAPI/nucleus.
Sustained impairment of skeletal muscle regeneration in Nrf2-null mice after post-AEES recovery
Since it is evident that AEES induced oxidative stress impaired the regeneration (PAX7) and differentiation (MyoD1) in aging skeletal muscle of Nrf2 deficient, we next sought to determine whether recovery from AEES normalize these regulatory markers. In the post-AEES recovery experiments, we determined transcript (Fig. 9A-B) and protein levels for PAX7 and MyoD1 (9C). We observed that AEES induced downregulation mRNA levels of PAX7 and MyoD1 were prolonged even after 1 week of recovery from AEES (Fig. 9A-B). However, the protein levels of PAX7 and MyoD1 in WT mice showed a trend of normalization in response to recovery from AEES (compare lane 1 vs 3; Fig. 9C). Notably, down regulation of these proteins were sustained in Nrf2-null mice versus WT (compare lane 3 vs lane 4; Fig. 9C) following recovery from AEES. Overall, these data suggest that abrogation of Nrf2 results in a delayed regeneration of the skeletal muscle.
Figure 9. Sustained impairment of skeletal muscle regeneration in Nrf2-null mice after post-AEES recovery.
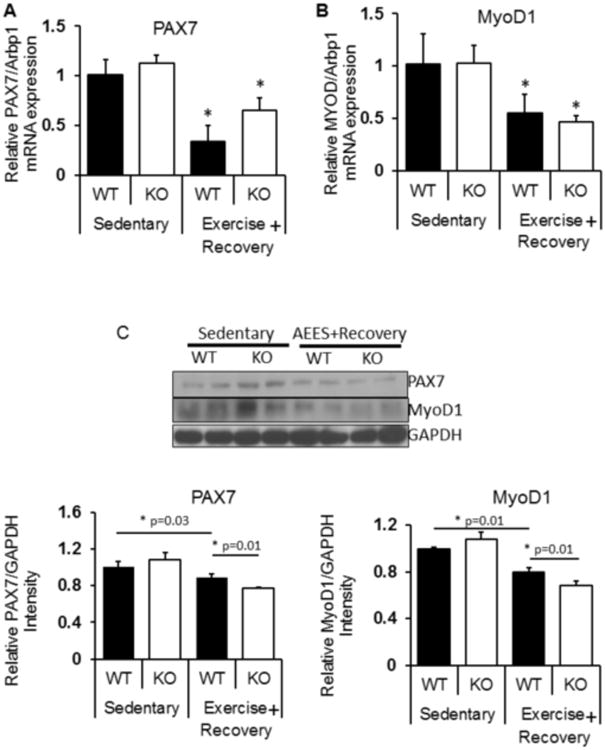
Effect of recovery from AEES on skeletal muscle regeneration. In the post-AEES recovery experiments, we determined transcript (Fig. 9A-B) and protein levels for PAX7 and MyoD1 (9C).
(A-B) RTPCR experiments showing downregulation of mRNA levels (PAX7 and MyoD1) were prolonged even after 1 week of recovery from AEES.
(C) Representative Immunoblots for PAX7 and MyoD1 protein levels in WT mice showed a trend of normalization in response to recovery from AEES (compare lane 1 vs 3; Fig. 9C). Notably, down regulation of these proteins were sustained in Nrf2-null mice versus WT (compare lane 3 vs lane 4; Fig. 9C) following recovery from AEES. (n=4/group; *p<0.05).
Discussion
Oxidative stress associated pathological processes in the skeletal muscles of aged humans has been an important research focus for decades. While Nrf2-Keap1 pathway is one of the critical regulators of intracellular redox homeostasis in skeletal muscle (9, 68, 69), the age-dependent effect of Nrf2 signaling in skeletal muscle is poorly understood. We recently demonstrated that either ablation or age-dependent decline of Nrf2 is coupled with increased oxidative stress and accelerated skeletal muscle degeneration through induction of apoptotic and ubiquitin mediated protein degradation pathways (9). However, the molecular cross talk between Nrf2-signaling and muscle differentiation or stem cell lineage has not been investigated previously in the context of acute stress/aging-mediated oxidative stress. In the present investigation, we demonstrate that (i) abrogation of Nrf2 induces oxidative stress in association with impaired antioxidant defense mechanisms upon aging; (ii) acute endurance exercise stress (AEES) in aged mice differentially regulate the transcription and translation of antioxidants; (iii) PGC1α-dependent mechanisms are activated in response to AEES and (iv) apoptotic signaling overrides PGC1α and AKT dependent cytoprotection/pro-survival associated with impaired stem cell activation and muscle differentiation in Nrf2-null mice. We also demonstrate that abrogation of Nrf2 in combination with oxidative stress aggravates age-associated muscle degeneration and impairs stem cell-based regenerative mechanisms.
Stress-mediated activation of PGC1α is an alternate mechanism to Nrf2 signaling, but it is not sufficient to protect myocytes from aging or oxidative stress-associated injury and apoptosis
Since the discovery of Nrf2 as a potential transcriptional regulator of antioxidant genes, it has been demonstrated that other transcription factors including AP1, PGC1α and NF-kB are also major redox responding factors responsible for transcription of antioxidant genes (53-55). In particular, it has been proposed that PGC1α is critical for the biogenesis of mitochondria and skeletal muscle function (57, 58). In this study, augmentation of PGC1α was observed in both WT and Nrf2-null mice in response to AEES, suggesting this factor responds to AEES independent of Nrf2. Furthermore, it is conceivable that either gain or loss of Nrf2 may not have a direct role on PGC1α function. The role of NF-kB-mediated transcription of antioxidant genes in the context of aging muscle regeneration under forced stress or Nrf2 ablation is not known. Here, we demonstrate that NFkB is not directly involved in antioxidant transcription as indicated by the activation of IkBα and IkBβ, repressors of NFkB in response to severe oxidative stress. The magnitude of increase in IkBα and IkBβ was higher in Nrf2-null mice upon AEES, indicating inhibition of NFkB-dependent cytoprotective mechanisms in skeletal muscles. Taken together, these results demonstrate that PGC1α, at least in part, is an alternative mechanism to Nrf2 signaling in response to severe oxidative stress, aging and AEES.
Apoptotic pathways override the AKT/pro-survival mechanisms under Nrf2 diminution or ablation
Recently, we demonstrated that loss of Nrf2 with aging is associated with increased ubiquitination, lipid peroxidation and activation of apoptotic cascades in the heart (8). Several reports have indicated that oxidative stress and other pathological conditions are strongly correlated with protein degradation and increased cell death (15, 16, 34, 35, 63, 70). However, there is limited information on Nrf2 signaling in skeletal muscle regeneration with aging. Using older (>23 months) WT and Nrf2-null mice, we investigated the molecular pathways involved in these events. We demonstrated that loss of Nrf2 activates pro-survival pathways under AEES, as shown by induction of AKT and pGSK3β, which was accompanied by activation of pp53 dependent cell cycle pathways. This increased AKT expression after AEES may reflect compensatory activation of pro-survival pathways due to stress. Further, the AEES-induced pro-survival mechanisms were greater in Nrf2-null compared to WT skeletal muscle. We expected that activation of antioxidant and pro-survival pathways in response to stress might be associated with diminished apoptotic signals. Therefore, we further analyzed apoptotic pathways under basal and stressed conditions in WT and Nrf2-null mice. At basal conditions, Nrf2-null mice exhibited increased apoptosis as shown by increased BAX, BAD, Caspase-3 and cleaved PARP. These effects were further magnified with AEES in Nrf2-null but not WT mice suggesting that abrogation of Nrf2 increased cell death. This likely account for the significant structure alterations we observed in Nrf2-null skeletal muscle and perhaps functional changes.
Nrf2 signaling is necessary to protect stem cell lineage and muscle differentiation under stress conditions
Effective regeneration of skeletal muscle could occur through activation and proliferation of satellite cells (7, 67, 71). Defects in activation or a decrease in number of satellite cells results in reduced muscle regeneration. It has been reported that redox-dependent regulation is involved in self-renewal of stem cells and myogenic differentiation (72-78). Upon skeletal muscle injury, activated satellite cells rapidly enter the cell cycle and proliferate, augmenting skeletal muscle in the presence of Pax7 (78-82). Further, activated satellite cells possess the ability to induce the downstream genes such as Myf5 and MyoD, which promote differentiation into myoblasts (83, 84). Pax7 expressing satellite cells are necessary for muscle regeneration (85). We postulated that chronic oxidative stress plays a role in stem cell renewal. Our results indicate that loss of Nrf2 signaling is associated with increased oxidative stress over time that facilitates a compensatory activation of stem cells as indicated by increased Pax7 positive cells. This suggests that moderate pro-oxidative states are able to promote expression of stem cell lineage cells even in the absence of Nrf2 signaling, indicating Pax7 can be regulated by an Nrf2-independent pathway. Interestingly, the observed increase in Pax7 and MyoD in WT skeletal muscle in response to AEES suggests that induction of moderate oxidative stress might have a key role in activation of satellite cells and muscle regeneration. However, more intense oxidative stress in Nrf2-null mice induced by AEES is associated with attenuation of Pax7 and MyoD expression. Interestingly, MyoD expression is tightly correlated with Pax7 expression in both Nrf2 abrogation and AEES conditions. Further, Nrf2-null mice displayed a delayed rescue of regeneration after withdrawal of AEES due to overwhelming oxidative stress that is a consequence of loss of Nrf2 and aging. Therefore, we suggest that Nrf2 signaling is necessary to protect stem cells by diminishing harmful oxidative stress conditions. Clearly, while mild oxidative stress favors stem cell and MyoD lineage, severe oxidative stress (Nrf2 abrogation) is detrimental to these mechanisms. Taken together, these findings implicate a role for Nrf2 in the regulation of stem cell lineage and muscle differentiation during aging.
Conclusions
In summary, we identify Nrf2 as a critical component in the transcriptional signaling of antioxidants to maintain skeletal muscle during AEES. Although Nrf2-independent pathways such as PGC1α and AKT are activated in response to AEES, this induction is insufficient to prevent subsequent onset of apoptosis in aging skeletal muscle. Furthermore, significant activation of apoptotic pathways overrides the PGC1α and AKT signaling and promotes cell protein degradation as indicated by increased ubiquitination and lipid peroxidation. In summary, suppression of survival and activation of apoptotic signals along with ubiquitination and oxidation of proteins impair skeletal muscle regeneration through decreasing Pax7 and MyoD lineage. This study supports Nrf2 as a key player in skeletal muscle protection and opens up new avenues for potential therapeutic treatment of aging skeletal muscle disorders.
References
- 1.Nilwik R, Snijders T, Leenders M, Groen BB, van Kranenburg J, Verdijk LB, van Loon LJ. The decline in skeletal muscle mass with aging is mainly attributed to a reduction in type II muscle fiber size. Experimental gerontology. 2013;48:492–498. doi: 10.1016/j.exger.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 2.Barbieri E, Sestili P. Reactive oxygen species in skeletal muscle signaling. Journal of signal transduction. 2012;2012:982794. doi: 10.1155/2012/982794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas DR. Loss of skeletal muscle mass in aging: examining the relationship of starvation, sarcopenia and cachexia. Clinical nutrition. 2007;26:389–399. doi: 10.1016/j.clnu.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Figueiredo PA, Powers SK, Ferreira RM, Appell HJ, Duarte JA. Aging impairs skeletal muscle mitochondrial bioenergetic function. The journals of gerontology Series A, Biological sciences and medical sciences. 2009;64:21–33. doi: 10.1093/gerona/gln048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fulle S, Di Donna S, Puglielli C, Pietrangelo T, Beccafico S, Bellomo R, Protasi F, Fano G. Age-dependent imbalance of the antioxidative system in human satellite cells. Experimental gerontology. 2005;40:189–197. doi: 10.1016/j.exger.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Fulle S, Protasi F, Di Tano G, Pietrangelo T, Beltramin A, Boncompagni S, Vecchiet L, Fano G. The contribution of reactive oxygen species to sarcopenia and muscle ageing. Experimental gerontology. 2004;39:17–24. doi: 10.1016/j.exger.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 7.Zaccagnini G, Martelli F, Magenta A, Cencioni C, Fasanaro P, Nicoletti C, Biglioli P, Pelicci PG, Capogrossi MC. p66(ShcA) and oxidative stress modulate myogenic differentiation and skeletal muscle regeneration after hind limb ischemia. The Journal of biological chemistry. 2007;282:31453–31459. doi: 10.1074/jbc.M702511200. [DOI] [PubMed] [Google Scholar]
- 8.Gounder SS, Kannan S, Devadoss D, Miller CJ, Whitehead KS, Odelberg SJ, Firpo MA, Paine R, 3rd, Hoidal JR, Abel ED, Rajasekaran NS. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PloS one. 2012;7:e45697. doi: 10.1371/journal.pone.0045697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller CJ, Gounder SS, Kannan S, Goutam K, Muthusamy VR, Firpo MA, Symons JD, Paine R, 3rd, Hoidal JR, Rajasekaran NS. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation. pathways in aged skeletal muscle. Biochimica et biophysica acta. 2012;1822:1038–1050. doi: 10.1016/j.bbadis.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. The Journal of biological chemistry. 2010;285:8463–8471. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Satoh T, Okamoto SI, Cui J, Watanabe Y, Furuta K, Suzuki M, Tohyama K, Lipton SA. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muthusamy VR, Kannan S, Sadhaasivam K, Gounder SS, Davidson CJ, Boeheme C, Hoidal JR, Wang L, Rajasekaran NS. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free radical biology & medicine. 2012;52:366–376. doi: 10.1016/j.freeradbiomed.2011.10.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes to cells : devoted to molecular & cellular mechanisms. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 14.Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, Jakel RJ, Johnson JA. Nrf2, a multi-organ protector? FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19:1061–1066. doi: 10.1096/fj.04-2591hyp. [DOI] [PubMed] [Google Scholar]
- 15.Aoi W, Sakuma K. Oxidative stress and skeletal muscle dysfunction with aging. Current aging science. 2011;4:101–109. doi: 10.2174/1874609811104020101. [DOI] [PubMed] [Google Scholar]
- 16.Gianni P, Jan KJ, Douglas MJ, Stuart PM, Tarnopolsky MA. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Experimental gerontology. 2004;39:1391–1400. doi: 10.1016/j.exger.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Warner HR, Hodes RJ, Pocinki K. What does cell death have to do with aging? Journal of the American Geriatrics Society. 1997;45:1140–1146. doi: 10.1111/j.1532-5415.1997.tb05981.x. [DOI] [PubMed] [Google Scholar]
- 18.Cataldi A, Zara S, Rapino M, Zingariello M, di Giacomo V, Antonucci A. p53 and telomerase control rat myocardial tissue response to hypoxia and ageing. European journal of histochemistry : EJH. 2009;53:e25. doi: 10.4081/ejh.2009.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldspink DF, Burniston JG, Tan LB. Cardiomyocyte death and the ageing and failing heart. Experimental physiology. 2003;88:447–458. doi: 10.1113/eph8802549. [DOI] [PubMed] [Google Scholar]
- 20.Zara S, Rapino M, Centurione L, di Giacomo V, Petruccelli G, Cataldi A. Inducible nitric oxide synthase-activated mitochondrial apoptotic pathway in hypoxic and aged rat hearts. Gerontology. 2010;56:544–552. doi: 10.1159/000299105. [DOI] [PubMed] [Google Scholar]
- 21.Bertoni-Freddari C, Fattoretti P, Giorgetti B, Solazzi M, Balietti M, Meier-Ruge W. Role of mitochondrial deterioration in physiological and pathological brain aging. Gerontology. 2004;50:187–192. doi: 10.1159/000076779. [DOI] [PubMed] [Google Scholar]
- 22.Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2004;58:39–46. doi: 10.1016/j.biopha.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Kaufmann JA, Bickford PC, Taglialatela G. Oxidative-stress-dependent up-regulation of Bcl-2 expression in the central nervous system of aged Fisher-344 rats. Journal of neurochemistry. 2001;76:1099–1108. doi: 10.1046/j.1471-4159.2001.00118.x. [DOI] [PubMed] [Google Scholar]
- 24.Marks N, Berg MJ. Recent advances on neuronal caspases in development and neurodegeneration. Neurochemistry international. 1999;35:195–220. doi: 10.1016/s0197-0186(99)00061-3. [DOI] [PubMed] [Google Scholar]
- 25.Santos RX, Correia SC, Zhu X, Smith MA, Moreira PI, Castellani RJ, Nunomura A, Perry G. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer's disease. Antioxidants & redox signaling. 2013;18:2444–2457. doi: 10.1089/ars.2012.5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chopard A, Hillock S, Jasmin BJ. Molecular events and signalling pathways involved in skeletal muscle disuse-induced atrophy and the impact of countermeasures. Journal of cellular and molecular medicine. 2009;13:3032–3050. doi: 10.1111/j.1582-4934.2009.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dirks AJ, Hofer T, Marzetti E, Pahor M, Leeuwenburgh C. Mitochondrial DNA mutations, energy metabolism and apoptosis in aging muscle. Ageing research reviews. 2006;5:179–195. doi: 10.1016/j.arr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Marzetti E, Hwang JC, Lees HA, Wohlgemuth SE, Dupont-Versteegden EE, Carter CS, Bernabei R, Leeuwenburgh C. Mitochondrial death effectors: relevance to sarcopenia and disuse muscle atrophy. Biochimica et biophysica acta. 2010;1800:235–244. doi: 10.1016/j.bbagen.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siu PM. Muscle apoptotic response to denervation, disuse, and aging. Medicine and science in sports and exercise. 2009;41:1876–1886. doi: 10.1249/MSS.0b013e3181a6470b. [DOI] [PubMed] [Google Scholar]
- 30.Tamilselvan J, Jayaraman G, Sivarajan K, Panneerselvam C. Age-dependent upregulation of p53 and cytochrome c release and susceptibility to apoptosis in skeletal muscle fiber of aged rats: role of carnitine and lipoic acid. Free radical biology & medicine. 2007;43:1656–1669. doi: 10.1016/j.freeradbiomed.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 31.Anversa P, Hiler B, Ricci R, Guideri G, Olivetti G. Myocyte cell loss and myocyte hypertrophy in the aging rat heart. Journal of the American College of Cardiology. 1986;8:1441–1448. doi: 10.1016/s0735-1097(86)80321-7. [DOI] [PubMed] [Google Scholar]
- 32.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free radical biology & medicine. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 33.Csiszar A, Podlutsky A, Wolin MS, Losonczy G, Pacher P, Ungvari Z. Oxidative stress and accelerated vascular aging: implications for cigarette smoking. Frontiers in bioscience : a journal and virtual library. 2009;14:3128–3144. doi: 10.2741/3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goi G, Massaccesi L, Baquero Herrera CJ, Musetti C, Ciurlino D, Cusi D, Bertoli S. Oxidative stress in elderly chronic renal failure patients: effects of renal replacement therapies on cell membrane fluidity. Journal of nephrology. 2009;22:630–636. [PubMed] [Google Scholar]
- 35.Hermann J, Gulati R, Napoli C, Woodrum JE, Lerman LO, Rodriguez-Porcel M, Sica V, Simari RD, Ciechanover A, Lerman A. Oxidative stress-related increase in ubiquitination in early coronary atherogenesis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17:1730–1732. doi: 10.1096/fj.02-0841fje. [DOI] [PubMed] [Google Scholar]
- 36.Jejurikar SS, Henkelman EA, Cederna PS, Marcelo CL, Urbanchek MG, Kuzon WM., Jr Aging increases the susceptibility of skeletal muscle derived satellite cells to apoptosis. Experimental gerontology. 2006;41:828–836. doi: 10.1016/j.exger.2006.06.053. [DOI] [PubMed] [Google Scholar]
- 37.Tatsumi R, Liu X, Pulido A, Morales M, Sakata T, Dial S, Hattori A, Ikeuchi Y, Allen RE. Satellite cell activation in stretched skeletal muscle and the role of nitric oxide and hepatocyte growth factor. American journal of physiology Cell physiology. 2006;290:C1487–1494. doi: 10.1152/ajpcell.00513.2005. [DOI] [PubMed] [Google Scholar]
- 38.Young AP, Wagers AJ. Pax3 induces differentiation of juvenile skeletal muscle stem cells without transcriptional upregulation of canonical myogenic regulatory factors. Journal of cell science. 2010;123:2632–2639. doi: 10.1242/jcs.061606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kassar-Duchossoy L, Giacone E, Gayraud-Morel B, Jory A, Gomes D, Tajbakhsh S. Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes & development. 2005;19:1426–1431. doi: 10.1101/gad.345505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lagha M, Rocancourt D, Relaix F. Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Medecine sciences : M/S. 2005;21:801–803. doi: 10.1051/medsci/20052110801. [DOI] [PubMed] [Google Scholar]
- 41.Relaix F, Montarras D, Zaffran S, Gayraud-Morel B, Rocancourt D, Tajbakhsh S, Mansouri A, Cumano A, Buckingham M. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. The Journal of cell biology. 2006;172:91–102. doi: 10.1083/jcb.200508044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435:948–953. doi: 10.1038/nature03594. [DOI] [PubMed] [Google Scholar]
- 43.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochemical and biophysical research communications. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 44.Hoene M, Lehmann R, Hennige AM, Pohl AK, Haring HU, Schleicher ED, Weigert C. Acute regulation of metabolic genes and insulin receptor substrates in the liver of mice by one single bout of treadmill exercise. The Journal of physiology. 2009;587:241–252. doi: 10.1113/jphysiol.2008.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Safdar A, Abadi A, Akhtar M, Hettinga BP, Tarnopolsky MA. miRNA in the regulation of skeletal muscle adaptation to acute endurance exercise in C57Bl/6J male mice. PloS one. 2009;4:e5610. doi: 10.1371/journal.pone.0005610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajasekaran NS, Varadharaj S, Khanderao GD, Davidson CJ, Kannan S, Firpo MA, Zweier JL, Benjamin IJ. Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxidants & redox signaling. 2011;14:957–971. doi: 10.1089/ars.2010.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varadharaj S, Watkins T, Cardounel AJ, Garcia JG, Zweier JL, Kuppusamy P, Natarajan V, Parinandi NL. Vitamin C-induced loss of redox-dependent viability in lung microvascular endothelial cells. Antioxidants & redox signaling. 2005;7:287–300. doi: 10.1089/ars.2005.7.287. [DOI] [PubMed] [Google Scholar]
- 48.Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, Barry WH, Loscalzo J, Odelberg SJ, Benjamin IJ. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 31:90–99. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annual review of pharmacology and toxicology. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 51.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. The Journal of clinical investigation. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. The Journal of experimental medicine. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clark J, Simon DK. Transcribe to survive: transcriptional control of antioxidant defense programs for neuroprotection in Parkinson's disease. Antioxidants & redox signaling. 2009;11:509–528. doi: 10.1089/ars.2008.2241. [DOI] [PubMed] [Google Scholar]
- 54.Marmolino D, Manto M, Acquaviva F, Vergara P, Ravella A, Monticelli A, Pandolfo M. PGC-1alpha down-regulation affects the antioxidant response in Friedreich's ataxia. PloS one. 2010;5:e10025. doi: 10.1371/journal.pone.0010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1996;10:709–720. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 56.Prigione A, Adjaye J. Modulation of mitochondrial biogenesis and bioenergetic metabolism upon in vitro and in vivo differentiation of human ES and iPS cells. The International journal of developmental biology. 2010;54:1729–1741. doi: 10.1387/ijdb.103198ap. [DOI] [PubMed] [Google Scholar]
- 57.Chen SD, Yang DI, Lin TK, Shaw FZ, Liou CW, Chuang YC. Roles of Oxidative Stress, Apoptosis, PGC-1alpha and Mitochondrial Biogenesis in Cerebral Ischemia. International journal of molecular sciences. 2011;12:7199–7215. doi: 10.3390/ijms12107199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santos JM, Tewari S, Goldberg AF, Kowluru RA. Mitochondrial biogenesis and the development of diabetic retinopathy. Free radical biology & medicine. 2011;51:1849–1860. doi: 10.1016/j.freeradbiomed.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cai Y, Wang Q, Ling Z, Pipeleers D, McDermott P, Pende M, Heimberg H, Van de Casteele M. Akt activation protects pancreatic beta cells from AMPK-mediated death through stimulation of mTOR. Biochemical pharmacology. 2008;75:1981–1993. doi: 10.1016/j.bcp.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 60.Gao M, Liang J, Lu Y, Guo H, German P, Bai S, Jonasch E, Yang X, Mills GB, Ding Z. Site-specific activation of AKT protects cells from death induced by glucose deprivation. Oncogene. 2013 doi: 10.1038/onc.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li B, Desai SA, MacCorkle-Chosnek RA, Fan L, Spencer DM. A novel conditional Akt ‘survival switch’ reversibly protects cells from apoptosis. Gene therapy. 2002;9:233–244. doi: 10.1038/sj.gt.3301641. [DOI] [PubMed] [Google Scholar]
- 62.Wang J, Chen Y, Zhang W, Zheng G, Meng S, Che H, Ke T, Yang J, Chen J, Luo W. Akt activation protects liver cells from apoptosis in rats during acute cold exposure. International journal of biological sciences. 2013;9:509–517. doi: 10.7150/ijbs.5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramanathan M, Hassanain M, Levitt M, Seth A, Tolman JS, Fried VA, Ingoglia NA. Oxidative stress increases ubiquitin--protein conjugates in synaptosomes. Neuroreport. 1999;10:3797–3802. doi: 10.1097/00001756-199912160-00014. [DOI] [PubMed] [Google Scholar]
- 64.Cao C, Li Y, Leng Y, Li P, Ma Q, Kufe D. Ubiquitination and degradation of the Arg tyrosine kinase is regulated by oxidative stress. Oncogene. 2005;24:2433–2440. doi: 10.1038/sj.onc.1208454. [DOI] [PubMed] [Google Scholar]
- 65.Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, Marquez VE, Valente S, Mai A, Forcales SV, Sartorelli V, Puri PL. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell stem cell. 2010;7:455–469. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Machida S, Booth FW. Increased nuclear proteins in muscle satellite cells in aged animals as compared to young growing animals. Experimental gerontology. 2004;39:1521–1525. doi: 10.1016/j.exger.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 67.Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–3656. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- 68.Safdar A, deBeer J, Tarnopolsky MA. Dysfunctional Nrf2-Keap1 redox signaling in skeletal muscle of the sedentary old. Free radical biology & medicine. 2010;49:1487–1493. doi: 10.1016/j.freeradbiomed.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 69.Whitman SA, Long M, Wondrak GT, Zheng H, Zhang DD. Nrf2 modulates contractile and metabolic properties of skeletal muscle in streptozotocin-induced diabetic atrophy. Experimental cell research. 2013 doi: 10.1016/j.yexcr.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart (British Cardiac Society) 2007;93:903–907. doi: 10.1136/hrt.2005.068270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 2011;138:3625–3637. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxidative medicine and cellular longevity. 2010;3:23–34. doi: 10.4161/oxim.3.1.10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hosokawa K, Arai F, Yoshihara H, Nakamura Y, Gomei Y, Iwasaki H, Miyamoto K, Shima H, Ito K, Suda T. Function of oxidative stress in the regulation of hematopoietic stem cell-niche interaction. Biochemical and biophysical research communications. 2007;363:578–583. doi: 10.1016/j.bbrc.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 74.Kobayashi CI, Suda T. Regulation of reactive oxygen species in stem cells and cancer stem cells. Journal of cellular physiology. 2012;227:421–430. doi: 10.1002/jcp.22764. [DOI] [PubMed] [Google Scholar]
- 75.Nagano O, Okazaki S, Saya H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene. 2013 doi: 10.1038/onc.2012.638. [DOI] [PubMed] [Google Scholar]
- 76.Ogasawara MA, Zhang H. Redox regulation and its emerging roles in stem cells and stem-like cancer cells. Antioxidants & redox signaling. 2009;11:1107–1122. doi: 10.1089/ars.2008.2308. [DOI] [PubMed] [Google Scholar]
- 77.Pervaiz S, Taneja R, Ghaffari S. Oxidative stress regulation of stem and progenitor cells. Antioxidants & redox signaling. 2009;11:2777–2789. doi: 10.1089/ars.2009.2804. [DOI] [PubMed] [Google Scholar]
- 78.Kuang S, Gillespie MA, Rudnicki MA. Niche regulation of muscle satellite cell self-renewal and differentiation. Cell stem cell. 2008;2:22–31. doi: 10.1016/j.stem.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 79.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, Tempst P, Rudnicki MA, Beier DR, Spiegelman BM. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kuang S, Rudnicki MA. The emerging biology of satellite cells and their therapeutic potential. Trends in molecular medicine. 2008;14:82–91. doi: 10.1016/j.molmed.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 81.Rudnicki MA, Le Grand F, McKinnell I, Kuang S. The molecular regulation of muscle stem cell function. Cold Spring Harbor symposia on quantitative biology. 2008;73:323–331. doi: 10.1101/sqb.2008.73.064. [DOI] [PubMed] [Google Scholar]
- 82.Tedesco FS, Dellavalle A, Diaz-Manera J, Messina G, Cossu G. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. The Journal of clinical investigation. 2010;120:11–19. doi: 10.1172/JCI40373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dey BK, Gagan J, Dutta A. miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Molecular and cellular biology. 2011;31:203–214. doi: 10.1128/MCB.01009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Collins CA, Gnocchi VF, White RB, Boldrin L, Perez-Ruiz A, Relaix F, Morgan JE, Zammit PS. Integrated functions of Pax3 and Pax7 in the regulation of proliferation, cell size and myogenic differentiation. PloS one. 2009;4:e4475. doi: 10.1371/journal.pone.0004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McCarthy JJ, Mula J, Miyazaki M, Erfani R, Garrison K, Farooqui AB, Srikuea R, Lawson BA, Grimes B, Keller C, Van Zant G, Campbell KS, Esser KA, Dupont-Versteegden EE, Peterson CA. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development. 2011;138:3657–3666. doi: 10.1242/dev.068858. [DOI] [PMC free article] [PubMed] [Google Scholar]