

Abstract
Adenosine receptors (ARs) and P2Y receptors for purine and pyrimidine nucleotides have widespread distribution and regulate countless physiological processes. Various synthetic ligands are in clinical trials for treatment of inflammatory diseases, pain, cancer, thrombosis, ischemia, and other conditions. The methanocarba (bicyclo[3.1.0]hexane) ring system as a rigid substitution for ribose, which maintains either a North (N) or South (S) conformation, tends to preserve or enhance the potency and/or selectivity for certain receptor subtypes. This review summarizes recent developments in the synthetic approaches to these biologically important nucleoside and nucleotide analogues.
Introduction
G protein-coupled receptors (GPCRs) serve as the mechanistic target of a large fraction of the pharmaceuticals in current use. Among the numerous members of this superfamily, i.e. corresponding to ~800 human genes including olfactory receptors, are two families consisting of a total of twelve GPCR subtypes that are activated by endogenous nucleosides and nucleotides present in the extracellular medium.1,2 These include four subtypes of adenosine receptors (ARs) and the eight subtypes of P2Y receptors (P2YRs) for purine and pyrimidine nucleotides. The production and release of these small molecule modulators that contain D-ribose, such as adenosine, ATP and UTP, are widespread in the body and involved in countless physiological processes. Numerous studies of the structure activity relationships (SARs) at these receptors have been performed, and in many cases agonists and/or antagonists that are selective for a given subtype have been reported.2–5 At ARs, the subtype selectivity is largely determined by the substitution at the adenine N6 and C2 positions and at ribose. Some of these ligands are currently in clinical trials or preclinical testing for the treatment of inflammatory diseases, pain, cancer, thrombosis, ischemia, and other conditions.1
A general approach to restricting the recognition of these nucleoside and nucleotide derivatives by specific receptor subtypes, as a means of enhancing selectivity, is to rigidify the ribose ring in the form of a bicyclo[3.1.0]hexane (also known as “methanocarba”), which is designed to maintain a receptor-preferred conformation of the ribose ring. The methanocarba ring system can maintain either a North (N) or South (S) conformation, depending on the position of fusion of the cyclopentane and cyclopropane rings. This approach to rigidify the normally flexible ribose moiety was introduced and first applied to antiviral agents by Marquez and coworkers.6a Fig. 1 shows the (N) and (S)-envelope conformations of methanocarba ring systems (P = −12° or 192°, respectively) on the pseudorotational cycle, in relation to the pure (N) and (S)-conformations of P = 0° or 180°, respectively.7 Conformationally locked methanocarba nucleosides and nucleotides have applicability to probing structure–function of nucleic acids in general, as well the specific focus on GPCRs. This substitution has produced great selectivity in substrates and inhibitors of various enzymes, such as polymerases, DNA methyltransferase, and HIV-RT, and when incorporated into DNA.6a Other constrained ring systems, such as locked nucleic acids (LNAs), have been utilized for this purpose,8 but the methanocarba ring system has displayed a generally higher success rate in preserving or enhancing the potency at a given receptor subtype.
Fig. 1.
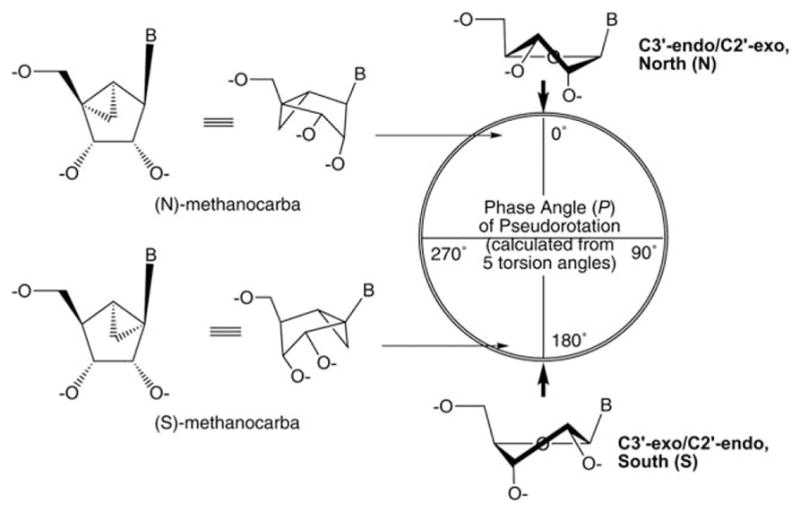
Relationship between the (N)- and (S)-envelope conformations that are maintained in the methanocarba nucleosides and nucleotides and the possible conformations of the ribose ring. The pseudorotational cycle is a mathematical construct derived from all of the torsion angles of ribose to describe the full range of ring twist conformations. The degree of deviation from planarity of the 5-membered ring corresponds to the distance from the center of the cycle. In nature, the observed ribose conformations cluster around the (N) and (S) poles. Thus, the two methanocarba ring systems approximately mimic the favored natural states of ribose. B = nucleobase.
Nucleoside and nucleotide analogues containing a novel methanocarba ring system in place of ribose ring have been systematically explored as ligands for the ARs and P2YRs. In general, the (N)-methanocarba modification enhances affinity of adenosine derivatives at the A3 subtype of ARs and of adenine nucleotide derivatives at the P2Y1 subtype of P2YRs. The A3AR (activated by endogenous adenosine and inosine) is coupled to inhibition of adenylate cyclase, and the P2Y1R (activated by ADP) is coupled to stimulation of phospholipase C. At the A2AAR and A2BAR, which are coupled to stimulation of adenylate cyclase, and certain other P2YRs, the introduction of (N)- methanocarba tends to reduce affinity. Therefore, this modification has led to agonist and antagonists of exceptionally high selectivity at the A3 and P2Y1 subtypes. At the A1AR, P2Y2R and P2Y4R, the (N)-methanocarba-substituted analogues are similar in potency to the native ligands. The only receptor within these families found to prefer the (S) conformation using a methanocarba substitution is the P2Y6R, at which the (N)-methanocarba analogue of the native ligand (UDP) is inactive. At all three of the P2YR subtypes of the P2Y12-like family, which are coupled to inhibition of adenylate cyclase, we have not yet identified the preferred conformation of ribose. Modification of the native ligands in each case with either (N)-methanocarba or (S)-methanocarba rings greatly reduces the potency. Thus, the use of this pair of bicyclic modifications of nucleosides and nucleotides has been effective in the design of selective receptor ligands. This review summarizes recent developments in the synthetic approaches to methanocarba derivatives in the context of their use as definitive pharmacological probes of various ARs and P2Ys.
Synthetic approaches to methanocarba nucleosides
General synthetic methods for the preparation of carbanucleosides, including methanocarba ribonucleosides and 2′-deoxyribonucleosides have been reviewed.6
a (N)-Methanocarba derivatives
The first methanocarba nucleoside was designed7,9 as a 2′,3′-dideoxy nucleoside for conformational comparison with the more planar antiviral agent neplanocin C.10 Marquez and coworker reported a simple method for the synthesis of a series of racemic (N)-methanocarba dideoxynucleosides.7,9 A hydroxyl-directed cyclopropanation of benzyloxy cyclopentenol 1 via a samarium(II) carbenoid intermediate provided the pseudosugar 2 (Scheme 1). Nucleobases were incorporated via a Mitsunobu coupling reaction, following a convergent strategy. Mitsunobu coupling of 2 with 6-chloropurine afforded a mixture of N9–N7 isomers in a 3 : 1 ratio favoring the N9 isomer 4. The pyrimidine derivatives 6 were obtained using the same Mitsunobu conditions. With N-benzoylthymine, a separable 1 : 1 mixture of N- and O-alkylated products 6 and 7 (R=CH3) was obtained, while in the case of N-benzoyluracil (R=H) no O-alkylated product was detected. Compound 4 was transformed into an adenosine derivative by heating with methanolic ammonia followed by catalytic debenzylation to give racemic compound 5. Treatment of compound 6 with BCl3 resulted in removal of the benzyl and benzoyl groups to give racemic pyrimidine derivatives of (N)-methanocarba nucleosides 8.
Scheme 1.

Earliest reported synthesis of methanocarba nucleosides (2′,3′-dideoxy), from cyclopentenol 1 by Marquez and coworkers.7,9 Reagents and conditions: (a) CH2ICl, Sm, HgCl2, THF; (b) 6-chloropurine or N-benzoyluracil, Ph3P, DEAD; (c) (i) NH3/MeOH; (ii) NH4COOH, Pd/C, MeOH; (d) BCL3, CH2Cl2.
Marquez et al. also reported11,12 an enantioselective route for the synthesis of (N)-methanocarba-riboadenosine analogues (Scheme 2). Treatment of (−)-5-O-benzyl-2,3-O-isopropylidene-D-ribonolactone 9 with lithium dimethyl methylphosphonate in THF provided hemiketal 10. Subsequently, the hemiketal group was opened with sodium methoxide, which upon oxidation with a modified Collins reagent provided the diketo phosphonate 11. Intramolecular cyclization of compound 11 in the presence of potassium carbonate and 18-crown-6 ether afforded the desired cyclopentenone 12. A synthesis of intermediate 12 was reported by Chu and coworkers,13a and a later synthesis of the same intermediate by Jeong and coworkers was based on a ring closing metathesis reaction.13b,c Treatment of compound 12 with sodium borohydride in the presence of cerium(III) chloride yielded a cyclopentenol derivative, which underwent a stereoselective Simmon–Smith cyclopropanation to give the key precursor 13. Mesylation of alcohol 13 followed by adenine condensation and deprotection of benzyl and isopropylidine groups provided the enantiomerically pure (N)-methanocarba-adenine derivative 14. (N)-Methanocarba-adenosine derivatives, including those with 3′-amino substitution, were also synthesized from D-ribose using ring closing metathesis.14,15 The most efficient synthesis currently of (N)-methanocarba-ribonucleosides involves ring closing metathesis, as described by the groups of Jeong and Strazewski.13,15
Scheme 2.
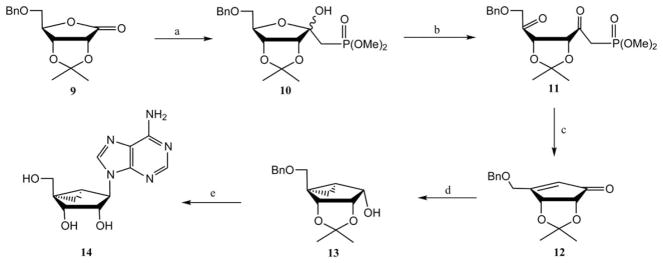
Earliest reported synthesis of (N)-methanocarba-adenosine, from a D-ribonolactone derivative 9.7,9 Reagents and conditions: (a) LiCH2P(O)(OMe)2; (b), (i) NaOMe, MeOH; (ii) CrO3, pyridine; (c) K2CO3, 18-crown-6; (d), (i) NaBH4, CeCl3; (ii) Et2Zn, CH2I2, CH2Cl2; (iii) MsCl, pyridine; (e), (i) adenine, K2CO3, 18-crown-6, DMF; (ii) Pd black, HCOOH, MeOH.
A novel intramolecular cyclopropanation approach for the synthesis of (N)-methanocarba-adenosine analogues was explored in several laboratories.16,17 L-Ribose 15a was converted to an isopropylidene derivative, which upon treatment with iodine in the presence of triphenylphosphine and imidazole provided the iodo derivative 15b (Scheme 3a). The reductive elimination of iodine of compound 15b in the presence of zinc and acetic acid generated the unstable aldehyde 15c, which was immediately treated with ethyl diazoacetate in the presence of tin(II) chloride to give compound 15d. The ketoester 15d was reacted with tosyl azide to afford the diazo derivative 15e, which underwent a thermally induced intramolecular cyclopropanation reaction to give the bicyclo[3.1.0]hexane derivatives 16a and 16b in a 4 : 1 ratio. After chromatographic separation, the reduction of compound 16a with sodium borohydride followed by isomerization of alcohol 17a in the presence trifluoromethane sulfonic acid afforded the glycosyl donor 17b. After isolation by crystallization, compound 17b was subjected to a Mitsunobu condensation with 2,6-dichloropurine to give the nucleoside derivative 18. This intermediate was aminated with 3-chlorobenzylamine followed by 5′-amidation and isopropylidene deprotection to provide the enantiomerically pure (N)-methanocarba-adenine derivative 19.
Scheme 3.

(a) Synthesis starting from L-ribose of a (N)-methanocarba-adenosine analogue 19 that was optimized for binding to the A3AR.16,17 Reagents and conditions: (a) (i) acetone, MeOH; (ii) Ph3P, I2, imidazole (b) Zn, AcOH; (c) N2CHCOOEt, SnCl2 CH2Cl2 (d) TsN3, Et3N, MeCN; (e) CuI, toluene; (f) NaBH4, MeOH; (g) TfOH, CH2Cl2; (h) 2,6-dichloropurine, Ph3P, DIAD; (i) (i) 3-Cl-BnNH2, Et3N; (ii) MeNH2, MeOH; (iii) TFA–H2O, MeOH. (b) Introduction of a 2-arylethynyl substituent, which further increased for binding selectivity at the A3AR.18 Reagents and conditions: (a) (i) 3-Cl-BnNH2, Et3N; (ii) MeNH2, MeOH (b) (i) phenylacetylene, PdCl2(Ph3P)2, CuI, Et3N; (ii) 10%TFA, MeOH. (c) Synthesis starting from D-ribose of 5′-truncated analogues of the (N)-methanocarba-adenosine derivatives that were selective for the A3AR. This modification tended to decrease efficacy of receptor activation, but not binding affinity at this subtype.19 Reagents and conditions: (a) (i) NaBH4, Ce(III)Cl3·7H2O; (ii) Et2Zn, CH2I2; (b) purine derivatives, DIAD, Ph3P; (c) (i) R1NH2, Et3N; (ii) TFA–H2O.
Our group has recently reported a novel class of (N)-methanocarba derivatives having a phenylacetylene moiety at the C2 position that are highly potent and selective agonists of the A3AR.18 This class of compounds was synthesized by using an extension of the methodology shown in Scheme 3a. Compound 20a was derived from (N)-methanocarba sugar 17b through a Mitsunobu condensation with 6-chloro-2-iodo-purine. Treatment of compound 20a with 3-chlorobenzylamine followed by amination of the resulting intermediate gave the amide derivative 20b (Scheme 3b). A Sonogashira coupling of 20b with phenylacetylene and subsequent acid hydrolysis of the resulting compound afforded the C2-phenylacetylene methanocarba derivative 21.
Truncation of adenosine derivatives at the ribose C4′ has variable effects on the pharmacological properties of potency and efficacy at the ARs, and the SAR of truncated methanocarba nucleosides has been explored.21 Extensive modification of this class of molecules provided various lead molecules to achieve selectivity for either the A1AR or the A3AR.19,20 The cyclopentone derivative 22b was synthesized from D-ribose 22a in six steps (Scheme 3c),22a and the glycosyl sugar 22c was formed upon the stereoselective reduction of 22b by sodium borohydride in the presence of cerium(III) chloride followed by a Simmons–Smith cyclopropanation of the resulting alcohol. An alternate synthesis of intermediate 22b involving ring closing metathesis reaction and starting with D-isoascorbic acid had a lower yield and a volatile intermediate.22b Mitsunobu condensation of the compound 22c with various purine bases gave the intermediates of the general formula 23, which upon amination with various amines at the N6 position, and subsequent acid hydrolysis provided truncated methanocarba nucleosides 24.19,20 Alternately, group R of 24 was an arylethynyl substitution, which despite its steric bulk was recognized at the A3AR, especially with small R1 groups.19
Lowary and Li devised a novel synthetic route for the synthesis of (N)-methanocarba equivalents of β-arabinofuranosyl and α-galactofuranosyl amino sugar intermediates (Scheme 4).23 The key step of the synthetic strategy was the base-promoted ring contraction of an epoxy ketone. Thus, the ketal–allylic alcohol 25 was hydrolysed with oxalic acid followed by protection of the hydroxyl group as a methoxymethyl (MOM) ether, and subsequent epoxidation provided the keto epoxy compound 26. The epoxy ketone 26 underwent a ring contraction in the presence of sodium hydroxide, which afforded the methanocarba derivative 27 along with the undesired isomer. Compound 27 was then converted to enone derivative 28 by sequential benzoylation, α-selenation and selenoxide elimination. Enone derivative 28 was epoxidised in the presence of alkaline hydrogen peroxide to give compound 29, which was elaborated to compound 31 by a sequence of reactions. Treatment of compound 31 with O-acetyl-(S)-mandelic acid followed by hydrolysis of the ketal gave the easily separable diastereomers 32a and 32b. Periodate cleavage of the diol, subsequent ester hydrolysis and reduction provided the (N)-methanocarba β-arabinofuranosyl 33a and α-galactofuranosyl 33b amino sugars.
Scheme 4.

Novel synthetic route to the (N)-methanocarba equivalents of β-arabinofuranosyl and α-galactofuranosyl 1-amino sugars.23 Reagents and conditions: (a) (i) oxalic acid, acetone–H2O; (ii) (MeO)2CH2, LiBr, p-TsOH; (iii) m-CPBA; (b) NaOH, EtOH; (c) (i) BzOH, Ph3P, DIAD; (ii) PhSeCl, HCl; (iii) NaIO4; (d)H2O2, NaOH; (e) (i) H2SO4, (ii) BzCl, pyridine; (iii) NaBH4; (f) (i) (PhO)2PON3, Ph3P, DIAD; (ii) MeONa, MeOH; (iii) (MeO)2CMe2, p-TsOH; (g) (i) O-acetyl-(S)-mandelic acid, DCC, DMP; (ii) AcOH–H2O; (h) (i) NaIO4; (ii) MeONa; (iii) NaBH4; (iv) H2, Pd/C.
(N)-Methanocarba-2′-deoxy purine analogue 40 was synthesized by a convergent approach from compound 34 (derived from 12) as depicted in Scheme 5.24 Regioselective opening of the isopropylidene group in the presence of trimethylaluminum and selective silylation of the allylic alcohol gave compound 35. Treatment of compound 35 with carbon disulfide in the presence of sodium hydride gave xanthate ester 36, which underwent deoxygenation in the presence of TBTH and AIBN under Barton’s condition and subsequent desilylation to provide the precursor 37. The unmasked hydroxyl derivative 37 was cyclopropanated in the presence of Sm–HgCl2 and chloro-iododo-methane to give the glycosyl donor 38, which was directly coupled with 6-chloropurine under Mitsunobu conditions to afford the protected nucleoside intermediate 39. Ammonolysis of compound 39 and simultaneous removal of benzyl and tert-butyl groups provided the (N)-methanocarba-2′-deoxy adenine derivative 40.
Scheme 5.
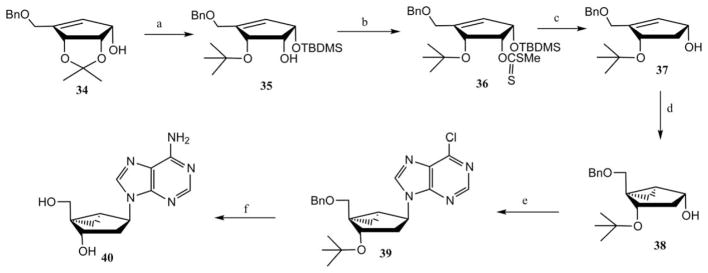
A convergent approach to the synthesis of (N)-methanocarba-2′-deoxyadenosine.24 Reagents and conditions: (a) (i) AlMe3, CH2Cl2; (ii) TBDMSCl, imidazole; (b) CS2, NaH, MeI; (c) (i) TBTH, AIBN; (ii) TBAF; (d) Sm, HgCl2, CH2ICl; (e) 6-chloropurine, Ph3P, DEAD; (f) (i) NH4OH, dioxane; (ii) BCl3, CH2Cl2.
Marquez and coworkers have also developed another simple and efficient route for the synthesis of (N)-methanocarba-2′-deoxy nucleoside derivatives25 from an inexpensive starting material, ethyl acetoacetate (Scheme 6), with intramolecular olefin-ketocarbene cycloaddition and lipase-catalysed resolution as the key steps. Thus, compound 41 was treated with LDA in the presence of acrolein, and the resulting alcohol was silylated with TBDPSCl to give silyl derivative 42. Treatment of compound 42 with p-toluenesulfonyl azide provided the carbene precursor 43. Intramolecular cyclopropanation of 43, which presumably proceeds via a copper-carbenoid intermediate under thermolysis, generated a chromatographically separable mixture of compounds (±)-44 and (±)-45. Sequential reduction of compound 45 with sodium borohydride followed by lithium aluminium hydride afforded compound 46. Surprisingly, the simultaneous reduction of both keto and ester groups of compound 45 with diisobutyl aluminium hydride (DIBAL) resulted a mixture of epimeric alcohols. Treatment of compound (±)-46 with a commercially available Lipase PS-C “Amano” I from Pseudomonas cepacia, in the presence of a large excess of vinyl acetate was able to resolve the two enantiomers giving (−)-47 as a monoacetate and (+)-48 as a diacetate. Deblocking of the silyl group of compound (+)-48 with (NH4)HF2 gave (+)-49 having high optical purity (99% e.e.). A Mitsunobu condensation under standard conditions of compound (+)-49 with 2-acetamido-6-chloropurine afforded (+)-50, which was converted to a (N)-methanocarba-2′-deoxyguanosine derivative (+)-51 by heating with 2 N HCl at 80 °C. Chiral resolution of (N)-methanocarba-2′-deoxy pyrimidine nucleosides was also accomplished using the same synthetic strategy.
Scheme 6.

Synthesis from ethyl acetoacetate 41 of enantiomerically pure (N)-methanocarba-2′-deoxyguanosine 51 using an enzymatic resolution.25 Reagents and conditions: (a) (i) acrolein, LDA; (ii) TBDPSCl, imidazole; (b) TsN3, Et3N; (c) CuSO4, cyclohexane; (i) NaBH4, CH2Cl2–MeOH; (ii) LiAlH4, Et2O (e) vinyl acetate, Lipase PS-C; (f) (NH4)HF2, DMF–H2O; (g) 2-acetamide-6-chloropurine, Ph3P, DEAD; (h) 2N HCl, THF, 80 °C.
b (S)-Methanocarba derivatives
The synthesis of (S)-methanocarba nucleosides is more challenging than (N)-methanocarba because the nucleobase has to be attached directly to a tertiary carbon of the bicyclo[3.1.0]hexane moiety. This structural template ruled out a convergent strategy, and the synthesis must proceed from an appropriately protected bicylic amine. Altmann and coworkers have reported26 the first synthesis of a (S)-methanocarba nucleoside, which was a (S)-methanocarba-2′-deoxythymidine 57. Thus, the chiral bicylic lactone 52 was opened to give the γ-bromo ester in the presence of TMSBr, and subsequent protection as a TBDMS ether gave compound 53 (Scheme 7). Treatment of compound 53 with potassium tert-butoxide in tert-butanol created the cyclopropane ring derivative 54, which gave the acid derivative 55 upon saponification. Compound 55 underwent a one-pot three-step procedure involving formation of a carboxylic acid azide. This consisted of an in situ Curtius rearrangement followed by quenching of the ensuing isocyanate with benzyl alcohol, to provide a benzyloxycarbonyl-protected amine, which was hydrogenated in the presence of palladium-carbon to afford the key amino sugar derivative 56. Construction of the thymine nucleobase was accomplished by reaction of 56 with β-methoxy α-methacryloyl-isocyanate and subsequent cyclization of the resulting acryloyl-urea and deprotection of the silyl group simultaneously under acidic condition to furnish (S)-methanocarba- 2′-deoxythymidine 57.
Scheme 7.
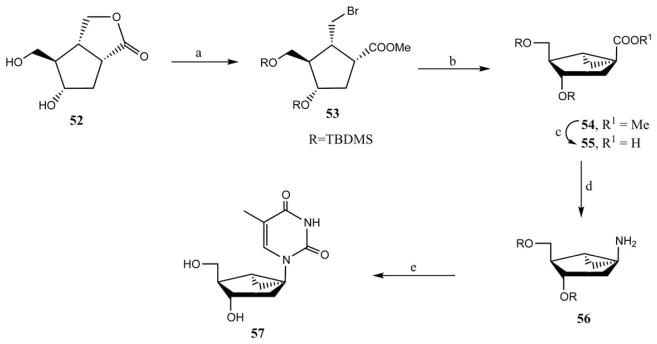
Synthesis of (S)-methanocarba-2′-deoxythymidine 57, which has been utilized for enzymatic, nonreceptor targets.26 Reagents and conditions: (a) (i) TMSBr, ZnBr2, MeOH; (ii) N-TBDMS, N-methylacetamide, DMF; (b) tBuOK, tBuOH; (c) KOH, EtOH; (d) (i) DPPA, Et3N, toluene, heat; (ii) BnOH, heat; (iii) H2, Pd/C, toluene; (e) (i) MeOCH=C(Me)C(O)NCO, CH2Cl2; (ii) 0.2N HCl/EtOH–H2O.
Marquez and coworkers utilized an intramolecular olefinketocarbene cycloaddition strategy for the synthesis of an (S)-methanocarba- riboadenosine analogue27 as shown in Scheme 8. Esterfication of (E)-butanol 58 and p-methoxybenzyloxyacetic acid in the presence of DCC gave the glycolate ester 59, which underwent an enolate Claisen-Ireland rearrangement in the presence of LDA and TMSCl to afford the desired γ,δ-unsaturated acid diastereomer 60 mixed with its isomer in a 95 : 5 ratio. The favorable outcome was explained in term of a chair-like transition state that was controlled by the trans-geometry of the starting olefin and the local geometry of the enolate induced by the coordinated metal. A Dieckmann condensation of methyl 2-lithioacetate with the carbonyldiimidazole-activated acid 60, produced the β-ketoester 61, which upon diazo transfer with tosyl azide in the presence of triethylamine gave the diazo derivative 62. Copper acetylacetonate-catalysed thermolysis of compound 62 resulted in an intramolecular olefin keto-carbene cycloaddition and provided a separable mixture of bicylo[3.1.0]hexane derivatives 63 and 64 in a 2 : 1 ratio. Sodium borohydride reduction of compound 64 gave exclusively the alcohol 65, which upon the simultaneous deprotection of the PMB group and protection of the diol as an acetonide in the presence of copper(II) sulfate and a catalytic amount of sulfuric acid afforded the precursor 66. Saponification of the ester of 66 and a one-pot three-step Curtius rearrangement as in Scheme 7 gave the amine derivative 67, which was elaborated to the racemic (S)-methanocarba riboadenosine derivative 68 by a sequence of reactions.27
Scheme 8.

Synthesis from a trans-1,4-butenediol derivative 58 of racemic (S)-methanocarba-adenosine 67.27 Reagents and conditions: (a) PMBOCH2COOH, DCC, DMAP; (b) (i) LDA, THF; (ii) TMSCl, Et3N; (c) (i) carbonyldiimidazole, THF; (ii) LiCH2COOMe, THF; (d) TsN3, Et3N; (e) Cu(AcAc)2, cyclohexane; (f) NaBH4, MeOH–CH2Cl2; (g) CuSO4, cat. H2SO4, acetone.
In collaboration with the Marquez group, we recently reported a synthesis of enantiomerically pure (S)-methanocarba-uridine and its nucleotide derivatives (Scheme 9).28 The intermediate 70 was synthesized from D-ribose 69 in seven steps, following the same method as for its enantiomer 17b from L-ribose 15a as presented in Scheme 3a. Treatment of compound 70 with trfluoromethanesulfonic anhydride (Tf2O) in the presence of pyridine gave the triflate derivative 71, which was immediately converted to a cyano analogue 72 by an SN2 reaction with LiCN. A controlled catalytic hydrogenation of the nitrile in the presence of palladium in aqueous acetic acid provided the unstable aldehyde 73, which immediately underwent a sodium borohydride reduction and subsequent silyl protection of the resulting alcohol to afford silyl ether 74. Compound 74 was transformed to the key amine derivative 75, which was further elaborated to (S)-methanocarba-uridine 76 following the same method as in Scheme 7.
Scheme 9.
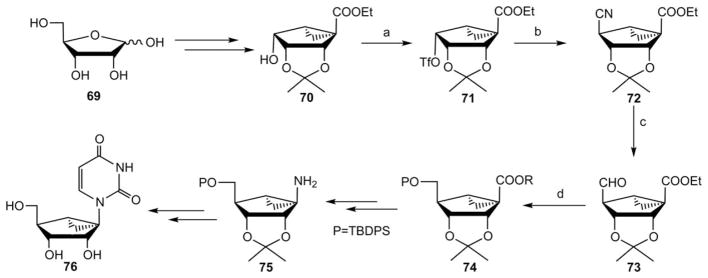
Synthesis from a D-ribose 69 of enantiomerically pure (S)-methanocarba-uridine 76.28 Reagents and conditions: (a) Tf2O, pyridine, CH2Cl2; (b) LiCN, DMF; (c) H2/Pd, H2O–AcOH; (d) (i) NaBH4, MeOH; (ii) TBDPSCl, imidazole, DMF.
Application of methanocarba templates to AR and P2YR ligands
Ring-constrained nucleosides have been used to define conformational preferences of adenine nucleosides at the ARs and adenine and uracil nucleotides at the P2YRs. Medicinal chemists frequently utilize the approach of conformationally constraining otherwise flexible molecules to probe the active conformation and to increase ligand affinity by overcoming the energy barriers needed to attain this preferred conformation. The focus on conformational factors of the ribose or ribose-like moiety allows the introduction of general modifications that lead to enhanced potency and selectivity at certain subtypes of these receptors.
Jacobson and coworkers were the first to report29 that a methanocarba modification of the ribose moiety to incorporate ring constraints is a general approach to the design of AR agonists having favorable pharmacodynamic properties. While simple carbocyclic substitution of adenosine derivatives often diminishes potency at ARs, methanocarba-adenosine analogue have preserved or enhanced pharmacological properties as agonists at two AR subtypes. In such methanocarba analogues, the fused cyclopropane moiety constrains the pseudosugar ring of nucleoside to either a Northern (N)- or Southern (S)-conformation, which helps to define the role of sugar puckering in stabilizing the active AR-bound conformation, and thereby allows identification of a favoured isomer.
In the binding assays at A1, A2A and A3ARs, (N)-methanocarba-adenosine had a higher affinity than the (S)-analogue, particularly at human A3AR [(N)/(S)—affinity ratio of 150]. (N)-methanocarba analogues of various N6 substituted adenosine derivatives, including N6-cyclopentyl and N6-(3-iodobenzyl), in which parent compounds are potent agonists at either A1 or A3ARs, receptively, were synthesized (Fig. 2). The N6-cyclopentyl derivatives were A1AR selective and maintained high efficacy at recombinant human, but not rat brain A1ARs, indicated in a functional assay based on stimulation of binding of [35S]GTP-γ-S. The 2-chloro analogue 80 (MRS 1761), was a selective full agonist at the human A1AR. (N)-Methanocarba-N6-(3-iodobenzyl) adenosine 81 and its 2-chloro derivative 82 (MRS1760) had equilibrium inhibition constant (Ki) values of 4.1 and 2.2 nM at the A3AR, respectively, and were highly selective partial agonists. Partial agonism combined with high functional potency at A3ARs (EC50 < 1 nM) may lead to tissue selectivity.
Fig. 2.

N6 and C2 substituted adenosine derivatives that act as prototypical AR ligands in the ribose (77, 78) and (N)-methanocarba (79–82) series.
After finding the enhancement of potency and selectivity in ligand binding to ARs by a substitution of the ribose ring with a methanocarba moiety, a series of new (N)-methanocarba nucleoside was designed and synthesized30 by modifying the 5′-position of the methanocarba sugar along with N6 position of the purine as shown in Fig. 3. The N6-methyl derivatives 83 and 84 having 4′-hydroxymethyl were selective in binding to the A3AR in comparison to the A1 by 12- and 39-fold, respectively, while compound 85 having a 5′-uronamide displayed 4-fold increased A3AR affinity over compound 84 and consequently 130-fold selectivity in comparison to the A1AR. The 5′-uronamide modified N6-(3-halobenzyl) analogues 19 (MRS3558), 89 (MRS1898) and 91 maintained the approximate affinities associated with the corresponding 9-ribosides at A1 and A3ARs and therefore selectivity for the A3AR. The corresponding 5′-truncated N6-(3-halobenzyl) analogues 88 and 90 (general formula 24) maintained affinity at the A3AR but had reduced efficacy. The 3-bromo derivatives 90 (MRS5147) and 91 (MRS3581) were reported as [87Br]-labeled ligands for in vivo imaging of the A3AR.31 The 2′-deoxynucleosides derivatives 92 and 93 having an N6-cyclopentyl group bound weakly to ARs and were nonselective, while compounds 86 (MRS3630) and 87 having a 5′-uronamide and a N6-cyclopentyl or bicycloalkyl group had mixed A1 and A3AR selectivity.32 This clearly indicated that a 2′-hydroxy group or 5′-uronamide are important for receptor recognition, and 2′-deoxy analogues tend to be partial A3AR agonists. All of the N6-modified (N)-methanocarba-adenosine-5′-uronamide derivatives had full A3AR agonist efficacy.
Fig. 3.

Second-generation adenosine derivatives in the (N)-methanocarba series that were explored as AR ligands.
The (N)-methanocarba modification was introduced into known purine receptor ligands containing a 6-oxo substitution and studied further at the ARs by Jacobson and coworkers.30 The (N)-methanocarba purine nucleosides 94–98 (Fig. 4) and nucleotides 103–108 (Fig. 5) containing alternate nucleobases to adenine were synthesized by conventional methods. While inosine itself displayed a measurable affinity at the A3AR and was even suggested to be an endogenous ligand for this AR, the corresponding (N)-methanocarba analogue 94 bound weakly. However introduction of a 5′-methyluronamide group 95 slightly increased the affinity. The truncated (N)-methanocarba purine analogues 99 (MRS5474) and 100 (MRS5719) showed selectivity as A1AR full agonist and partial agonist, respectively. 20 The 2-phenylalkynyl derivative 21 in the truncated series showed selectivity as an A3AR antagonist,19,21 and the related 5′-methyluronamides 101 (MRS5697) and 102 (MRS5698) were highly selective agonists at both human and mouse A3ARs.18
Fig. 4.

Purine nucleoside derivatives in the (N)-methanocarba series that were explored as AR ligands. Both nonadenosine (94–98) and adenosine derivatives (99–102) are shown.
Fig. 5.

Purine nucleotide derivatives in the (N)-methanocarba series that were explored as AR and P2YR ligands. Nonadenine purines (103–98), adenine derivatives (109–111) and a uracil derivative 112 are shown.
Fig. 5 shows various nucleotides that have increased potency and or selectivity, either at P2YRs or at ARs, based on the presence of a (N) or (S) methanocarba ring. The inosine-5′-monophosphate derivative 103 was completely inactive at the A3AR, while the dinucleotide 105 displayed binding at A3AR with a Ki value of 22 μM.33 Dimerization of an inactive ligand might increase the affinity by bridging two sites on a receptor. It is unusual for such a highly anionic molecule to bind to an AR, which may be the result of an enhancing effect of the (N)-methanocarba ring on A3AR affinity. The (N)-methanocarbaguanosine analogue 96 bound very weakly at the A3AR, and its corresponding triphosphate derivative 106 was completely inactive. The 1,3-dibutylxanthine-7-riboside 97 was equipotent at A1 and A3 AR and inactive at A2AAR, whereas introduction of 5′-methyluronamide 98 decreased A1AR affinity by 8-fold and increased A3AR affinity by 2-fold. The xanthine-7-riboside-5′-triphosphate analogue 107 was inactive at the A3AR. (N)-Methanocarba nucleotide derivatives provide high selectivity for P2Y1Rs, as in antagonists 109 (MRS2279) and 110 (MRS2500) or agonist 111 (MRS2365), and maintain affinity at certain other P2YRs, such as P2Y2, while a South (S) conformation of UDP and its analogues increases potency with respect to the native riboside at the P2Y6R (as in potent agonist MRS2795 112).34
Conclusions
ARs and P2YRs have become important pharmaceutical targets for treating diverse chronic and acute diseases. The methanocarba (bicyclo[3.1.0]hexane) ring system, as a substitution for ribose, may maintain a receptor-preferred conformation, and thereby preserve or enhance the potency and/or selectivity for certain receptor subtypes. North (N) methanocarba derivatives provide selectivity for the A3 and P2Y1Rs and maintain affinity at the A1AR, while a South (S) conformation is highly preferred at the P2Y6R. No receptor ligands that are methanocarba analogues are yet in clinical trials, but some analogues are widely used as pharmacological probes. Although the synthetic approaches to these important nucleoside and nucleotide analogues require many steps, methods are being developed to make these feasible for drug development.
Acknowledgments
Support from the NIDDK Intramural Research Program is acknowledged.
Abbreviations
- AIBN
Azobisisobutyronitrile
- AR
Adenosine receptor
- DCC
Dicyclohexylcarbodiimide
- DIBAL
Diisobutyl aluminium hydride
- DPPA
Diphenylphosphoryl azide
- GPCR
G protein-coupled receptor
- LDA
Lithium diisopropylamide
- MOM
Methoxymethyl
- MRS1760
(1′S,2′R,3′S,4′R,5′S)-4′-{2-Chloro-6-[(3-iodophenylmethyl)amino]purin-9-yl}-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol
- MRS1761
2-Chloro-(N)-methanocarba-N6-(cyclopentyl) adenosine
- MRS2279
(1′R,2′S,4′S,5′S)-4-(2-Chloro-6-methylaminopurin- 9-yl)-1-[(phosphato)-methyl]-2-(phosphato)-bicyclo[3.1.0]hexane
- MRS2500
(1′R,2′S,4′S,5′S)-4-(2-Iodo-6-methylaminopurin- 9-yl)-1-[(phosphato)-methyl]-2- (phosphato)-bicyclo[3.1.0]hexane
- MRS3558
(1′S,2′R,3′S,4′R,5′S)-4′-{2-Chloro-6-[(3- chlorophenylmethyl)amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo[3.1.0]hexane- 2,3-diol
- P2YR
P2Y receptor
- PMB
p-Methoxybenzyl
- TBTH
tri-n-butyltin hydride
- TBDPS
tert-Butyldiphenylsilyl
- THF
tetrahydrofuran
- TBDMS
tert-Butyldimethylsilyl
- TMS
Trimethylsilyl
Biographies

Dilip K. Tosh, Ph.D. is a Visiting Fellow at the Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, NIH in Bethesda, Maryland, USA. He received his Ph.D. in Organic Chemistry from the Indian Institute of Technology, Bombay, under the supervision of Prof. V. K. Singh. Subsequently he joined as a postdoc in the laboratory of Prof. Lak Shin Jeong, at Ewha Womans University, Seoul, Korea, where he worked on Medicinal Chemistry and discovery employing nucleoside chemistry. Currently he works on drug discovery based on G protein-coupled receptors (GPCRs), in particular for adenosine and P2Y receptors under the direction of Dr Kenneth A. Jacobson.

Kenneth A. Jacobson, Ph.D. is Chief of the Laboratory of Bioorganic Chemistry and the Molecular Recognition Section at the National Institute of Diabetes and Digestive and Kidney Diseases, NIH in Bethesda, Maryland, USA. He is a medicinal chemist with interests in the structure and pharmacology of G protein-coupled receptors (GPCRs), in particular receptors for adenosine and for purine and pyrimidine nucleotides. He was inducted into the Medicinal Chemistry Hall of Fame of the American Chemical Society in 2009. Several adenosine agonists from his laboratory are in advanced clinical trials for autoimmune inflammatory diseases and cancer.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. Pharmacol Rev. 2011;63:1. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.von Kügelgen I, Harden TK. Adv Pharmacol. 2011;61:373. doi: 10.1016/B978-0-12-385526-8.00012-6. [DOI] [PubMed] [Google Scholar]
- 3.Müller CE, Jacobson KA. Biochim Biophys Acta, Biomembr. 2011;1808:1290. doi: 10.1016/j.bbamem.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baraldi PG, Preti D, Borea PA, Varani K. J Med Chem. 2012;55:5676. doi: 10.1021/jm300087j. [DOI] [PubMed] [Google Scholar]
- 5.Brunschweiger A, Müller CE. Curr Med Chem. 2006;13:289. doi: 10.2174/092986706775476052. [DOI] [PubMed] [Google Scholar]
- 6.(a) Marquez VE. In: Modified Nucleosides: In Biochemistry, Biotechnology and Medicine. Herdewijn P, editor. ch 12. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2008. pp. 307–341. [Google Scholar]; (b) Agrofoglio LA, Challand SR. Acyclic, Carbocyclic and L-nucleosides. Kluwer Academic; Dordrecht: 1998. p. 174. [Google Scholar]; (c) Agrofoglio LA, Suhas E, Farese A, Condom R, Challand SR, Earl RA, Guedj R. Tetrahedron. 1994;50:10611. [Google Scholar]; (d) Crimmins MT. Tetrahedron. 1998;54:9229. [Google Scholar]
- 7.Rodriguez JB, Marquez VE, Nicklaus MC, Mitsuya H, Barchi JJ. J Med Chem. 1994;37:3389. doi: 10.1021/jm00046a024. [DOI] [PubMed] [Google Scholar]
- 8.Ravn J, Qvortrup K, Rosenbohm C, Koch T. Bioorg Med Chem. 2007;15:5440. doi: 10.1016/j.bmc.2007.05.056. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez JB, Marquez VE, Nicklaus MC, Barchi JJ. Tetrahedron Lett. 1993;34:6233. [Google Scholar]
- 10.Kinoshita K, Yaginuma S, Hayashi M, Nakatsu K. Nucleosides Nucleotides. 1985;4:661. [Google Scholar]
- 11.(a) Marquez VE, Lim MI, Tseng CHK, Markovac A, Priest MA, Khan MS, Kaskar B. J Org Chem. 1988;53:5709. doi: 10.1021/jm00117a004. [DOI] [PubMed] [Google Scholar]; (b) Lim MI, Marquez VE. Tetrahedron Lett. 1983;24:5559. [Google Scholar]; (c) Altenbach HJ, Holzapfel W, Smerat G, Finkler SH. Tetrahedron Lett. 1985;26:6329. [Google Scholar]
- 12.Jeong LS, Marquez VE, Yuan CS, Borchardt RT. Heterocycles. 1995;12:2651. [Google Scholar]
- 13.(a) Wang P, Agrofoglio LA, Newton MG, Chu CK. J Org Chem. 1999;64:4173. [Google Scholar]; (b) Choi WJ, Moon HR, Kim HO, Yoo BN, Lee JA, Shin DH, Jeong LS. J Org Chem. 2004;69:2634. doi: 10.1021/jo0356762. [DOI] [PubMed] [Google Scholar]; (c) Lee JA, Kim HO, Tosh DK, Moon HR, Kim S, Jeong LS. Org Lett. 2006;8:5081. doi: 10.1021/ol061959f. [DOI] [PubMed] [Google Scholar]
- 14.Lee K, Cass C, Jacobson KA. Org Lett. 2001;3:597. doi: 10.1021/ol006999c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michel BY, Strazewski P. Chem–Eur J. 2009;15:6244. doi: 10.1002/chem.200802629. [DOI] [PubMed] [Google Scholar]
- 16.Joshi BV, Melman A, Mackman RL, Jacobson KA. Nucleosides, Nucleotides Nucleic Acids. 2008;27:279. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Gallos JK, Koftis TV, Massen ZS, Dellios CC, Mourtzinos IT, Coutouli-Argyropoulou E, Koumbis AK. Tetrahedron. 2002;58:8043. [Google Scholar]; (b) Joshi BV, Moon HR, Fettinger JC, Marquez VE, Jacobson KA. J Org Chem. 2005;70:439. doi: 10.1021/jo0487606. [DOI] [PubMed] [Google Scholar]
- 18.Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. J Med Chem. 2012;55:4847. doi: 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tosh DK, Paoletta S, Phan K, Gao ZG, Jacobson KA. ACS Med Chem Lett. 2012;3:596. doi: 10.1021/ml300107e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Tosh DK, Paoletta S, Deflorian F, Phan K, Moss SM, Gao ZG, Jiang X, Jacobson KA. J Med Chem. 2012;55:8075. doi: 10.1021/jm300965a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tosh DK, Phan K, Deflorian F, Wei Q, Gao ZG, Jacobson KA. ACS Med Chem Lett. 2011;2:626. doi: 10.1021/ml200114q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Tosh DK, Chinn M, Yoo KL, Kang LS, Luecke H, Gao ZG, Jacobson KA. Bioorg Med Chem. 2010;18:508. doi: 10.1016/j.bmc.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Melman A, Wang B, Joshi BV, Gao ZG, de Castro S, Heller CL, Kim SK, Jeong LS, Jacobson KA. Bioorg Med Chem. 2008;16:8546. doi: 10.1016/j.bmc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Moon HR, Choi WJ, Kim HO, Jeong LS. Tetrahedron: Asymmetry. 2002;13:1189. [Google Scholar]; (b) Choi WJ, Park JG, Yoo SJ, Kim HP, Moon HR, Chun MW, Jung YH, Jeong LS. J Org Chem. 2001;66:6490. doi: 10.1021/jo015733w. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Lowary TL. Org Lett. 2008;10:881. doi: 10.1021/ol703041y. [DOI] [PubMed] [Google Scholar]
- 24.(a) Siddiqui MA, Ford H, George C, Marquez VE. Nucleosides, Nucleotides Nucleic Acids. 1996;15:235. [Google Scholar]; (b) Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang J, Wagner RW, Matteucci MD. J Med Chem. 1996;39:3739. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- 25.(a) Yoshimura Y, Moon HR, Choi Y, Marquez VE. J Org Chem. 2002;67:5938. doi: 10.1021/jo020249u. [DOI] [PubMed] [Google Scholar]; (b) Moon HR, Ford H, Marquez VE. Org Lett. 2000;2:3739. doi: 10.1021/ol000238s. [DOI] [PubMed] [Google Scholar]
- 26.Altmann KH, Imwinkelrled R, Kesselring R, Rihs G. Tetrahedron Lett. 1994;35:7625. [Google Scholar]
- 27.Shin KJ, Moon HR, George C, Marquez VE. J Org Chem. 2000;65:2172. doi: 10.1021/jo9917691. [DOI] [PubMed] [Google Scholar]
- 28.Melman A, Zhong M, Marquez VE, Jacobson KA. J Org Chem. 2008;73:8085. doi: 10.1021/jo801224j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobson KA, Ji XD, Li AH, Melman N, Siddiqui MA, Shin KJ, Marquez VE, Ravi RG. J Med Chem. 2000;43:2196. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee K, Ravi G, Ji XD, Marquez VE, Jacobson KA. Bioorg Med Chem Lett. 2001;11:1333. doi: 10.1016/s0960-894x(01)00213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiesewetter DO, Lang L, Ma Y, Bhattacharjee AK, Gao ZG, Joshi BV, Melman A, de Castro S, Jacobson KA. Nucl Med Biol. 2009;36:3. doi: 10.1016/j.nucmedbio.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobson KA, Gao ZG, Tchilibon S, Duong HT, Joshi BV, Sonin D, Liang BT. J Med Chem. 2005;48:8103. doi: 10.1021/jm050726b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ravi G, Lee K, Ji XD, Kim HS, Soltysiak K, Marquez VE, Jacobson KA. Bioorg Med Chem Lett. 2001;11:2295. doi: 10.1016/s0960-894x(01)00450-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg AK, Erlinge D, Malmsjö M, Boyer JL, Harden TK, Jacobson KA. J Med Chem. 2002;45:208. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim HS, Ohno M, Xu B, Kim HO, Choi Y, Ji XD, Maddileti S, Marquez VE, Harden TK, Jacobson KA. J Med Chem. 2003;46:4974. doi: 10.1021/jm030127+. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Maruoka H, Barrett MO, Ko H, Tosh DK, Melman A, Burianek LE, Balusubramanian R, Berk B, Costanzi X, Harden TK, Jacobson KA. J Med Chem. 2010;53:4488. doi: 10.1021/jm100287t. [DOI] [PMC free article] [PubMed] [Google Scholar]