

Abstract
4-Alkyloxyimino derivatives of pyrimidine nucleotides display high potency as agonists of certain G protein-coupled P2Y receptors (P2YRs). In an effort to functionalize a P2Y6R agonist for fluorescent labeling, we probed two positions (N4 and γ-phosphate of cytidine derivatives) with various functional groups, including alkynes for click chemistry. Functionalization of extended imino substituents at the 4 position of the pyrimidine nucleobase of CDP preserved P2Y6R potency generally better than γ-phosphoester formation in CTP derivatives. Fluorescent Alexa Fluor 488 conjugate 16 activated the human P2Y6R expressed in 1321N1 human astrocytoma cells with an EC50 of 9 nM, and exhibited high selectivity for this receptor over other uridine nucleotide-activated P2Y receptors. Flow cytometry detected specific labeling with 16 to P2Y6R-expressing but not to wild-type 1321N1 cells. Additionally, confocal microscopy indicated both internalized 16 (t1/2 of 18 min) and surface-bound fluorescence. Known P2Y6R ligands inhibited labeling. Theoretical docking of 16 to a homology model of the P2Y6R predicted electrostatic interactions between the fluorophore and extracellular portion of TM3. Thus, we have identified the N4-benzyloxy group as a structurally permissive site for synthesis of functionalized congeners leading to high affinity molecular probes for studying the P2Y6R.
Introduction
The P2Y6 receptor (P2Y6R) belongs to a family of eight G protein-coupled receptors (GPCRs) activated by extracellular nucleotides.1 The P2Y6R, which is both cytoprotective and proinflammatory, promotes inositol lipid signaling through Gq mediated activation of phospholipase C-β (PLC-β) isozymes and is found in intestinal epithelial, endocrine, skeletal muscle, bone, neuronal, vascular, immune and other cells.1–10 Pharmacological modulation of the P2Y6R has been proposed to be useful in treatment of osteoporosis, neurodegeneration, gout, ocular hypertension, glaucoma, inflammation, intestinal disorders, diabetes, and other diseases.2–10
Uridine 5′-diphosphate (UDP 1) is the endogenous P2Y6R agonist (EC50 = 0.30 μM), and various structure activity relationship (SAR) studies have identified sites for modification of UDP that improve its potency and/or selectivity (Chart 1).11–17 Potent P2Y6R agonists (EC50 values, nM) include 3-phenacyl-UDP 2 (70),11 derivatives of the 5 position 3 (15) and 4 (300),12,13 a bicyclic analogue 5 (MRS2795, 42) of UDP that maintains a South (S) conformation of the ribose-like ring,14 N4-benzyloxy-CDP 6 (MRS2964, 26) and various dinucleoside triphosphates, 14–16 including Up3U 7 (270), N4-methoxy-Cp3U 8 (MRS2957, 12), and INS48823 9 (130). Here, we considered structural extensions of UDP that would both preserve P2Y6R agonist potency and permit the coupling of reporter groups or other large chemical carrier moieties. A functionalized congener approach to ligand derivatization has been explored for a variety of GPCRs, leading to fluorescent probes,18 affinity labels, cross-linking ligands, and specialized radioactive probes.39 SAR analyses suggested that the steric constraints of the pharmacophoric binding site of the receptor likely could be circumvented by chain elongation through two specific sites in P2Y6R agonists. Thus, we introduced a 4-alkyloxyimino group in the nucleobase, and the terminal phosphate group was extended in γ-ester derivatives of UTP. The potent N4-benzyloxy derivative 6, which was 82- and 44-fold P2Y6R-selective vs. P2Y2 and P2Y4Rs, respectively,14 served as the lead compound for a series of 4-oxyimino analogues of cytidine 5′-diphosphate in the present study. Similarly, several O-alkyl and aryl ester derivatives14 of the γ-phosphate of UTP suggested extension in that region as a possible tethering approach for P2Y6R agonists.
Chart 1.

Structures of prototypical agonist ligands for studying P2Y6Rs.
This work has a two-fold goal: (1) the exploration of SAR of pyrimidine nucleotides at the P2YRs leading to a successful strategy for tethering large reporter groups to the pharmacophore, and (2) the characterization of a new fluorescent probe as a useful tool for studying the P2Y6R. We chose to study a fluorescent probe initially using flow cytometry (FCM) and confocal microscopy. Similar to previous studies of fluorescent agonists of the A2A and A3 adenosine receptors,18a,34 there was significant internalization of this fluorescent GPCR agonist in P2Y6R-expressing cells.
Results and discussion
Novel nucleotide derivatives for testing at P2YRs (Table 1) were prepared by the synthetic routes shown in Schemes 1 and 2. The nucleotide products for biological testing were purified by HPLC to assure high purity (>95%). The types of nucleotide modifications include: CTP and CDP analogues containing a N4-alkoxy group (Schemes 1 and 2); Cp3 analogues containing a substituted γ-alkyl group at the terminal phosphate (Scheme 1). Click chemistry, especially [2 + 3] cycloaddition of acetylene and azide groups,19 is often used as a linking reaction for preparing conjugates of biologically active small molecules, and therefore, we chose to incorporate alkynyl groups in the nucleotide analogues.
Table 1.
Potency of a series of pyrimidine nucleotide derivatives at three subtypes of hP2YRs
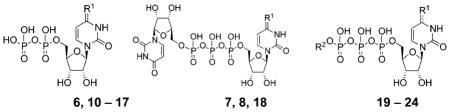
| |||||
|---|---|---|---|---|---|
| No. | Structure, R1 = | Potency, EC50, nM or % activation
|
|||
| P2Y6a | P2Y2a | P2Y4a | |||
| Diphosphates | |||||
| 6b | =N–O–Bn | 26 ± 2 | 2130 ± 640 | 1150 ± 150 | |
| 10 | =N–O–(3-Br–Bn) | 39 ± 2.2 | 6200 ± 2100 | >10 000 | |
| 11 | =N–O-(3-I–Bn) | 19 ± 5 | 2500 ± 800 | 352 ± 62 | |
| 12 | =N–O–(4-Br–Bn) | 32 ± 4.1 | 5200 ± 90 | 7440 ± 500 | |
| 13 | =N–O–(4-I–Bn) | 148 ± 37 | NE | <50%c | |
| 14 | =N–O–(4-NO2–Bn) | 41 ± 2.0 | 3300 ± 510 | 2200 ± 320 | |
| 15 | =N–O–(3-(HC≡C(CH2)2C≡C)–Bn) | 100 ± 11 | 3900 ± 1200 | 952 ± 205 | |
| 16 | =N–O–(3-(AlexaFluor–NH–(CH2)6-triazole–(CH2)2–C≡C)–Bn) | 9 ± 1.9 | 2500 ± 660 | <50%c | |
| 17 | =N–O–(3-(Cy5–NH–(CH2)3–triazole–(CH2)2–C≡C)–Bn) | ND | ND | ND | |
| Dinucleoside triphosphates | |||||
| 7b,d | =O | 270 ± 70 | 1310 ± 210 | 870 ± 110 | |
| 8b | =N–O–Me | 12 ± 3 | 170 ± 40 | 790 ± 120 | |
| 18 | =N–O–Bn | 126 ± 17 | 1700 ± 500 | 636 ± 42 | |
| Triphosphates and γ-phosphoesters | |||||
| Compd | R1 | R2 | P2Y6a | P2Y2a | P2Y4a |
| 19b | =N–O–Me | H | 130 ± 21 | 28 ± 4 | 25 ± 3 |
| 20 | =N–O–Me | 2-Naphthyl | 731 ± 28 | 2900 ± 200 | <50%c |
| 21b | =N–O–Bn | H | 230 ± 37 | 620 ± 75 | 97 ± 14 |
| 22 | =N–O–Bn | HC≡C(CH2)2 | 1750 ± 280 | <50%c | <50%c |
| 23 | =N–O–Bn | HC≡C(CH2)3 | >10 000 | NE | <50%c |
| 24 | =N–O–Bn | CH3CONH–(CH2)2–triazole(CH2)2 | 879 ± 32 | 3800 ± 690 | 3300 ± 1200 |
Functional assays were conducted with 1321N1 astrocytoma cells expressing recombinant hP2Y2, hP2Y4, or hP2Y6. Values are expressed as the mean ± SEM. ND, not determined.
Percent of maximal activation by full agonist at 10 μM.
7, Up3U, 10, MRS4141; 16, MRS4129; 17, MRS4137; 42, MRS4158 (5′-monophosphate derivative of 16).
NE – no effect. ND – not determined.
Scheme 1.

Synthesis of various pyrimidine ribonucleoside 5′-tri and 5′-diphosphates.
Scheme 2.

Synthesis of fluorescent conjugates 16 and 17 click-linked through the N4 position and a model compound 24 click-linked through the β-phosphate.
The synthesis of N4-alkoxycytidines 32–38 from cytidine was performed using corresponding alkoxyamines 25–31. The resulting N4-alkoxycytidines were phosphorylated by standard methods14,16,20 to give the desired N4-alkoxycytidine 5′-diphosphates 10–15 (Scheme 1). N4-3-(1,5-Hexadiynyl)-benzyloxycytidine 39 was prepared from N4-3-iodobenzyloxycytidine 35 using a palladium-catalyzed cross coupling Sonogashira reaction. 21 In each case, the unprotected nucleoside was first treated with phosphorous oxychloride, and after 2 h the reaction mixture was treated with phosphoric acid to produce 5′-diphosphates. The desired N4-alkoxycytidine 5′-triphosphates 19 and 21 were synthesized from corresponding N4-alkoxycytidines 32 and 33 after treatment with phosphorous oxychloride followed by bis(tri-n-butylammonium) pyrophosphate. The γ-phosphoester derivatives 20, 22, and 23 were prepared by the condensation of corresponding alkyl monophosphoric acid with the corresponding N4-alkoxycytidine 5′-diphosphate in the presence of N,N′-diisopropylcarbodiimide (DIC).
The fluorescent 5′-diphosphates (N4-(3-(AlexaFluor-NH-(CH2)6-triazole-(CH2)2-C≡C)-benzyloxy))-CDP 16 and N4-(3-(Cy5-NH-(CH2)6-triazole-(CH2)2-C≡C)-benzyloxy)-CDP 17 and model compound γ-(CH3CONH-(CH2)2-triazole-(CH2)2)-N4-benzyloxy-CTP 24 were synthesized from alkynes 15 and 22 using copper-catalyzed click cycloaddition reactions (Scheme 2).22 The 5′-monophosphates 42 and 44 corresponding to the two fluorescent analogues 16 and 17, respectively, were also isolated as byproducts of the reaction.
Functional assays of the nucleotide analogues (Table 1) consisted of measuring potency for activation of phospholipase C (PLC) in 1321N1 human astrocytoma cells stably expressing the human (h) P2Y6R (1321N1-P2Y6R cells),23 and selectivity was assessed by quantification of PLC activation in 1321N1 cells stably expressing the hP2Y2R or hP2Y4R. Previously reported data for 5′-diphosphate analogue 6, dinucleotides 7 and 8, and 5′-triphosphate analogues 19 and 21 were included for comparison (Table 1).14
High potency at the P2Y6R was achieved with N4-benzyloxycytidine 5′-diphosphates, including various benzyl-substituted analogues. Halogenated analogues 10–13 were compared with 14, which has a strong electron withdrawing nitro group and is highly selective for the P2Y6R. 3-Bromo derivative 10 was 160- and >260-fold selective toward the P2Y6R, in comparison to the P2Y2R and P2Y4R, respectively. N4-(3-iodobenzyloxy)-CDP (11, EC50 19 nM) was more potent than the corresponding 4-iodo isomer 13. This finding led to the introduction at the N4 position in 15 of a terminal alkyne for tethering a sterically bulky group by click chemistry. P2Y6R potency was largely retained in this 3-dialkyne, with an EC50 value of 100 nM and at least one order of magnitude subtype selectivity. In general, the 5′-diphosphate derivatives containing various N4-benzyloxy substitutions at the m- or p-position were potent and selective at the P2Y6R, suggesting that this region of the ligand protrudes outside of the steric and electronic constraints of the principal binding site. The P2Y6R selectivity of fluorescent agonist 16 in comparison to the P2Y2R and P2Y4R was 280- and >1100-fold, respectively. This was especially striking, and its potency surpassed that of the immediate precursor 15 lacking the fluorophore by 11-fold. However, among dinucleoside triphosphate derivatives, the enlargement of the N4-methyloxy in 8 to N4-benzyloxy in 18 reduced both potency and selectivity at the P2Y6R.
UDP and its 5′-diphosphate derivatives are known to potently activate the P2Y14R.40 However, CDP analogues containing a N4-alkoxy group were shown to be only weakly active at this subtype.14 Consistently, 16 at concentrations up to 10 μM did not activate the hP2Y14R to inhibit forskolin-stimulated adenylate cyclase (in C6 rat glioma cells stably expressing the receptor,40 Fig. S1†). Thus, the Alexa Fluor 488 conjugate 16 promises to be a potent and relatively selective ligand probe of the P2Y6R.
Derivatization at the γ-phosphate moiety of 5′-triphosphates was less favorable than at the N4 position for retention of potency and selectivity at the P2Y6R. A naphthyl ester of the γ-phosphate moiety 20 failed to enhance potency at the P2Y6R in comparison to 19. Several alkynyl γ-phosphoester model compounds exhibited substantially reduced P2Y6R potency in comparison to the corresponding 5′-triphosphate 21. A terminal alkynyl ester 22 was significantly more potent than the higher homologue 23, which was nearly inactive at the P2Y6R. A model click product, triazole 24 with an amide-containing terminal chain, had improved potency compared to precursor 23 but still had an EC50 value of only 879 nM.
Thus, the pyrimidine N4 position of CDP was thought to provide a better prospect than the γ-phosphate moiety of CTP derivatives, for chain extension with retention of potency at the P2Y6R. N4-linked fluorescent conjugates, such as the Alexa Fluor 488 conjugate 16, were considered for use as fluorescent probes in assays using 1321N1-P2Y6R cells. Compound 16 proved to be a useful fluorescent probe for FCM, a technique that has proven effective for studying other GPCRs in intact cells.18 The fluorescent dye Alexa Fluor 488 is a suitable fluorophore for FCM binding assays and live cell imaging.18,34
The visible fluorescence spectrum of aqueous 16 displayed absorption and emission maxima at 494 and 518 nm, respectively (ESI, Fig. S2†). This fluorescent probe was initially tested in FCM after a long incubation period to allow internalization. A FCM histogram of the total binding and nonspecific binding to 1321N1-P2Y6R cells using 400 nM 16 after 3 h incubation is shown in Fig. 1A. The peak for total binding was significantly higher than the autofluorescence and was located within the optimal window for analysis, but it was not completely separated from the peak for auto-fluorescence. However, the corresponding fluorescent 5′-monophosphate 42 at the same concentrations did not label the cells during a 60 min incubation (Fig. 1B–E and S3†). This is consistent with the inactivity of 5′-monophosphate derivatives in general at the P2Y6R. There was no specific labeling by 16 or 42 of control 1321N1 cells. Also, we observed no specific labeling by 16 of 1321N1 cells expressing the hP2Y1R (data not shown).
Fig. 1.

(A) FCM histogram following 3 h incubation of 1321N1 astrocytoma cells expressing the P2Y6R in 400 nM 16 (MRS4129), showing the total binding (red), autofluorescence (blue) and nonspecific binding (green). The histogram represents data of one of the three independent experiments. (B–E) FCM histograms following 60 min incubation of: (B) control 1321N1 astrocytoma cells with various concentrations of P2Y6R agonist 16; (C) control 1321N1 astrocytoma cells with various concentrations of inactive 5′-monophosphate derivative 42; (D) 1321N1 astrocytoma cells expressing the P2Y6R with various concentrations of P2Y6R agonist 16; (E) 1321N1 astrocytoma cells expressing the P2Y6R with various concentrations of inactive 5′-monophosphate derivative 42.
Increasing the concentration of 16 from 10 nM to 10 μM increased fluorescent labeling of 1321N1-P2Y6R cells during a 60 min incubation, and a comparison by FCM of specific and nonspecific binding shows a clear separation over a wide concentration range (10 nM to 1.5 μM, 3 h incubation at 37 °C, Fig. 2A). The fluorescence values were converted to molecules of equivalent soluble fluorochrome (MESF).18 Cells expressing the P2Y6R in the absence of 16 were used to measure cellular autofluorescence. Nonspecific binding of 16 was measured in the presence of 100 μM UDP, and the ratio of specific to nonspecific binding was high (~4 : 1 at 500 nM 16). Fluorescent labeling of the cells with 16 approached saturation. The measured cell fluorescence is a composite of the surface-bound and the internalized fluorescence due to the agonist nature of 16. Therefore, it was not possible to calculate an apparent equilibrium binding constant (Kd) from this curve, which represents a multistep process. Also, the metabolic stability of these nucleotide derivatives was not studied; it is possible that hydrolysis of the diphosphate during the incubations affects the fluorescent labeling of whole cells.
Fig. 2.
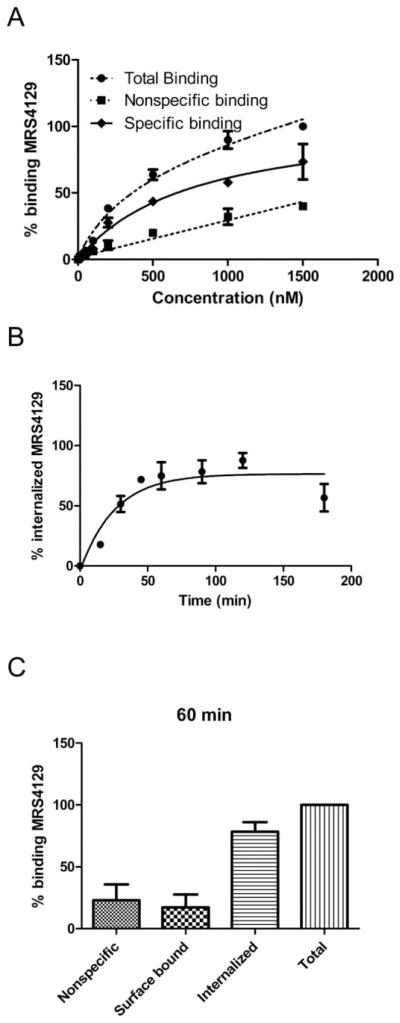
(A) Saturation binding experiment with Alexa Fluor 488 conjugate 16 at the P2Y6R expressed on 1321N1 astrocytoma cells using FCM (3 h incubation). Nonspecific binding was measured in the presence of 100 μM UDP. (B) Internalization kinetics of 400 nM P2Y6R-bound 16 at 37 °C. Internalized amount (%int) was calculated as the acid insensitive fluorescence at x time point (MESFx) is compared to total MESF (MESFtotal) corrected with the nonspecific binding (MESFnonspec): %int=(MESFx − MESFnonspec)/(MESFtotal − MESFnonspec). Fluorescence of internalized 16 was measured by FCM after removing cell-surface bound ligand by 3 × 5 min acid wash (pH 3.5). The t1/2 of the internalization was 18 min. (C) Comparison of the nonspecifically bound, surface bound, internalized and total bound 16 (sum of surface bound and internalized) after 60 min incubation. In this plot, nonspecific has been subtracted from surface bound, internalized and total bound values. 1321N1-P2Y6R was incubated with 400 nM fluorescent ligand 16 at 37 °C. Nonspecific binding was measured in the presence of 100 μM UDP, the surface bound fraction was measured with incubation of 0.4 M sucrose-containing media, and the internalized ligand was measured as the acid-insensitive fraction following acid wash. After a 60 min incubation period, the 17% of the total binding was found on the cell surface, 77% was internalized and 23% was nonspecifically bound.
Another fluorophore (Cy5) with a longer emission wave-length than Alexa Fluor 488 was introduced in 17 at the same position as Alexa Fluor 488 in 16. Unfortunately, the labeling of 1321N1 astrocytoma cells with 17 (100 nM to 1 μM) was independent of the expression of the P2Y6R, and it could not be blocked by 100 μMUDP (Fig. S4†). Therefore, this conjugate was not suitable for further studies of P2Y6R labeling.
Kinetics of cell labeling by 16 was also determined using a FCM assay (Fig. 2B). Binding of 400 nM 16 to 1321N1-P2Y6R cells approached a maximum slowly, as observed previously with a fluorescent antagonist of the hA3 adenosine receptor.18 An association rate constant (k1) was determined to be 0.019 min−1 (Fig. S5†). Nonspecific P2Y6R binding was measured in the presence of 100 μM UDP, the surface bound fraction was measured with incubation of 0.4 M sucrose-containing media, and the internalized ligand was measured as the acid-insensitive fraction following acid wash. The internalization of the P2Y6R (determined by the comparison of the acid-insensitive fraction and the total binding) occurred with a t1/2 of 18 min. The fraction of fluorescence associated with the internalized label was much greater (77% of the total binding) than the fraction on the cell surface (17% of the total binding) at the end of the 60 min incubation period (Fig. 2C).
In confocal microscopy, after 60 min incubation at 37 °C, the cell-associated fluorescence of compound 16 (2 μM) in 1321N1-P2Y6R cells was receptor-dependent and mainly intracellular, providing further evidence of P2Y6R internalization upon agonist binding (Fig. 3). Also, the addition of hyperosmolaric sucrose to the medium clearly left the fluorescent labeling on the cell surface and prevented its internalization. The appearance of the labeling on the cell surface in Fig. 3C is punctate, suggesting that the receptor is organized into concentrated regions on the cell surface rather than being evenly distributed. Micrographs in Fig. 4 illustrate the ability of known P2Y6R ligands, agonist 8 and diisothiocyanate antagonist MRS2578, to inhibit fluorescent labeling in 1321N1-P2Y6R cells by compound 16. Several known P2Y6R ligands were compared for the ability to inhibit the fluorescent binding of 16 to 1321N1-P2Y6R cells were compared after a 30 min pre-incubation followed by a brief treatment with the fluorescent ligand 16 (2 min). Agonists UDP (10 μM) and MRS2957 (0.5 μM) and antagonist MRS2578 (1 μM) caused a significant reduction of labeling determined using FCM (Fig. S6†).
Fig. 3.
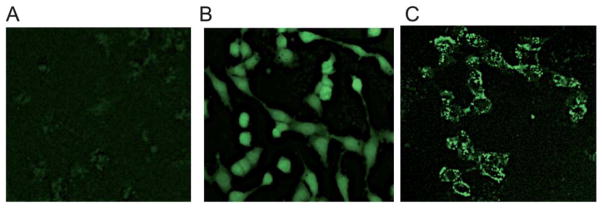
Fluorescent micrographs using a Zeiss LSM 700 confocal microscope of P2Y6R-expressing 1321N1 astrocytoma cells exposed to the fluorescent agonist 16 (2 μM, 60 min incubation at 37 °C). (A) Control cells in the absence of 16; (B) incubation with 1 μM 16 at 37 °C for 60 min in medium without sucrose; (C) incubation with 1 μM 16 at 37 °C for 60 min in medium containing 0.4 M sucrose.
Fig. 4.
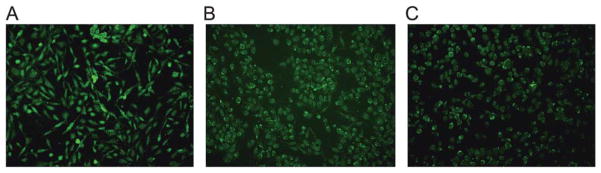
Inhibition of fluorescent labeling by known P2Y6R ligands, using a Keyence BZ-9000 fluorescent microscope equipped with filters for green fluorescence with excitation at 495 nm and emission at 519 nm. Fluorescent micrographs of P2Y6R-expressing 1321N1 astrocytoma cells exposed to (A) the fluorescent agonist 16 (500 nM, 60 min incubation at 37 °C) in the absence of inhibitor or sucrose; (B) same conditions, except a 30 min preincubation with P2Y6R agonist MRS2957 (8, 500 nM) was performed at 37 °C; (C) same conditions, except a 30 min preincubation with P2Y6R antagonist MRS2578 (500 nM) was performed at 37 °C P2Y6R-expressing 1321N1 astrocytoma cells in the absence of 16 showed a lack of fluorescence, similar to Fig. 3A. Control experiments using 1321N1 astrocytoma cells not expressing a P2YR showed no increase in fluorescence when incubated with 16.
Other fluorescent agonist ligands for GPCRs have been characterized, such as adenosine receptor agonists,24,34 which internalized in a similar fashion.34 It has been shown that a fluorescently labeled A2A adenosine receptor agonist Alexa488-APEC internalized upon agonist stimulation via clathrin-coated pit endocytosis, promoting a localization of the receptor in Rab5-positive early endosomes in both CHO and HEK293 cells.34 It was also demonstrated that desensitization of the P2Y6R is delayed compared to the P2Y4R,35 and that prolonged incubation with UDP caused a loss of surface P2Y6R, and rapid recovery of surface P2Y6R did not occur following removal of the agonist.36 The desensitization and internalization of two other Gq-coupled receptors, P2Y2R and P2Y4R, have also been described.36–38 Nevertheless, in this study the fluorescent agonist 16 was clearly internalized in P2Y6R-expressing cells.
Molecular modeling was used to probe the structural basis for the significantly enhanced potency and selectivity of the Alexa Fluor conjugate 16. To generate a hypothesis of the interactions of the fluorescent ligand 16 with the receptor, we constructed a new P2Y6R model based on our recently published homology model of the P2Y4R, constructed using the CXCR4 chemokine receptor structure as a template.20,25 As we explained recently,20,26 among the GPCRs structures that have been solved crystallographically, the CXCR4 receptor appears to be a suitable template for the modeling of the P2YRs, since it is relatively close in sequence and shares common structural features with P2YRs. It was first necessary to align the sequences of the two receptors (Fig. S7†). After the construction of the model, we subjected the fluorescent ligand 16 to a Monte Carlo Multiple Minima (MCMM) conformational search within the binding cavity of the P2Y6R, granting full flexibility to this ligand as well as all of the surrounding amino acid residues. The conformational search suggested that while the nucleotide portion of the fluorescent ligand bound rigidly to the receptor, the large N4 substituent retains some flexibility (Fig. 5A). Specifically, it suggested that the nucleotide portion of the fluorescent ligand bound to the receptor with a mode consistent with that seen in our previous models of the P2YRs (Fig. 5B).12,20,27–29 In particular, the 5′-diphosphate moiety of the ligand interacts, in our model, with three cationic residues located in TM3, TM6 and TM7: Arg1033.29, Lys2596.55 and Arg2877.39 (using a standard numbering convention30). These residues are conserved as cationic residues in all subtypes of the P2Y1-like subfamily of the P2YRs and, according to mutagenesis data gathered for the P2Y1 and P2Y2Rs, are fundamental for ligand recognition.31–33 A fourth cationic residue located in TM7 and conserved as a Lys in all the subtypes of the P2Y1-like receptors that bind uracil nucleotides, namely Lys2847.36 in the P2Y6R, interacted in the model with the phosphate as well as with the pyrimidine base of the ligand.
Fig. 5.

Molecular model of the lowest energy conformation of the hP2Y6R complexed with the Alexa Fluor 488-labeled agonist 16, as obtained after a fully flexible MCMM conformational search, (A) viewed from the plane of the phospholipid bilayer (with other conformations of 16 obtained), or (B) and (C) from the extracellular side. (C) shows the molecular surface of 16 (mesh) and the residues that surround its N4 substituent (solid), colored by electrostatic potential (red: negative; blue: positive). The schematic representation of the backbone of the receptor is colored by residue number (TM1: orange; TM2: dark yellow; TM3: yellow; TM4 light green; TM5: green; TM6: blue; TM7: purple/blue). The ligand is shown as a ball and stick structure, colored by element (carbon atoms colored in gray). The residues located in proximity to the ligand are labeled and shown as sticks, colored by residue number. The fluorophore is located near the outer segment of TM3.
The conformational search also suggested that the large N4 substituent of 16 protruded from the interhelical binding cavity of the receptor toward the extracellular space to wrap around the second extracellular loop and to end in proximity of the extracellular portion of TM3, interacting with Asp963.22 and Arg1003.26 (Fig. 5C). Notably, a visualization of the molecular surface of the residues that surround the extended N4 substituent, colored according to their electrostatic potential, suggested a substantial steric and electrostatic complementarity between the distal portions of the ligand and the receptor. A sulfonate group of the Alexa Fluor 488 forms an electrostatic interaction with R100, and an amino group is in proximity to Asp96 (both in TM3 near EL2). By analogy, the enhanced affinity of a fluorescent probe for the A3 adenosine receptor also correlated with predicted charged interactions of a bulky Alexa Fluor 488 moiety with EL2.18
Conclusions
In an effort to functionalize a P2Y6R agonist for fluorescent labeling, we probed two positions of pyrimidine nucleotides by introducing various functional groups, including alkynes for click chemistry. Functionalization of extended imino substituents at the N4 position of the pyrimidine nucleobase of CDP retained P2Y6R potency to a higher degree than γ-phosphoester formation in UTP derivatives. Thus, we have identified a pyrimidine N4-alkoxy group as a site for tethering P2Y6R agonists to a fluorescent label. The bifunctional conjugate 16 containing a fluorophore (Alexa Fluor 488) and spacer chain constructed through click chemistry, was potent and selective agonist at the P2Y6R. It was suitable for the study of binding and internalization kinetics on whole cells using FCM and microscopy. The binding of 16 to purified membranes of cells overexpressed the P2Y6R remains to be characterized. Thus, we introduce the first high affinity fluorescent ligand specific for a P2YR, although additional pharmacological characterization to define the complex cell labeling is needed.
Based on the bulk tolerance of some of these nucleotide derivatives, the functionalized chain appears to be accessing the extracellular regions of the receptor. Although only one fluorescent conjugate 16 showed high potency at the P2Y6R, this site on the nucleotide promises to be a general site for derivatization with bulky substituents, including other fluorophores and specialized reporter groups for receptor detection and characterization.39 A molecular modeling study was carried out to explore the recognition of the agonist 16 in binding to the P2Y6R to highlight the key putative interactions between charged groups of a large fluorophore moiety and the outer regions of TM3 in the P2Y6R, which evidently enhance the potency of 16. In conclusion, we have identified the N4-benzyloxy group as a structurally permissive region for synthesis of a family of functionalized congeners as fluorescent conjugates and other pharmacological probes for studying the P2Y6R. This will enable further drug discovery related to this receptor.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases and a grant (GM38213) from the National Institute of General Medical Sciences. We thank Noel Whittaker (NIDDK) for mass spectral determinations. EK thanks the Hungarian-American Enterprise Scholarship Foundation (HAESF) for financial support. LS thanks the University of Florence, Italy for financial support.
Abbreviations
- 1321N1-P2Y6R cells
1321N1 Human astrocytoma cells expressing the human P2Y6 receptor
- CDP
Cytidine, 5′-diphosphate
- CTP
Cytidine 5′-triphosphate
- DCC
N,N′-Dicyclohexylcarbodiimide
- DIC
N,N′-Diisopropylcarbodiimide
- DMF
N,N-Dimethylformamide
- EDC
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide
- FCM
Flow cytometry
- GPCR
G protein-coupled receptor
- HBSS
Hank's balanced salt solution
- HEPES
N-2-Hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- HPLC
High performance liquid chromatography
- MCMM
Monte Carlo Multiple Minima
- MESF
Molecules of equivalent soluble fluorochrome
- MRS2957
P1-(Uridine 5′-)-P4-(N4-methoxycytidine 5′-) triphosphate
- MRS2578
N,N′-1,4-butanediylbis[N′-(3-isothiocyanatophenyl)thiourea
- P2YR
P2Y receptor
- PLC
Phospholipase C
- SAR
Structure activity relationship
- TBAP
Tetrabutylammonium dihydrogenphosphate
- TEAA
Triethylammonium acetate
- THF
Tetrahydrofuran
- TLC
Thin layer chromatography
- TM
Transmembrane helical domain
- UDP
Uridine 5′-diphosphate
- UTP
Uridine 5′-triphosphate
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c3md00132f
Notes and references
- 1.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vieira RP, Müller T, Grimm M, von Gernler V, Vetter B, Dürk T, Cicko S, Ayata CK, Sorichter S, Robaye B, Zeiser R, Ferrari D, Kirschbaum A, Zissel G, Virchow JC, Boeynaems JM, Idzko M. Am J Respir Crit Care Med. 2011;184:215. doi: 10.1164/rccm.201011-1762OC. [DOI] [PubMed] [Google Scholar]
- 3.Mamedova LK, Wang R, Besada P, Liang BT, Jacobson KA. Pharmacol Res. 2008;58:232. doi: 10.1016/j.phrs.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balasubramanian R, Ruiz de Azua I, Wess J, Jacobson KA. Biochem Pharmacol. 2010;79:1317. doi: 10.1016/j.bcp.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orriss IR, Wang N, Burnstock G, Arnett TR, Gartland A, Robaye B, Boeynaems JM. Endocrinology. 2011;152:3706. doi: 10.1210/en.2011-1073. [DOI] [PubMed] [Google Scholar]
- 6.Koizumi S, Shigemoto-Mogam Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, Inoue K. Nature. 2007;446:1091. doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grbic DM, Degagné E, Larrive JF, Bilodeau MS, Vinette V, Arguin G, Stankova V, Gendron FP. Inflammatory Bowel Dis. 2012;18:1456. doi: 10.1002/ibd.21931. [DOI] [PubMed] [Google Scholar]
- 8.Markovskaya A, Crooke A, Guzmán-Aranguez AI, Peral A, Ziganshin AU, Pintor J. Eur J Pharmacol. 2008;579:93. doi: 10.1016/j.ejphar.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 9.Bar I, Guns PJ, Metallo J, Wilkin F, Cammarata D, Boeynaems JM, Bult H, Robaye B. Mol Pharmacol. 2008;74:777. doi: 10.1124/mol.108.046904. [DOI] [PubMed] [Google Scholar]
- 10.Uratsuji H, Tada Y, Kawashima T, Kamata M, Hau CS, Asano Y, Sugaya M, Kadono T, Asahina A, Sato S, Tamaki K. J Immunol. 2012;188:436. doi: 10.4049/jimmunol.1003746. [DOI] [PubMed] [Google Scholar]
- 11.El-Tayeb A, Qi A, Müller CE. J Med Chem. 2006;49:7076. doi: 10.1021/jm060848j. [DOI] [PubMed] [Google Scholar]
- 12.Besada P, Shin DH, Costanzi S, Ko HJ, Mathé C, Gagneron J, Gosselin G, Maddileti S, Harden TK, Jacobson KA. J Med Chem. 2006;49:5532–5543. doi: 10.1021/jm060485n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ginsburg-Shmuel T, Haas M, Schumann M, Reiser G, Kalid O, Stern N, Fischer B. J Med Chem. 2010;53:1673. doi: 10.1021/jm901450d. [DOI] [PubMed] [Google Scholar]
- 14.Maruoka H, Barrett MO, Ko H, Tosh DK, Melman A, Burianek LE, Balasubramanian R, Berk B, Costanzi S, Harden TK, Jacobson KA. J Med Chem. 2010;53:4488. doi: 10.1021/jm100287t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaver SR, Rideout JL, Pendergast W, Douglass JG, Brown EG, Boyer JL, Patal RI, Redick CC, Jones AC, Picher M, Yerxa BR. Purinergic Signalling. 2005;1:183. doi: 10.1007/s11302-005-0648-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko H, Carter RL, Cosyn L, Petrelli R, de Castro S, Besada P, Zhou Y, Cappellacci L, Franchetti P, Grifantini M, Van Calenbergh S, Harden TK, Jacobson KA. Bioorg Med Chem. 2008;16:6319. doi: 10.1016/j.bmc.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Tayeb A, Qi A, Nicholas RA, Müller CE. J Med Chem. 2011;54:2878. doi: 10.1021/jm1016297. [DOI] [PubMed] [Google Scholar]
- 18.(a) Kozma E, Kumar TS, Federico S, Phan K, Balasubramanian R, Gao ZG, Paoletta S, Moro S, Spalluto G, Jacobson KA. Biochem Pharmacol. 2012;83:1552. doi: 10.1016/j.bcp.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kozma E, Gizewski ET, Tosh DK, Squarcialupi L, Auchampach JA, Jacobson KA. Biochem Pharmacol. 2013;85:1171. doi: 10.1016/j.bcp.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moses JE, Moorhouse AD. Chem Soc Rev. 2007;36:1249. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]
- 20.Maruoka H, Jayasekara MPS, Barrett MO, Franklin DA, de Castro S, Kim N, Costanzi S, Harden TK, Jacobson KA. J Med Chem. 2011;54:4018. doi: 10.1021/jm101591j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chinchilla R, Nájera C. Chem Rev. 2007;107:874. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 22.Tosh DK, Yoo LS, Chinn M, Hong K, Kilbey SM, Barrett MO, Fricks IP, Harden TK, Gao ZG, Jacobson KA. Bioconjugate Chem. 2010;21:372. doi: 10.1021/bc900473v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourdon DM, Wing MR, Edwards EB, Sondek J, Harden TK. Methods Enzymol. 2006;406:489. doi: 10.1016/S0076-6879(06)06037-X. [DOI] [PubMed] [Google Scholar]
- 24.Dale CL, Hill SJ, Kellam B. Med Chem Commun. 2012;3:333. [Google Scholar]
- 25.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Science. 2010;330:1066. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deflorian F, Jacobson KA. J Comput-Aided Mol Des. 2011;25:329. doi: 10.1007/s10822-011-9423-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costanzi S, Mamedova L, Gao ZG, Jacobson KA. J Med Chem. 2004;47:5393. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costanzi S, Joshi B, Maddileti S, Mamedova L, Gonzalez-Moa M, Marquez V, Harden TK, Jacobson KA. J Med Chem. 2005;48:8108. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobson KA, Costanzi S, Ivanov A, Tchilibon S, Besada P, Gao Z, Maddileti S, Harden TK. Biochem Pharmacol. 2006;71:540. doi: 10.1016/j.bcp.2005.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ballesteros JA, Weinstein H. Methods Neurosci. 1995;25:366. [Google Scholar]
- 31.Hillmann P, Ko GY, Spinrath A, Raulf A, von Kügelgen I, Wolff SC, Nicholas RA, Kostenis E, Höltje HD, Müller CE. J Med Chem. 2009;52:2762. doi: 10.1021/jm801442p. [DOI] [PubMed] [Google Scholar]
- 32.Moro S, Guo D, Camaioni E, Boyer JL, Harden TK, Jacobson KA. J Med Chem. 1998;41:1456. doi: 10.1021/jm970684u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang Q, Guo D, Lee BX, Van Rhee AM, Kim YC, Nicholas RA, Schachter JB, Harden TK, Jacobson KA. Mol Pharmacol. 1997;52:499. doi: 10.1124/mol.52.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brand F, Klutz A, Jacobson KA, Fredholm BB, Schulte G. Eur J Pharmacol. 2008;590:36. doi: 10.1016/j.ejphar.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robaye B, Boeynaems JM, Communi D. Eur J Pharmacol. 1997;329:231. [PubMed] [Google Scholar]
- 36.Brinson AE, Harden TK. J Biol Chem. 2001;276:11939. doi: 10.1074/jbc.M009909200. [DOI] [PubMed] [Google Scholar]
- 37.Flores RV, Hernández-Pérez MG, Aquino E, Garrad RC, Weisman GA, Gonzalez FA. Mol Cell Biochem. 2005;280:35. doi: 10.1007/s11010-005-8050-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sromek SM, Harden TK. Mol Pharmacol. 1998;54:485. doi: 10.1124/mol.54.3.485. [DOI] [PubMed] [Google Scholar]
- 39.Jacobson KA. Bioconjugate Chem. 2009;20:1816. doi: 10.1021/bc9000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carter RL, Fricks IP, Barrett MO, Burianek LE, Zhou Y, Ko H, Das A, Jacobson KA, Lazarowski ER, Harden TK. Mol Pharmacol. 2009;76:1341. doi: 10.1124/mol.109.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.