

Abstract
The response to exogenous pathogens leads to activation of innate immunity through the release of pathogen-associated molecular patterns (PAMPs) and their binding to pattern recognition receptors. A classic example is septic shock where Toll receptor 4 recognizes PAMPs. Although well accepted, this concept does not explain the activation of innate immunity and inflammation occurs with transplantation, autoimmunity, or trauma. Increasingly recognized is that endogenous molecules released by dying cells (damage-associated molecular patterns; DAMPs) activate cellular receptors leading to downstream inflammation. Thus endogenous danger signals and exogenous PAMPs elicit similar responses through seemingly similar mechanisms. Also emerging is our understanding that normal repair processes benefit from dampening the immune response to these endogenous danger molecules. Here we focus on the role of DAMPs and their putative receptors in the pathogenesis of acute and chronic kidney diseases.
The inflammatory response to acute or chronic tissue injury engages the immune system. What are the initial activators of injury or disease? How does the immune system discriminate between live versus dead cells and know whether to respond? What factors regulate the inflammatory response to clear injury without causing excessive tissue damage and then initiate repair? We now recognize that the well-known activation of the immune system in response to foreign pathogens is recapitulated in an immune response to endogenous molecules released from necrotic, and perhaps apoptotic, cells after tissue injury or trauma related to hypoxia, ischemia, mechanical stress, or pathogen-induced inflammation.
Matzinger1 originally proposed the danger model to clarify exceptions to Janeway's model2 of the immune response to foreign antigens, which did not at the time explain autoimmunity or the immune response to transplantation. The danger model suggests that damaged or dying cells release endogenous molecules called damage/danger-associated molecular patterns (DAMPs) that activate the immune system in a fashion analogous to pathogen-associated molecular patterns (PAMPs), molecules released by pathogenic bacteria or viruses. These endogenous or self-molecules (extracellular matrix proteins (ECM), calcium-binding proteins, and structural proteins) typically function in normal cell homeostasis but are also recognized as danger signals when released into the extracellular space3 exposing hydrophobic portions of the molecules that are normally hidden in healthy living cells.4 Matzinger4–6 and others have extended the danger model as more has been learned about the role of danger signals in tissue injury7,8 and diseases such as arthritis9 and cancer10,11 and the body's need for mechanisms that dampen the immune response and initiate repair. Here we focus on the role of DAMPs and ligands of Toll receptors (TLRs) in renal disease12–15 and expand recent interest to a broader view of the currently identified classes of DAMPs and their putative receptors.
DANGER-ASSOCIATED MOLECULAR PATTERNS: ENDOGENOUS DANGER SIGNALS
A consistent terminology has not been adopted for the endogenous molecules that convey a danger signal to the immune system. Some DAMPs that stimulate an immune response have been called adjuvant molecules to distinguish them from DAMPs that produce only acute pro-inflammatory effects,3 sometimes referred to as alarmins.16,17 PAMPs and alarmins have been grouped together as subcategories of a large family of DAMPs,16,17 whereas others consider alarmins and DAMPs to be related molecules that are clearly distinguished from PAMPs.9 Here we use the term DAMPS to describe endogenous danger molecules as a group that is separate from pathogen-derived PAMPs (Table 1 and Figure 1); their classification is predicated on direct evidence of involvement in the immune response to injury with a clear absence of confounding effects from potential bacterial contaminants, such as LPS.3,16
Table 1.
DAMPs and receptors for DAMPs
| DAMP | Putative Receptors | References |
|---|---|---|
| Ligands of RAGE | 40,42 | |
| AGEs | RAGE | 39–42 |
| HMGB1 | TLR2, TLR4, TLR9, CD44, | 18–28 |
| RAGE | ||
| S100 proteins/calgranulins | RAGE, TLR4 | 29–33 |
| amyloid-β | RAGE, NLRP3 | 160 |
| HSPs | CD14, CD91, TLR2, TLR4, CD40 | 34–38 |
| Chromatin and DNA | TLR9 | 43–47 |
| Uric acid (MSU) crystals | TLR2, TLR4, CD14 | 48–52 |
| Neutrophil-derived alarmins | 16,17 | |
| cathelicidins | TLR7, TLR9, FPRL1, FPR2 | 54 |
| defensins | TLR4, CCR6 | 53 |
| lactoferrin | TLR4 | 55 |
| Extracellular matrix proteins | 3,56,57 | |
| proteoglycans | ||
| hyaluronan | TLR2, TLR4, NLRP3 | |
| biglycan | TLR2, TLR4, NLRP3 | |
| versican | TLR2 | |
| fibrinogen | TLR4, Integrins | |
| heparan sulfate | TLR4 | |
| fibronectin extra domain A | TLR4 | |
| laminin | Integrins | |
| elastin-derived peptides | Integrins | |
| collagen-derived peptides | CXCR2 | |
| Galectins | Unknown | 58,59 |
| Thioredoxin | Unknown | 65,66 |
| Adenosine; ATP | P1, P2X, and P2Y receptors | 60–64 |
| IL-33 | ST2/IL-1R | 67 |
| Tamm-Horsfall glycoprotein | TLR4 | 68–73 |
CXCR2, CXC-chemokine receptor 2; FPR2, formyl peptide receptor 2; FPRL1, formyl peptide receptor-like receptor 1; MSU, monosodium uric acid; NLRP3, NLR family, pyrin domain-containing 3.
Figure 1.

Danger and stranger models. Infections of pathogenic bacteria or viruses cause release of PAMPs that bind to pattern recognition receptors (PRRs), such as TLRs, on immune cells and stimulate an innate immune response that is accompanied by inflammation, activation of adaptive immunity, and eventually processes to resolve the infection and allow for tissue repair. The danger model recognizes that similar events occur when cells are stressed or injured and that necrotic cells release molecules that are normally hidden within the cell. In the extracellular space these DAMPs can bind to PRRs or to specialized DAMP receptors to elicit an immune response by promoting release of pro-inflammatory mediators and recruiting immune cells to infiltrate the tissue. The immune cells that participate in these processes include, for example, APC, such as dendritic cells and macrophages, as well as T cells and neutrophils (PMN). DAMPs may also stimulate adaptive immunity and participate in autoimmune responses and tissue repair. A wide variety of intracellular and extracellular molecules function as DAMPs when released from cells (Table 1). The functions of such a diverse group of molecules may not yet be fully elucidated; it is unknown whether different DAMPs have specific roles, whether specific functions are elicited in different cell types or conditions, or even whether immune responses to DAMPs can be distinguished from those of PAMPs.
High-mobility Group Box 1 Protein
Perhaps the most well characterized DAMP, high-mobility group box 1 (HMGB1) protein, is a ubiquitously expressed nonhistone DNA-binding protein that regulates chromosomal stability, stabilizes nucleosomes, and regulates transcription.18–22 As an extracellular DAMP after secretion or passive release,23 HMGB1 is a late-phase pro-inflammatory mediator in sepsis24 and in sterile inflammation, such as hepatic ischemia-reperfusion injury (IRI).18,25 HMGB1 is chemotactic for immune cells and stimulates dendritic cell (DC) maturation and migration.26 HMGB1 binds to the receptor for advanced glycation end products (RAGE)27 and stimulates NFκB-induced transcription through interactions with TLR2, TLR4,28 and RAGE.
S100 Protein Family
The S100 proteins, a large family of calcium-binding proteins, are implicated in the inflammation or fibrosis associated with cancer or diseases of the kidney, heart, joints, and lungs.29 When functioning extracellularly as DAMPs after release from phagocytes30 and other cells in response to cell stress, S100 proteins bind to RAGE31 and other receptors and produce earlier pro-inflammatory effects like HMGB1.32,33 S100A8 and S100A9 release from activated phagocytes activates TLR4 and amplifies lethal endotoxin-induced shock.33
Heat-shock Proteins
In normal healthy cells, heat-shock proteins (HSPs) are intracellular protein chaperones that guide newly synthesized polypeptide chains to prevent aggregation and misfolding. During cell stress, induction and secretion of HSPs causes pro-inflammatory cytokine and chemokine release and activation and maturation of antigen-presenting cells (APCs) to produce a robust innate immune response.34–36 Indeed, HSPs extend their role as chaperones intracellularly by binding and presenting antigens to cell surface MHC class I molecules.37,38 Complexes of extracellular HSPs and antigens are taken up by APCs and presented to MHC I to activate T cells through cross-presentation.36,37
Other Ligands of RAGE
A diverse group of ligands (including HMGB1, S100 proteins, and HPSs) bind to RAGE. Advanced glycation end products (AGE) and related molecules are glycation and oxidation products of proteins and lipids that are formed in hyperglycemic states by oxidative stress, such as hypoxia and ischemia/reperfusion, and in a number of diseases.39–42
Genomic Double-stranded DNA
Microbial DNA is a ligand for TLR9, but TLR9 can also recognize self-DNA from injured mammalian cells.43 Release of DNA from dying mammalian cells initiates an innate immune response by activation of TLR9 and the NALP3 (cryopyrin) inflammasome, release of activated IL-1β and IL-18,44 and induction of DC maturation.45 In addition to genomic DNA, injury-induced release of mitochondrial DNA can also cause inflammation by conserved pathogenic PAMP sequences.46,47
Uric Acid
The accumulation of uric acid in tissues has long been known to cause gout, but soluble uric acid released by injured cells also acts as a danger signal.48 The active moiety, monosodium urate crystals, forms in the extracellular space where it stimulates an immune response by activating the NALP3 inflammasome49–51 and by stimulating DC maturation and T-cell responses.52
Neutrophil-derived Alarmins
Neutrophils, an early leukocyte to infiltrate tissue after injury or infection, undergo degranulation and release immune modulatory proteins and polypeptides. Neutrophil-derived alarmins (such as α-defensins,53 cathelicidin,54 and lactoferrin55), named for their response to danger signals, link innate and adaptive immunity by recruiting leukocytes and inducing maturation of APCs, thereby bridging neutrophil and DC function.17
Extracellular DAMPS
Damaged or dying cells can also generate danger signals by cleaving molecules that are structural components of the ECM. Cleaved ECM glycoproteins, such as fibronectin and fibrinogen, and proteoglycans, such as hyaluronan, biglycan, and versican, function as DAMPs by signaling through TLRs, the NLRP3 inflammasome, and other receptors such as CD44, promoting a vigorous immune response3,56 and contributing to kidney disease.57
Other DAMPs
Other candidate DAMPs include galectins58,59; ATP and adenosine60–64; thioredoxin, a ubiquitous antioxidant enzyme65,66; the intranuclear cytokine, IL-3367; and Tamm-Horsfall protein (THP; or uromodulin), a glycoprotein expressed in the thick ascending limb that is excreted into the urine after proteolytic cleavage. THP is the most abundant protein in urine, but its function remains unclear68; it has been implicated as a biomarker of kidney disease and allograft rejection69,70 and may be a damage signal in renal tubular injury. THP promotes cytokine release71 and immune cell activation, including DC maturation,72 but may also have a protective anti-inflammatory role.73
RECEPTORS
DAMPs are recognized by pattern-recognition receptors such as certain TLRs, NOD-like receptors (NLRs, including the NLRP inflammasomes), and RLRs (RIG-I-like receptors), thereby overlapping with PAMPs,74,75 and by specialized receptors such as RAGE.76 Common signaling pathways shared by DAMPs and PAMPs may have an evolutionary basis.47 Injury-induced release of bacterial-related DAMPs from mitochondria, evolutionary symbiotic descendents of bacteria, causes inflammation.46 Other DAMPs receptors include CD91, scavenger receptors, CD2, integrins, chemokine receptors, and CD44.3,11,16,74
RAGE is a multiligand receptor that binds AGE, some S100 proteins, amyloid protein, and HMGB1. It amplifies immune responses39 and is implicated in diseases such as diabetes, Alzheimer's disease, cancer, and various inflammatory conditions.40,76,77 A soluble form of RAGE (sRAGE) found in human and mouse plasma78 may act as a ligand decoy to bind danger molecules and check the immune response. Circulating sRAGE and a splice variant (endogenous secretory RAGE) may be clinical indicators or biomarkers for the severity of disease.41,79
Inflammasomes, large cytosolic complexes of NOD-like receptors, adaptor protein, and caspase-1, link pathogenic and endogenous danger signals to activation of caspase-1, and proteolytic processing and release of the proinflammatory cytokines, IL-1β, and IL-18.50,80,81 Inflammasomes are important in gout,82 and their role in disease has been linked to several DAMPs, including the ECM molecule, biglycan.57
CHECKS AND BALANCES
With such a vast array of stranger and danger signals and some crossover in their receptors, how does the immune system distinguish PAMPs from DAMPs? Furthermore, once an immune response is initiated, what mechanisms are engaged to prevent rampant tissue damage and begin the repair process? Although PAMPs and DAMPs bind to some of the same receptors, interaction with different coreceptors may account nt for a divergencefor a divergence in downstream effects83 allowing for discrimination between PAMPs and DAMPs84,85 and selective suppression of DAMP signaling (Figure 2).85–87
Figure 2.
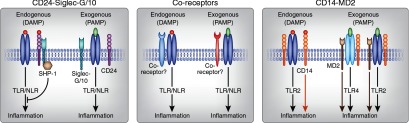
Proposed mechanisms for discriminating DAMPS from PAMPs and limiting the immune response. Several groups have recently proposed mechanisms that share a common theme involving coreceptors that pair with receptors for DAMPs and PAMPs to allow cells to distinguish between these related molecules and perhaps elicit downstream immune responses that may be specific for the ligand. For simplicity, these examples are illustrated only with inflammation as the target response, but selective signaling mechanisms may also result in varied inflammatory responses and provide specificity that may or may not include activation of adaptive immunity and tissue repair processes. (1) The CD24-Siglec pathway can distinguish DAMPs from PAMPs and can suppress DAMP signaling to prevent an unrestrained immune response and excessive collateral tissue damage.85–87 By interacting with sialic acid-binding Ig-like lectins (Siglec-G (mouse) or Siglec-10 (human)),157 CD24, a glycosylphosphatidylinositol (GPI)-anchored molecule (also known as heat-stable antigen) with diverse T-cell homeostatic functions,158 negatively regulates the TLR- or NLR-mediated immune response to HMGB1 and heat-shock proteins but not PAMPs (LPS or poly(I:C)) perhaps by facilitating association with phosphatases, like SHP-1.85 (2) Others suggest that although PAMPs and DAMPs bind to some of the same receptors, interaction with different coreceptors may account for a divergence in downstream effects.83 (3) A mechanism for discriminating DAMPs from PAMPs has also been demonstrated for two other cell surface proteins, CD14 (which recognizes DAMPs in the absence of TLR2 and promotes both TLR2-DAMP and TLR2-PAMP responses) and myeloid differentiation protein 2 (MD2; responding to and enhancing only exogenous PAMP responses in complexes with CD14-TLR2 or CD14-TLR4), that form complexes with Toll receptors.84 (4) Similarly, biglycan binding and induction of a multireceptor complex with TLR2/4 and purinergic P2×4 or P2×7 receptors activates the NLRP3 inflammasome,57 and this complex may regulate immune cell infiltration and tissue injury in kidneys (not illustrated here).56
Some mechanisms control damage-inducing immune responses through DAMPs inactivation. Anti-HMGB1 antibodies88 and sRAGE reduce HMGB1 bioavailability.40 Extracellular redox conditions may regulate the immune response by balancing early-stage reducing environment-promoting pro-inflammatory conditions with late-stage oxidation-induced DAMPs inactivation; disruption of this balance may contribute to chronic inflammation.19,89,90 BCL2 proteins, a family of pro- or antiapoptotic moieties, may be tissue protective when released extracellularly in IRI.91
Some endogenous molecules contribute to the resolution of inflammation and initiation of renal repair.92 Weibel-Palade bodies are organelles of endothelial cells that contain bioactive substances such as von Willebrand factor, IL-8, P-selectin, angiopoietin-2, and eotaxin93 that participate in hemostasis and inflammation. Uric acid release after IR mobilizes stem cells, protects kidneys from injury,94,95 and through exocytosis of Weibel-Palade bodies and release of their constituent molecules may promote postischemic repair.96 Some DAMPs, including HMGB1 and some ECM proteins, such as hyaluronan and heparin sulfate, initiate immune responses but are also necessary for recovery and healing.19
DAMPS AND KIDNEY DISEASE
Despite significant advances in understanding the important contribution of inflammation and immune mechanisms to the pathogenesis of a variety of kidney diseases, few specific or efficacious therapies exist. Understanding the role of candidate DAMPs released from somatic kidney or immune cells could reveal novel drug targets for inhibiting the inflammatory response or promoting repair processes in acute and chronic kidney disease (CKD). DAMPs contribute to multiple diseases. TLRs are important in IRI and various forms of glomerulonephritis including lupus nephritis.12–15 Advanced glycation end products, DAMPs that bind to RAGE rather than TLRs, are increased in renal failure and are involved in the progression of CKD in diabetic and nondiabetic kidney disease.97,98
Acute Kidney Injury
Acute kidney injury (AKI) results most commonly from IR, and although there are many causes, a common pathway leading to proximal tubule injury is activation of innate and adaptive immunity leading to inflammation.99 Injury elicits release of pro-inflammatory DAMPs, which may be signals that engender the inflammatory response to IRI by binding to receptors such as TLRs and RAGE.100 The best-characterized DAMPs in AKI are HSPs and HMGB1. Although their role in mediating renal injury in acute ischemic events is well documented, some studies do not establish that they function as DAMPs to stimulate the immune system. Nevertheless, we include them as viable DAMPs candidates on the basis of their role as DAMPs in other pathologies. In ischemic kidneys or hypoxic renal epithelial cells, stimulation of the TLR2-mediated ERK pathway is regulated by the HSP, gp96.101 Geldanamycin, an inhibitor of Hsp90 and gp96, protects mouse kidneys from IRI.102 Hsp70 and Hsp27 are up-regulated in rat kidneys after IRI103; however, tissue-protective104 and pro-inflammatory105 roles for Hsp27 have also been reported in kidneys after IRI. Ethyl pyruvate, an inhibitor of HMGB1 release, protects kidneys from IRI and reduces the increase in TNFα in rat kidneys subjected to IRI.106 TLR4- and MyD88-deficient mice showed protection from kidney IRI; HMGB1, hyaluronan, and biglycan expression increased in these null mice, suggesting that these DAMPs may be ligands for the observed role of TLR4 in IRI.107
High concentrations of uric acid accumulate in ischemic tissues and precipitate to form monosodium urate crystals that elicit an immune response. However, a single treatment with uric acid or the enzyme uricase to produce an acute rise in blood levels of uric acid mobilized endothelial progenitor cells and protected kidneys from IRI; this effect was absent with hyperuricemia.95
Kidney injury molecule-1 (Kim-1/Tim-1), one of a growing list of biomarkers of AKI and CKD, is a multifunction receptor protein expressed in proximal tubules that is released into the urine of patients with kidney disease.108 In addition to imparting phagocytic properties to tubule epithelial cells,109 Kim-1/Tim-1 may inhibit development of autoimmune responses and promote resolution of inflammation after kidney injury,108 therefore suggesting its role as a possible DAMP molecule or receptor.
Diabetic Nephropathy and Nondiabetic Glomerular Diseases
There is expansive literature on the involvement of AGE and RAGE in podocytes, diabetes, and diabetic nephropathy.42,76,97,110–117 It is presumed but not always demonstrated that these molecules are important as DAMPs because of the involvement of inflammation and the immune system in diabetes.42,115,118–120 RAGE, functioning as an endothelial adhesion receptor, is implicated in a novel pathway for leukocyte recruitment in inflammatory disorders and diabetic mice, and this process is enhanced by S100, a proinflammatory RAGE ligand.121 Experiments using RAGE null or overexpressing mice, neutralizing RAGE antibodies, or sRAGE implicate RAGE in the pathophysiology of diabetic nephropathy and chronic inflammation.76,112,122 In db/db mice, increased RAGE and S100 expression in podocytes associates both with renal pathology and with increased infiltration of mononuclear phagocytes to glomeruli; these effects are blocked by anti-RAGE antibody.112 Serum RAGE levels, kidney damage, and inflammatory mediators in diabetic animals and humans are inversely related to levels of AGE receptor-1,123,124 suggesting an anti-inflammatory protective role for AGE receptor-1 in diabetes.123
AGE, RAGE,97,117 and other DAMPs are also strongly implicated in the pathogenesis of nondiabetic glomerular diseases. Podocyte injury and adriamycin-induced glomerulosclerosis are accompanied by production of RAGE ligands; RAGE-deficient or sRAGE-treated mice are protected from glomerular injury.125 Increased AGE and RAGE levels, macrophage infiltration, fibrosis, and inflammation are found in the kidneys of mice that develop glomerular disease from an atherogenic high-fat diet; this lipid-induced renal injury is regulated in part by galectin-3.126
TLR4 is up-regulated in mouse models of cryoglobulinemic membranoproliferative glomerulonephritis and localizes to podocytes of nephritic glomeruli; stimulation of podocytes with either TLR4 ligands or fibrinogen results in similar patterns of enhanced chemokine expression, suggesting that TLR4 functions as a DAMPs receptor for endogenous ligands and may mediate glomerular injury by stimulating innate immunity.127 In nephrotoxic nephritis, a model of rapidly progressive glomerulonephritis, Hsp60 is released from kidneys and excreted in urine, and administration of Hsp60 exacerbates disease in a T cell-dependent manner.128
Expression of S100 proteins increases in kidneys in anti-Thy-1 antibody-induced glomerulonephritis, a rat model of mesangial proliferative glomerulonephritis.129 Patients with glomerulonephritis have HMGB1 in serum, interstitial mononuclear cells, and glomeruli.130 Two S100 proteins, myeloid-related proteins MRP8 and MRP14, are detected in macrophages in renal biopsies from patients with glomerulonephritis; the presence of these proteins and their heterodimeric complexes in macrophages infiltrating glomeruli correlates with the severity of the acute inflammatory process, and chronic inflammation associates with MRP8/MRP14 infiltrates without complex formation in the renal interstitium.131
Fibrosis
Expression of Hsp27, phospho-Hsp27,132 RAGE,133 and calreticulin134 is up-regulated in an animal model of tubulointerstitial fibrosis (unilateral ureteral obstruction [UUO]) and TGFβ-induced epithelial-to-mesenchymal transition in proximal tubular epithelial cells in vitro. The ECM protein, biglycan, increases in tubule epithelial cells 4 days after UUO and later in infiltrating and interstitial cells.135 Biglycan is a pro-inflammatory DAMP in UUO-induced kidney injury; activation of the NLRP3 inflammasome and increases in levels of pro-inflammatory IL-1β after UUO are reduced in biglycan null mice.57 Reactive oxygen species, oxidative stress, S100A4, and Hsp47 are involved in fibrosis after IRI, but their role in immune function has not been investigated.136
Lupus Nephropathy and Autoimmune Disease
HMGB1 has been linked to the pathogenesis of a variety of proinflammatory and autoimmune diseases, including systemic lupus erythematosus (SLE).137 Circulating HMGB1 levels increase in SLE patients and in mice,138,139 and HMGB1 may be involved in antibody-induced kidney damage in SLE.140 In lupus-prone MRL-Fas(lpr) mice, p38 MAPK activation-induced infiltration and maturation of DCs and secretion of HMGB1 from DCs have been implicated in autoimmune kidney disease.141 Inflammatory and immune responses in SLE, and particularly in lupus nephritis, can be induced by HMGB1-nucleosome complexes,138,142 but necrotic nucleosomes also contain double-stranded DNA, which has long been recognized as a key mediator of lupus nephritis. Chromatin fragments can participate in the pathogenesis of kidney disease in SLE by stimulating the innate immune system and by engaging the adaptive immune response to produce antichromatin and anti-double-stranded DNA antibodies leading to glomerular deposits of immune complexes, which are the hallmark and likely the critical initiating events in lupus nephritis.143
Transplantation
The contribution of DAMPs to IRI is likely also pertinent in kidney transplantation, because donor kidneys are susceptible to delayed graft function resulting from ischemia followed by reperfusion. Release of DAMPs, such as HMGB1, ATP, uric acid, and IL-1α, from allografts may induce pro-inflammatory effects and adaptive immune responses.144 TLRs mediate the effects of some these endogenous molecules in transplant by signaling through type 1 interferons.145 In human kidney transplants, increased expression of TLR4 and HMGB1 is found in deceased donor kidneys, and kidneys from patients with a TLR4 loss-of-function mutation have lower levels of inflammatory markers and improved graft function post-transplant.146 Moreover, TLRs and their endogenous ligands may be important in chronic allograft dysfunction. Expression of DAMPs, including biglycan, HSPs, fibrinogen, and HMGB1, increases in the acute and chronic phases after kidney transplantation.147 Kidney transplants to mice lacking TLR2, TLR4, or the adaptor proteins, MyD88 and TRIF, show improved graft function and morphology and decreased leukocyte infiltration and expression of fibrotic markers, cytokines, and chemokines.147
Higher levels of S100A8 and S100A9 in renal biopsies taken in the acute phase after transplant are predictive of favorable graft outcome (patients that later had stable graft function compared with those progressing to chronic allograft nephropathy)148; these S100 proteins are typically pro-inflammatory, but their beneficial role in wound repair may predominate in transplantation and rejection. Glycosylation of Tamm-Horsfall glycoprotein is altered, and its various immunomodulatory functions are diminished in renal transplant patients at least in part because of altered NF-κB p52 nuclear translocation.149
Nondiabetic Chronic Kidney Disease
Many of the DAMPs molecules associated with AKI are also important in CKD. Increased HMGB1 levels correlate with pro-inflammatory markers and declining kidney function in CKD.150 Endothelial dysfunction in CKD, which associates with elevated serum levels of AGE, may be due to AGE-induced suppression of endothelial NOS.151 RAGE is proinflammatory in hemodialysis patients152; sRAGE and endogenous secretory RAGE levels inversely correlate with renal function and inflammatory state in hemodialysis patients.153,154 Elevated TLR2 expression in patients with CKD and in mice with obstructive nephropathy associates with inflammation and increased expression of biglycan and HMGB1, but TLR2 may not be necessary for chronic kidney injury.155 By contrast Tamm-Horsfall protein activates innate and adaptive immunity through TLR472 and may play an inflammatory role in progression of CKD.71 Levels of HSPs, anti-HSP antibodies, and inflammatory markers are higher in children and young adults with CKD.156
CONCLUSIONS
Processes that contribute to the pathogenesis of acute and chronic kidney disease involve the activation of innate and adaptive immunity. Release of endogenous molecules from dying cells leads to activation of innate immunity and downstream inflammation. By bridging to adaptive immunity, DAMPs also contribute to progressive injury or to attenuation of injury. As our understanding of these molecules and pathways evolves, information on precise therapeutic targets will likely lead to improved outcomes.
DISCLOSURES
This work was supported in part by grants from the National Institutes of Health (R01DK062324 and R01DK056223) and Genzyme (GRIP).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
REFERENCES
- 1.Matzinger P: Tolerance, danger, and the extended family. Annu Rev Immunol 12: 991–1045, 1994 [DOI] [PubMed] [Google Scholar]
- 2.Janeway CA, Jr: Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54: 1–13, 1989 [DOI] [PubMed] [Google Scholar]
- 3.Kono H, Rock KL: How dying cells alert the immune system to danger. Nat Rev Immunol 8: 279–289, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matzinger P: Friendly and dangerous signals: Is the tissue in control? Nat Immunol 8: 11–13, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Matzinger P: The danger model: A renewed sense of self. Science 296: 301–305, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Seong SY, Matzinger P: Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 4: 469–478, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Peter C, Wesselborg S, Lauber K, Krysko DV, Vandenabeele P: Role of attraction and danger signals in the uptake of apoptotic and necrotic cells and its immunological outcome. In: Phagocytosis of Dying Cells: From Molecular Mechanisms to Human Diseases, Berlin, Springer Science+ Business Media BV, 2009, pp 63–101 [Google Scholar]
- 8.Xiang M, Fan J: Pattern recognition receptor-dependent mechanisms of acute lung injury. Mol Med 16: 69–82, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foell D, Wittkowski H, Roth J: Mechanisms of disease: A “DAMP” view of inflammatory arthritis. Nat Clin Pract Rheumatol 3: 382–390, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P: Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim Biophys Acta 1805: 53–71, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Srikrishna G, Freeze HH: Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia 11: 615–628, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eleftheriadis T, Lawson BR: Toll-like receptors and kidney diseases. Inflamm Allergy Drug Targets 8: 191–201, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Robson MG: Toll-like receptors and renal disease. Nephron Exp Nephrol 113: e1–e7, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Shirali AC, Goldstein DR: Tracking the toll of kidney disease. J Am Soc Nephrol 19: 1444–1450, 2008 [DOI] [PubMed] [Google Scholar]
- 15.Gluba A, Banach M, Hannam S, Mikhailidis DP, Sakowicz A, Rysz J: The role of Toll-like receptors in renal diseases. Nat Rev Nephrol 6: 224–235, 2010 [DOI] [PubMed] [Google Scholar]
- 16.Bianchi ME: DAMPs PAM: Ps and alarmins: All we need to know about danger. J Leukocyte Biol 81: 1–5, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Yang D, de la RG, Tewary P, Oppenheim JJ: Alarmins link neutrophils and dendritic cells. Trends Immunol 30: 531–537, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang H, Tracey KJ: Targeting HMGB1 in inflammation. Biochim Biophys Acta 1799: 149–156, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, Devera ME, Liang X, Tor M, Billiar T: The grateful dead: Damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev 220: 60–81, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Bianchi ME, Manfredi AA: High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev 220: 35–46, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A: HMGB1: Endogenous danger signaling. Mol Med 14: 476–484, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bianchi ME: HMGB1 loves company. J Leukocyte Biol 86: 573–576, 2009 [DOI] [PubMed] [Google Scholar]
- 23.Scaffidi P, Misteli T, Bianchi ME: Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418: 191–195, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ: HMGB-1 as a late mediator of endotoxin lethality in mice. Science 285: 248–251, 1999 [DOI] [PubMed] [Google Scholar]
- 25.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR: The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 201: 1135–1143, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ: High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukocyte Biol 81: 59–66, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, Morser J, Stern D, Schmidt AM: The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem 270: 25752–25761, 1995 [DOI] [PubMed] [Google Scholar]
- 28.Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E: High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol 290: C917–C924, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Schneider M, Hansen JL, Sheikh SP: S100A4: A common mediator of epithelial-mesenchymal transition, fibrosis and regeneration in diseases? J Mol Med 86: 507–522, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Foell D, Wittkowski H, Vogl T, Roth J: S100 proteins expressed in phagocytes: A novel group of damage-associated molecular pattern molecules. J Leukocyte Biol 81: 28–37, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM: RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 97: 889–901, 1999 [DOI] [PubMed] [Google Scholar]
- 32.Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA: Proinflammatory activities of S100: Proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol 170: 3233–3242, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Ehrchen JM, Sunderkotter C, Foell D, Vogl T, Roth J: The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukocyte Biol 86: 557–566, 2009 [DOI] [PubMed] [Google Scholar]
- 34.van Eden W, van der Zee R, Prakken B: Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol 5: 318–330, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Srivastava P: Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol 2: 185–194, 2002 [DOI] [PubMed] [Google Scholar]
- 36.Tsan MF, Gao B: Heat shock proteins and immune system. J Leukocyte Biol 85: 905–910, 2009 [DOI] [PubMed] [Google Scholar]
- 37.Li Z, Menoret A, Srivastava P: Roles of heat-shock proteins in antigen presentation and cross-presentation. Curr Opin Immunol 14: 45–51, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Ishii T, Udono H, Yamano T, Ohta H, Uenaka A, Ono T, Hizuta A, Tanaka N, Srivastava PK, Nakayama E: Isolation of MHC class I-restricted tumor antigen peptide and its precursors associated with heat shock proteins hsp70, hsp90, and gp96. J Immunol 162: 1303–1309, 1999 [PubMed] [Google Scholar]
- 39.Schmidt AM, Yan SD, Yan SF, Stern DM: The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108: 949–955, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramasamy R, Yan SF, Schmidt AM: RAGE: Therapeutic target and biomarker of the inflammatory response: The evidence mounts. J Leukocyte Biol 86: 505–512, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Yan SF, Ramasamy R, Schmidt AM: Soluble RAGE: Therapy and biomarker in unraveling the RAGE axis in chronic disease and aging. Biochem Pharmacol 79: 1379–1386, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan SF, Ramasamy R, Schmidt AM: Receptor for AGE (RAGE) and its ligands: Cast into leading roles in diabetes and the inflammatory response. J Mol Med 87: 235–247, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamphier MS, Sirois CM, Verma A, Golenbock DT, Latz E: TLR9 and the recognition of self and non-self nucleic acids. Ann N Y Acad Sci 1082: 31–43, 2006 [DOI] [PubMed] [Google Scholar]
- 44.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ: Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest 119: 305–314, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H, Kohn LD, Klinman DM: Genomic DNA released by dying cells induces the maturation of APCs. J Immunol 167: 2602–2607, 2001 [DOI] [PubMed] [Google Scholar]
- 46.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ: Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Calfee CS, Matthay MA: Clinical immunology: Culprits with evolutionary ties. Nature 464: 41–42, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi Y, Evans JE, Rock KL: Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425: 516–521, 2003 [DOI] [PubMed] [Google Scholar]
- 49.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Martinon F, Mayor A, Tschopp J: The Inflammasomes: Guardians of the Body. Annu Rev Immunol 27: 229–265, 2009 [DOI] [PubMed] [Google Scholar]
- 51.Liu-Bryan R: Intracellular innate immunity in gouty arthritis: Role of NALP3 inflammasome. Immunol Cell Biol 88: 20–23, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi Y, Galusha SA, Rock KL: Cutting edge: Elimination of an endogenous adjuvant reduces the activation of CD8 T lymphocytes to transplanted cells and in an autoimmune diabetes model. J Immunol 176: 3905–3908, 2006 [DOI] [PubMed] [Google Scholar]
- 53.Ganz T, Selsted ME, Szklarek D, Harwig SS, Daher K, Bainton DF, Lehrer RI: Defensins: Natural peptide antibiotics of human neutrophils. J Clin Invest 76: 1427–1435, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zaiou M, Gallo R: Cathelicidins, essential gene-encoded mammalian antibiotics. J Mol Med 80: 549–561, 2002 [DOI] [PubMed] [Google Scholar]
- 55.Masson PL, Heremans JF, Schonne E: Lactoferrin, an iron-binding protein in neutrophilic leukocytes. J Exp Med 130: 643–658, 1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaefer L: Extracellular matrix molecules: Endogenous danger signals as new drug targets in kidney diseases. Curr Opin Pharmacol 10: 185–190, 2010 [DOI] [PubMed] [Google Scholar]
- 57.Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, Bruckner P, Pfeilschifter J, Schaefer RM, Grone HJ, Schaefer L: Biglycan, a danger signal that activates the NLRP3 inflammasome via Toll and P2X receptors. J Biol Chem 284: 24035–24048, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sato S, St-Pierre C, Bhaumik P, Nieminen J: Galectins in innate immunity: Dual functions of host soluble beta-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs). Immunol Rev 230: 172–187, 2009 [DOI] [PubMed] [Google Scholar]
- 59.Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, Hirashima M, Liu FT: Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol 165: 2156–2164, 2000 [DOI] [PubMed] [Google Scholar]
- 60.Fredholm BB: Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 14: 1315–1323, 2007 [DOI] [PubMed] [Google Scholar]
- 61.Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC: Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 112: 358–404, 2006 [DOI] [PubMed] [Google Scholar]
- 62.Trautmann A: Extracellular ATP in the immune system: More than just a “danger signal.” Sci Signal 2: pe6, 2009 [DOI] [PubMed] [Google Scholar]
- 63.Di Virgilio F: Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol Sci 28: 465–472, 2007 [DOI] [PubMed] [Google Scholar]
- 64.Blackburn MR, Vance CO, Morschl E, Wilson CN: Adenosine receptors and inflammation. Handb Exp Pharmacol: 215–269, 2009 [DOI] [PubMed] [Google Scholar]
- 65.Schenk H, Vogt M, Droge W, Schulze- Osthoff K: Thioredoxin as a potent costimulus of cytokine expression. J Immunol 156: 765–771, 1996 [PubMed] [Google Scholar]
- 66.Bertini R, Howard OM, Dong HF, Oppenheim JJ, Bizzarri C, Sergi R, Caselli G, Pagliei S, Romines B, Wilshire JA, Mengozzi M, Nakamura H, Yodoi J, Pekkari K, Gurunath R, Holmgren A, Herzenberg LA, Ghezzi P: Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med 189: 1783–1789, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haraldsen G, Balogh J, Pollheimer J, Sponheim J, Knchler AM: Interleukin-33 - cytokine of dual function or novel alarmin? Trends Immunol 30: 227–233, 2009 [DOI] [PubMed] [Google Scholar]
- 68.Serafini-Cessi F, Malagolini N, Cavallone D: Tamm-Horsfall glycoprotein: Biology and clinical relevance. Am J Kidney Dis 42: 658–676, 2003 [DOI] [PubMed] [Google Scholar]
- 69.Kistler AD, Mischak H, Poster D, Dakna M, Wuthrich RP, Serra AL: Identification of a unique urinary biomarker profile in patients with autosomal dominant polycystic kidney disease. Kidney Int 76: 89–96, 2009 [DOI] [PubMed] [Google Scholar]
- 70.Ling XB, Sigdel TK, Lau K, Ying L, Lau I, Schilling J, Sarwal MM: Integrative Urinary Peptidomics in Renal Transplantation Identifies Biomarkers for Acute Rejection. J Am Soc Nephrol 21: 646–653, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prajczer S, Heidenreich U, Pfaller W, Kotanko P, Lhotta K, Jennings P: Evidence for a role of uromodulin in chronic kidney disease progression. Nephrol Dial Transplant 25: 1896–1903, 2010 [DOI] [PubMed] [Google Scholar]
- 72.Saemann MD, Weichhart T, Zeyda M, Staffler G, Schunn M, Stuhlmeier KM, Sobanov Y, Stulnig TM, Akira S, von GA, von AU, Horl WH, Zlabinger GJ: Tamm-Horsfall glycoprotein links innate immune cell activation with adaptive immunity via a Toll-like receptor-4-dependent mechanism. J Clin Invest 115: 468–475, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.El-Achkar TM, Wu XR, Rauchman M, McCracken R, Kiefer S, Dagher PC: Tamm-Horsfall protein protects the kidney from ischemic injury by decreasing inflammation and altering TLR4 expression. Am J Physiol Renal Physiol 295: F534–F544, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsan MF, Gao B: Endogenous ligands of Toll-like receptors. J Leukocyte Biol 76: 514–519, 2004 [DOI] [PubMed] [Google Scholar]
- 75.Miyake K: Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol 19: 3–10, 2007 [DOI] [PubMed] [Google Scholar]
- 76.Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP: Understanding RAGE, the receptor for advanced glycation end products. J Mol Med 83: 876–886, 2005 [DOI] [PubMed] [Google Scholar]
- 77.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ: HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 28: 367–388, 2010 [DOI] [PubMed] [Google Scholar]
- 78.Harashima A, Yamamoto Y, Cheng C, Tsuneyama K, Myint KM, Takeuchi A, Yoshimura K, Li H, Watanabe T, Takasawa S, Okamoto H, Yonekura H, Yamamoto H: Identification of mouse orthologue of endogenous secretory receptor for advanced glycation end-products: Structure, function and expression. Biochem J 396: 109–115, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Santilli F, Vazzana N, Bucciarelli LG, Davi G: Soluble forms of RAGE in human diseases: Clinical and therapeutical implications. Curr Med Chem 16: 940–952, 2009 [DOI] [PubMed] [Google Scholar]
- 80.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G: The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10: 241–247, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lamkanfi M, Dixit VM: Inflammasomes: Guardians of cytosolic sanctity. Immunol Rev 227: 95–105, 2009 [DOI] [PubMed] [Google Scholar]
- 82.Schroder K, Zhou R, Tschopp J: The NLRP3 inflammasome: A sensor for metabolic danger? Science 327: 296–300, 2010 [DOI] [PubMed] [Google Scholar]
- 83.Iwasaki A, Medzhitov R: Regulation of adaptive immunity by the innate immune system. Science 327: 291–295, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chun KH, Seong SY: CD14 but not MD2 transmit signals from DAMP. Int Immunopharmacol 10: 98–106, 2010 [DOI] [PubMed] [Google Scholar]
- 85.Chen GY, Tang J, Zheng P, Liu Y: CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 323: 1722–1725, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu Y, Chen GY, Zheng P: CD24-Siglec G/10 discriminates danger from pathogen-associated molecular patterns. Trends Immunology 30: 557–561, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bianchi ME, Manfredi AA: Immunology: Dangers in and out. Science 323: 1683–1684, 2009 [DOI] [PubMed] [Google Scholar]
- 88.Urbonaviciute V, Furnrohr BG, Weber C, Haslbeck M, Wilhelm S, Herrmann M, Voll RE: Factors masking HMGB1 in human serum and plasma. J Leukocyte Biol 81: 67–74, 2007 [DOI] [PubMed] [Google Scholar]
- 89.Rubartelli A, Lotze MT: Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol 28: 429–436, 2007 [DOI] [PubMed] [Google Scholar]
- 90.Carta S, Castellani P, Delfino L, Tassi S, Vene R, Rubartelli A: DAMPs and inflammatory processes: The role of redox in the different outcomes. J Leukocyte Biol 86: 549–555, 2009 [DOI] [PubMed] [Google Scholar]
- 91.Iwata A, Morgan-Stevenson V, Schwartz B, Liu L, Tupper J, Zhu X, Harlan J, Winn R: Extracellular BCL2 proteins are danger-associated molecular patterns that reduce tissue damage in murine models of ischemia-reperfusion injury. PLoS ONE 5: e9103, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Goligorsky MS: Immune system in renal injury and repair: Burning the candle from both ends? Pharmacol Res 58: 122–128, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Metcalf DJ, Nightingale TD, Zenner HL, Lui-Roberts WW, Cutler DF: Formation and function of Weibel-Palade bodies. J Cell Sci 121: 19–27, 2008 [DOI] [PubMed] [Google Scholar]
- 94.Kuo MC, Patschan D, Patschan S, Cohen-Gould L, Park HC, Ni J, Addabbo F, Goligorsky MS: Ischemia-induced exocytosis of Weibel-Palade bodies mobilizes stem cells. J Am Soc Nephrol 19: 2321–2330, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patschan D, Patschan S, Gobe GG, Chintala S, Goligorsky MS: Uric Acid Heralds Ischemic Tissue Injury to Mobilize Endothelial Progenitor Cells. J Am Soc Nephrol 18: 1516–1524, 2007 [DOI] [PubMed] [Google Scholar]
- 96.Goligorsky MS, Patschan D, Kuo MC: Weibel-Palade bodies: Sentinels of acute stress. Nat Rev Nephrol 5: 423–426, 2009 [DOI] [PubMed] [Google Scholar]
- 97.Bohlender JM, Franke S, Stein G, Wolf G: Advanced glycation end products and the kidney. Am J Physiol Renal Physiol 289: F645–F659, 2005 [DOI] [PubMed] [Google Scholar]
- 98.Semba RD, Ferrucci L, Fink JC, Sun K, Beck J, Dalal M, Guralnik JM, Fried LP: Advanced glycation end products and their circulating receptors and level of kidney function in older community-dwelling women. Am J Kidney Dis 53: 51–58, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li L, Okusa MD: Blocking the immune response in ischemic acute kidney injury: The role of adenosine 2A agonists. Nat Clin Pract Nephrol 2: 432–444, 2006 [DOI] [PubMed] [Google Scholar]
- 100.Lu CY, Hartono J, Senitko M, Chen J: The inflammatory response to ischemic acute kidney injury: A result of the “right stuff” in the “wrong place”? Curr Opin Nephrol Hypertens 16: 83–89, 2007 [DOI] [PubMed] [Google Scholar]
- 101.Mkaddem SB, Werts C, Goujon JM, Bens M, Pedruzzi E, Ogier-Denis E, Vandewalle A: Heat shock protein gp96 interacts with protein phosphatase 5 and controls Toll-like receptor 2 (TLR2)-mediated activation of extracellular signal-regulated kinase (ERK) 1/2 in post-hypoxic kidney cells. J Biol Chem 284: 12541–12549, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Harrison EM, Sharpe E, Bellamy CO, McNally SJ, Devey L, Garden OJ, Ross JA, Wigmore SJ: Heat shock protein 90-binding agents protect renal cells from oxidative stress and reduce kidney ischemia-reperfusion injury. Am J Physiol Renal Physiol 295: F397–F405, 2008 [DOI] [PubMed] [Google Scholar]
- 103.Zhang PL, Lun M, Schworer CM, Blasick TM, Masker KK, Jones JB, Carey DJ: Heat shock protein expression is highly sensitive to ischemia-reperfusion injury in rat kidneys. Annals Clin Lab Sci 38: 57–64, 2008 [PubMed] [Google Scholar]
- 104.Park SW, Chen SWC, Kim M, D'Agati VD, Lee HT: Human heat shock protein 27-overexpressing mice are protected against acute kidney injury after hepatic ischemia and reperfusion. Am J Physiol Renal Physiol 297: F885–F894, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen SWC, Kim M, Kim M, Song JH, Park SW, Wells D, Brown K, Belleroche JD, D'Agati VD, Lee HT: Mice that overexpress human heat shock protein 27 have increased renal injury following ischemia reperfusion. Kidney Int 75: 499–510, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chung KY, Park JJ, Kim YS: The role of high mobility group box-1 in renal ischemia and reperfusion injury and the effect of ethyl pyruvate. Transplant Proc 40: 2136–2138, 2008 [DOI] [PubMed] [Google Scholar]
- 107.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ: TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest 117: 2847–2859, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bonventre JV: Kidney injury molecule-1 (KIM-1): A urinary biomarker and much more. Nephrol Dial Transplant 24: 3265–3268, 2009 [DOI] [PubMed] [Google Scholar]
- 109.Ichimura T, Asseldonk EJP, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV: Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest 118: 1657–1668, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tanji NOZO, Markowitz GS, Fu CAIF, Kislinger THOM, Taguchi AKIH, Pischetsrieder MONI, Stern DAVI, Schmidt AM, D'Agati VD: Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol 11: 1656–1666, 2000 [DOI] [PubMed] [Google Scholar]
- 111.Heidland A, Sebekova K, Schinzel R: Advanced glycation end products and the progressive course of renal disease. Am J Kidney Dis 38: S100–S106, 2001 [DOI] [PubMed] [Google Scholar]
- 112.Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, Bucciarelli LG, Rong LL, Moser B, Markowitz GS, Stein G, Bierhaus A, Liliensiek B, Arnold B, Nawroth PP, Stern DM, D'Agati VD, Schmidt AM: RAGE Drives the Development of Glomerulosclerosis and Implicates Podocyte Activation in the Pathogenesis of Diabetic Nephropathy. Am J Pathol 162: 1123–1137, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cooper ME: Importance of advanced glycation end products in diabetes-associated cardiovascular and renal disease. Am J Hypertens 17: 31S–38S, 2004 [DOI] [PubMed] [Google Scholar]
- 114.Myint KM, Yamamoto Y, Doi T, Kato I, Harashima A, Yonekura H, Watanabe T, Shinohara H, Takeuchi M, Tsuneyama K, Hashimoto N, Asano M, Takasawa S, Okamoto H, Yamamoto H: RAGE control of diabetic nephropathy in a mouse model: Effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes 55: 2510–2522, 2006 [DOI] [PubMed] [Google Scholar]
- 115.Tan AL, Forbes JM, Cooper ME: AGE, RAGE, and ROS in diabetic nephropathy. Semin Nephrol 27: 130–143, 2007 [DOI] [PubMed] [Google Scholar]
- 116.Sourris KC, Forbes JM: Interactions between advanced glycation end-products (AGE) and their receptors in the development and progression of diabetic nephropathy: Are these receptors valid therapeutic targets? Curr Drug Targets 10: 42–50, 2009 [DOI] [PubMed] [Google Scholar]
- 117.D'Agati V, Yan SF, Ramasamy R, Schmidt AM: RAGE, glomerulosclerosis and proteinuria: Roles in podocytes and endothelial cells. Trends Endocrinol Metab 21: 50–56, 2010 [DOI] [PubMed] [Google Scholar]
- 118.Coughlan MT, Mibus AL, Forbes JM: Oxidative stress and advanced glycation in diabetic nephropathy. Ann NY Acad Sci 1126: 190–193, 2008 [DOI] [PubMed] [Google Scholar]
- 119.Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, Tan AL, Fukami K, Thallas-Bonke V, Nawroth PP, Brownlee M, Bierhaus A, Cooper ME, Forbes JM: RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol 20: 742–752, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen SC, Guh JY, Hwang CC, Chiou SJ, Lin TD, Ko YM, Huang JS, Yang YL, Chuang LY: Advanced glycation end-products activate extracellular signal-regulated kinase via the oxidative stress-EGF receptor pathway in renal fibroblasts. J Cell Biochem 109: 38–48, 2010 [DOI] [PubMed] [Google Scholar]
- 121.Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, Nawroth PP: The Pattern Recognition Receptor (RAGE) Is a Counterreceptor for Leukocyte Integrins: A Novel Pathway for Inflammatory Cell Recruitment. J Exp Med 198: 1507–1515, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yamamoto Y, Kato I, Doi T, Yonekura H, Ohashi S, Takeuchi M, Watanabe T, Yamagishi S-i, Sakurai S, Takasawa S, Okamoto H, Yamamoto H: Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J Clin Invest 108: 261–268, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cai W, He JC, Zhu L, Lu C, Vlassara H: Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. Proc Natl Acad Sci U S A 103: 13801–13806, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vlassara H, Uribarri J, Cai W, Striker G: Advanced glycation end product homeostasis: exogenous oxidants and innate defenses. Ann N Y Acad Sci 1126: 46–52, 2008 [DOI] [PubMed] [Google Scholar]
- 125.Guo J, Ananthakrishnan R, Qu W, Lu Y, Reiniger N, Zeng S, Ma W, Rosario R, Yan SF, Ramasamy R, D'Agati V, Marie Schmidt A: RAGE Mediates Podocyte Injury in Adriamycin-induced Glomerulosclerosis. J Am Soc Nephrol 19: 961–972, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Iacobini C, Menini S, Ricci C, Scipioni A, Sansoni V, Mazzitelli G, Cordone S, Pesce C, Pugliese F, Pricci F, Pugliese G: Advanced lipoxidation end-products mediate lipid-induced glomerular injury: Role of receptor-mediated mechanisms. J Pathol 218: 360–369, 2009 [DOI] [PubMed] [Google Scholar]
- 127.Banas MC, Banas B, Hudkins KL, Wietecha TA, Iyoda M, Bock E, Hauser P, Pippin JW, Shankland SJ, Smith KD, Stoelcker B, Liu G, Grone HJ, Kramer BK, Alpers CE: TLR4 links podocytes with the innate immune system to mediate glomerular injury. J Am Soc Nephrol 19: 704–713, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lang A, Benke D, Eitner F, Engel D, Ehrlich S, Breloer M, Hamilton-Williams E, Specht S, Hoerauf A, Floege J, von BA, Kurts C: Heat shock protein 60 is released in immune-mediated glomerulonephritis and aggravates disease: In vivo evidence for an immunologic danger signal. J Am Soc Nephrol 16: 383–391, 2005 [DOI] [PubMed] [Google Scholar]
- 129.Sadlier DM, Ouyang X, McMahon B, Mu W, Ohashi R, Rodgers K, Murray D, Nakagawa T, Godson C, Doran P, Brady HR, Johnson RJ: Microarray and bioinformatic detection of novel and established genes expressed in experimental anti-Thy1 nephritis. Kidney Int 68: 2542–2561, 2005 [DOI] [PubMed] [Google Scholar]
- 130.Sato F, Maruyama S, Hayashi H, Sakamoto I, Yamada S, Uchimura T, Morita Y, Ito Y, Yuzawa Y, Maruyama I, Matsuo S: High mobility group box chromosomal protein 1 in patients with renal diseases. Nephron Clinical Practice 108: c194–c201, 2008 [DOI] [PubMed] [Google Scholar]
- 131.Frosch M, Vogl T, Waldherr R, Sorg C, Sunderkotter C, Roth J: Expression of MRP8 and MRP14 by macrophages is a marker for severe forms of glomerulonephritis. J Leukocyte Biol 75: 198–206, 2004 [DOI] [PubMed] [Google Scholar]
- 132.Vidyasagar A, Reese S, Acun Z, Hullett D, Djamali A: HSP27 is involved in the pathogenesis of kidney tubulointerstitial fibrosis. Am J Physiol Renal Physiol 295: F707–F716, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lange-Sperandio B, Schimpgen K, Rodenbeck B, Chavakis T, Bierhaus A, Nawroth P, Thornhill B, Schaefer F, Chevalier RL: Distinct roles of Mac-1 and its counter-receptors in neonatal obstructive nephropathy. Kidney Int 69: 81–88, 2006 [DOI] [PubMed] [Google Scholar]
- 134.Kypreou KP, Kavvadas P, Karamessinis P, Peroulis M, Alberti A, Sideras P, Psarras S, Capetanaki Y, Politis PK, Charonis AS: Altered expression of calreticulin during the development of fibrosis. Proteomics 8: 2407–2419, 2008 [DOI] [PubMed] [Google Scholar]
- 135.Schaefer L, Macakova K, Raslik I, Micegova M, Grone HJ, Schonherr E, Robenek H, Echtermeyer FG, Grassel S, Bruckner P, Schaefer RM, Iozzo RV, Kresse H: Absence of decorin adversely influences tubulointerstitial fibrosis of the obstructed kidney by enhanced apoptosis and increased inflammatory reaction. Am J Pathol 160: 1181–1191, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim J, Seok YM, Jung KJ, Park KM: Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. Am J Physiol Renal Physiol 297: F461–F470, 2009 [DOI] [PubMed] [Google Scholar]
- 137.Pan HF, Wu GC, Li WP, Li XP, Ye DQ: High mobility group box 1: A potential therapeutic target for systemic lupus erythematosus. Mol Biol Rep 37: 1191–1195, 2010 [DOI] [PubMed] [Google Scholar]
- 138.Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De MF, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, Kalden JR, Schett G, Rovere-Querini P, Herrmann M, Voll RE: Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: Implications for the pathogenesis of SLE. J Exp Med 205: 3007–3018, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jiang W, Pisetsky DS: Expression of high mobility group protein 1 in the sera of patients and mice with systemic lupus erythematosus. Annals Rheum Dis 67: 727–728, 2008 [DOI] [PubMed] [Google Scholar]
- 140.Qing X, Pitashny M, Thomas DB, Barrat FJ, Hogarth MP, Putterman C: Pathogenic anti-DNA antibodies modulate gene expression in mesangial cells: Involvement of HMGB1 in anti-DNA antibody-induced renal injury. Immunol Lett 121: 61–73, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Iwata Y, Furuichi K, Sakai N, Yamauchi H, Shinozaki Y, Zhou H, Kurokawa Y, Toyama T, Kitajima S, Okumura T, Yamada S, Maruyama I, Matsushima K, Kaneko S, Wada T: Dendritic cells contribute to autoimmune kidney injury in MRL-Faslpr mice. J Rheumatol 36: 306–314, 2009 [DOI] [PubMed] [Google Scholar]
- 142.Mortensen ES, Fenton KA, Rekvig OP: Lupus nephritis: The central role of nucleosomes revealed. Am J Pathol 172: 275–283, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mortensen ES, Rekvig OP: Nephritogenic potential of anti-DNA antibodies against necrotic nucleosomes. J Am Soc Nephrol 20: 696–704, 2009 [DOI] [PubMed] [Google Scholar]
- 144.Rao DA, Pober JS: Endothelial injury, alarmins, and allograft rejection. Crit Rev Immunol 28: 229–248, 2008 [DOI] [PubMed] [Google Scholar]
- 145.Alegre ML, Goldstein DR, Chong AS: Toll-like receptor signaling in transplantation. Curr Opin Organ Transplant 13: 358–365, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kruger B, Krick S, Dhillon N, Lerner SM, Ames S, Bromberg JS, Lin M, Walsh L, Vella J, Fischereder M, Kramer BK, Colvin RB, Heeger PS, Murphy BT, Schroppel B: Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci U S A 106: 3390–3395, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang S, Schmaderer C, Kiss E, Schmidt C, Bonrouhi M, Porubsky S, Gretz N, Schaefer L, Kirschning CJ, Popovic ZV, Grone HJ: Recipient Toll-like receptors contribute to chronic graft dysfunction by both MyD88- and TRIF-dependent signaling. Dis Model Mech 3: 92–103, 2010 [DOI] [PubMed] [Google Scholar]
- 148.Eikmans M, Roos-van Groningen MC, Sijpkens YW, Ehrchen J, Roth J, Baelde HJ, Bajema IM, de Fijter JW, de HE, Bruijn JA: Expression of surfactant protein-C, S100A8, S100A9, and B cell markers in renal allografts: investigation of the prognostic value. J Am Soc Nephrol 16: 3771–3786, 2005 [DOI] [PubMed] [Google Scholar]
- 149.Wu TH, Hsieh SC, Li KJ, Wu CH, Yu CL, Yang AH, Tsai CY: Altered glycosylation of Tamm-Horsfall glycoprotein derived from renal allograft recipients leads to changes in its biological function. Transpl Immunol 18: 237–245, 2008 [DOI] [PubMed] [Google Scholar]
- 150.Bruchfeld A, Qureshi AR, Lindholm B, Barany P, Yang L, Stenvinkel P, Tracey KJ: High Mobility Group Box Protein-1 correlates with renal function in chronic kidney disease (CKD). Mol Med 14: 109–115, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Linden E, Cai W, He JC, Xue C, Li Z, Winston J, Vlassara H, Uribarri J: Endothelial dysfunction in patients with chronic kidney disease results from advanced glycation end products (AGE)-mediated inhibition of endothelial nitric oxide synthase through RAGE activation. ClinJ Am Soc Nephrol 3: 691–698, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rodriguez-Ayala E, Anderstam B, Suliman ME, Seeberger A, Heimburger O, Lindholm B, Stenvinkel P: Enhanced RAGE-mediated NFkappaB stimulation in inflamed hemodialysis patients. Atherosclerosis 180: 333–340, 2005 [DOI] [PubMed] [Google Scholar]
- 153.Kalousova M, Jachymova M, Mestek O, Hodkova M, Kazderova M, Tesar V, Zima T: Receptor for advanced glycation end products–soluble form and gene polymorphisms in chronic haemodialysis patients. Nephrol Dial Transplant 22: 2020–2026, 2007 [DOI] [PubMed] [Google Scholar]
- 154.Kalousova M, Hodkova M, Kazderova M, Fialova J, Tesar V, Dusilova-Sulkova S, Zima T: Soluble receptor for advanced glycation end products in patients with decreased renal function. Am J Kidney Dis 47: 406–411, 2006 [DOI] [PubMed] [Google Scholar]
- 155.Leemans JC, Butter LM, Pulskens WP, Teske GJ, Claessen N, van der PT, Florquin S: The role of Toll-like receptor 2 in inflammation and fibrosis during progressive renal injury. PLoS One 4: e5704, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Musial K, Szprynger K, Szczepanska M, Zwolinska D: The heat shock protein profile in children with chronic kidney disease. Peritoneal Dialysis International 30: 227–232, 2010 [DOI] [PubMed] [Google Scholar]
- 157.Crocker PR, Paulson JC, Varki A: Siglecs and their roles in the immune system. Nat Rev Immunol 7: 255–266, 2007 [DOI] [PubMed] [Google Scholar]
- 158.Liu Y, Zheng P: CD24: A genetic checkpoint in T cell homeostasis and autoimmune diseases. Trends Immunol 28: 315–320, 2007 [DOI] [PubMed] [Google Scholar]
- 159.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT: The NALP3 inflammasome is involved in the innate immune response to amyloid-[beta]. Nat Immunol 9: 857–865, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]