

Abstract
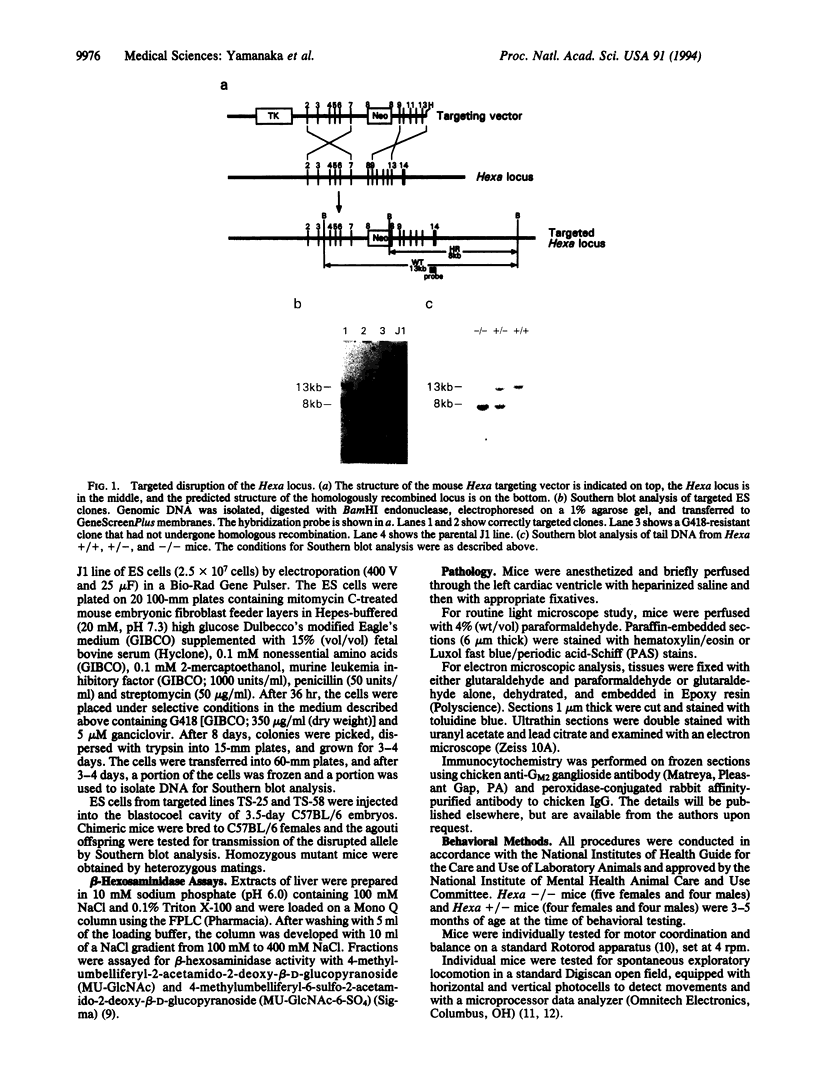
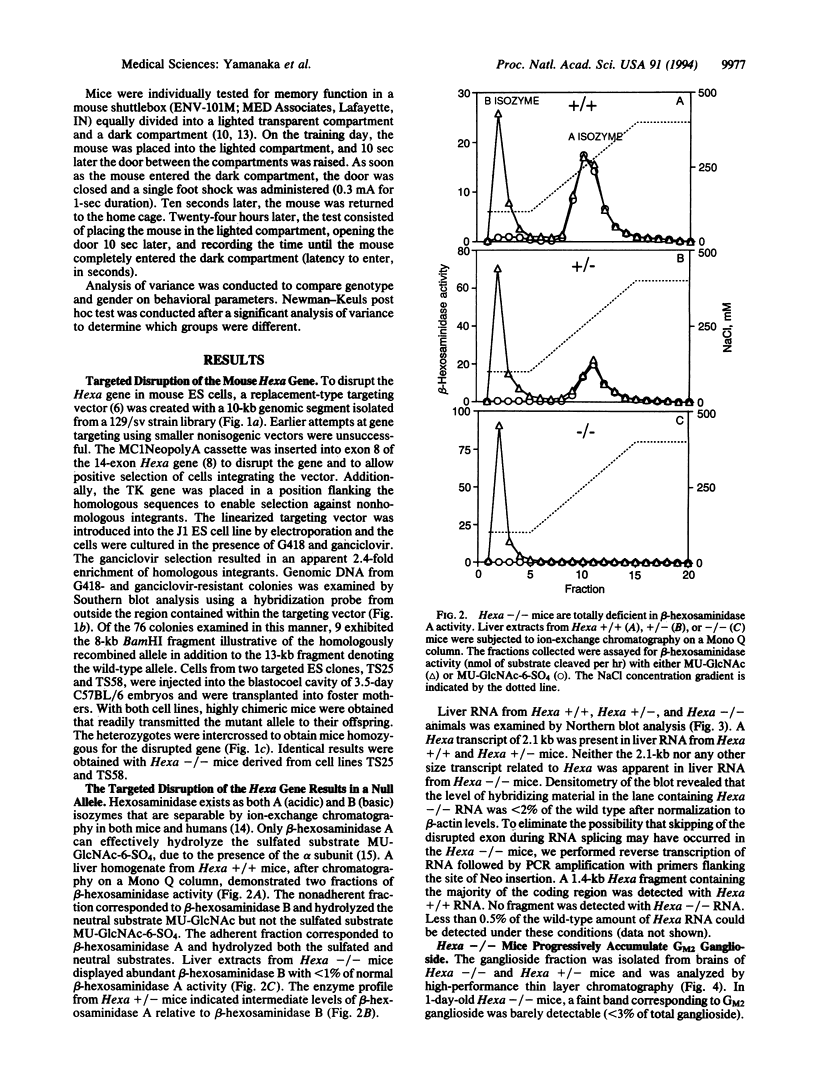
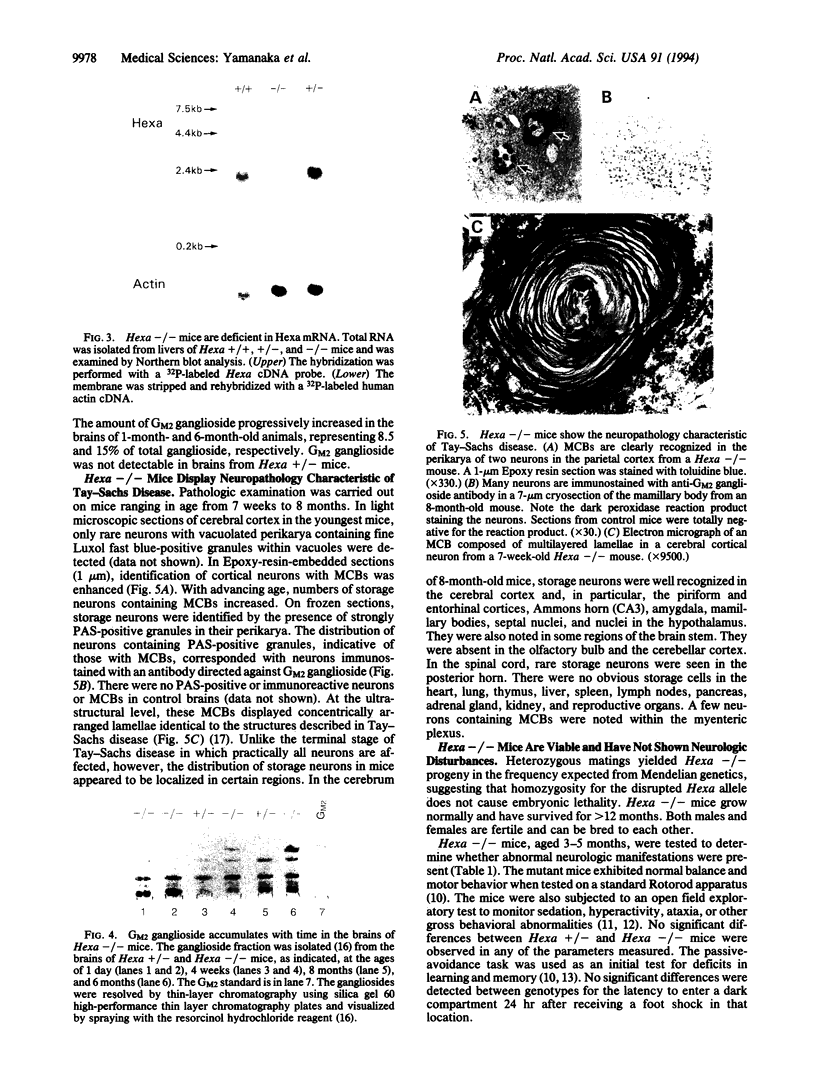
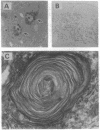
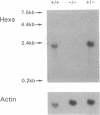
Tay-Sachs disease, the prototype of the GM2 gangliosidoses, is a catastrophic neurodegenerative disorder of infancy. The disease is caused by mutations in the HEXA gene resulting in an absence of the lysosomal enzyme, beta-hexosaminidase A. As a consequence of the enzyme deficiency, GM2 ganglioside accumulates progressively, beginning early in fetal life, to excessive amounts in the central nervous system. Rapid mental and motor deterioration starting in the first year of life leads to death by 2-4 years of age. Through the targeted disruption of the mouse Hexa gene in embryonic stem cells, we have produced mice with biochemical and neuropathologic features of Tay-Sachs disease. The mutant mice displayed < 1% of normal beta-hexosaminidase A activity and accumulated GM2 ganglioside in their central nervous system in an age-dependent manner. The accumulated ganglioside was stored in neurons as membranous cytoplasmic bodies characteristically found in the neurons of Tay-Sachs disease patients. At 3-5 months of age, the mutant mice showed no apparent defects in motor or memory function. These beta-hexosaminidase A-deficient mice should be useful for devising strategies to introduce functional enzyme and genes into the central nervous system. This model may also be valuable for studying the biochemical and pathologic changes occurring during the course of the disease.
Full text
PDF

Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Adachi M., Schneck L., Volk B. W. Ultrastructural studies of eight cases of fetal Tay-Sachs disease. Lab Invest. 1974 Jan;30(1):102–112. [PubMed] [Google Scholar]
- Beccari T., Orlacchio A., Stirling J. L. Identification of beta-N-acetylhexosaminidase A in mouse tissues with the fluorigenic substrate 4-methylumbelliferyl-beta-N-acetylglucosamine 6-sulphate. Biochem J. 1988 Jun 1;252(2):617–620. doi: 10.1042/bj2520617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg J., Banerjee A., Conzelmann E., Sandhoff K. Activating proteins for ganglioside GM2 degradation by beta-hexosaminidase isoenzymes in tissue extracts from different species. Hoppe Seylers Z Physiol Chem. 1983 Jul;364(7):821–829. doi: 10.1515/bchm2.1983.364.2.821. [DOI] [PubMed] [Google Scholar]
- Capecchi M. R. Altering the genome by homologous recombination. Science. 1989 Jun 16;244(4910):1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- Conzelmann E., Nehrkorn H., Kytzia H. J., Sandhoff K., Macek M., Lehovský M., Elleder M., Jirásek A., Kobilková J. Prenatal diagnosis of GM2 gangliosidosis with high residual hexosaminidase A activity (variant B1; pseudo AB variant). Pediatr Res. 1985 Nov;19(11):1220–1224. doi: 10.1203/00006450-198511000-00022. [DOI] [PubMed] [Google Scholar]
- Conzelmann E., Sandhoff K. Biochemical basis of late-onset neurolipidoses. Dev Neurosci. 1991;13(4-5):197–204. doi: 10.1159/000112160. [DOI] [PubMed] [Google Scholar]
- Cork L. C., Munnell J. F., Lorenz M. D., Murphy J. V., Baker H. J., Rattazzi M. C. GM2 ganglioside lysosomal storage disease in cats with beta-hexosaminidase deficiency. Science. 1977 May 27;196(4293):1014–1017. doi: 10.1126/science.404709. [DOI] [PubMed] [Google Scholar]
- Crawley J. N. Subtype-selective cholecystokinin receptor antagonists block cholecystokinin modulation of dopamine-mediated behaviors in the rat mesolimbic pathway. J Neurosci. 1992 Sep;12(9):3380–3391. doi: 10.1523/JNEUROSCI.12-09-03380.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFries J. C., Gervais M. C., Thomas E. A. Response to 30 generations of selection for open-field activity in laboratory mice. Behav Genet. 1978 Jan;8(1):3–13. doi: 10.1007/BF01067700. [DOI] [PubMed] [Google Scholar]
- Decker M. W., McGaugh J. L. Effects of concurrent manipulations of cholinergic and noradrenergic function on learning and retention in mice. Brain Res. 1989 Jan 16;477(1-2):29–37. doi: 10.1016/0006-8993(89)91391-7. [DOI] [PubMed] [Google Scholar]
- Dobrenis K., Joseph A., Rattazzi M. C. Neuronal lysosomal enzyme replacement using fragment C of tetanus toxin. Proc Natl Acad Sci U S A. 1992 Mar 15;89(6):2297–2301. doi: 10.1073/pnas.89.6.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller B. H., Smithies O. Altering genes in animals by gene targeting. Annu Rev Immunol. 1992;10:705–730. doi: 10.1146/annurev.iy.10.040192.003421. [DOI] [PubMed] [Google Scholar]
- Kytzia H. J., Sandhoff K. Evidence for two different active sites on human beta-hexosaminidase A. Interaction of GM2 activator protein with beta-hexosaminidase A. J Biol Chem. 1985 Jun 25;260(12):7568–7572. [PubMed] [Google Scholar]
- Ledeen R. W., Yu R. K. Gangliosides: structure, isolation, and analysis. Methods Enzymol. 1982;83:139–191. doi: 10.1016/0076-6879(82)83012-7. [DOI] [PubMed] [Google Scholar]
- Neuwelt E. A., Johnson W. G., Blank N. K., Pagel M. A., Maslen-McClure C., McClure M. J., Wu P. M. Characterization of a new model of GM2-gangliosidosis (Sandhoff's disease) in Korat cats. J Clin Invest. 1985 Aug;76(2):482–490. doi: 10.1172/JCI111997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhr S. T., Gage F. H. Gene therapy for neurologic disease. Arch Neurol. 1993 Nov;50(11):1252–1268. doi: 10.1001/archneur.1993.00540110122012. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Neuropathology of late onset gangliosidoses. A review. Dev Neurosci. 1991;13(4-5):205–210. doi: 10.1159/000112161. [DOI] [PubMed] [Google Scholar]
- TERRY R. D., WEISS M. Studies in Tay-Sachs disease. II. Ultrastructure of the cerebrum. J Neuropathol Exp Neurol. 1963 Jan;22:18–55. doi: 10.1097/00005072-196301000-00003. [DOI] [PubMed] [Google Scholar]
- Yamanaka S., Johnson O. N., Norflus F., Boles D. J., Proia R. L. Structure and expression of the mouse beta-hexosaminidase genes, Hexa and Hexb. Genomics. 1994 Jun;21(3):588–596. doi: 10.1006/geno.1994.1318. [DOI] [PubMed] [Google Scholar]