

Abstract
Sensory deprivation can induce profound changes to central processing during developmental critical periods (CPs), and the recovery of normal function is maximal if the sensory input is restored during these epochs. Therefore, we asked whether mild and transient hearing loss (HL) during discrete CPs could induce changes to cortical cellular physiology. Electrical and inhibitory synaptic properties were obtained from auditory cortex pyramidal neurons using whole-cell recordings after bilateral earplug insertion or following earplug removal. Varying the age of HL onset revealed brief CPs of vulnerability for membrane and firing properties, as well as, inhibitory synaptic currents. These CPs closed 1 week after ear canal opening on postnatal day (P) 18. To examine whether the cellular properties could recover from HL, earplugs were removed prior to (P17) or after (P23), the closure of these CPs. The earlier age of hearing restoration led to greater recovery of cellular function, but firing rate remained disrupted. When earplugs were removed after the closure of these CPs, several changes persisted into adulthood. Therefore, long-lasting cellular deficits that emerge from transient deprivation during a CP may contribute to delayed acquisition of auditory skills in children who experience temporary HL.
Keywords: auditory, hearing loss, intrinsic property, recovery, sensitive period
Introduction
During development, primary sensory cortices display discrete epochs of heightened plasticity known as critical periods (CPs) (Hubel and Wiesel 1970; Van der Loos and Woolsey 1973; Knudsen et al. 1984; Simons et al. 1984; de Villers-Sidani et al. 2007; Sanes and Bao 2009; Popescu and Polley 2010; Kral and Sharma 2012). Sensory deprivation during CPs can cause permanent neuropathology in adults; however, a similar duration of deprivation leads to less severe effects when induced in adulthood (Dews and Wiesel 1970; LeVay, Wiesel, and Hubel 1980; Carvell and Simons 1996; Takesian, Kotak, and Sanes 2012). Consistent with these findings, the restoration of sensory function prior to, but not after, the closure of CPs can permit the development of normal cortical function (Dawson et al. 1992; Daw 1998; Kral et al. 2001, 2002; Scheiman et al. 2005; Putzar et al. 2007). The principles of CP plasticity are particularly relevant to understanding whether bouts of mild childhood hearing loss (HL), especially those induced by transient periods of otitis media, lead to persistent central auditory deficits (Whitton and Polley 2011). Furthermore, they inform the degree to which functional recovery occurs in deaf subjects implanted with cochlear prostheses (Kral et al. 2001, 2002; Nikolopoulos and Kiprouli 2004; Holt and Svirsky 2008; May-Mederake et al. 2010). If there are discrete CPs for mild HL, we predicted that brief epochs of sound attenuation would disrupt the maturation of cortical membrane and synaptic properties, but that function would return to normal if hearing was reinstated prior to CP closure.
CPs have been most thoroughly documented at the functional level using extracellular neurophysiological recordings to quantify sensory receptive field properties or maps. At a mechanistic level, many of these functional characteristics are thought to depend on the emergence of mature membrane and synaptic properties (e.g., visual system: Di Marco et al. 2009; somatosensory system: Shepherd et al. 2003; auditory system: Dorrn et al. 2010; Sun et al. 2010). For example, input resistance and resting potential contribute to neuronal excitability (Oswald and Reyes 2008), while inhibitory synaptic currents regulate stimulus selectivity (Isaacson and Scanziani 2011). In fact, our approach was motivated by recent studies that reveal early CPs of plasticity within the auditory cortex that open and close shortly after hearing onset (de Villers-Sidani et al. 2007; Barkat, et al. 2011; Polley et al. 2013). In order to explain such system-level changes, this study explored the cellular mechanisms that are sensitive to peripheral manipulation during brief CPs.
Although both moderate and severe HL can disrupt excitatory and inhibitory synaptic properties in auditory cortex (Kotak et al. 2005, 2008; Takesian et al. 2012), little is known about the potential for recovery. The HL-induced reduction of inhibitory current amplitude can be reversed by in vivo pharmacological intervention (Kotak et al. 2013). However, the capacity for cortical cellular deficits to recover following restoration of normal hearing has not been tested. Here, we first show that mild HL can induce significant deficits in membrane and inhibitory synaptic properties during distinct CPs that close by postnatal day (P) 18. When HL was reversed prior to CP closure, most membrane properties recover to normal values, but when HL was reversed just a few days after CP closure, the deficits persisted for several months. These findings imply that long-lasting deficits can emanate from transient bouts of developmental HL, and may contribute to behavioral delays following temporary bouts of HL during childhood (Whitton and Polley 2011).
Materials and Methods
Experimental Animals
This study used 132 Gerbils (Meriones unguiculatus), aged postnatal day (P) 11—91, born from breeding pairs (Charles River Laboratories). Animal care and maintenance were in accordance with the guidelines and rules of the institutional care and use committee, New York University approved by the Office of Laboratory Animal Welfare, Office of Extramural Research, US National Institutes of Health.
Mild Hearing Loss Induced with Bilateral Earplugs
Mild HL was induced by inserting a malleable plug (BlueStik Adhesive Putty, RPM International Inc.) into the opening of each ear canal. This form of HL elevates auditory thresholds by ∼27.5 dB (Fig. 1B). The postnatal day on which earplugs were inserted varied from P11 to P23. Using a stereomicroscope (Olympus), the animal's head was positioned so that the ear canal was fully visible and curved mouse-toothed microtweezers were used to insert the earplug. Earplugs were inserted as round balls and then formed around the unique curvature of each ear canal opening to make a seal. Earplugs were checked every day in the morning and, if necessary, the plug was readjusted or a larger size plug was inserted. The animals acclimated to handling over the first few days of earplug insertion without the need for anesthetics. On the day of recording, earplugs were verified to be in place. Postmortem dissection was used to confirm that the tympanic membranes were intact, and no cases of earplug-induced damage were observed. When earplugs were removed, the tympanic membrane was visualized to confirm that no adhesive putty remained.
Figure 1.
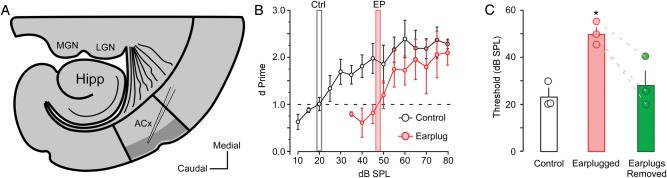
Thalamocortical slice preparation and auditory thresholds. (A) Diagrammatic sketch illustrates the thalamocortical slice preparation and L2/3 sampling region where whole-cell recordings were conducted (gray area). MGN: medial geniculate nucleus, LGN: lateral geniculate nucleus, Hipp: Hippocampus, and ACx: primary auditory cortex. (B) Line plot shows behaviorally assessed auditory thresholds for control animals (gray bar; n = 3) and animals that received earplugs on P11 (red bar; n = 3). Dashed line represents a threshold criterion of d′ = 1. (C) Bar graph shows the group average and individual auditory threshold for control animals, and earplug animals before and after earplug removal. * = P < 0.05.
Behavioral Training and Testing
All animals (3 controls, 3 earplugs) were trained on an operant conditioning task, as previously described (Sarro and Sanes 2010). Animals were placed on controlled water access, and body weight was monitored daily to ensure that it remained >80% of the initial value. Furthermore, animals were allowed to drink until sated on each day of training or testing. Animals were first trained to obtain water from the spout, and were then trained to withdraw from the spout when an acoustic cue (4 kHz pure tone, 1-s duration) was presented. To train the withdrawal response, a low AC current (0.5–1.0 mA, 300 ms; Lafayette Instruments) was delivered through the waterspout immediately after the 4 kHz training (80 dB sound pressure level [SPL]) signal, until animals achieved a criterion of 80% hit rate over 3 consecutive days.
Each trial was 2500 ms in total length. Warning trials contained a 1000 ms 4 kHz pure tone, and the tone was followed immediately by the unconditioned stimulus. To determine whether the animal detected the warning stimulus, contact with the spout was monitored during the final 100 ms of the warn stimulus. For warn trials, a contact time of <50 ms was scored as a hit. For safe trials, the entire 2500 ms duration consisted of silence. A contact time of <50 ms during the corresponding period in the trial was scored as a false alarm (FA). Warn trials always occurred at the end of a block of 2–4 safe trials, randomized to avoid temporal conditioning. All animals finished testing before P60.
After reaching criterion, animals were tested each day to assess audiometric thresholds. On the first day of testing, an array of 4 kHz tones at 5 intensities (80, 75, 70, 65, and 60 dB SPL) were presented in a descending order that repeated throughout the session. A performance value, d′ = z(hit) − z(false alarm), was obtained for z scores that corresponded to the right-tail P values (Swets 1973; Yanz 1984), and was calculated for each SPL. When the d′ for the middle SPL value was >1 on 2 consecutive days, the animal was graduated to a new array of stimuli that represented a 5 dB step down (e.g., 75, 70, 65, 60, and 55 dB SPL). Each animal progressed through testing sessions until they reached a dB SPL range in which they could no longer achieve a d′ of at least 1 for the middle value over 5 consecutive days. A d′ = 1 was defined as the auditory threshold.
There was a main effect of earplugging on auditory threshold [F1, 4 = 6.88, P < 0.05]. Earplugging induced a 27.5-dB increase in behaviorally revealed auditory thresholds (Fig. 1B). After earplug removal, animals were tested for 6 additional days. The thresholds recovered to within 5 dB of the pre-earplug values, and were no longer significantly elevated (Fig. 1C). Since the status of the audiometric threshold for each animal was not quantified prior to in vitro physiology, it is possible that normal peripheral thresholds were not restored even after reversal of the HL manipulation. Long-term earplugging can result in abnormal morphology (Moore et al. 1989), although full peripheral recovery is possible following earplug removal (Moore et al. 1999).
Thalamocortical Brain Slice Preparation
Thalamocortical brain slices (500 µm) were generated as described previously (Kotak et al. 2005). The brain was sectioned peri-horizontally to preserve the ventral medial geniculate (MGv) and its ascending pathways to the auditory cortex (Cruikshank et al. 2002). The slices were incubated in artificial cerebral spinal fluid (ACSF) at 32° ± 1°C for 30 min, then at room temperature for 60 min, and were transferred to a recording chamber continuously superfused (3 mL/min) with ACSF at 32° ± 1°C. The ACSF contained (in mM) 125 NaCl, 4 KCl, 1.2 KH2PO4, 1.3 MgSO4, 24 NaHCO3, 15 glucose, 2.4 CaCl2, and 0.4 l-ascorbic acid (pH 7.3) when bubbled with 95%O2–5%CO2). Before each whole-cell recording, the auditory cortex was identified by extracellular field responses to MGv stimulation.
Whole-Cell Current Clamp Recordings
Current clamp recordings (n = 475) were obtained from pyramidal neurons in supragranular auditory cortex as described previously (Takesian et al. 2010). Each neuron was verified as thalamo-recipient by recording an MG-evoked response, and all data were collected within layer 2/3 (Fig. 1A, dark gray area). Recording electrodes were fabricated from borosilicate glass microcapillaries (outer diameter, 1.5 mm) with a micropipette puller (Model P-97; Sutter Instruments, Novato, CA). The current clamp internal solution contained (in mM) 5 KCl, 127.5 K-gluconate, 10 HEPES, 2 MgCl2, 0.6 EGTA, 2 ATP, 0.3 GTP, and 5 phosphocreatine (pH 7.2 with KOH). The tip resistance of the patch electrode filled with internal solution was 5–10 MΩ. Access resistance was 15–30 MΩ, and was compensated by about 70%.
Neurons visually identified as pyramidal cell bodies under IR-DIC were selected. Passive membrane and intrinsic firing properties were evaluated on the basis of responses to current pulses (1500 ms). To determine action potential (AP) threshold, incremental steps of current (10 pA steps) were delivered at 0.2 Hz until a spike was evoked. To investigate frequency–current (F–I) curves, cells received depolarizing current injections from 100 to 600 pA in 100 pA steps.
Whole-Cell Voltage Clamp Recordings
Voltage clamp recordings (n = 67) (PC-501A; Warner Instruments, Hamden, CT) were obtained from a separate group of pyramidal neurons in supragranular auditory cortex to assess synaptic inhibition. The internal solution contained (in mM) 100 KCl, 40 K-gluconate, 8 NaCl, 10 HEPES, 2 MgCl2, 0.1 EGTA, 2 adenosine 5′-triphosphate disodium salt (ATP), 0.3 guanosine 5′-triphosphate sodium salt (GTP), and 5 lidocaine derivative QX-314 (pH 7.2 with KOH). This internal solution contained a high chloride concentration to obtain inward inhibitory postsynaptic currents (IPSCs) at a holding potential of −60 mV. Neurons visually identified as pyramidal cell bodies under IR-DIC were selected. It was not possible to characterize cells based on discharge properties because intracellular QX-314 blocks sodium channels. However, after breaking into the cell, it was possible to verify the presence of an acceptable resting potential (< −55 mV) and an overshooting AP. IPSCs were recorded in the presence of the ionotropic glutamate receptor blockers 6,7-dinitroquinoxaline-2,3-dione (DNQX; 20 µM; Sigma, St. Louis, MO) and 2-amino-5-phosphopentanoate (AP-5; 50 µM; Tocris Cookson, Ballwin, MO), added to the superfusing ACSF. The drugs were applied for 15 min before IPSCs were recorded. Spontaneous (s) IPSCs were recorded by acquiring 5–10 sweeps of 30-s duration for each cell. To assess evoked IPSCs, synaptic responses were elicited with electrical stimuli delivered via a stimulus isolator (model BSI-9501; Dagan, Minneapolis, MN) to a bipolar stimulating electrode (MX211EWDS2; Matrix Microelectrode) placed in layer 4. All stimuli were 100 µs in duration. Incremental stimulus intensities were delivered at 0.05 Hz until an evoked IPSC was discernible from failures (Kotak et al. 2008; Takesian et al. 2012). Minimum-evoked (me-)IPSCs were then collected using a stimulus intensity that evoked about 50% failures.
Data Acquisition and Analysis
All data were acquired at a sampling rate of 10 kHz using a custom-designed IGOR (version 4.08; WaveMetrics, Lake Oswego, OR) macro on an iMac (Apple, Cupertino, CA). A second IGOR macro was used for offline analysis. Pyramidal neuron intrinsic membrane and firing properties were analyzed offline. Resting membrane potential (RMP), voltage threshold to action potential (AP threshold), AP half-width (HW), and AP amplitude (AP Amp), were quantified by averaging values for the first 6 traces after reaching action potential threshold. RMP was calculated based on the 20 ms prestimulus baseline. AP threshold was calculated from the difference between the RMP and the inflection point to the first action potential. AP HW was calculated by measuring the time in milliseconds halfway between the peak of the action potential and the AP threshold. AP Amp was calculated by measuring the difference in millivolts between the RMP and the AP peak. The change in membrane voltage per 10 pA step (ΔmV) was calculated by averaging the change in membrane voltage to each 10 pA step prior to threshold. The input resistance (Rin) and membrane time constant (τ) were quantified from traces in which a −30 pA hyperpolarizing current was injected. The sag potential was quantified by measuring the difference between the peak hyperpolarization in response to a −100 pA current and the subsequent asymptotic potential due to membrane depolarization. Firing rates (Hz) were quantified for each depolarizing current injection trace (100–600 pA) by counting the spikes in each trace and dividing by the duration of the current pulse (1500 ms).
For spontaneous inhibitory synaptic currents (sIPSCs), amplitudes were determined from the peak of the sIPSC to baselines continuously identified during the 30-s traces using slope thresholds. A 10 pA amplitude threshold was used to detect sIPSCs from the baseline noise. sIPSC decay time constants were measured from single exponential fits of individual sIPSCs and were excluded if a subsequent IPSC occurred within 250 ms. me-IPSC amplitudes were measured from a baseline averaged for 5 ms prior to stimulus onset.
All data are presented as means ± SEM. Statistical tests were performed using statistical software (JMP; SAS Institute, Cary, NC). All experimental data were compared with age-matched control data using one-way analysis of variance (ANOVA). In cases where multiple group comparisons were made to a single age-matched control group, the Tukey's Honestly Significant Different (HSD) test was used as a conservative post hoc analysis to account for between group variance and unequal sample sizes. For F–I curves, a 2-way mixed-model ANOVA was used to verify a main effect of earplugging on evoked firing rate during incremental current injection steps.
Results
Brief Critical Periods are Property-Specific and Close by P18
To determine whether CPs of sensitivity to mild HL exist for membrane and firing properties, the age of onset of HL was systematically increased from the day of ear canal opening (∼P11) until significant effects were no longer observed. CPs were defined as postnatal days during which membrane and firing properties in layer 2/3 cortical pyramidal neurons (n = 475) of bilaterally deprived animals were significantly different from age-matched control animals.
This influence of HL was assessed as a function of HL age of onset, HL duration, and age of earplug removal. To assess HL age of onset, cortical membrane properties were recorded from animals that had bilateral earplugs (EP) inserted at postnatal day (P) 11, 12, 13, 14, 15, 16, 17, 18, 19, or 23 (Fig. 2A). For EP insertion ages from P11 to P17, recordings were obtained between P21 and P23, and for EP insertion ages from P18 to P23, recordings were obtained between P29 and P35. Controls recordings were obtained from age-matched animals (P21–23 or P29-35).
Figure 2.

Membrane properties display a CP for EP onset between P11 and P13. (A) A schematic illustrating experimental design to investigate the age of hearing loss onset. (B) Representative example traces of the effect of earplugging between P11 and P23 on membrane properties in response to depolarizing current injection 50 pA above firing threshold or hyperpolarizing current injection −30 pA from resting potential. (C–H) Bar graphs showing the CP for action potential amplitude, action potential HW, RMP, input resistance, membrane time constant, and action potential threshold. *P < 0.05, **P < 0.01, ***P < 0.001.
Each earplug group was compared with an age-matched control group using the Tukey's HSD test. This analysis revealed significant differences, as well as, discrete developmental periods of sensitivity to HL (Table 1—grey shaded boxes). Based on these observations, further analyses were performed on data grouped from P11 to 13, and P14 to 17. As shown in Figures 2 and 3, all membrane and firing properties were sensitive to HL when earplugs were inserted on P11, P12, or P13. These properties included RMP (means ± SEM; Ctrl −64.5 ± 0.7 mV vs. −59.6 ± 0.3 mV, q = 2.36, P < 0.0001), AP amplitude (means ± SEM; Ctrl 97.5 ± 1.4 mV vs. 86.9 ± 1.6 mV, q = 2.36, P < 0.0001), AP HW (means ± SEM; Ctrl 1.17 ± 0.04 ms vs. 1.48 ± 0.03 ms, q = 1.97, P < 0.0001), Rin (means ± SEM; Ctrl 141 ± 13 MΩ vs. 194 ± 8 MΩ, q = 2.36, P < 0.0001), and τ (means ± SEM; Ctrl 11.5 ± 0.7 ms vs. 20.3 ± 0.9 ms, q = 2.36, P < 0.0001). Therefore, for these properties, sensitivity to mild HL terminated abruptly at P13.
Table 1.
CPs for membrane properties
| n | RMP (mV) | AP Amp (mV) | AP Thresh (mV) | Rin (MΩ) | τ (ms) | AP width (ms) | Sag (mV) | ΔmV (mV/10 pA) | |
|---|---|---|---|---|---|---|---|---|---|
| Ctrl (P23) | 24 | −64.5 ± 0.6 | 97.5 ± 2.2 | −31.8 ± 1.2 | 141 ± 12 | 11.5 ± 1.2 | 1.2 ± 0.1 | 1.4 ± 0.1 | 1.7 ± 0.1 |
| EP11-23 | 31 | −58.7 ± 0.5*** | 88.4 ± 1.9* | −26.5 ± 1.1* | 179 ± 10 | 19.1 ± 1.0 | 1.4 ± 0.0* | 2.1 ± 0.1** | 2.5 ± 0.3 |
| EP12-23 | 16 | −59.1 ± 0.7*** | 88.2 ± 2.7* | −26.1 ± 1.5 | 198 ± 14* | 24.0 ± 1.5** | 1.7 ± 0.1** | 2.4 ± 0.0*** | 3.0 ± 0.2** |
| EP13-23 | 19 | −61.1 ± 0.7* | 85.2 ± 2.4** | −28.7 ± 1.4 | 218 ± 13** | 19.2 ± 1.3 | 1.5 ± 0.1* | 2.5 ± 0.1*** | 2.9 ± 0.5** |
| EP14-23 | 28 | −63.5 ± 0.5 | 95.1 ± 2.4 | −32.4 ± 1.3 | 125 ± 11 | 8.3 ± 1.1 | 1.2 ± 0.1 | 0.8 ± 0.1 | 1.3 ± 0.1 |
| EP15-23 | 27 | −64.6 ± 0.6 | 96.4 ± 2.8 | −31.1 ± 1.3 | 144 ± 11 | 11.1 ± 0.6 | 1.3 ± 0.1 | 0.7 ± 0.0** | 1.4 ± 0.1 |
| EP16-23 | 10 | −65.0 ± 0.8 | 96.7 ± 2.0 | −31.4 ± 0.9 | 116 ± 14 | 10.8 ± 1.8 | 1.0 ± 0.1 | 0.8 ± 0.9* | 1.3 ± 0.1 |
| EP17-23 | 11 | −66.2 ± 0.8 | 95.1 ± 1.7 | −31.8 ± 0.6 | 106 ± 11 | 14.0 ± 1.4 | 1.1 ± 0.1 | 0.6 ± 0.6* | 1.2 ± 0.1 |
| Ctrl (P35) | 24 | −64.2 ± 0.5 | 99.1 ± 1.3 | −32.5 ± 1.0 | 118 ± 9 | 8.4 ± 0.8 | 1.0 ± 0.0 | 1.3 ± 0.1 | 1.5 ± 0.0 |
| EP18-35 | 25 | −66.4 ± 0.8 | 98.7 ± 2.0 | −32.5 ± 2.4 | 87 ± 6 | 11.1 ± 0.6 | 1.0 ± 0.1 | 1.3 ± 0.2 | 1.6 ± 0.2 |
| EP19-35 | 27 | −66.7 ± 0.7 | 106.3 ± 1.4 | −34.9 ± 1.2 | 134 ± 11 | 10.8 ± 1.8 | 1.1 ± 0.0 | 1.3 ± 0.0 | 1.6 ± 0.2 |
| EP23-35 | 44 | −65.0 ± 0.4 | 100.7 ± 1.4 | −33.4 ± 0.8 | 124 ± 7 | 8.5 ± 0.6 | 1.0 ± 0.0 | 1.3 ± 0.1 | 1.5 ± 0.0 |
RMP, resting membrane potential; AP amp, action potential amplitude; AP threshold, voltage threshold to spike; Rin, input resistance; τ, membrane time constant; AP width, action potential HW; Sag, depolarizing sag; ΔmV, change in membrane voltage per 10 pA step. EP, earplug insertion days.
*P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3.

Specific membrane properties display a CP for EP onset for both P11 to P13 and P14 to P17. (A) A schematic illustrating experimental design to investigate the age of hearing loss onset. (B) Top, representative example traces of the effect of EP between P11 and P23 on the membrane response to hyperpolarizing current injection of −100 pA/1500 ms. Bottom, bar graph shows the 2 CPs of development for depolarizing sag emanating after peak hyperpolarization. (C) Top, Representative traces of the effect of earplugging between P11 and P23 on the response to depolarizing current injection between resting potential and action potential threshold. Bottom, bar graph shows the 2 CPs of development for membrane depolarization in response to a +10 pA current step. (D) Top, representative traces of current-evoked discharge in response to depolarizing current injection of 600 pA. Bottom, Line plots displaying the impact of earplugging from P11 to P13 (red) and P14 to P17 (blue) on firing rates (left), and the impact of earplugging between P18 and P23 on firing rates (right). *P < 0.05, **P < 0.01, ***P < 0.001.
A subset of cellular properties displayed a unique effect when HL was initiated on P14, P15, P16, or P17 (Fig. 3). The hyperpolarization-evoked depolarization (sag potential) was enhanced when EPs were inserted from P11 to P13 (Ctrl 1.38 ± 0.09 mV vs. 2.2 ± 0.1 mV, q = 2.36, P < 0.0001), but were diminished when EPs were inserted from P14 to P17 (Ctrl 1.38 ± 0.09 mV vs. 0.73 ± 0.04 mV, q = 2.36, P < 0.0001). While the change in membrane voltage to depolarizing current (ΔmV/10pA) was significantly increased when EPs were inserted from P11 to P13 (Ctrl 1.7 ± 0.009 mV vs. 2.8 ± 0.002 mV, q = 2.36, P < 0.0001), there was a significantly lower sensitivity to current injection when earplugging occurred between P14 and P17 (Ctrl 1.7 ± 0.009 mV vs. 1.3 ± 0.005 mV, q = 2.36, P < 0.05). This unique effect was observed for firing properties as well. When EP insertion was induced on P11, P12, or P13, firing rates were unaffected for depolarizing currents up to 300 pA, but were severely depressed at higher currents [F1,88 = 34.6, P < 0.0001]. In contrast, when EP insertion began on P14, P15, P16, or P17, firing rate was depressed at all injected currents [F1,98 = 29.5, P < 0.0001]. The insertion of EPs beginning on P18, P19, or P23 did not influence any of the membrane or firing properties measured in this study (Table 1). Therefore, for this subset of properties, sensitivity to mild HL opened at P11 and ended abruptly at P18.
The Critical Period for Inhibitory Synaptic Transmission Closes by P19
The strength of cortical inhibition is reduced after severe or moderate forms of developmental HL (Kotak et al. 2008; Takesian et al. 2012). To determine whether inhibition displayed a similar dependency on HL age of onset, we recorded spontaneous (sIPSCs) and minimum-evoked inhibitory postsynaptic currents (me-IPSCs) from layer 2/3 auditory cortical pyramidal neurons (n = 67) in animals that received EP insertion during the CPs of membrane property development (P11, P15), or after the CPs for membrane properties had closed (P19). As shown in Figure 4, sIPSC amplitude (means ± SEM; Ctrl, −32.4 ± 3.0 vs. EP11, −23.2 ± 1.8, P < 0.05; EP15, −21.1 ± 1.3, P < 0.01; EP19, −38.2 ± 2.6, P > 0.1), sIPSC time constant (means ± SEM; Ctrl, 13.0 ± 2.6 vs. EP11, 27.0 ± 4.9, P < 0.05; EP15, 24.6 ± 3.0, P < 0.05; EP19, 14.5 ± 0.7, P > 0.1), and me-IPSC amplitude (means ± SEM; Ctrl, −9.9 ± 0.9 vs. EP11, −4.6 ± 0.6, P < 0.001; EP15, −5.5 ± 0.4, P < 0.001; EP19, −9.3 ± 0.8, P > 0.1) were significantly reduced when EPs were inserted at either P11 or 15, but not at P19. This suggests that inhibitory synapse function displays a CP that spans the temporal window observed for membrane and firing properties.
Figure 4.
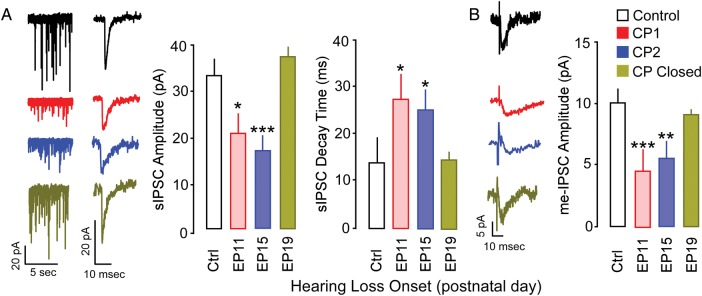
CP for the influence of EP onset on the development of inhibitory synaptic transmission. (A) Effect of earplugging on spontaneous IPSC amplitude and decay time constant. Left: representative recordings of sIPSC amplitudes. Individual sIPSC displays decay time constant. Right; bar graph showing the quantitative effect of earplugging on sIPSC amplitudes and decay time constant. (B) Effect of earplugging on intracortically-evoked minimum IPSC amplitude. Left: representative example recordings of me-IPSCs. Right: bar graph showing the quantitative effect of earplugging on me-IPSC amplitudes. All recordings were carried out in the presence of DNQX and AP-5. *P < 0.05, **P < 0.01, ***P < 0.001.
The Effects Induced by Earplugs Display Property-Specific Time-Courses
Environmental manipulations during development can produce changes to response properties or topography that persist into adulthood. Therefore, we investigated the time course over which sensory deprivation alters the developmental trajectory of cellular properties. We first characterized the development of intrinsic membrane properties for pyramidal neurons (n = 148) from the time of ear canal opening (P11) to P35, and found that mature function was achieved by P21–23 (Fig. 5A–H, black circles). To determine the impact of deprivation duration, animals received EPs on P11, and recordings were subsequently obtained between P14 and P35. To determine whether the influence of EP insertion at P11 was evident prior to completion of maturation, recordings were carried out between P14 and P17. With the exception of RMP, all measured properties were significantly different from age-matched controls at this early time point (Fig. 5A–H, red circles). The cumulative effect of these membrane properties was reflected in the current-evoked firing rates (Fig. 5I). For all current levels above 200 pA, the firing rate was significantly diminished [F1,43 = 284.3, P < 0.001].
Figure 5.
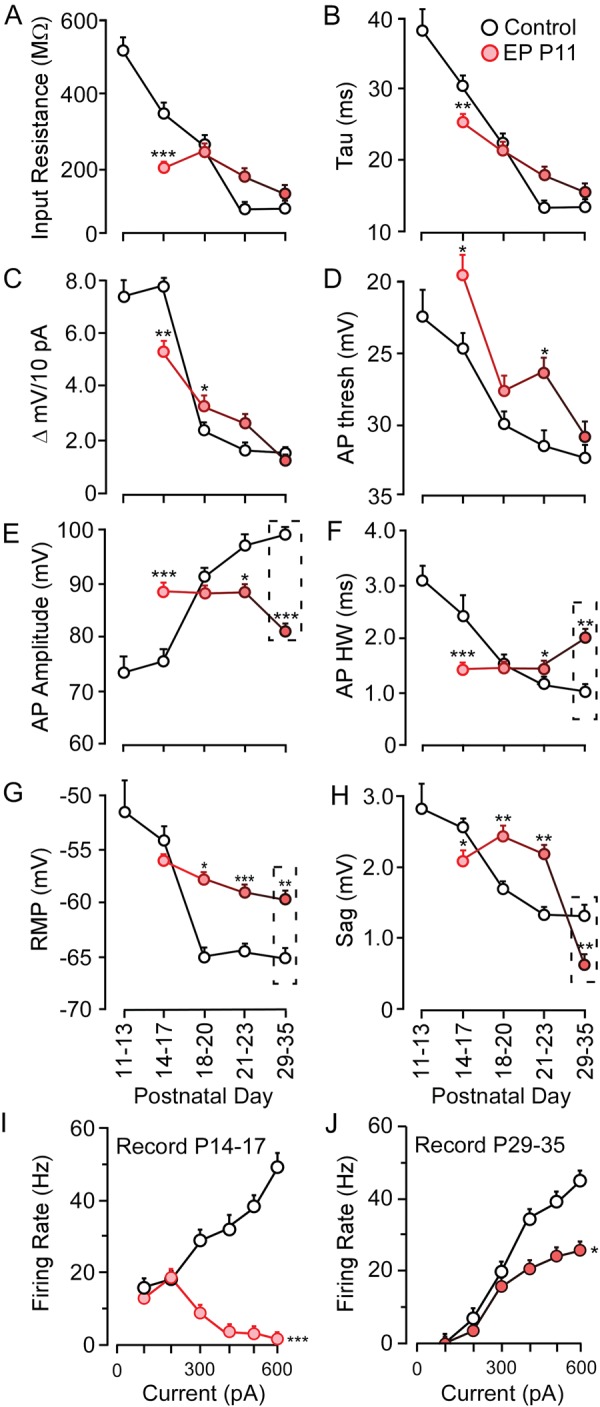
The impact of auditory deprivation duration on membrane property development. Line plots display the normal development (black) and effect of EP duration (red) for (A) input resistance, (B) time constant, (C) ΔmV, (D) action potential threshold, (E) action potential amplitude, (F) action potential HW, (G) RMP, (H) depolarizing sag, (I, J) firing rate. Dashed rectangles highlight properties that remain significantly different at P29-35. *P < 0.05, **P < 0.01, ***P < 0.001.
As the duration of HL progressed, and the animals continued to develop, it became apparent that only some membrane properties remained impaired, whereas others reached control values (Fig. 5). One set of membrane property deficits persisted when EPs remained in place through P35. Specifically, AP amplitude remained smaller (means ± SEM; Ctrl 99.5 ± 0.7 mV vs. 81.6 ± 0.3 mV, q = 2.89, P < 0.01), AP width remained longer, (means ± SEM; Ctrl 0.86 ± 0.1 ms vs. 1.25 ± 0.1 ms, q = 2.89, P < 0.01), the RMP remained depolarized (means ± SEM; Ctrl −64.2 ± 0.7 mV vs. −59.4 ± 0.3 mV, q = 2.89, P < 0.01), and the sag potential became significantly smaller (means ± SEM; Ctrl 1.33 ± 0.7 mV vs. 0.66 ± 0.3 mV, q = 2.89, P < 0.01). Furthermore, firing rate remained significantly diminished at all current levels beyond 300 pA [F1,31 = 8.2, P < 0.05]. In contrast, control values were reached by P35 for all remaining properties including Rin,τ, ΔmV/10 pA, and AP threshold (Fig. 5A–D).
Critical Period for Recovery of Normal Function is Property-Specific
To determine whether an earlier age of hearing restoration would permit better recovery of cellular function, we inserted bilateral EPs at P11, and removed them before (P17) or after (P23) the closure of the CPs described above (Figs 2 and 3). When the EPs were removed at P17, most neuronal membrane properties recovered to control values by the age of recording, P23 (Fig. 6B–F, Table 2). However, the current-evoked firing rate remained significantly depressed [Fig. 6G, F1,49 = 19.7, P < 0.0001]. When earplugs were removed at P23 and membrane properties recorded at P35, cells displayed greater impairment. While RMP, AP amplitude, AP threshold, Rin, and τ did recover, AP width (means ± SEM; Ctrl 1.0 ± 0.1 ms vs. 1.3 ± 0.1 ms, q = 2.89, P < 0.05), sag potential (means ± SEM; Ctrl 1.30 ± 0.1 mV vs. 0.9 ± 0.1 mV, q = 2.89, P < 0.05), epolarization to positive current injection (means ± SEM; Ctrl 1.5 ± 0.1 mV vs. 0.98 ± 0.1 mV, q = 2.89, P < 0.05) and firing rate [Fig. 6H, F1,39 = 10.5, P < 0.01] were significantly different from control values (Table 2). This suggests that EP removal prior to CP closure led to better recovery than EP removal after CP closure. However, diminished firing rates persisted even when EPs were removed before the closure of the second CP at P18.
Figure 6.

The CP for recovery of membrane and firing properties following EP removal. (A) A schematic illustrates the experimental design used to investigate recovery from hearing loss. (B–F) Bar graphs showing the effect of P11 earplugging on action potential amplitude, RMP, action potential HW, depolarizing sag, and ΔmV when earplugs are removed on P17 followed by short-term recovery (P23), earplug removal at P23 followed by short-term recovery (P35), or earplug removal at P23 followed by long-term recovery (P86). (G–I) Line plots displaying the impact of P11 earplugging on firing rates when earplugs are removed at P17 followed by short-term recovery (P23), earplug removal at P23 followed by short-term recovery (P35), or earplug removal at P23 followed by long-term recovery (P86). *P < 0.05, **P < 0.01, ***P < 0.001.
Table 2.
Effect of hearing reinstatement on membrane properties
| n | RMP | AP Amp | AP thresh | Rin | τ | AP width | Sag | ΔmV | |
|---|---|---|---|---|---|---|---|---|---|
| (mV) | (mV) | (mV) | (MΩ) | (ms) | (ms) | (mV) | (mV) | ||
| Ctrl P23 | 24 | –64.5 ± 0.6 | 97.5 ± 2.2 | –31.8 ± 1.2 | 141 ± 12 | 11.5 ± 1.2 | 1.2 ± 0.1 | 1.4 ± 0.1 | 1.7 ± 0.1 |
| EP11-17 | 27 | –65.6 ± 0.6 | 97.9 ± 2.1 | –30.0 ± 1.2 | 142 ± 11 | 12.1 ± 1.3 | 1.2 ± 0.1 | 1.6 ± 0.2 | 1.7 ± 0.0 |
| Ctrl P35 | 24 | –64.2 ± 0.5 | 99.1 ± 1.3 | –32.5 ± 1.0 | 118 ± 9 | 8.4 ± 0.8 | 1.0 ± 0.0 | 1.3 ± 0.1 | 1.5 ± 0.0 |
| EP11-23 | 17 | –65.1 ± 0.6 | 99.1 ± 1.7 | –32.4 ± 1.4 | 107 ± 11 | 7.0 ± 1.0 | 1.3 ± 0.1* | 0.9 ± 0.2* | .98 ± 0.0* |
| Ctrl P86 | 22 | –64.5 ± 0.5 | 99.7 ± 1.8 | –28.4 ± 1.0 | 59 ± 10 | 7.5 ± 0.8 | 0.8 ± 0.1 | 0.6 ± 0.1 | 1.0 ± 0.1 |
| EP11-23 | 23 | –63.6 ± 0.6 | 98.6 ± 2.2 | –28.8 ± 1.9 | 60 ± 5 | 6.6 ± 0.5 | 1.0 ± 0.1** | 0.4 ± 0.0** | 0.7 ± 0.1* |
RMP, resting membrane potential; AP amp, action potential amplitude; AP threshold, voltage threshold to spike; Rin, input resistance; τ, membrane time constant; AP width, action potential HW; Sag, depolarizing sag; ΔmV, change in membrane voltage per 10 pA step. EP, earplug insertion days.
*P < 0.05, **P < 0.01, ***P < 0.001.
Based on these results we predicted that functional deficits would persist through adulthood if reinstatement of hearing did not occur early enough. Therefore, we examined the effect of a long duration of recovery following EP removal. Animals had EPs inserted on P11 and removed just a few days after the closure of the CPs (P23); however, whole-cell recordings were not carried out until after sexual maturity (P86–P91). As reported in other studies functional properties continue to develop during this time in gerbils (Takesian et al. 2012; Rosen et al. 2012), requiring a separate set of recordings from age-matched controls. Despite the continued maturation of many properties, the same deficits that we observed at P35 were still present several months after EP removal (Fig. 6D–F, H/I, Table 2). Action potential HW (means ± SEM; Ctrl 0.8 ± 0.1 ms vs. 1.0 ± 0.1 ms, q = 2.01, P < 0.01), sag potential (means ± SEM; Ctrl 0.6 ± 0.1 mV vs. 0.4 ± 0.1 mV, q = 2.01, P < 0.01), depolarization to positive current injection (means ± SEM; Ctrl 1.0 ± 0.1 mV vs. 0.7 ± 0.1 mV, q = 2.01, P < 0.05) and firing rate [Fig. 6I, F1,43 = 5.06, P < 0.05] were significantly different from control values. Therefore, for these membrane and firing properties, the CP of recovery from mild HL closes prior to P23.
Discussion
The central nervous system is particularly vulnerable to the disruption of sensory activity during discrete intervals prior to adulthood, referred to as CPs. If sensory deprivation occurs throughout a CP, then the specific neural properties that are sensitive to the loss of input display a slow or negligible recovery, even if sensory input is later restored (Wiesel and Hubel, 1965; Hubel and Wiesel 1970; Blakemore et al. 1978; LeVay et al. 1980; Knudsen et al. 1984). Furthermore, an earlier restoration of sensory activity following developmental blindness or HLHL in humans is associated with better behavioral performance (vision: Lewis and Maurer 2005; Jain et al. 2010; hearing: Sharma et al. 2002; Dettman et al. 2007; Miyamoto et al. 2008; May-Mederake 2010). Here, we report mechanistic bases for these system-level effects: firing rate, sag potential, AP width, and membrane depolarization to current injection failed to recover several months after transient developmental HL (Fig. 6, Table 2). Therefore, our results demonstrate that the restoration of peripheral input prior to CP closure is essential for the normal development of a specific subset of cellular properties. These impaired properties could form the mechanistic basis for system-level effects that have been reported by others (e.g., Popescu and Polley 2010). Taken together, these findings provide a theoretical framework in which a limited set of cellular properties account for persistent deficits to perceptual skills following CP deprivation (Whitton and Polley 2011).
Cortical Sensitivity to Age of Deprivation Onset
One characteristic of CP plasticity is that the system's sensitivity to deprivation is determined by the age-of-onset. In the rodent visual system, monocular deprivation (MD) for the first 3 days after eye opening leads to an increase in excitability in deprived cortex, while MD between P21 and P35 leads to a decrease in excitability as assessed by the strength of miniature EPSCs (Maffei and Turrigiano 2008).
In the rodent somatosensory system, deprivation during the first 3 postnatal days produces persistent deficits in behavior, whisker-evoked responses, and cortical cytoarchitecture (Van der Loos and Woolsey 1973; Popescu and Ebner 2010; Chu et al. 2013). After this developmental window these properties become less sensitive to the same duration or severity of peripheral deprivation (Van der Loos and Woolsey 1973; Fox 1992; Lee et al. 2009); however, the developing system remains plastic and displays additional CPs (Glazewski, and Fox 1996; Maravall et al. 2004; Wen and Barth 2011).
In the rat auditory system, cortical tonotopy can be altered during a 3-day CP after the onset of hearing (de Villers-Sidani et al. 2007; Barkat et al. 2011). Furthermore, multiple CPs for binaural integration have been reported during which auditory cortex is highly sensitive to unilateral deprivation up to 1 week after hearing onset (Polley et al. 2013). However, other central properties are vulnerable to acoustic manipulations during separate developmental epochs, suggesting that CPs may extend late into development for perceptual features such as frequency modulation selectivity (Razak et al. 2008; Insanally et al. 2009) or binaural processing (Popescu and Polley 2010). In addition, the adult nervous system displays many forms of plasticity in response to deprivation. However, adult forms of plasticity may require longer periods of deprivation to induce a neural change, and a slower recovery to control levels when the deprivation is reversed (visual system: Spolidoro et al. 2009; somatosensory—Godde et al. 2002; auditory system: Caspary et al. 2008; Eggermont 2013).
If the principles discussed above apply to cellular mechanisms, then the age at which deprivation is induced should determine how their development is altered. In the current study, membrane properties were sensitive to HL from P11 to P13 (Fig. 2, Table 1). However, immediately thereafter, a subset of these membrane properties remained vulnerable to EP insertion (Fig. 3, Table 1).
It is important to note that the form of HL used in this study is considerably milder than that used in generating our previous results (Kotak et al. 2005, 2008; Takesian et al. 2010, 2012). Many overlapping CPs have been described in the auditory system throughout development (de Villers-Sidani et al. 2007; Razak et al. 2008; Insanally et al. 2009; Popescu and Polley 2010; Barkat et al. 2011; Polley et al. 2013). These CPs were revealed with different forms of unilateral and bilateral changes to peripheral input. Therefore, it is possible that increasing the severity of HL may induce more profound changes, or reveal different CPs. Regardless; our results suggest the presence of at least 2 CPs of sensitivity to mild HL (P11–P13 and P14–P17). In contrast, inhibitory synaptic strength was susceptible to EPs from P11–17 (Fig. 4). Therefore, the common principle is that CPs for both membrane and inhibitory properties terminated at P18. Our data suggest that the inhibitory CP does not regulate the termination of all cellular properties. That is, the CP for RMP and Rin closed at P14, yet the inhibitory CP did not close until P18. The earliest membrane property CP occurred contemporaneously to the CP for tonotopic map formation (de Villers-Sidani et al. 2007; Barkat et al. 2011). However, the CP for firing rate closed at P18, and could have been regulated by the inhibitory CP, broadly consistent with the idea that inhibition regulates cortical excitatory synapse plasticity (Hensch 2005; Hensch and Fagiolini 2005).
The Impact of Deprivation Duration
A second characteristic of CP plasticity is that certain neural deficits induced by long-term deprivation can persist into adulthood. In the rodent visual system, dark rearing from birth leads to weak visually evoked responses that are unselective for orientation and direction (Fagiolini et al. 1994). In the rodent somatosensory system, whisker trimming from birth leads to permanent abnormalities for whisker-evoked responses recorded in the adult somatosensory barrel cortex (Simons and Land 1987). In the auditory system, rodents reared with permanent conductive hearing loss (CHL) exhibit reduced inhibitory synaptic currents when recorded in adulthood (Takesian et al. 2012). In each of these examples, the sensory-deprived adult neurons display immature functional characteristics, suggesting that development is delayed. A second line of research demonstrates that permanent deficits to adult coding properties arise when deprivation occurs throughout development even if sensory inputs are restored (Knudsen et al. 1984; Fagiolini et al. 1994; Carvel and Simons 1996; Fox et al. 1996).
If the principles discussed above apply to mild HL that extends through the CP, then the initial cellular deficits should persist with a longer duration of deprivation. In contrast, our results demonstrated that that the cellular response to sensory deprivation is induced shortly after earplugging begins (P14) and many changes were no longer different from controls by P18. At the same time these changes continue to evolve between P18 and P35. For one subset of properties (e.g., RMP), the deficits did persist (Fig. 5E–H, J), and may serve as a mechanistic basis for permanent changes to coding properties that are observed in system's level studies. However, a second set of properties were delayed in maturation, but eventually resolved to control values (Fig. 5A–D).
The cellular changes described here are proposed as basic mechanisms for HL-induced alteration of auditory coding properties. However, these mechanisms will have a biophysical and molecular explanation at the level of ion channels. The fact that certain properties display short-term changes 3 days after EP insertion at P11 suggests an increase in leak conductance (e.g., decreased input resistance), as well as, heightened sodium and potassium channel function (e.g., increased spike height, decreased spike width). However, there are further changes to membrane properties within 7 days of EP insertion that strongly suggest compensatory mechanisms for the peripheral deprivation (e.g., depolarized RMP). Furthermore, our results share many similarities with studies that manipulate key ion channels during development (e.g., Na+, Komai et al. 2006). Most importantly, the F–I curves display a continuous transition that correlates with EP duration. A candidate that may account for the sequential changes in current-evoked discharge is the family of voltage-sensitive potassium channels (Kv) that regulate membrane properties, including the discharge pattern (Kasten et al. 2007). Thus, CP auditory deprivation in birds (cochlear removal) has been shown to have a significant effect on the expression of the Kv1.1 and Kv3.1 potassium channels within the cochlear nucleus (Lu et al. 2004). A change like this occurring in the cochlear nucleus after milder forms of HL could systemically reduce activity levels throughout the auditory sensory neuraxis.
At EP durations of 3–12 days, the discharge rate evoked by smaller currents was normal, but the large currents resulted in a near-complete suppression of discharge, that is, a switch to nonmonotonic input–output function (Figs 3D and 5I). Therefore, EPs may lead to an increase in potassium conductances that are activated at higher membrane potentials. At EP durations of 18–24 days, the discharge rate evoked by all current injection levels becomes smaller than normal (Fig. 5J). This result suggests that both potassium conductances activated near threshold and well above threshold remain larger than in controls.
The Critical Period for Recovery From Deprivation
A third characteristic of developmental plasticity is that CP closure is itself, an experience-dependent process. Complete deprivation can induce global delays in development (e.g., Ghoshal et al. 2009), and extend CP closure by limiting the maturation of excitation and inhibition (Hensch 2004, 2005). This suggests that bilateral HL may delay CP closure, thereby prolonging the system's sensitivity to recovery. In humans, an earlier age of restoration of hearing or eyesight is positively correlated with the recovery of behavioral skills (Svirsky et al. 2004; Lewis and Maurer 2005; May-Mederake 2010). In contrast, partial deprivation allows an intact input to gain preferential access to the neuraxis (Fagiolini et al. 1994; Frenkel and Bear 2004; Popescu and Ebner 2010; Popescu and Polley 2010). Thus, when animals are reared with bilateral visual deprivation, the CP for ocular dominance plasticity is prolonged well past its expected closure (Cynader 1983).
We tested these principles by assessing cellular properties following deprivation reversal at 2 ages. EP removal before CP closure at P17 led to a near-complete recovery of intrinsic properties, whereas EP removal after CP closure at P23 was associated with a greater number of deficits (Fig. 6, Table 2). These findings are consistent with the idea that auditory deficits are more profound when restoration of hearing occurs at later ages (Svirsky et al. 2004; Sharma and Dorman 2002). The persistence of these deficits into adulthood also demonstrates that the amount of recovery is significantly reduced when deprivation extends beyond the original CP (P18). Special emphasis should be placed on the changes to input–output functions shown in the F–I curves, as neuronal firing and discharge patterns are crucial for perception and coding in both developing and adult systems. Aberrant firing patterns that persist in adults (see Fig. 6), may contribute to coding deficits recorded in the auditory cortex of awake gerbils in response to slow amplitude-modulated auditory inputs (Rosen et al. 2012). Such deficits can then explain the poor AM detection threshold tested during these behavioral tasks.
Taken together these results suggest that CP closure is delayed for some properties (e.g., AP amplitude), but not others. In fact, we have found that the decrease in inhibitory strength caused by bilateral HL can be rescued by enhancing GABAergic transmission (zolpidem) long after the inhibitory CP should have closed (Kotak et al. 2013). This suggests that the closure of the CP for inhibitory synaptic transmission may be extended as well. Therefore, our results are generally consistent with the principle that bilateral deprivation prevents the CP closure, at least for some properties.
Summary
This study provides a theoretical framework in which the age of onset of HL, the duration of HL, and the age of recovery from HL differentially alter the cellular basis of functional deficits reported in the primary auditory cortex of juveniles and adults. The age of onset experiments suggest that the cellular basis for persistent changes seen in adult cortex occur shortly after sensory onset begins. The duration experiments suggest that while membrane property development is significantly delayed, only a subset of persistently affected properties likely provide the cellular basis for deficits seen in adult cortex. Finally, our recovery experiments underscore the importance of recovery prior to CP closure and the possibility that very early experience is vital to normal development. Therein these findings provide important insight into the cellular basis of deficits reported in system-level studies using animal models (Popescu and Polley 2010) and human populations that demonstrate long-term deficits to auditory processing and perception following transient HL (Pillsbury et al. 1991; Hall and Grose 1994; Hall et al. 1995, 1998; Hogan et al. 1996; Hogan and Moore 2003; Whitton and Polley 2011).
Funding
This work was supported by National Institutes of Health R01DC011284 (D.H.S. and V.C.K.) and F32DC013482 (T.M.M.).
Notes
We thank Melissa Caras for manuscript review and Kristina Penikis for data collection and manuscript review. Conflict of Interest: None declared.
References
- Barkat TR, Polley DB, Hensch TK. 2011. A critical period for auditory thalamocortical connectivity. Nat Neurosci. 14:1189–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore C, Garey LJ, Vital-Durand F. 1978. The physiological effects of monocular deprivation and their reversal in the monkey's visual cortex. J Physiol. 283:223–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvell GE, Simons DJ. 1996. Abnormal tactile experience early in life disrupts active touch. J Neurosci. 16:2750–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary DM, Ling L, Turner JG, Hughes LF. 2008. Inhibitory neurotransmission, plasticity and aging in the mammalian central auditory system. J Exp Biol. 211:1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu YF, Yen CT, Lee LJ. 2013. Neonatal whisker clipping alters behavior, neuronal structure and neural activity in adult rats. Behav Brain Res. 238:124–133. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ. 1990. Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci. 13(3):99–104. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Rose HJ, Metherate R. 2002. Auditory thalamocortical synaptic transmission in vitro. J Neurophysiol. 87:361–384. [DOI] [PubMed] [Google Scholar]
- Cynader M. 1983. Prolonged sensitivity to monocular deprivation in dark-reared cats: effects of age and visual exposure. Brain Res. 284:155–164. [DOI] [PubMed] [Google Scholar]
- Daw NW. 1998. Critical periods and amblyopia. Arch Ophthalmol. 116:502–505. [DOI] [PubMed] [Google Scholar]
- Dawson PW, Blamey PJ, Rowland LC, Dettman SJ, Clark GM, Busby PA, Brown AM, Dowell RC, Rickards FW. 1992. Cochlear implants in children, adolescents, and prelinguistically deafened adults: speech perception. J Speech Hear Res. 35:401–417. [DOI] [PubMed] [Google Scholar]
- Dettman SJ, Pinder D, Briggs RJ, Dowell RC, Leigh JR. 2007. Communication development in children who receive the cochlear implant younger than 12 months: risks versus benefits. Ear Hear. 28:11S–18S. [DOI] [PubMed] [Google Scholar]
- de Villers-Sidani E, Chang EF, Bao S, Merzenich MM. 2007. Critical period window for spectral tuning defined in the primary auditory cortex (A1) in the rat. J Neurosci. 27:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dews PB, Wiesel TN. 1970. Consequences of monocular deprivation on visual behaviour in kittens. J Physiol. 206:437–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco S, Nguyen VA, Bisti S, Protti DA. 2009. Permanent functional reorganization of retinal circuits induced by early long-term visual deprivation. J Neurosci. 29:13691–13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrn AL, Yuan K, Barker AJ, Schreiner CE, Froemke RC. 2010. Developmental sensory experience balances cortical excitation and inhibition. Nature. 465:932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont JJ. 2013. On the similarities and differences of non-traumatic sound exposure during the critical period and in adulthood. Front Syst Neurosci. 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Pizzorusso T, Berardi N, Domenici L, Maffei L. 1994. Functional postnatal development of the rat primary visual cortex and the role of visual experience: dark rearing and monocular deprivation. Vision Res. 34:709–720. [DOI] [PubMed] [Google Scholar]
- Fox K. 1992. A critical period for experience-dependent synaptic plasticity in rat barrel cortex. J Neurosci. 12:1826–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K, Glazewski S, Chen CM, Silva A, Li X. 1996. Mechanisms underlying experience-dependent potentiation and depression of vibrissae responses in barrel cortex. J Physiol Paris. 90:263–269. [DOI] [PubMed] [Google Scholar]
- Frenkel M, Bear M. 2004. How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron. 44:917–923. [DOI] [PubMed] [Google Scholar]
- Ghoshal A, Pouget P, Popescu M, Ebner F. 2009. Early bilateral sensory deprivation blocks the development of coincident discharge in rat barrel cortex. J Neurosci. 29:2384–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazewski S, Fox K. 1996. Time course of experience-dependent synaptic potentiation and depression in barrel cortex of adolescent rats. J Neurophysiol. 75:1714–1729. [DOI] [PubMed] [Google Scholar]
- Godde B, Berkefeld T, David-Jürgens M, Dinse HR. 2002. Age-related changes in primary somatosensory cortex of rats: evidence for parallel degenerative and plastic-adaptive processes. Neurosci Biobehav Rev. 26:743–752. [DOI] [PubMed] [Google Scholar]
- Gouma P, Mallis A, Daniilidis V, Gouveris H, Armenakis N, Naxakis S. 2011. Behavioral trends in young children with conductive hearing loss: a case–control study. Eur Arch Otorhinolaryngol. 268:63–66. [DOI] [PubMed] [Google Scholar]
- Hall JW, Grose JH. 1994. The effect of otitis media with effusion on comodulation masking release in children. J Speech Hear Res. 37:1441–1449. [DOI] [PubMed] [Google Scholar]
- Hall J, Grose J, Dev M, Drake A, Pillsbury HC. 1998. The effect of otitis with effusion on complex masking tasks in children. Arch Otolaryngol Head Neck Surg. 124:892–896. [DOI] [PubMed] [Google Scholar]
- Hall JW, Grose JH, Pillsbury HC. 1995. Long-term effects of chronic otitis media on binaural hearing in children. Arch Otolaryngol Head Neck Surg. 121:847–852. [DOI] [PubMed] [Google Scholar]
- Hensch TK. 2004. Critical period regulation. Annu Rev Neurosci. 27:549–579. [DOI] [PubMed] [Google Scholar]
- Hensch TK. 2005. Critical period mechanisms in developing visual cortex. Curr Top Dev Biol. 69:215–237. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M. 2005. Excitatory–inhibitory balance and critical period plasticity in developing visual cortex. Prog Brain Res. 147:115–124. [DOI] [PubMed] [Google Scholar]
- Hogan SC, Meyer SE, Moore DR. 1996. Binaural unmasking returns to normal in teenagers who had otitis media in infancy. Audiol Neurootol. 1:104–111. [DOI] [PubMed] [Google Scholar]
- Hogan SC, Moore DR. 2003. Impaired binaural hearing in children produced by a threshold level of middle ear disease. J Assoc Res Otolaryngol. 4:123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt RF, Svirsky MA. 2008. An exploratory look at pediatric cochlear implantation: is earliest always best?. Ear Hear. 29:492–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN. 1970. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol. 206:419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insanally MN, Köver H, Kim H, Bao S. 2009. Feature-dependent sensitive periods in the development of complex sound representation. J Neurosci. 29:5456–5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Scanziani M. 2011. How inhibition shapes cortical activity. Neuron. 72:231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Ashworth J, Biswas S, Lloyd IC. 2010. Duration of form deprivation and visual outcome in infants with bilateral congenital cataracts. J AAPOS. 14:31–34. [DOI] [PubMed] [Google Scholar]
- Kasten MR, Rudy B, Anderson MP. 2007. Differential regulation of action potential firing in adult murine thalamocortical neurons by Kv3.2, Kv1, and SK potassium and N-type calcium channels. J Physiol. 584:565–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen EI, Knudsen PF, Esterly SD. 1984. A critical period for the recovery of sound localization accuracy following monaural occlusion in the barn owl. J Neurosci. 4:1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai S, Licznerski P, Cetin A, Waters J, Denk W, Brecht M, Osten P. 2006. Postsynaptic excitability is necessary for strengthening of cortical sensory responses during experience-dependent development. Nat Neurosci. 9:1125–1133. [DOI] [PubMed] [Google Scholar]
- Kotak VC, Fujisawa S, Lee FA, Karthikeyan O, Aoki C, Sanes DH. 2005. Hearing loss raises excitability in the auditory cortex. J Neurosci. 25:3908–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Takesian AT, McKenzie TM, Sanes DH. 2013. Rescue of inhibitory synapse strength following developmental hearing loss. PLoS One. 8(1):e53438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Takesian AE, Sanes DH. 2008. Hearing loss prevents the maturation of GABAergic transmission in the auditory cortex. Cereb Cortex. 18:2098–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kral A, Hartmann R, Tillein J, Heid S, Klinke R. 2001. Delayed maturation and sensitive periods in the auditory cortex. Audiol Neurootol. 6:346–362. [DOI] [PubMed] [Google Scholar]
- Kral A, Hartmann R, Tillein J, Heid S, Klinke R. 2002. Hearing after congenital deafness: central auditory plasticity and sensory deprivation. Cereb Cortex. 12:797–807. [DOI] [PubMed] [Google Scholar]
- Kral A, Sharma A. 2012. Developmental neuroplasticity after cochlear implantation. Trends Neurosci. 35:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LJ, Chen WJ, Chuang YW, Wang YC. 2009. Neonatal whisker trimming causes long-lasting changes in structure and function of the somatosensory system. Exp Neurol. 219:524–532. [DOI] [PubMed] [Google Scholar]
- LeVay S, Wiesel TN, Hubel DH. 1980. The development of ocular dominance columns in normal and visually deprived monkeys. J Comp Neurol. 191:1–51. [DOI] [PubMed] [Google Scholar]
- Lewis TL, Maurer D. 2005. Multiple sensitive periods in human visual development: evidence from visually deprived children. Dev Psychobiol. 46:163–183. [DOI] [PubMed] [Google Scholar]
- Lu Y, Monsivais P, Tempel BL, Rubel EW. 2004. Activity-dependent regulation of the potassium channel subunits Kv1.1 and Kv3.1. J Comp Neurol. 470:93–106. [DOI] [PubMed] [Google Scholar]
- Maffei A, Turrigiano GG. 2008. Multiple modes of network homeostasis in visual cortical layer 2/3. J Neurosci. 28:4377–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravall M, Stern EA, Svoboda K. 2004. Development of intrinsic properties and excitability of layer 2/3 pyramidal neurons during a critical period for sensory maps in rat barrel cortex. J Neurophysiol. 92:144–156. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. 2004. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 5:793–807. [DOI] [PubMed] [Google Scholar]
- May-Mederake B, Kuehn H, Vogel A, Keilmann A, Bohnert A, Mueller S, Witt G, Neumann K, Hey C, Stroele A, et al. 2010. Evaluation of auditory development in infants and toddlers who received cochlear implants under the age of 24 months with the LittlEARS auditory questionnaire. Int J Pediatr Otorhinolaryngol. 74:1149–1155. [DOI] [PubMed] [Google Scholar]
- Metherate R, Aramakis VB. 1999. Intrinsic electrophysiology of neurons in thalamorecipient layers of developing rat auditory cortex. Brain Res Dev Brain Res. 115:131–144. [DOI] [PubMed] [Google Scholar]
- Moore DR, Hine JE, Jiang ZD, Matsuda H, Parsons CH, King AJ. 1999. Conductive hearing loss produces a reversible binaural hearing impairment. J Neurosci. 19:8704–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DR, Hutchings ME, King AJ, Kowalchuk NE. 1989. Auditory brain stem of the ferret: some effects of rearing with a unilateral ear plug on the cochlea, cochlear nucleus, and projections to the inferior colliculus. J Neurosci. 9:1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto RT, Hay-McCutcheon MJ, Kirk KI, Houston DM, Bergeson-Dana T. 2008. Language skills of profoundly deaf children who received cochlear implants under 12 months of age: a preliminary study. Acta Otolaryngol. 128:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolopoulos TP, Kiprouli K. 2004. Cochlear implant surgery in challenging cases. Cochlear Implants Int. 1:56–63. [DOI] [PubMed] [Google Scholar]
- Oswald AM, Reyes AD. 2008. Maturation of intrinsic and synaptic properties of layer 2/3 pyramidal neurons in mouse auditory cortex. J Neurophysiol. 99:2998–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillsbury HC, Grose JH, Hall JW. 1991. Otitis media with effusion in children: binaural hearing before and after corrective surgery. Arch Otolaryngol Head Neck Surg. 117:718–723. [DOI] [PubMed] [Google Scholar]
- Polley DB, Thompson JH, Guo W. 2013. Brief hearing loss disrupts binaural integration during two early critical periods of auditory cortex development. Nat Commun. 4:2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu MV, Ebner FF. 2010. Neonatal sensory deprivation and the development of cortical function: unilateral and bilateral sensory deprivation result in different functional outcomes. J Neurophysiol. 104:98–107. [DOI] [PubMed] [Google Scholar]
- Popescu MV, Polley DB. 2010. Monaural deprivation disrupts development of binaural selectivity in auditory midbrain and cortex. Neuron. 65:718–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzar L, Goerendt I, Lange K, Rösler F, Röder B. 2007. Early visual deprivation impairs multisensory interactions in humans. Nat Neurosci. 10:1243–1245. [DOI] [PubMed] [Google Scholar]
- Razak KA, Richardson MD, Fuzessery ZM. 2008. Experience is required for the maintenance and refinement of FM sweep selectivity in the developing auditory cortex. Proc Natl Acad Sci USA. 105:4465–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MJ, Sarro EC, Kelly JB, Sanes DH. 2012. Diminished behavioral and neural sensitivity to sound modulation is associated with moderate developmental hearing loss. PLoS One. 7:e41514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagopan S, Wang X. 2010. Contribution of inhibition to stimulus selectivity in primary auditory cortex of awake primates. J Neurosci. 30:7314–7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes DH, Bao S. 2009. Tuning up the developing auditory CNS. Curr Opin Neurobiol. 19:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarro EC, Kotak VC, Sanes DH, Aoki C. 2008. Hearing loss alters the subcellular distribution of presynaptic GAD and postsynaptic GABAA receptors in the auditory cortex. Cereb Cortex. 18:2855–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarro EC, Sanes DH. 2010. Prolonged maturation of auditory perception and learning in gerbils. Dev Neurobiol. 70:636–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiman MM, Hertle RW, Beck RW, Edwards AR, Birch E, Cotter SA, Crouch ER, Jr, Cruz OA, Davitt BV, Donahue S, et al. 2005. Pediatric Eye Disease Investigator Group Randomized trial of treatment of amblyopia in children aged 7 to 17 years. Arch Ophthalmol. 123:437–447. [DOI] [PubMed] [Google Scholar]
- Schorr EA, Fox NA, van Wassenhove V, Knudsen EI. 2005. Auditory-visual fusion in speech perception in children with cochlear implants. Proc Natl Acad Sci USA. 102:18748–18750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Dorman MF, Spahr AJ. 2002. A sensitive period for the development of the central auditory system in children with cochlear implants: implications for age of implantation. Ear Hear. 23:532–539. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Pologruto TA, Svoboda K. 2003. Circuit analysis of experience-dependent plasticity in the developing rat barrel cortex. Neuron. 38:277–289. [DOI] [PubMed] [Google Scholar]
- Simons DJ, Durham D, Woolsey TA. 1984. Functional organization of mouse and rat SmI barrel cortex following vibrissal damage on different postnatal days. Somatosens Res. 1:207–245. [DOI] [PubMed] [Google Scholar]
- Simons DJ, Land PW. 1987. Early experience of tactile stimulation influences organization of somatic sensory cortex. Nature. 326:694–697. [DOI] [PubMed] [Google Scholar]
- Spolidoro M, Sale A, Berardi N, Maffei L. 2009. Plasticity in the adult brain: lessons from the visual system. Exp Brain Res. 192:335–341. [DOI] [PubMed] [Google Scholar]
- Sun YJ, Wu GK, Liu BH, Li P, Zhou M, Xiao Z, Tao HW, Zhang LI. 2010. Fine-tuning of pre-balanced excitation and inhibition during auditory cortical development. Nature. 465:927–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svirsky MA, Teoh SW, Neuburger H. 2004. Development of language and speech perception in congenitally, profoundly deaf children as a function of age at cochlear implantation. Audiol Neurootol. 9:224–233. [DOI] [PubMed] [Google Scholar]
- Swets JA. 1973. The relative operating characteristic in psychology. Science. 182:990–1000. [DOI] [PubMed] [Google Scholar]
- Takesian AE, Kotak VC, Sanes DH. 2012. Age-dependent effect of hearing loss on cortical inhibitory synapse function. J Neurophysiol. 107:937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takesian AE, Kotak VC, Sanes DH. 2010. Presynaptic GABA(B) receptors regulate experience-dependent development of inhibitory short-term plasticity. J Neurosci. 30:2716–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Loos H, Woolsey TA. 1973. Somatosensory cortex: structural alterations following early injury to sense organs. Science. 179:395–398. [DOI] [PubMed] [Google Scholar]
- Wen JA, Barth AL. 2011. Input-specific critical periods for experience-dependent plasticity in layer 2/3 pyramidal neurons. J Neurosci. 31:4456–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitton JP, Polley DB. 2011. Evaluating the perceptual and pathophysiological consequences of auditory deprivation in early postnatal life: a comparison of basic and clinical studies. J Assoc Res Otolaryngol. 12:535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel TN, Hubel DH. 1965. Extent of recovery from the effects of visual deprivation in kittens. J Neurophysiol. 28:1060–1072. [DOI] [PubMed] [Google Scholar]
- Yang EJ, Lin EW, Hensch TK. 2012. Critical period for acoustic preference in mice. Proc Natl Acad Sci USA. 109:17213–17220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanz JL. 1984. The application of the theory of signal detection to the assessment of speech perception. Ear Hear. 5:64–71. [DOI] [PubMed] [Google Scholar]