

Abstract
Elizabethkingia anophelis is an emerging pathogen that can cause life-threatening infections in neonates, severely immunocompromised and postoperative patients. The lack of genomic information on E. anophelis hinders our understanding of its mechanisms of pathogenesis. Here, we report the first complete genome sequence of E. anophelis NUHP1 and assess its response to oxidative stress. Elizabethkingia anophelis NUHP1 has a circular genome of 4,369,828 base pairs and 4,141 predicted coding sequences. Sequence analysis indicates that E. anophelis has well-developed systems for scavenging iron and stress response. Many putative virulence factors and antibiotic resistance genes were identified, underscoring potential host–pathogen interactions and antibiotic resistance. RNA-sequencing-based transcriptome profiling indicates that expressions of genes involved in synthesis of an yersiniabactin-like iron siderophore and heme utilization are highly induced as a protective mechanism toward oxidative stress caused by hydrogen peroxide treatment. Chrome azurol sulfonate assay verified that siderophore production of E. anophelis is increased in the presence of oxidative stress. We further showed that hemoglobin facilitates the growth, hydrogen peroxide tolerance, cell attachment, and biofilm formation of E. anophelis NUHP1. Our study suggests that siderophore production and heme uptake pathways might play essential roles in stress response and virulence of the emerging pathogen E. anophelis.
Keywords: Elizabethkingia anophelis, genome, transcriptome, iron siderophore, heme, oxidative stress response
Introduction
Elizabethkingia is a genus of highly resistant, Gram-negative bacillus that is ubiquitous in the environment. Among the Elizabethkingia spp., E. meningoseptica (formerly known as Flavobacterium meningosepticum) is well documented as a deadly infectious agent. Elizabethkingia meningoseptica thrives in humid habitats and hospital settings such as water supplies and is responsible for neonatal meningitis and sepsis (Dooley et al. 1980; Frank et al. 2013) as well as nosocomial outbreaks of pneumonia among intubated patients in the intensive care unit (Weaver et al. 2010; Jean et al. 2014). Treatment of Elizabethkingia infections is notoriously difficult due to their inherent resistance to a wide range of antimicrobial agents (Teo et al. 2014). The mortality rates of pneumonia and meningitis caused by E. meningoseptica can be more than 50% (Bloch et al. 1997; Weaver et al. 2010; da Silva and Pereira 2013; Teo et al. 2013). However, E. meningoseptica’s pathogenesis mechanism remains largely unexplored due to the lack of complete genome information and gene expression data.
Recently, Elizabethkingia spp. have been reported to be a dominant resident of gut microbiota of the malaria mosquito Anopheles gambiae (Wang et al. 2011; Boissiere et al. 2012). A new species of Elizabethkingia spp. with 98.6% 16S rRNA gene sequence similarity to E. meningoseptica ATCC 13253, Elizabethkingia anophelis, was isolated from the midgut of the A. gambiae (Kampfer et al. 2011). Similar to E. meningoseptica, E. anophelis is also able to cause neonatal meningitis (Frank et al. 2013) and nosocomial outbreaks (Teo et al. 2013). Comparative genomic analysis indicated that E. meningoseptica and E. anophelis have different genome contents and predicted metabolic capacities (Teo et al. 2014). In accordance with this work, another study suggested that E. anophelis might have specific antioxidant genes that provide protection against the oxidative stress generated by A. gambiae blood digestion (Kukutla et al. 2014). The resistance to oxidative stress might also help E. anophelis to survive the human host’s innate immune response.
To gain a deep understanding of the antimicrobial resistance and virulence mechanisms of Elizabethkingia, we completely sequenced the genome of the E. anophelis isolate NUHP1 obtained from a patient, which is the first complete genome of the Elizabethkingia genus. Furthermore, we used RNA-sequencing (RNA-Seq)-based transcriptomic analysis to investigate the oxidative stress response of E. anophelis NUHP1.
Genome Characterization
The genome of E. anophelis NUHP1 strain consists of a circular chromosome comprising 4,369,828 bp and average GC content of 35.62% (fig. 1). A total of 4,141 genes are predicted from the NUHP1 genome, where 4,074 of which are protein-coding genes and 67 of which are RNA-coding genes (52 tRNAs, 10 rRNAs, and 5 other RNAs). A total of 2,489 of the protein-coding genes can be assigned to a putative function with the remaining annotated as hypothetical proteins.
Fig. 1.—

Circular representation of sequence conservation between E. anophelis NUHP1 and 11 Elizabethkingia spp. strains for identify genome regions with high flexibility. Circles are numbered from 1 (outermost circle) to 14 (innermost circle). Circle 1: E. meningoseptica ATCC13253 (OSU). Circle 2: E. meningoseptica ATCC13253 (NITE). Circle 3: E. meningoseptica 502. Circle 4: E. anophelis R26. Circle 5: E. anophelis Ag1. Circle 6: E. anophelis NUH11. Circle 7: E. anophelis NUH6. Circle 8: E. anophelis NUH4. Circle 9: E. anophelis NUH1. Circle 10: E. anophelis NUHP3. Circle 11: E. anophelis NUHP2. Circle 12: GC skew (positive GC skew, green; negative GC skew, violet). Circle 13: GC content. Circle 14: scale of NUHP1 genome.
Alignment of the draft genomes of available Elizabethkingia spp. (Teo et al. 2014) with the NUHP1 complete genome showed that there are several strain-specific genomic regions with low sequence identity (fig. 1). To identify the cause of these strain-specific genomic regions, we predicted the genomic islands (GIs) and visualized their distribution in the NUHP1 genome by using the IslandViewer server (Langille and Brinkman 2009). A total of 14 GIs were identified by either the SIGI-HMM or the IslandPath-DIMOB method used by the IslandViewer server (fig. 2). The distribution of GIs colocalized well with the strain-specific genomic regions among the different genomes of Elizabethkingia spp. (figs. 1 and 2). The functional annotation of genes carried by these predicted GIs is listed in supplementary table S1, Supplementary Material online. A large number of genes from the GIs encode products involved in generating transposons, virulence, efflux pumps, and capsule polysaccharides (supplementary table S1, Supplementary Material online), which emphasizes the potential importance of these GIs on survival of Elizabethkingia spp. under stressful conditions. A striking feature of the GIs is the existence of two large size conjugative DNA-transfer (Tra) regions at two different GIs (close to 0.5 and 4M of the genome) (fig. 2 and supplementary table S1, Supplementary Material online), indicating the importance of this mobile genetic element on modifying the genome content of E. anophelis NUHP1.
Fig. 2.—

Positions of GIs as predicted by IslandViewer program. Blue: GIs predicted by IslandPath-DIMOB approach. Orange: GIs predicted by SIGI-HMM approach. Red: Integrated GIs predicted by both approaches.
Antibiotic Resistance
One major reason for the failure of treatments for Elizabethkingia spp. infections is the lack of an appropriate treatment regimen (Hsu et al. 2011; Teo et al. 2014). The NUHP1 genome was searched against the Comprehensive Antibiotic Resistance Database (McArthur et al. 2013) to identify antibiotic resistant genes, with a 40% identity threshold assigned when performing the BLASTP search. This threshold was chosen because the genome of NUHP1 is very new and not likely to be included in any of the existing databases. A total of 14 antibiotic resistant genes were identified from the NUHP1 genome, including genes conferring resistance to aminoglycosides, beta-lactamases, macrolides, chloramphenicol, tetracycline, and trimethoprim (supplementary table S2, Supplementary Material online). The presence of these genes correlates well with the antibiotic resistant profiles of NUHP1 obtained by minimal inhibitory concentration (MIC) assay (supplementary table S3, Supplementary Material online). Among all our tested antibiotics, only ciprofloxacin gave a relatively low MIC (15.6 µg/ml), which is still much higher than the MIC of ciprofloxacin against other nosocomial Gram-negative pathogens, such as Pseudomonas aeruginosa (0.25 µg/ml) and Escherichia coli (0.0625 µg/ml). These antibiotic resistance genes might partially explain the difficulty in treating E. anophelis nosocomial infections (Dooley et al. 1980; Teo et al. 2013), which is also suggested by Kukutla et al. (2014).
Virulence Mechanisms Revealed by Genome Analysis
To identify potential virulence mechanisms for colonization of E. anophelis NUHP1 in the Anopheles malaria vector host as well as the patient, we did a Basic Local Alignment Search Tool (BLAST) search of the NUHP1 genome against the Virulence Factors of Pathogenic bacteria Database (VFDB) (Chen et al. 2012) and predicted 55 genes conferring to the virulence of this bacterium, including genes involved in capsule polysaccharide biosynthesis, iron siderophore synthesis, heavy metal resistance, and oxidative stress response (supplementary table S4, Supplementary Material online). Elizabethkingia anophelis species carries a siderophore synthesis operon (ORF2289–ORF2298), which is predicted to encode a siderophore similar to that produced by Yesinia spp. (denoted yersiniabactin) (supplementary fig. S1, Supplementary Material online).
Transcriptomic Analysis of Hydrogen Peroxide Stress Response of E. anophelis
Generation of reactive oxygen species is one of the major bactericidal mechanisms from the host innate immune system. Bacterial pathogens have evolved oxidative stress response mechanisms for surviving host colonization (Witko-Sarsat et al. 2000). Elizabethkingia anophelis showed a striking capacity for oxidative stress resistance since hydrogen peroxide (H2O2) is routinely used to clean the hospital sinks where E. anophelis NUHP1 was isolated (Teo et al. 2014). The MIC of H2O2 is 38 mM for E. anophelis NUHP1 cultivated in lysogeny broth (LB) medium under our lab conditions. We exposed exponential growth phase NUHP1 cultures to a sublethal concentration (20 mM) of H2O2 and then used RNA-seq-based transcriptomic analysis to examine potentially important genes for oxidative stress response. RNA-seq analysis indicated that out of the 4,076 predicted E. anophelis genes, 142 displayed statistically significant mRNA level changes of ≥4-fold (adjusted P value < 0.01): 104 of them displaying increased transcript levels (supplementary table S5, Supplementary Material online) and 38 of them displaying decreased transcript levels (supplementary table S6, Supplementary Material online) (fig. 3). Quantitative reverse transcriptase PCR (qRT-PCR) analysis confirmed the expression of 18 highly induced or repressed genes according to RNA-seq analysis. The values obtained by qRT-PCR have a good correspondence with the results of the RNA-seq analysis (table 1).
Fig. 3.—

Heat map of 142 genes whose mRNA level significantly changed. The differentially expressed genes (fold-change > 4, adjusted P value < 0.01) between H2O2-treated and nontreated NUHP1 cells were identified by performing a negative binomial test using the DESeq package of R/Bioconductor. The full list of genes that were differentially expressed between H2O2-treated and nontreated NUHP1 cells is provided in supplementary tables S5 and S6, Supplementary Material online.
Table 1.
Top Induced and Reduced Genes Determined by RNA-Seq and by qRT-PCR in H2O2-Treated Cells
| Locus Tag | Fold Change (by RNA-Seq) | Fold Change (by RT-PCR) | Gene Product Description |
|---|---|---|---|
| BD94_0071 | 291.1 | 2,976.5 | TonB-dependent receptor; outer membrane receptor for ferrienterochelin and colicins |
| BD94_1839 | 108.2 | 295.8 | Methionine aminopeptidase (EC 3.4.11.18) |
| BD94_2005 | 126.8 | 5.9 | TonB-dependent receptor; outer membrane receptor for ferrienterochelin and colicins |
| BD94_2298 | 195.9 | 578.0 | Siderophore biosynthesis l-2,4-diaminobutyrate decarboxylase |
| BD94_2299 | 156.3 | 64.4 | Siderophore biosynthesis protein, monooxygenase |
| BD94_2301 | 200.3 | 46.6 | Desferrioxamine E biosynthesis protein DesD |
| BD94_2937 | 88.8 | 21.8 | ABC-type hemin transport system, ATPase component |
| BD94_2938 | 126.1 | 680.5 | Hemin ABC transporter, permease protein |
| BD94_2939 | 100.3 | 19.8 | Periplasmic hemin-binding protein |
| BD94_1092 | −10.9 | −26.3 | Cytochrome c oxidase subunit CcoN (EC 1.9.3.1)/ cytochrome c oxidase subunit CcoO (EC 1.9.3.1) |
| BD94_1093 | −11.8 | −8.9 | Cytochrome c oxidase subunit CcoP (EC 1.9.3.1) |
| BD94_1333 | −4.5 | −1.5 | ATP phosphoribosyltransferase (EC 2.4.2.17) |
| BD94_2835 | −11.1 | −2.3 | Mg(2+) transport ATPase, P-type (EC 3.6.3.2) |
| BD94_2836 | −13.8 | −7.7 | Probable Co/Zn/Cd efflux system membrane fusion protein |
| BD94_2837 | −16.1 | −74.3 | Cobalt-zinc-cadmium resistance protein CzcA; Cation efflux system protein CusA |
| BD94_2838 | −15.5 | −50.7 | Heavy metal RND efflux outer membrane protein, CzcC family |
| BD94_3514 | −13.4 | −6.9 | Probable cytochrome-c peroxidase (EC 1.11.1.5) |
Treatment of E. anophelis NUHP1 with H2O2 resulted in a dramatically decreased expression of genes involved in heavy metal efflux systems such as Mg2+ transport ATPase (BD94_2835), a probable Co/Zn/Cd efflux system membrane fusion protein (BD94_2836), a cation efflux system protein CusA (BD94_2837), and the heavy metal RND efflux outer membrane protein (BD94_2838) (table 1). In contrast, homolog proteins of three well-known antioxidative proteins, manganese superoxide dismutase (Aguirre and Culotta 2012) (BD94_2310), catalase (BD94_1895) and nonspecific DNA-binding protein Dps (Martinez and Kolter 1997) (BD94_3681), were induced 40 -, 3.8 - and 14.4-fold by H2O2 treatment, respectively (supplementary table S5, Supplementary Material online). The expression of a large set of iron uptake related genes such as the siderophore receptor (BD94_0071, BD94_2005), yersiniabactin-like siderophore biosynthesis (BD94_2298, BD94_2299, BD94_2301), and hemin utilization genes (BD94_2937, BD94_2938, BD94_2939) was highly induced by H2O2 treatment. The above result suggests that iron uptake mechanisms might play an essential role in oxidative stress response for E. anophelis.
Siderophore Production Is Enhanced by Oxidative Stress in E. anophelis
We used the chrome azurol sulfonate (CAS) liquid assay, a universal siderophore detection method (Schwyn and Neilands 1987), to measure the siderophore produced by E. anophelis. The color of CAS assay solution changes from blue to yellow when siderophores in sample solutions chelate iron from CAS, which also leads to decrease in absorbance at 630 nm. The freshly prepared CAS solution was first mixed with a commercially available iron siderophore, deferoxamine, which gave a dose-dependent decrease of the absorbance at 630 nm (fig. 4A). Supernatants of the E. anophelis cultivated in LB contained siderophore activity, which is able to decrease the absorbance at 630 nm to a level close to 20 μM of deferoxamine (fig. 4B). In accordance with the transcriptomic analysis, E. anophelis cultivated in the presence of sublethal H2O2 concentration (20 mM) produced a significantly larger quantity of siderophore compared with E. anophelis cultivated in medium alone (fig. 4B). We noted that the presence of H2O2 will lead to certain interference of the CAS assay of the LB medium control (fig. 4B). However, the concentration of H2O2 in the E. anophelis overnight cultures should be rather low due to the neutralization of secreted catalase.
Fig. 4.—
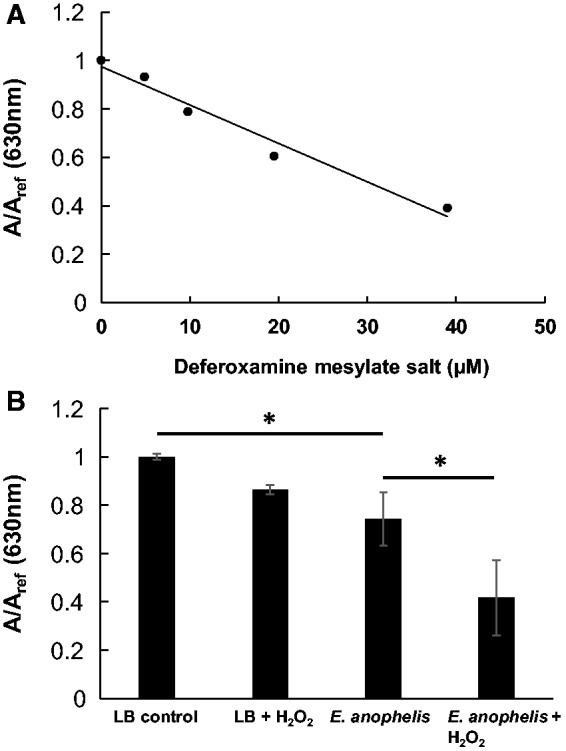
Standard curve for the determination of siderophore (deferoxamine) concentration using a CAS solution (A). Siderophore production by E. anophelis NUHP1 cultivated with and without the presence of H2O2 (B). Means and SD from triplicate experiments are shown. *P < 0.05, Student’s t-test.
The yersiniabactin-like siderophores are produced by Yesinia spp., certain strains of Klebsiella pneumonia (Bachman et al. 2011) and E. coli (Brumbaugh et al. 2015). Yersiniabactin-like siderophores could not be recognized by the host siderophore-binding protein lipocalin-2 and are thus essential for the virulence of Yersinia pestis, Yersinia pseudotuberculosis, Yersinia entercolitica, K. pneumonia, and E.coli (Bearden et al. 1997; Bachman et al. 2011; Brumbaugh et al. 2015). Yersiniabactin-like siderophores are able to reduce the respiratory oxidative stress response of innate immune cells (Paauw et al. 2009) and facilitate the survival of K. pneumoniae during pulmonary infection (Bachman et al. 2011). Yersiniabactin-like siderophores could also sequester heavy metals to prevent their toxicity (Chaturvedi et al. 2012; Bobrov et al. 2014). More interestingly, the copper–yersiniabactin complexes were shown to act as superoxide dismutase mimics, detoxifying the oxygen radicals (Chaturvedi et al. 2012). Further genetic analysis is required to confirm the production of yersiniabactin by E. anophelis under oxidative stress condition as the CAS assay is a general siderophore assay and not specific to the yersiniabactin.
Hemoglobin Enhances Growth, Hydrogen Peroxide Tolerance, Cell Attachment, and Biofilm Formation of E. anophelis
The blood meal feeding to the A. gambiae drastically reduced the microbial community diversity and favored the growth of Elizabethkingia spp. in its gut (Wang et al. 2011). Thus, we hypothesized that the heme uptake was essential for the growth and oxidative stress response of Elizabethkingia spp. We then compared the impact of ferric iron and the blood-associated iron source, hemoglobin (Hb), on the growth of E. anophelis NUHP1. Surprisingly, addition of ferric iron did not promote the growth of E. anophelis NUHP1 (fig. 5A). This may due to the fact that ferric iron does not have a good solubility in the minimal ABTGC medium we used. Hb enhanced the growth of E. anophelis NUHP1 in a dose-dependent manner (fig. 5B). Moreover, E. anophelis NUHP1 growing in the presence of Hb had a higher level of H2O2 tolerance compared with growth in the presence of ferric iron. The MIC values of H2O2 for E. anophelis NUHP1 grown in minimal medium containing 40 µM Hb or 40 µM FeCl3 were 20 mM and 150 µM, respectively. To test whether it is the utilization or presence of Hb causes enhanced H2O2 resistance, we performed a H2O2 time-kill assay of precultivated E. anophelis NUHP1 cultures with and without the supplementation of 10 µM Hb. We found that supplementation of 10 µM Hb to precultivated E. anophelis NUHP1 cultures enhanced the H2O2 resistance compared with the control cultures (fig. 5C).
Fig. 5.—

Growth curves of E. anophelis NUHP1 in ABTGC medium supplemented with different concentrations of FeCl3 (A) and Hb (B). Means and SD from duplicate experiments are shown. Molar concentration is for the Fe element only. Time-kill curves of E. anophelis NUHP1 by 20 mM H2O2 in ABTGC medium with and without supplementation of 10 µM Hb (C). Confocal images of 7,228.4 -μm2 substratum area of 24 h E. anophelis NUHP1 and E. meningoseptica ATCC13253 biofilms grown in iron free ABTGC medium and ABTGC medium supplemented with different iron sources (D). Representative confocal images from triplicate experiments are shown for each condition. Scale bars: 20 μm.
It has been proposed that Elizabethkingia spp. biofilm formation might contribute to the infections it causes (Jacobs and Chenia 2011). We thus also examined the impact of ferric iron and Hb on Elizabethkingia spp. cell attachment and biofilm formation. Interestingly, E. anophelis NUHP1 could not attach firmly on the glass substratum in ABTGC medium with no iron or with 10 μM ferric iron, whereas addition of 10 μM Hb enhanced the attachment of E. anophelis NUHP1 to the substratum (supplementary videos S1–S3, Supplementary Material online). The average biovolume of biofilm formed per 7,228.4 μm2 of three technical replicates was calculated using the Imaris software package (Bitplane, AG). In accordance with the attachment result, E. anophelis NUHP1 formed biofilms with a biovolume of 1,160.1 ± 196.7 μm3 when supplemented with ferric iron and 3,708.7 ± 621.4 μm3 when supplemented with Hb (fig. 5D). On average, biovolume increased 3.2-fold, with a range of 2.28 - to 4.50-fold when Hb was used as the iron source. In contrast, iron source has no obvious effect on the cell attachment and biofilm formation of E. meningoseptica ATCC13253 (NITE) (data not shown). Elizabethkingia meningoseptica ATCC13253 (NITE) formed biofilms with an average biovolume of 4,740.3 ± 553.2 μm3 when supplemented with ferric iron and 6,395.5 ± 1,724.3 μm3 when supplemented with Hb (fig. 5D). Here, there is an average 1.35-fold increase in biovolume, with a range of 0.88- to 1.94-fold when Hb was used as the iron source.
In summary, we report the first complete genome sequence of E. anophelis NUHP1 and assess its response to oxidative stress by transcriptomic analysis. Elizabethkingia anophelis NUHP1 has well-developed systems for stress response and encodes many putative virulence factors and antibiotic resistance products. Our study showed that production of a potential yersiniabactin-like iron siderophore and heme uptake mechanism might play essential roles in oxidative stress response of E. anophelis. We also showed E. anophelis NUHP1 and E. meningoseptica ATCC13253 have distinct response toward iron sources during cell attachment and biofilm formation. Further study will be carried to investigate the roles of yersiniabactin-like iron siderophore during infections caused by E. anophelis.
Materials and Methods
Bacterial Growth Condition
LB medium (Bertani 1951) was used to cultivate Elizabethkingia spp. strains. Batch cultivation of Elizabethkingia spp. was carried out at 37 °C with shaking. ABTGC medium (Chua et al. 2013) supplemented with FeCl3 or Hb was used to investigate the impact of the different iron sources on growth, tolerance to hydrogen peroxide (H2O2), and biofilm formation. Susceptibility of the E. anophelis NUHP1 to H2O2 was determined by serial dilution assays.
Genome Characterization
The whole-genome DNA of E. anophelis NUHP1 cultures was purified using QIAamp DNA Mini Kit (QIAgen) and the genome sequence of E. anophelis NUHP1 was determined using the Illumina HiSeq 2000 sequencing platform. 604 Mb data were produced for Tube for the 500-bp library, and 303 Mb data for the 2,000-bp library, 301 Mb data for the 6,000-bp library. The paired-end reads were de novo assembled with SOAPdenovo v. 1.03 software (BGI) (http://soap.genomics.org.cn/soapdenovo.html, last accessed June 9, 2015), and the reads were assembled into eight large scaffolds. Gaps between contigs were closed by custom primer walks or by long-distance PCR amplification followed by DNA sequencing. Genome annotation was performed using the RAST Server (http://rast.nmpdr.org, last accessed June 9, 2015). The metabolite synthesis operon was predicted by using the antiSMASH server (http://antismash.secondarymetabolites.org/, last accessed June 9, 2015).
The genomic sequence comparisons between the reference genome of E. anophelis NUHP1 and another ten available draft genomes of Elizabethkingia spp. (accession numbers: AVCQ00000000, AHHG00000000, ASAN00000000, ASYH01000000, ASYI01000000, ASYJ01000000, ASYK01000000, ASYF01000000, ASYG01000000, and ANIW01000000) were performed as follows. Scaffolds from the draft sequences were initially oriented and joined by aligning them to the reference genome of E. anophelis NUHP1 using the custom perl script scaffolding.pl (available at https://github.com/flauro/3tck_comparative, last accessed June 9, 2015; Lauro et al. 2014). The junctions between each adjacent scaffold were filled with the six-frame stop-codon spacer “NNNNCACACACTTAATTAATTAAGTGTGTGNNNN” resulting in contiguous pseudomolecules. Each pseudomolecule was then compared by BLAST searches against the reference genome of E. anophelis NUHP1 and visualized using the BLAST Ring Image Generator v0.95 (Alikhan et al. 2011).
The E. anophelis NUHP1 genome was compared against the Comprehensive Antibiotic Resistance Database (McArthur et al. 2013) using the BLAST search tool, and VFDB (Chen et al. 2012) to identify antibiotic resistant genes and virulence genes by using Bio-Edit (Ibis Biosciences, Carlsbad, CA; http://www.mbio.ncsu.edu/bioedit/bioedit.html, last accessed June 9, 2015) (minimum 40% identity with E value less than 1 e−5). Stress response genes were predicted from the E. anophelis NUHP1 genome using the RAST server (Aziz et al. 2008). GIs were predicted and visualized from the NUHP1 genome using the IslandViewer server (Langille and Brinkman 2009).
Biofilm Assay
Elizabethkingia anophelis NUHP1 and E. meningoseptica ATCC13253 (NITE) biofilms were grown in μ-Slide eight-well microscopy chambers (ibidi, München, Germany) under static conditions at 37 °C and in iron free ABTGC medium and ABTGC medium supplemented with 10 μM FeCl3 or 10 μM Hb, respectively. Biofilms were live/dead stained with SYTO62 (Invitrogen, CA) and PI (Invitrogen). Confocal images of 18 biofilms were captured (ZEISS LSM780 Confocal System) and analyzed using the Imaris software package (Bitplane).
Transcriptomic Analysis
To gain insights into the early transcriptional response of E. anophelis to oxidative stress, we performed a comparative transcriptomic analysis between H2O2-treated E. anophelis NUHP1 cells and untreated controls by using RNA-Seq. Elizabethkingia anophelis NUHP1 overnight culture was inoculated into LB medium with a starting optical density of 600 nm (OD600) = 0.01 and incubated at 37 °C with 200 rpm shaking. H2O2 was added to the cultures at mid-log phase (OD600 = 0.5) such that the final concentration for treatment was 20 mM. After 10-min H2O2 treatment, both treated and untreated NUHP1 cells were harvested by centrifugation. Total RNA was extracted with RNeasy Protect Bacteria Mini Kit with on-column DNase digestion as previously described (Chua et al. 2014).
RNA-Seq was conducted for three biological replicates of each sample. Libraries were produced using an Illumina TruSeq RNA Sample Prep Kit. The libraries were sequenced using the Illumina HiSeq 2000 platform with a paired-end protocol and read lengths of 100 nt. Each replicate generated around 50–59 million reads. Analysis of the RNA-Seq data was performed as previous described (Chua et al. 2014). Briefly, the sequence reads were mapped onto the E. anophelis NUHP1 genome using the RNA-Seq and expression analysis application of CLC genomics Workbench 6.0 (CLC Bio, Aarhus, Denmark). The transcript count table was subjected to DESeq package (Anders and Huber 2010) of R/Bioconductor (Gentleman et al. 2004) for statistical analysis. The transcript counts were normalized to the effective library size. The differentially expressed genes were identified by performing a negative binomial test. Transcripts were stringently determined as differentially expressed when having a fold change greater than 4 and an adjusted P value less than 0.01. Hierarchical clustering analysis was performed and a heatmap was produced for the differentially expressed genes, using heatmap.2 package of R/Bioconductor (Gentleman et al. 2004).
qRT-PCR Analysis
Total RNA was extracted using RNeasy Mini Kit (Qiagen) with on-column DNase digestion. The purity and concentration of the RNA were determined by NanoDrop spectrophotometry, and the integrity of RNA was measured using an Agilent 2200 TapeStation System. The elimination of contaminating DNA was confirmed through the real-time PCR amplification of the 16 s rRNA gene using total RNA as the template.
qRT-PCR was performed using a two-step method with primers listed in supplementary table S7, Supplementary Material online. First-strand cDNA was synthesized from total RNA using SuperScript III First-Strand Synthesis SuperMix kit (Cat. No. 18080-400; Invitrogen). The cDNA was used as a template for qRT-PCR with an SYBR Select Master Mix kit (Cat. No. 4472953; Applied Biosystems by Life Technologies) on an Applied Biosystems StepOnePlus Real-Time PCR System. The 16 s rRNA gene was used as an endogenous control. Melting curve analyses were employed to verify the specific single-product amplification.
CAS Liquid Assay
The CAS liquid assay was performed as previously described, with modifications (Schwyn and Neilands 1987). All solutions and dilutions were made with 1 mM PIPES (pH 5.6) unless otherwise stated. The water LC-MS CHROMASOLV (Fluka, Singapore) was used to prepare the PIPES solution. Six milliliters of 10 mM HDTMA solution was placed in a 100-ml volumetric flask and diluted with 14 ml 1 mM PIPES. A mixture of 1.5 ml ferric iron solution (1 mM FeCl3, 10 mM HCl) and 7.5 ml 2 mM aqueous CAS (Tokyo Chemical Industry, Singapore) solution was slowly added under stirring. A 4.307 g quantity of anhydrous piperazine was dissolved in 20 ml PIPES solution to which 6.25 ml of 12 M HCl was carefully added. This buffer solution (pH = 5.6) was washed into the 100-ml volumetric flask which was then filled with 1 mM PIPES to make up 100 ml of CAS assay solution.
To demonstrate the efficiency of the CAS assay solution, a standard curve was prepared with purified siderophore deferoxamine mesylate salt (Sigma-Aldrich, Singapore). Hundred microliters of different concentrations of deferoxamine mesylate salt was mixed with equal volume of CAS assay solution in 96-well plate and the absorbance measured at 630 nm after reaching equilibrium.
To determine the siderophore produced by E. anophelis, E. anophelis NUHP1 overnight culture was inoculated into LB medium with a starting optical density of OD600 = 0.01 and incubated at 37 °C with 200 rpm shaking. When the cultures reached mid-log phase (OD600 = 0.5), H2O2 was added so a final concentration of 20 mM was achieved for treatment. After incubating overnight (16–18 h), both H2O2-treated and untreated cultures were centrifuged and filtered through the 0.22-µm filters. A 0.5-ml aliquot of culture supernatants or the control LB medium (treated or untreated with H2O2) was mixed with 0.5 ml CAS assay solution. After reaching equilibrium the absorbance was measured at 630 nm. Experiments were performed in triplicate, and the results are shown as the mean ± standard deviation (SD). Student’s t-test was used to determine the significance of the differences.
Time-Kill Assay
Elizabethkingia anophelis strain NUHP1 was cultivated in iron-free ABTGC medium without addition of iron at 37 °C with 200 rpm shaking. Overnight cultures of NUHP1 were adjusted to 109 CFU/ml in fresh ABTGC medium with and without the supplementation of 10 μM Hb. H2O2 was added to the Hb-supplemented and control NUHP1 cultures at a final concentration of 20 mM, after which samples were harvested at time 5, 15, 30 min and diluted as necessary and plated in LB agar plates for viable counts. Experiments were performed in triplicate, and the results are shown as the mean ± SD.
Nucleotide Sequence Accession Numbers
The genome sequence of E. anophelis NUHP1 has been deposited at the GenBank under the accession of CP007547. The RNA-Seq data have been deposited in the NCBI Short Read Archive (SRA) database with accession code SRP043452.
Supplementary Material
Supplementary videos S1–S3, tables S1–S7, and figure S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This research was supported by the National Research Foundation and Ministry of Education Singapore under its Research Centre of Excellence Program, the Start-up Grants (M4330002.C70) from Nanyang Technological University, and the AcRF Tier 2 Grant (MOE2014-T2-2-172) from the Ministry of Education (Singapore) to L.Y. F.M.L. was partially supported by fellowship from the Australian Research Council (DE120102610). The authors acknowledge Dr Damien Keogh for his guidance on the siderophore CAS assay.
Literature Cited
- Aguirre JD, Culotta VC. 2012. Battles with iron: manganese in oxidative stress protection. J Biol Chem. 287:13541–13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. 2011. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz RK, et al. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman MA, et al. 2011. Klebsiella pneumoniae yersiniabactin promotes respiratory tract infection through evasion of lipocalin 2. Infect Immun. 79:3309–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden SW, Fetherston JD, Perry RD. 1997. Genetic organization of the yersiniabactin biosynthetic region and construction of avirulent mutants in Yersinia pestis. Infect Immun. 65:1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol. 62:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch KC, Nadarajah R, Jacobs R. 1997. Chryseobacterium meningosepticum: an emerging pathogen among immunocompromised adults. Report of 6 cases and literature review. Medicine 76:30–41. [DOI] [PubMed] [Google Scholar]
- Bobrov AG, et al. 2014. The Yersinia pestis siderophore, yersiniabactin, and the ZnuABC system both contribute to zinc acquisition and the development of lethal septicaemic plague in mice. Mol Microbiol. 93:759–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissiere A, et al. 2012. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 8:e1002742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumbaugh AR, et al. 2015. Blocking Yersiniabactin import attenuates extraintestinal pathogenic Escherichia coli in cystitis and pyelonephritis and represents a novel target to prevent urinary tract infection. Infect Immun. 83:1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi KS, Hung CS, Crowley JR, Stapleton AE, Henderson JP. 2012. The siderophore yersiniabactin binds copper to protect pathogens during infection. Nat Chem Biol. 8:731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xiong Z, Sun L, Yang J, Jin Q. 2012. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 40:D641–D645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua SL, et al. 2013. Bis-(3′-5′)-cyclic dimeric GMP regulates antimicrobial peptide resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 57:2066–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua SL, et al. 2014. Dispersed cells represent a distinct stage in the transition from bacterial biofilm to planktonic lifestyles. Nat Commun. 5:4462. [DOI] [PubMed] [Google Scholar]
- da Silva PS, Pereira GH. 2013. Elizabethkingia meningoseptica: emergent bacteria causing pneumonia in a critically ill child. Pediatr Int. 55:231–234. [DOI] [PubMed] [Google Scholar]
- Dooley JR, Nims LJ, Lipp VH, Beard A, Delaney LT. 1980. Meningitis of infants caused by Flavobacterium meningosepticum: report of a patient and analysis of 63 infections. J Trop Pediatr. 26:24–30. [DOI] [PubMed] [Google Scholar]
- Frank T, et al. 2013. First case of Elizabethkingia anophelis meningitis in the Central African Republic. Lancet 381:1876. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu MS, et al. 2011. Clinical features, antimicrobial susceptibilities, and outcomes of Elizabethkingia meningoseptica (Chryseobacterium meningosepticum) bacteremia at a medical center in Taiwan, 1999-2006. Eur J Clin Microbiol Infect Dis. 30:1271–1278. [DOI] [PubMed] [Google Scholar]
- Jacobs A, Chenia HY. 2011. Biofilm formation and adherence characteristics of an Elizabethkingia meningoseptica isolate from Oreochromis mossambicus. Ann Clin Microbiol Antimicrob. 10:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean SS, Lee WS, Chen FL, Ou TY, Hsueh PR. 2014. Elizabethkingia meningoseptica: an important emerging pathogen causing healthcare-associated infections. J Hosp Infect. 86:244–249. [DOI] [PubMed] [Google Scholar]
- Kampfer P, et al. 2011. Elizabethkingia anophelis sp. nov., isolated from the midgut of the mosquito Anopheles gambiae. Int J Syst Evol Microbiol. 61:2670–2675. [DOI] [PubMed] [Google Scholar]
- Kukutla P, et al. 2014. Insights from the genome annotation of Elizabethkingia anophelis from the malaria vector Anopheles gambiae. PLoS One 9:e97715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MG, Brinkman FS. 2009. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25:664–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauro FM, et al. 2014. Ecotype diversity and conversion in Photobacterium profundum strains. PLoS One 9:e96953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Kolter R. 1997. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J Bacteriol. 179:5188–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur AG, et al. 2013. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 57:3348–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paauw A, Leverstein-van Hall MA, van Kessel KP, Verhoef J, Fluit AC. 2009. Yersiniabactin reduces the respiratory oxidative stress response of innate immune cells. PLoS One 4:e8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwyn B, Neilands JB. 1987. Universal chemical assay for the detection and determination of siderophores. Anal Biochem. 160:47–56. [DOI] [PubMed] [Google Scholar]
- Teo J, et al. 2013. First case of E anophelis outbreak in an intensive-care unit. Lancet 382:855–856. [DOI] [PubMed] [Google Scholar]
- Teo J, et al. 2014. Comparative genomic analysis of malaria mosquito vector-associated novel pathogen Elizabethkingia anophelis. Genome Biol Evol. 6:1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gilbreath TM, 3rd, Kukutla P, Yan G, Xu J. 2011. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One 6:e24767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver KN, et al. 2010. Acute emergence of Elizabethkingia meningoseptica infection among mechanically ventilated patients in a long-term acute care facility. Infect Control Hosp Epidemiol. 31:54–58. [DOI] [PubMed] [Google Scholar]
- Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L. 2000. Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest. 80:617–653 [DOI] [PubMed] [Google Scholar]