

Abstract
PURPOSE
To investigate the relationships between clinical and full-field electroretinographic (ERG) findings and progressive loss of visual function in Stargardt disease.
DESIGN
Retrospective cohort study.
METHODS
We performed a retrospective review of data from 198 patients with Stargardt disease. Measures of visual function over time, including visual acuity, quantified Goldmann visual fields, and full-field ERG data were recorded. Data were analyzed using SAS statistical software. Subgroup analyses were performed on 148 patients with ERG phenotypic data, 46 patients with longitudinal visual field data, and 92 patients with identified ABCA4 mutations (46 with 1 mutation, and 47 with 2 or more mutations).
RESULTS
Of 46 patients with longitudinal visual field data, 8 patients with faster central scotoma progression rates had significantly worse scotopic B-wave amplitudes at their initial assessment than 20 patients with stable scotomata (P = .014) and were more likely to have atrophy beyond the arcades (P = .047). Overall, 47.3% of patients exhibited abnormal ERG results, with rod–cone dysfunction in 14.2% of patients, cone–rod dysfunction in 17.6% of patients, and isolated cone dysfunction in 15.5% of patients. Abnormal values in certain ERG parameters were associated significantly with (maximum-stimulation A- and B-wave amplitudes) or tended toward (photopic and scotopic B-wave amplitudes) a higher mean rate of central scotoma progression compared with those patients with normal ERG values. Scotoma size and ERG parameters differed significantly between those with a single mutation versus those with multiple mutations.
CONCLUSIONS
Full-field ERG examination provides clinically relevant information regarding the severity of Stargardt disease, likelihood of central scotoma expansion, and visual acuity deterioration. Patients also may exhibit an isolated cone dystrophy on ERG examination.
Mutations in the adenosine 5′-triphosphate–binding cassette transporter gene ABCA4 are known to result in several phenotypic patterns of retinal degeneration. These include Stargardt disease, an autosomal recessive form of juvenile-onset macular dystrophy first described in 1905 by Karl Stargardt, who presented cases with foveal atrophy, paramacular yellow deposits, and fleck. Other phenotypic presentations of ABCA4 mutations include progressive cone–rod degenerations with rapidly expanding central scotomata, often called inverse retinitis pigmentosa, and rarely typical retinitis pigmentosa.1–8 The most common clinical manifestation of 2 or more ABCA4 mutations is Stargardt disease, which involves progressive loss of central vision, with near normal peripheral visual fields. Symptom onset of Stargardt disease typically occurs during the first and second decades of life, although there is a subgroup of patients who demonstrate symptoms later in life.9 Funduscopic findings include pisciform-shaped yellow deposits (or flecks) and macular involvement, which can manifest as either a beaten-bronze sheen, macular granularity, a bull’s-eye lesion, or retinal pigment epithelium depigmentation with atrophy.10 Our experience has demonstrated that the disease course in mutation-proven individuals can vary greatly from patient to patient, with variable age of onset as well as differential rates of symptomatic progression with respect to visual acuity and visual fields.
Full-field electroretinography (ERG) is one of the methods that has been used to describe the severity of Stargardt disease.11–20 The extent of full-field ERG abnormality seems to be related to disease severity and has been used to classify the type of photoreceptor dysfunction present in Stargardt disease (cone dysfunction vs rod–cone dysfunction vs cone–rod dysfunction). The presence of normal full-field ERG results often accompanies a clinical diagnosis of Stargardt disease.8,14,21 However, in a limited number of patients with more advanced disease, Fishman and associates reported a higher percentage of abnormal full-field ERG values.14 More recently, Simonelli and associates used full-field ERG values to group patients with ABCA4-associated disease into mild and severe phenotypes.20 In contrast, Oh and associates were unable to correlate clinical appearance with full-field ERG characteristics in a large cohort of patients.19
Given the variability seen in full-field electroretinographic patterns and Stargardt disease progression rates, we sought to examine the prognostic value of full-field ERG results and their association with visual field changes in our large cohort of Stargardt disease patients. We focused on defining groups based on the clinical progression over time of the central scotoma size.
METHODS
The design of this study was retrospective, and approval from the University of Michigan Institutional Review Board was granted to perform a retrospective study. All patients signed informed consent forms for genetic testing as well as use of their clinical data for research purposes.
A retrospective review was performed of patients with a diagnosis of Stargardt disease who were seen at the Kellogg Eye Center, University of Michigan (UM; n = 131) and at the Jules Stein Eye Institute at the University of California, Los Angeles (UCLA; n = 67) by the same senior investigator (J.H.). The study was approved by the UM Institutional Review Board and the UCLA Human Subject Protection Committee. Patients qualified for the study if they had received a clinical diagnosis of Stargardt disease, had a pedigree consistent with autosomal recessive inheritance, and met one of the following inclusion criteria: (1) 2 causative mutations in ABCA4; (2) 1 causative mutation in ABCA4 and 1 of the following clinical findings: a dark choroid sign, documented pisciform flecks, macular atrophy, reduced central acuity, or the presence of central scotoma on Goldmann visual field testing; or (3) a clinical diagnosis based on the combined presence of fundus flecks and a dark choroid on fluorescein angiography, with mutational analysis pending. It should be noted that finding only 1 causative mutation in ABCA4 patients is not unusual.21–24 We did not include patients who had no mutations identified in ABCA4 after sequencing of all 50 exons.
All patients underwent comprehensive ocular examinations, Goldmann perimetry (every visit), standardized full-field ERG following International Society for Clinical Electrophysiology of Vision guidelines,25 fundus photography, and fluorescein angiography. ABCA4 mutational screenings were performed at UM, the University of Iowa Carver Lab, the University Medical Centre Nijmegen, or through the eyeGENE Research Project at the National Eye Institute.
Clinical data collected included: age of symptom onset, family history, best-corrected visual acuity, the area of central scotomata for standardized isopters (I4e, III4e, IV4e) on Goldmann visual field testing measured by digital planimetry (Placom 45C digital planimeter; Koizumi Sokki Mfg Co., Nagaoka Nagata, Japan), retinal distribution of flecks and atrophy on fundus photography, and the presence or absence of a dark choroid sign. Snellen visual acuities were transformed into logarithm of the minimal angle of resolution (logMAR) notation for the purposes of analysis.26
STAGES
The clinical phenotype was evaluated by examining all patient charts and color fundus photographs, which were graded by 2 retinal dystrophy specialists (J.H., K.T.J.). Fundus flecks and atrophy were scored according to their location on the fundus: presence or absence in the macula lutea in the posterior pole.3 These retinal features were used to stratify the patients into 3 distinct phenotypes (see Supplemental Figure). Stage I involved flecks limited to the macula, whereas stage II included flecks limited to the posterior pole with foveal atrophy. Stage III patients had advanced disease and were defined by macular atrophy and diffuse flecks throughout the fundus. Stage III in this study is essentially an amalgamation of stages III and IV as described previously by Fishman.27 Goldmann visual field data (ie, peripheral field, central scotoma, and physiologic blind spot areas) were obtained using digital planimetry. Data were recorded from standardized Goldmann visual field sheets (Haag-Streit, Bern, Switzerland) and entered by isopters I4e, III4e, and IV4e.
Patients also were separated into 4 distinct groups based on their full-field ERG results at presentation. Of note, given different protocols and normal ranges for full-field ERG values at UCLA and UM (there were 2 sets of normal reference values at UM depending on when the full-field ERG results were obtained), the normal means were derived for each center separately (eg, scotopic B-wave amplitude mean: UM1–274 μV, UM2–325.36μV, UCLA–375 μV; photopic B-wave amplitude mean: UM1–166 μV, UM2–125.36μV, UCLA–169 μV). Patient full-field ERG data were expressed as a percentage of the respective mean for each center to facilitate analysis of data compiled from the 2 centers. For the purposes of our study, abnormal was defined as 2 standard deviations less than the normal mean values for amplitude measures and 2 standard deviations more than the normal mean values for latency measures, which were derived separately from control patients seen at UM and UCLA. Patients with abnormal photopic and scotopic full-field ERG values whose scotopic deficiency was more severe were considered to have rod–cone dysfunction. Patients with abnormal photopic and scotopic full-field ERG values with a more severe photopic deficiency were considered to have cone–rod dysfunction. Patients with abnormal photopic full-field ERG values in the presence of normal scotopic full-field ERG values were considered to have cone dysfunction. Finally, patients with normal photopic and scotopic B-wave amplitudes were considered to have a normal full-field ERG phenotype.
STATISTICAL ANALYSES
Statistical analyses were performed using SAS software version 9.2 (SAS Institutes, Cary, North Carolina, USA). Statistical comparisons between groups were made using t tests or analyses of variance for continuous variables and chi-square or Fisher exact tests for categorical variables. Linear regression was used to estimate the slope of scotoma progression. The data for the right eye of each patient were included in statistical analyses, given that Stargardt disease is a dystrophy that affects eyes in a nearly symmetrical fashion (Tegins EO, et al. IOVS 2011;52:ARVO E-Abstract 5005).
RESULTS
The 198 patients with stargardt disease we studied were on average 25.6 years of age (standard deviation [SD], 15.1 years) when symptoms first presented and were on average 34.5 years of age (SD, 16.2 years) when first seen at either UM or UCLA, and 59.1% (n = 117) were female. Follow-up was available on subjects for an average of 3.5 years (SD, 5.9 years) after their initial visit from a mean of 2.8 visits (SD, 4.1 visits). When first seen, 15.8% (n = 31) of subjects had stage I Stargardt disease, 54.4% (n = 108) had stage II Stargardt disease, and 29.9% (n = 59) had stage III Stargardt disease. Baseline and follow-up patient data are summarized briefly in Table 1.
TABLE 1.
Baseline and Follow-up Data for Patients with Stargardt Disease
| Continuous Variables | Mean (SD) | Minimum, Maximum | Median |
|---|---|---|---|
| Age (y) | |||
| At first visit (n = 196) | 34.5 (16.2) | 7.2, 79.7 | 32.5 |
| At symptom onset (n = 167) | 25.7 (15.0) | 5.0, 72.0 | 22.0 |
| Follow-up (y, n = 198) | 3.5 (5.9) | 0.0, 29.7 | 0.5 |
| No. of visits (n = 198) | 2.8 (4.1) | 1.0, 38.0 | 2.0 |
| BCVA (logMAR) | |||
| At first visit (n = 179) | 0.71 (0.54) | 0.00, 2.30 | 0.60 |
| At last visit (n = 184) | 0.81 (0.57) | −0.12, 2.30 | 1.00 |
| I4e scotoma size (cm2) | |||
| At first visit (n = 160) | 6.4 (12.0) | 0.0, 76.8 | 1.6 |
| At last visit (n = 160) | 10.5 (16.7) | 0.0, 86.5 | 3.5 |
|
| |||
| Categorical Variables | Percent | ||
|
| |||
| Sex (n = 198) | |||
| Male | 40.9 | ||
| Female | 59.1 | ||
| Disease stage at presentation (n = 184) | |||
| I | 15.8 | ||
| II | 54.4 | ||
| III | 29.9 | ||
BCVA = best-corrected visual acuity; logMAR = logarithm of the minimal angle of resolution; SD = standard deviation.
VISUAL FIELD ANALYSIS: DIFFERENCES IN SCOTOMA PROGRESSION
We performed a subgroup analysis for those patients for whom we could describe rates of central scotoma progression for the I4e isopter using Goldmann visual fields (n = 46). All patients with at least 3 Goldmann visual field data points were included, and a slope of scotoma progression was calculated for each patient. Based on their rates of scotoma progression, we divided these patients into 3 distinct groups. Faster progressors were defined as those patients with a rate of scotoma size progression of more than 2 cm2/year (n = 8; mean number of scotoma measurements, 6.1; median number of scotoma measurements, 5; range number of scotoma measurements, 3 to 13; mean follow-up, 9.5 years; median follow-up, 6.4 years; follow-up range, 0.6 to 21.2 years). One outlying fast progressor exhibited a scotoma progression rate of 7.8 cm2/year over the course of 4.1 years after initial presentation at age 32 years. Nonprogressors exhibited a rate of scotoma size progression of less than 1 cm2/year (n = 27; mean number of scotoma measurements, 5.2; median number of scotoma measurements, 4.0; range number of scotoma measurements, 3 to 15; mean follow-up, 8.3 years, median follow-up, 6.9 years; follow-up range, 1.0 to 24.1 years). The rate of central scotoma progression using the I4e isopter was plotted against patient age at first reported scotoma to illustrate these 2 groups and the broad range of ages at first scotoma (Figure 1). Comparing available full-field ERG data from these 2 groups, the faster-progressing patients (n = 8) had significantly worse scotopic B-wave amplitudes at initial presentation than the slowly progressing patients (n = 20; percent of mean, 53% and 83%, respectively; P = .014, t test). Of note, another group of patients progressed at an intermediate rate of 1 to 2 cm2/year (n = 11) and exhibited an intermediate mean for scotopic B-wave amplitude (percent mean, 66%). Interestingly, patients with faster clinical progression were more likely to have atrophic-appearing retinal pigment epithelium beyond the vascular arcades than slower progressors (2 of 8 vs 0 of 27, respectively; P = .047, Fisher exact test).
FIGURE 1.
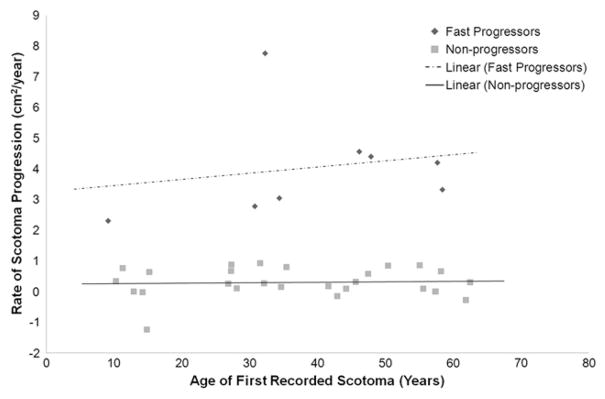
Graph showing scotoma progression rate in patients with Stargardt disease. Patients with at least 3 visits had scotoma sizes quantitated with planimetry. Scotomata progression of more than 2.0 cm2/year is depicted as dark grey diamonds, and patients with progression of less than 1.0 cm2/year are shown as light grey squares. Quickly progressing patients had an average progression rate of 4.04 cm2/year compared with the slowly progressing patients, who exhibited an average progression rate of 0.29 cm2/year.
Age of onset in fast progressors averaged 35.7 years, whereas that of nonprogressors averaged 29.8 years (P = .429, t test). There were minimal differences in clinical stage at presentation between progressors and non-progressors (P = .814, Fisher exact test). There was a statistically significant association between gender and scotoma progression. Specifically, all fast progressors were female (8/8; 100%), whereas 59% of slow progressors were female (16/27; 59%; P = 0.037, Fisher exact test). Average logMAR visual acuities in the fast progressors were somewhat worse than those in nonprogressors at the first visit (0.78 vs 0.54 logMAR units, respectively; P = .427, t test) and the last visit (1.19 vs 0.76 logMAR units, respectively; P = .139), although these differences were not statistically significant. There were statistically significant differences in scotoma size at the first visit (P = .030, t test) and the last visit (P = .004, t test), with the fast progressors exhibiting larger average central scotoma sizes at both time points.
FULL-FIELD ELECTRORETINOGRAPHY PHENOTYPE SUBGROUP ANALYSIS: CHARACTERIZING ELECTRORETI-NOGRAPHIC ABNORMALITIES
One hundred fifty-one patients had full-field ERG data, of which 148 had both scotopic and photopic B-wave amplitude data available for analysis. Of these patients, 47.3% (n = 70) had abnormalities of one or both waveforms. The remainder (52.7%; n = 78) had both rod and cone amplitudes within the normal range. We categorized the 70 patients with abnormal full-field ERG results into groups by the type of full-field ERG abnormality detected: 14.2% (21/148) of patients exhibited rod–cone dysfunction, 17.6% (26/148) had cone–rod dysfunction, and 15.5% (23/148) had isolated cone dysfunction with normal rod function. Full-field ERG phenotypic distribution and abnormalities in each full-field ERG parameter from all available data are shown in Figure 2 and Figure 3, respectively. The percentage of patients showing abnormal full-field ERG values ranged from 19.6% (scotopic B-wave latency) to 63.4% (photopic A-wave latency).
FIGURE 2.

Bar graph showing full-field electroretinography (ERG) phenotypes in patients with Stargardt disease from all available ERG data: 47.3% all Stargardt patients exhibited abnormal full-field ERG results.
FIGURE 3.
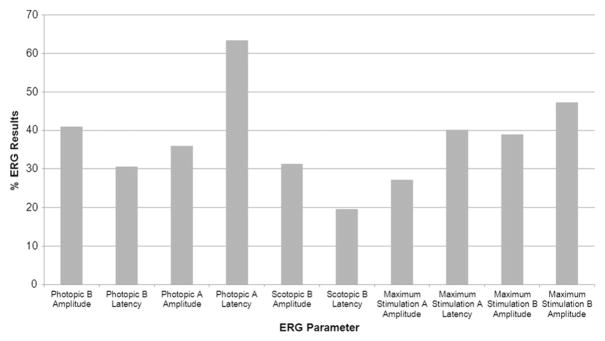
Bar graph showing the percent of abnormal results by full-field electroretinography (ERG) parameter in patients with Stargardt disease. Percentage of patients with abnormal full-field ERG parameters are shown. All values less than 2 standard deviations from the mean were considered abnormal for amplitude measures, and all values more than 2 standard deviations from the mean were considered abnormal for latency measures.
FULL-FIELD ELECTRORETINOGRAPHY AND SCOTOMA SIZE
Patients who exhibited abnormal photopic and scotopic B-wave amplitudes tended to have a higher average rate of central scotoma progression (1.88 cm2/year [n = 19; P = .066] and 2.05 cm2/year [n = 14; P = .064], respectively) compared with patients with respective normal amplitudes (0.78 cm2/year [n = 16] and 0.94 cm2/ year [n = 22], respectively), although these differences were not statistically significant. Patients with abnormal maximum stimulation A-wave amplitudes exhibited a higher average rate of scotoma progression when compared with patients with corresponding normal ERG values (2.56 cm2/year [n = 11] and 0.87 cm2/year [n = 23], respectively; P = .008, t test). Similarly, patients with abnormal maximum stimulation B-wave amplitudes exhibited a higher average rate of scotoma progression compared with patients with corresponding normal values (2.08 cm2/ year [n = 17] and 0.76 cm2/year [n = 17], respectively; P = .030, t test). See Figure 4 for a comparison of the average rate of scotoma progression between patients with abnormal and normal full-field ERG parameters.
FIGURE 4.

Bar graph showing full-field electroretinography (ERG) parameter versus scotoma progression rates in Stargardt disease. Patients with abnormal full-field ERG parameters tended toward exhibiting larger average scotoma progression rates, with abnormal photopic and scotopic B-wave amplitudes nearing statistical significance. Abnormal maximum stimulation A-wave and B-wave amplitudes were associated significantly with larger average scotoma progression rates.
FULL-FIELD ELECTRORETINOGRAPHY AND CLINICAL STAGE
When the various full-field ERG testing parameters were compared with the disease stage at presentation, patients with abnormal full-field ERG parameters were more likely to have more advanced stages of disease when compared with patients with normal full-field ERG values. The proportional differences were statistically significant for 7 particular full-field ERG parameters. These include photopic and scotopic B-wave amplitudes and latencies (P < .0001 for both photopic amplitude and latency; P = .045 and P = .011 for scotopic amplitude and latency, respectively), as well as maximum stimulation A-wave amplitude (P < .0001) and latency (P = .049) and maximum stimulation B-wave latency (P = .039). These data are shown in Figure 5.
FIGURE 5.

Bar graph showing the percent of patients with abnormal electroretinography (ERG) parameters by disease stage in Stargardt disease. P values correspond to the comparison of the distribution of disease stages with abnormal versus normal values of each full-field ERG parameter using an extended Fisher exact test. Abnormal full-field ERG parameters, in particular photopic and scotopic B-wave amplitudes and latencies, maximum stimulation A-wave amplitude and latency, and maximum stimulation B-wave latency, were statistically significantly associated with higher disease stages.
DIFFERENCES AMONG FULL-FIELD ELECTRORETINOGRAPHY PHENOTYPES
On average, patients with abnormal full-field ERG values tended to exhibit worse logMAR visual acuity values than patients with normal full-field ERG values. This difference was statistically significant for the photopic B-wave amplitude and latency, scotopic B-wave amplitude, maximal stimulation A-wave amplitude and latency, and maximal stimulation B-wave amplitude (P = .003, P = .014, P = .015, P = .001, P = .030, and P = .031, respectively; Table 2).
TABLE 2.
Comparison of Logarithm of the Minimal Angle of Resolution Visual Acuity in Patients with Stargardt Disease by Abnormal and Normal Full-Field Electroretinography Results
| Full-Field ERG Parameters in Patients with Stargardt Disease | Abnormal ERG Resultsa
|
Normal ERG Resultsa
|
P Valued | ||||
|---|---|---|---|---|---|---|---|
| No. | Mean LogMARb | SDc | No. | Mean LogMARb | SDc | ||
| Photopic ERG B amplitude | 57 | 0.88 | 0.64 | 86 | 0.58 | 0.46 | 0.003 |
| Photopic ERG B latency | 32 | 0.91 | 0.67 | 76 | 0.58 | 0.47 | 0.014 |
| Photopic ERG A amplitude | 49 | 0.79 | 0.61 | 86 | 0.62 | 0.50 | 0.090 |
| Photopic ERG A latency | 68 | 0.72 | 0.58 | 41 | 0.62 | 0.51 | 0.399 |
| Scotopic ERG B amplitude | 46 | 0.85 | 0.61 | 97 | 0.61 | 0.51 | 0.015 |
| Scotopic ERG B latency | 20 | 0.78 | 0.61 | 89 | 0.65 | 0.54 | 0.338 |
| Maximum stimulation A amplitude | 35 | 0.92 | 0.63 | 90 | 0.57 | 0.49 | 0.001 |
| Maximum stimulation A latency | 42 | 0.82 | 0.61 | 67 | 0.59 | 0.50 | 0.030 |
| Maximum stimulation B amplitude | 51 | 0.82 | 0.62 | 78 | 0.59 | 0.49 | 0.031 |
| Maximum stimulation B latency | 50 | 0.75 | 0.58 | 59 | 0.61 | 0.52 | 0.181 |
ERG = electroretinography; logMAR = logarithm of the minimal angle of resolution; SD = standard deviation.
Number of patients with abnormal or normal values for each full-field ERG parameter.
Mean logMAR visual acuity values (reported in logMAR units) for the normal versus abnormal groups for each full-field ERG parameter.
SD of logMAR visual acuity.
t test comparing mean logMAR visual acuity between patients with normal versus abnormal ERG results in each full-field ERG parameter.
Four full-field ERG phenotype groups (normal, cone–rod, rod–cone, and cone only) were contrasted for their logMAR visual acuity, central scotoma size, and fundus appearance at the time the first full-field ERG results were obtained. The various full-field ERG phenotypes exhibited significant differences in logMAR visual acuity and average central scotoma size (P = .003 and P < .0001, respectively, analysis of variance F test). A post hoc pairwise comparison using Tukey adjustment showed that the logMAR visual acuity was significantly worse in patients with cone–rod dysfunction (average, 1.05 logMAR units) compared with patients with normal full-field ERG results and patients with rod–cone dysfunction (average, 0.60 and 0.59 logMAR units, respectively; P < .05 in each case). Post hoc pairwise comparisons using Tukey adjustment showed that central scotoma size was significantly worse in patients with a cone–rod phenotype (average size, 17.7 cm2) compared with those with cone-only phenotypes (average size, 5.7 cm2) and normal full-field ERG phenotypes (average size, 2.9 cm2; all P < .05); patients with a rod–cone phenotype also showed significantly larger central scotoma size than those with normal full-field ERG phenotypes (12.6 vs 2.9 cm2, respectively; P < .05).
Funduscopic findings also were notably different between the different full-field ERG phenotypes. Wider macular atrophy (corresponding to higher stages of disease) was observed in 40.9% of cone–rod patients, in 30.0% of cone-only patients, in 31.3% of rod–cone patients, and in 12.7% of normal patients, whereas atrophy outside the vascular arcades was seen in 22.7% of code–rod patients, in 18.8% of rod–cone patients, in 10.0% of cone-only patients, and in 1.4% of normal patients. In particular, patients with a cone–rod full-field ERG phenotype were more likely to have wider macular atrophy and atrophy outside the vascular arcades (P = .023 and P = .002, respectively, extended Fisher exact test) than patients with rod–cone, cone-only, and normal full-field ERG phenotypes.
MUTATIONAL STATUS SUBGROUP ANALYSIS: 1 PROVEN MUTATION VERSUS 2 OR MORE PROVEN MUTATIONS
We evaluated differences between the 46 patients with 1 mutation and the 47 who had 2 or more mutations (36 patients with 2 mutations and 11 patients with 3 detected mutations). Average follow-up times for those who had 1 versus multiple mutations were comparable at 4.0 years each. Age of onset was significantly different between the groups. Patients with 2 or more mutations manifested symptoms at a younger age than those with 1 mutation (mean age, 20.6 vs 29.3 years, respectively; P = .009). The average rate of central scotoma size progression over time was somewhat greater in patients with multiple mutations (1.65 cm2/year) compared with those with 1 mutation (1.14 cm2/year), but this difference was not statistically significant (P = .480). On average, patients with 2 or more mutations had nonsignificantly larger central scotomata at their last visit than patients with 1 mutation (mean area, 12.1 and 8.4 cm2, respectively; P = .325). An evaluation of differences in full-field ERG parameters between the 2 mutation groups showed consistently fewer patients with aberrant photopic and scotopic B-wave amplitudes in those with 1 mutation, but the intergroup differences were not statistically significant (P = .191 and P = .353, respectively).
DISCUSSION
The variability in rates of clinical progression in patients with Stargardt disease poses multiple challenges for patients and ophthalmologists alike. Uncertainty regarding visual prognosis complicates patient counseling and makes therapeutic risk-benefit determinations more difficult. It also confuses the evaluation and eligibility criteria for clinical trials. Our study presents a meaningful prognostic relationship between full-field ERG testing and Stargardt disease progression rates and explores phenotypic differences in patients with 1 versus 2 proven mutations.
Although variability in progression between patients has been noted before, our study revealed that many patients do not exhibit progressive scotomata and that a smaller group progress rapidly. Although this is not a new finding, an interesting difference was found between these 2 groups. Patients who progressed rapidly (scotoma progression of more than 2 cm2/year by Goldmann testing) tended to have significantly worse scotopic B-wave amplitudes at presentation (P = .014). It is important to note that only a limited number of these patients had available data from at least 3 visits, which resulted in small subgroups; therefore, the true rate of progression may have been underestimated for this group of faster-progressing Stargardt disease patients. Further, although the rapid progressors span various ages at first presentation with scotoma, it may be possible that we happened to capture them at a time when their scotomata were maturing to their final size.
With respect to full-field ERG analysis, it is a commonly held view that normal full-field ERG results provide supportive evidence in making a clinical diagnosis of Stargardt disease.8,14,21 Our findings do not support this view. A large proportion of our cohort (47.3%) exhibited a full-field ERG abnormality that we categorized into distinct abnormal phenotypes. In fact, the mean scotoma progression rates tended to be higher for patients with abnormalities in 1 of the full-field ERG parameters when compared with Stargardt disease patients with normal full-field ERG results. Specifically, abnormal photopic and scotopic B-wave amplitudes had borderline significant associations with higher scotoma progression rates, whereas abnormal maximum stimulation A-wave and B-wave amplitudes were associated significantly with higher progression rates. Although the importance of this finding will be heightened if these abnormal full-field ERG parameters are found to be predictive of more rapid progression in a prospective study, these relationships may be useful to consider now, given the limited prognostic information available in the literature.
Our study also found a clinically relevant association between full-field ERG values at patient presentation and clinical stage with respect to funduscopic examination results. The data reveal that abnormal full-field ERG parameters are associated significantly with a more severe clinical stage, including photopic and scotopic B-wave amplitudes and latencies, maximum stimulation A-wave amplitude and latency, and maximum stimulation B-wave latency. Some previously published reports have not found any relationship between clinical stage and full-field Ganzfeld ERG values.18,19 However, our findings are consistent with the findings of Lois and associates, who reported a high frequency of patients with abnormal focal ERG findings, which led them to group patients based on the type of ERG abnormality found at the fovea.11 Finally, our results are consistent with those of other studies that have found that a substantial percentage of patients with Stargardt disease have abnormal full-field ERG values, which included patients exhibiting values reflecting isolated cone dysfunction usually suggestive of cone dystrophy.12,16,17 It is important to note that it is possible that the associations we have made between full-field ERG results at first examination and clinical phenotype, visual acuity, and progression rate, may just be a marker for more severe disease, rather than a marker for long-term prognosis. The clinical distinction between severity and prognosis is important to consider, especially given the retrospective nature of this study and the lack of long-term follow-up in some patients. This distinction can be explored properly only in a prospective study with longitudinal data for full-field ERG parameters, quantified scotomata, and visual acuity. Furthermore, given that multiple full-field ERG parameters were compared individually with different outcome measures, the possibility that significant associations are the result of chance alone may be increased.
Many of our patients exhibited only 1 mutation on ABCA4 mutation screening. Our comparisons of multiple parameters in patients with 1 mutation versus those with multiple mutations showed consistently more severe values in the latter group. This was significant for a younger age at onset in those with multiple mutations. Other insignificant findings, including the percentage with abnormal photopic and scotopic B-wave amplitudes on initial examination, the average rate of scotoma progression, and the central scotoma size, showed consistently less severe values in the 1-mutation group. Although variability in responses and limited power resulting from sample size may have contributed to the lack of statistical significance of these findings, a carefully designed, well-powered, prospective study re-examining the severity of phenotype based on the number of identified ABCA4 mutations may be fruitful.
Multiple other Stargardt disease genotype analyses also have reported finding only 1 mutation, or have failed to detect any mutations despite patients’ exhibiting Stargardt disease phenotypes.21,22,24,28 The reasons for this phenomenon are unclear, and could be attributed to an abundance of accumulated polymorphisms distorting protein structure in the opposite allele, deletions that may be undetectable by current methodologies, or mutations in introns.29 It is also possible these patients have mutations in genes that have not yet been associated with Stargardt disease. No patients found to have single ABCA4 mutations had any sign of dominant inheritance, suggesting that the other allele had a mutation undetectable by current methods. Given the retrospective nature of this study, it was not possible to perform segregation analysis for many of our families. It is possible that the phenotypic difference is caused by a hypomorphic allele (an allele exhibiting reduced levels of gene activity) that may alter the disease progression. As with any inherited disease, alternative genetic modifiers or environmental influences are putative causal factors that explain the observed differences. More mechanistic information likely can be found by exploring the cause and course of disease in this interesting subset of patients.
With gene-based therapies on the horizon for ophthalmic diseases, a better understanding of age of onset and scotoma progression rates may prove helpful in identifying mutations as more or less severe. These data are valuable for patient counseling and discussing prognosis, as well as for selecting and matching patients with controls for future gene therapy and pharmaceutical clinical trials. These include submacular injections of ABCA4 (StarGen; ClinicalTrials.gov identifier: NCT01367444) being performed as part of a phase I/IIa study, and pharmaceutical therapies such as Fenretinide, which is due to enter the clinical trial phase for Stargardt disease in the near future.30 For all these therapies, it will be important to identify the patients who would benefit most from the risks associated with intervention. Based on the revelations from our study, this group of patients may include those who progress most rapidly and who exhibit specific abnormal full-field ERG parameters.
There were some limiting factors in the study. Our cohort contained patients who had limited follow-up as well as many who had pending mutational analysis, which resulted in reduced numbers in our subgroup analyses. The full-field ERG results we assessed were obtained at different times in the patients’ clinical courses. Furthermore, although data analyzed were from 2 different ERG laboratories, they both used identical methods, and we adjusted the results as a percentage of the mean normal value for each laboratory for the analysis. Visual acuity was not measured using the Early Treatment Diabetic Retinopathy Study charts, and Goldmann visual fields were not performed by the same perimetrist, although standard isopters and testing methods were used in all patients. Many of the outcomes we measured, such as visual acuity, Goldmann visual fields, and full-field ERG amplitudes, are known to exhibit test–retest variability. These problems have the possibility of creating sufficient variability to limit the ability to uncover significant differences in this study. Limitations such as these are common in retrospective studies. However, the presence of certain abnormal full-field ERG parameters may serve as a useful tool to identify patients at risk of faster scotoma progression rates and may be an important stratification variable to consider in randomizing patients to treatment and control groups when testing novel therapies. A well-designed, standardized, prospective study will be invaluable in validating and expanding on the prognostic findings of this study. Such a study would be able to define more accurately disease severity and useful prognostic markers in Stargardt disease.
Supplementary Material
Acknowledgments
Publication of this article was supported in part by Core Center for Vision Research Grant EY007003 from the National Eye Institute, Bethesda, Maryland; Foundation Fighting Blindness; and Research to Prevent Blindness, Inc, New York, New York.
The authors thank Dr Edwin M. Stone and Dr Frans P. M. Cremers for their assistance in the mutational analysis Dr Steven Nusinowitz for his assistance with electroretinography testing at the University of California, Los Angeles.
Biographies

Sarwar Zahid is a fourth year medical student at the University of Michigan Medical School, Ann Arbor, Michigan. Given his immense interest in ophthalmology, he pursued a one-year research fellowship with the retinal dystrophy team at the Kellogg Eye Center. His long-term career goal is to further build on his research training in order to become an excellent clinical and academic ophthalmologist.

Thiran Jayasundera has a clinical practice caring for patients with inherited retinal degenerative diseases at the University of Michigan, Ann Arbor, Michigan, Ann Arbor, Michigan. His research program focuses on the genotype-structural-functional relationships of these diseases in order to better design clinical trials of therapies with the potential for preventing deterioration of visual function. Dr Jayasundera obtained dual fellowship training in retinal dystrophies/ electrophysiology (Michigan) and medical/surgical retina (McGill University) before joining as faculty at the University of Michigan in 2011.
Footnotes
Supplemental Material available at AJO.com.
ALL AUTHORS HAVE COMPLETED AND SUBMITTED THE ICMJE FORM FOR DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST and the following was reported. Dr Musch is a consultant for ReVision Therapeutics, Inc., for studies of fenretinide treatment for Stargardt disease. Dr Musch is a recipient of the Research to Prevent Blindness Lew R. Wasserman Merit Award. Involve din Design and conduct of study (K.T.J., K.B., J.R.H.); Collection, management, analysis, and interpretation of data (S.Z., K.T.J., W.R., N.K., L.M.N., D.C.M., J.R.H.); and Preparation, review, or approval of manuscript (S.Z., K.T.J., W.R., K.B., N.K., L.M.N., D.C.M., J.R.H.).
References
- 1.Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 2.Ayuso C, Garcia-Sandoval B, Najera C, Valverde D, Carballo M, Antinolo G. Retinitis pigmentosa in Spain. The Spanish Multicentric and Multidisciplinary Group for Research into Retinitis Pigmentosa. Clin Genet. 1995;48(3):120–122. [PubMed] [Google Scholar]
- 3.Blacharski P. Fundus flavimaculatus. In: Newsome DA, editor. Retinal Dystrophies and Degenerations. New York: Raven Press; 1988. pp. 135–159. [Google Scholar]
- 4.Martinez-Mir A, Bayes M, Vilageliu L, et al. A new locus for autosomal recessive retinitis pigmentosa (RP19) maps to 1p13–1p21. Genomics. 1997;40(1):142–146. doi: 10.1006/geno.1996.4528. [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Mir A, Paloma E, Allikmets R, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18(1):11–12. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 6.Rozet JM, Gerber S, Ghazi I, et al. Mutations of the retinal specific ATP binding transporter gene (ABCR) in a single family segregating both autosomal recessive retinitis pigmentosa RP19 and Stargardt disease: evidence of clinical heterogeneity at this locus. J Med Genet. 1999;36(6):447–451. [PMC free article] [PubMed] [Google Scholar]
- 7.Stargardt K. Über familiäre, progressive Degeneration in der Maculagegend des Auges. Graefes Arch Ophthalmol. 1909;(71):534–550. [Google Scholar]
- 8.Westerfeld C, Mukai S. Stargardt’s disease and the ABCR gene. Semin Ophthalmol. 2008;23(1):59–65. doi: 10.1080/08820530701745249. [DOI] [PubMed] [Google Scholar]
- 9.Noble KG, Carr RE. Stargardt’s disease and fundus flavimaculatus. Arch Ophthalmol. 1979;97(7):1281–1285. doi: 10.1001/archopht.1979.01020020023005. [DOI] [PubMed] [Google Scholar]
- 10.Glazer LC, Dryja TP. Understanding the etiology of Stargardt’s disease. Ophthalmol ClinNorth Am. 2002;15(1):93–100. viii. doi: 10.1016/s0896-1549(01)00011-6. [DOI] [PubMed] [Google Scholar]
- 11.Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001;119(3):359–369. doi: 10.1001/archopht.119.3.359. [DOI] [PubMed] [Google Scholar]
- 12.Aaberg TM. Stargardt’s disease and fundus flavimaculatus: evaluation of morphologic progression and intrafamilial co-existence. Trans Am Ophthalmol Soc. 1986;84:453–487. [PMC free article] [PubMed] [Google Scholar]
- 13.Armstrong JD, Meyer D, Xu S, Elfervig JL. Long-term follow-up of Stargardt’s disease and fundus flavimaculatus. Ophthalmology. 1998;105(3):448–457. doi: 10.1016/S0161-6420(98)93026-3. discussion 457–458. [DOI] [PubMed] [Google Scholar]
- 14.Fishman GA, Stone EM, Grover S, Derlacki DJ, Haines HL, Hockey RR. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol. 1999;117(4):504–510. doi: 10.1001/archopht.117.4.504. [DOI] [PubMed] [Google Scholar]
- 15.Hadden OB, Gass JD. Fundus flavimaculatus and Stargardt’s disease. Am J Ophthalmol. 1976;82(4):527–539. doi: 10.1016/0002-9394(76)90539-0. [DOI] [PubMed] [Google Scholar]
- 16.Lachapelle P, Little JM, Roy MS. The electroretinogram in Stargardt’s disease and fundus flavimaculatus. Doc Ophthalmol. 1989;73(4):395–404. doi: 10.1007/BF00154495. [DOI] [PubMed] [Google Scholar]
- 17.Moloney JB, Mooney DJ, O’Connor MA. Retinal function in Stargardt’s disease and fundus flavimaculatus. Am J Ophthalmol. 1983;96(1):57–65. doi: 10.1016/0002-9394(83)90455-5. [DOI] [PubMed] [Google Scholar]
- 18.Oh KT, Weleber RG, Oh DM, Billingslea AM, Rosenow J, Stone EM. Clinical phenotype as a prognostic factor in Stargardt disease. Retina. 2004;24(2):254–262. doi: 10.1097/00006982-200404000-00011. [DOI] [PubMed] [Google Scholar]
- 19.Oh KT, Weleber RG, Stone EM, Oh DM, Rosenow J, Billingslea AM. Electroretinographic findings in patients with Stargardt disease and fundus flavimaculatus. Retina. 2004;24(6):920–928. doi: 10.1097/00006982-200412000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Simonelli F, Testa F, Zernant J, et al. Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic Res. 2005;37(3):159–167. doi: 10.1159/000086073. [DOI] [PubMed] [Google Scholar]
- 21.Genead MA, Fishman GA, Stone EM, Allikmets R. The natural history of Stargardt disease with specific sequence mutation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2009;50(12):5867–5871. doi: 10.1167/iovs.09-3611. [DOI] [PubMed] [Google Scholar]
- 22.Jaakson K, Zernant J, Kulm M, et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22(5):395–403. doi: 10.1002/humu.10263. [DOI] [PubMed] [Google Scholar]
- 23.Lewis RA, Shroyer NF, Singh N, et al. Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet. 1999;64(2):422–434. doi: 10.1086/302251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maia-Lopes S, Aguirre-Lamban J, Castelo-Branco M, Riveiro-Alvarez R, Ayuso C, Silva ED. ABCA4 mutations in Portuguese Stargardt patients: identification of new mutations and their phenotypic analysis. Mol Vis. 2009;15:584–591. [PMC free article] [PubMed] [Google Scholar]
- 25.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M. ISCEV standard for full-field clinical electroretinography (2008 update) Doc Ophthalmol. 2009;118(1):69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 26.Schulze-Bonsel K, Feltgen N, Burau H, Hansen L, Bach M. Visual acuities ‘‘hand motion’’ and ‘‘counting fingers’’ can be quantified with the Freiburg visual acuity test. Invest Ophthalmol Vis Sci. 2006;47(3):1236–1240. doi: 10.1167/iovs.05-0981. [DOI] [PubMed] [Google Scholar]
- 27.Fishman GA. Fundus flavimaculatus. A clinical classification. Arch Ophthalmol. 1976;94(12):2061–2067. doi: 10.1001/archopht.1976.03910040721003. [DOI] [PubMed] [Google Scholar]
- 28.Ernest PJ, Boon CJ, Klevering BJ, Hoefsloot LH, Hoyng CB. Outcome of ABCA4 microarray screening in routine clinical practice. Mol Vis. 2009;15:2841–2847. [PMC free article] [PubMed] [Google Scholar]
- 29.Briggs CE, Rucinski D, Rosenfeld PJ, Hirose T, Berson EL, Dryja TP. Mutations in ABCR (ABCA4) in patients with Stargardt macular degeneration or cone-rod degeneration. Invest Ophthalmol Vis Sci. 2001;42(10):2229–2236. [PubMed] [Google Scholar]
- 30.Radu RA, Yuan Q, Hu J, et al. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following vitamin A supplementation. Invest Ophthalmol Vis Sci. 2008;49(9):3821–3829. doi: 10.1167/iovs.07-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.