

Abstract
The liver performs many key functions, the most prominent of which is serving as the metabolic hub of the body. For this reason, the liver is the focal point of many investigations aimed at understanding an organism’s toxicological response to endogenous and exogenous challenges. Because so many drug failures have involved direct liver toxicity or other organ toxicity from liver generated metabolites, the pharmaceutical industry has constantly sought superior, predictive in-vitro models that can more quickly and efficiently identify problematic drug candidates before they incur major development costs, and certainly before they are released to the public. In this broad review, we present a survey and critical comparison of in-vitro liver technologies along a broad spectrum, but focus on the current renewed push to develop “organs-on-a-chip”. One prominent set of conclusions from this review is that while a large body of recent work has steered the field towards an ever more comprehensive understanding of what is needed, the field remains in great need of several key advances, including establishment of standard characterization methods, enhanced technologies that mimic the in-vivo cellular environment, and better computational approaches to bridge the gap between the in-vitro and in-vivo results.
1. INTRODUCTION
The difficulties and increasing costs of drug development and testing faced by the pharmaceutical industry raise questions about the effectiveness and efficiency of current drug screening approaches. The cost of bringing a single compound to market is now estimated at almost a billion US dollars1–4. This high cost stems from the large number of failed drugs during both preclinical and clinical studies, where the two major factors for failure are a lack of efficacy and toxicity5. According to Adams and Brantner3 and a study conducted by the Boston Consulting group in 20016, a major portion of the drug development costs, 40–70% of the total development cost, is invested during the preclinical stages. This necessitates a closer examination of the preclinical screening studies in particular, where the efficacy and safety of new chemical entities in the pipeline are tested.
Animal testing is the most popular form of assessment used during the preclinical, and in some cases clinical, context. However, the success of animal studies in predicting the human physiological response in terms of both efficacy and toxicity is sometimes poor, and this practice has been increasingly questioned5,7,8. Moreover, animal models are also hampered by their poor ability to isolate cell-based mechanisms of action and pathways9. As a consequence, many drugs that are doomed to fail unnecessarily go through clinical trials, substantially increasing the overall cost of the drugs that make it through the certification processes. There is also a strong push to move away from animal models due to ethical concerns following the 3R approach, i.e. “Reduction, Refinement and Replacement” of animal studies10,11. One of the important aims of “Replacement” is to create alternative technologies and particularly in-vitro platforms that are less expensive, more predictive, and more time efficient than animal models. One example of this push was the 7th Amendment of the European Union, which banned all animal testing in safety evaluation of cosmetic products and commercial chemicals in 201312. Although the amendment did not include pharmaceuticals, it may be a step in that direction.
Among all organs, the liver plays the most central role in human-drug interactions and is also the most common target for drug-induced toxicity5,13. Liver toxicity results in costly, late stage drug failures as 25–40% of drugs are found to cause hepatic injuries by phase III clinical studies5,14. Moreover, despite our best efforts to ensure drug safety, a sizeable number of drugs are withdrawn from the market after approval. The primary reason for after-market release is hepatotoxicity15, which accounts for ~20–30% of all withdrawals in the US and EU over the last 30 years14,16. The FDA highlights the importance of liver toxicity and its severe risks during drug development with the following statement: “The presence of even a single case of liver injury from treatment in the premarketing clinical trials database is a signal of a high level of hepatotoxic risk”17.
Given the overwhelming importance of the liver in drug metabolism and toxicity, there have been a wide range of academic and commercial studies aimed at developing in-vitro models to predict liver toxicity associated with therapeutic drugs. These studies primarily examine the enzymatic and synthetic activities of drug uptake and metabolism, as well as drug-drug interactions that affect metabolism. The selection of in-vitro platforms ranges from microsomal18,19 and electrochemical assays20,21, suspension22–26 and plate cultures27–31 of primary cells and cell lines, and macroscopic flow culture systems32–39 to liver slices40–43 and whole perfused organs44. While liver slices and whole perfused organs provide the most physiologically realistic systems with intact tissue structure and cell proportions, their characterization and long-term maintenance have proven to be very difficult10. New technologies such as decellularized and repopulated liver slices45 and organs46 can alleviate some of these problems, but still lack the throughput and analytic flexibility for drug screening purposes. In this respect, a newer class of in-vitro tools that can potentially provide fine microscopic control of the cellular environment and dynamics, via microfabrication and tissue engineering methods has recently gained more attention. These “on-a-chip” tissue models may be able to mimic the architecture of small tissue sections and certain characteristics of the dynamic in-vivo flow environment, while also offering more precise spatial and temporal control of soluble factors. Moreover, unlike full organ or thick tissue sections, these models can be engineered to be imaging- and analysis-friendly for real-time/near real-time monitoring of the state of cells and their extracellular environment, which is crucial for determining cellular mechanisms of action. Another expected, but not yet fully realized, advantage is the large multiplexing capacity of these systems to make them amenable to high throughput screening approaches.
While there are a variety of other advances that may positively impact the drug development process (such as toxicogenomics, metabolomics, etc.), these microengineered in-vitro systems — “the organs-on-chips” (or more correctly defined, “tissues-on-a-chip”), with their improved physiological relevance may serve as an important platform for providing the data for such analytical approaches. While some researchers have been individually championing this idea for over a decade, with the publication of the US National Research Council (NRC)’s 2007 vision report titled “Toxicity Testing in the 21st Century”47,48, the push towards replacing traditional in-vitro models with these tissues on chips has been accelerated both in the US and EU. Notable EU initiatives along these lines are the SEURAT-149 and the Body-on-a-Chip (BoC)50. With a similar conviction and as set forth in the NRC report, in 2012, the NIH and the FDA, along with DARPA, also invested over one hundred million US dollars to create the “Tissue Chips for Drug Screening”51,52 program in response to these challenges and in line with the goal of reducing and/or replacing animal studies. This joint effort seeks to create robust, long-term 3D microengineered tissue systems that closely mimic the human physiological and pharmacological response. Cellular systems are required to be viable for at least 28 days to allow testing for chronic as well as acute drug toxicity. Eventually, the goal is to connect these organ systems in a physiologically relevant manner to create a “human-on-a-chip”. For example, our lab, in collaboration with investigators at the University of Pittsburgh, is creating a physiologically relevant “liver-on-a-chip” platform for enhanced prediction of the human liver response to exogenous and endogenous challenges. The scope beyond the liver chip involves building connections to other tissue systems (e.g. kidney, heart, gut, etc.) in a deliberate step-by-step fashion.
In this review, we highlight decades of work and ideas that inspired and guided the development of new microengineered tissue systems that focus on the liver. We start the review, in section 2, with a brief recap of the liver structure, importance, and toxicological intricacies. This is followed, in section 3, by a historical perspective on the static and perfused culture systems that paved the road for the current microscopic models that we review in the same section. We then discuss the gaps and opportunities in the field going forward in section 4.
Following this we give a brief review of the ongoing work in our lab towards building a more predictive liver model in section 4.5. We posit here that in order to have better predictive capabilities and better representation of the liver physiology, we should start with the notion of an underlying unit process/unit structure of the liver as opposed to arbitrarily matching one or several scaling parameters as others have done. Because the liver is a complex organ with an enormous array of functions, it is equally important to identify what functions we need to replicate to achieve our desired goals. This leads to a discussion of the cell types that are needed for the faithful recapitulation of those functions with the conviction that non-parenchymal cells constitute an important part of the liver response both in healthy and diseased states of the organ. We conclude with a summary and future directions, the challenges and opportunities that still need to be addressed, and the need for developing standardized criteria for testing the plethora of new in-vitro models in section 5.
2. LIVER
The liver, a complex organ with a multitude of functions, is the largest organ in the body53,54. It is located between the digestive tract and the upper body10 and plays a central role in homeostasis. It is a living factory responsible for synthesis, metabolism, storage, filtration, and removal of vital compounds along with some immune functions. Through the synthesis and secretion of bile, the liver enables digestion and absorption of fatty compounds55, and many waste products, such as bilirubin, are removed by secretion into bile. The liver, provides the body with nearly all the main plasma proteins including albumin, transferrin, prothrombin, fibrinogen, lipoproteins, and complement proteins54,56. One critical function of the liver is the maintenance of blood glucose levels via synthesis (glucogenesis) and storage of glucose (as glycogen). Of all the important liver functions, its metabolism — especially that of pharmaceutical drugs and other xenobiotics — is perhaps the most important. The liver is able to metabolically detoxify xenobiotics and modify them into either waste or non-toxic metabolites for further use. Accordingly, recapitulation of the liver drug metabolism in-vitro will be the central focus of this review.
2.1 Structure of the liver
The liver is comprised of a) a stroma that consists of connective tissue (septa), reticular fiber, and portal canals and b) the parenchyma that consists of the cells, the bile canaliculi, and sinusoidal and perisinusoidal spaces. The traditional view is that there are four major, but unequal, lobes identified by external demarcations57. Nevertheless, this lobular separation is incomplete56,58 and there are conflicting views on how best the liver can be segmented for surgical and radiological purposes57. We can, however, understand the structure and examine the smallest functional unit of the liver while attempting to create physiologically relevant in-vitro liver systems.
The liver has a highly vascularized structure that is perfused by a dual blood supply58–60: a) arterial blood via the hepatic artery and b) venous blood via the hepatic portal vein (Fig. 1a). While the arterial blood is supplied directly from the heart at systemic oxygen, hydrostatic pressure, and solute concentrations, the venous blood, which can be as much as 80% of the total supply57, has already circulated through the gastrointestinal tract and has been enriched with certain hormones, nutrients and toxins, but depleted of oxygen and pressure58. The hepatic artery and portal vein travel together and continuously branch into smaller veins in the portal tract to deliver and perfuse all of the cells in the liver. This perfused blood is eventually collected into the central vein, which is then returned to the systemic circulation. The excretion of bile by hepatocytes into bile canaliculi, which form a network independent of the blood circulation, forms a counter circulation where the bile eventually flows into the bile duct. Anatomically, each branch of the bile duct, the portal vein, and the hepatic artery forms a structure called the portal triad (Fig. 1a).
Figure 1.

Structure of the liver (a) A 2D cutout of the liver structure, showing the hexagonal organization of the theoretical classical lobules. At the center of each hexagon is a central vein that collects the all the blood from the sinusoids while the corners of the hexagon represent the main portal triad — comprised of the hepatic artery, portal vein and the bile duct. The acinus, the smallest functional unit of the liver, is usually described as the parallelogram whose corners consist of two neighboring central veins and the portal triads in between. (b) 3D illustration of half of the acinus structure. (c) Illustration of the liver sinusoid with the four prominent cells we discuss in this review.
One striking feature of the liver is the simplicity of its seemingly complex structure, wherein the basic vasculature and the circulatory network are repeated many times akin to fractal structures61–63. A unitary functional network structure has been deemed the hepatic acinus, which is shown in Fig. 1a,b by the parallelogram whose corners consist of two neighboring central veins and the portal triads in between. A larger unit in terms of blood circulation is the classical hepatic lobule (Fig. 1a). While the classical lobule has no morphological boundaries56,58, the center of each lobule is marked by the central vein where blood perfused through the liver mass is collected. This mass is hypothesized to be surrounded and fed by a highly vascularized diamond-like (with a hexagon like cross-section, see Fig. 1a) network of the portal tract system. A remarkable consequence of this organization (the repeating pattern of flow from the portal triads to the central veins), is the zonal gradation of oxygen, nutrients, and toxins and the resulting zones with different cellular specializations along the portal triads to the central veins (see Fig. 1a, section 2.3).
2.2 Cells of the liver
In each acinus structure, hepatocytes, the parenchymal cells of the liver, are organized into plates/laminae (Fig. 1b) forming a continuous 3D lattice56,58. While the apical surfaces on this hepatic matrix coordinate to form the bile canalicular system that exports bile into the bile ducts, the basal surfaces project microvilli that face the space of Disse64 (Fig. 1c). The space of Disse, a semi-fluidic space (thickness ~ 0.2–1 μm65) with a complex composition of extracellular matrix (ECM) proteins, is occupied by stellate cells66,67. Stellate cells are in close contact with both the hepatocytes and the liver sinusoidal endothelial cells (LSECs) that line the walls of the hepatic sinusoid (Fig. 1c). Hepatic sinusoids are the venous capillaries (diameter ~5–10 μm, length ~250–300 μm)68,69 between the plates of hepatocytes that are the principle site of exchange between the blood and the perisinusoidal space of Disse59.
The primary resident cells of the liver, i.e. hepatocytes, LSECs, and Kupffer and stellate cells, form a complex signaling and metabolic environment70–72 wherein these cells normally function in unison to fend off internal and external challenges, as well as to supply the rest of the body with useful metabolites and proteins. The cells perform liver functions directly and through autocrine and paracrine signaling. Below, we review each cell type and its contributions to liver function along with its importance in the context of toxicity; we summarize the important physical properties of the main liver cells in Table 1. While the liver houses other important cell types such as cholangiocytes, immune cells, and dendritic cells, we will not be reviewing them here, but instead refer readers to several excellent books57,58,65,73 for more information.
Table 1.
Approximate physical properties of important human liver cells (adapted from Ref. 69).
| Cell | Type | Diameter (μm) | Volume (% of total) | Number (% of total) |
|---|---|---|---|---|
| Parenchymal | ||||
| Hepatocytes | Epithelial | 20–30 | ~78 | ~60–65 |
| Non-parenchymal | ||||
| LSECs | Endothelial | 6.5–11 | 2.8 | 16 |
| Kupffer Cells | Macrophages | 10–13 | 2.1 | ~15 |
| Stellate Cells | Fibroblastic | 10.7–11.5 | 1.4 | 8 |
2.2.1 Parenchymal cells
Hepatocytes
Hepatocytes are highly differentiated epithelial cells31,74 that are responsible for a major portion of the complex liver functions31. They constitute the majority (~78% by mass/volume and 60–65% by number58,69,75,76 of cells in the liver. They are large (20–30 μm58,69), rhomboidal with many facets68, and highly polarized for specialized functions such as bile excretion and extraction from and secretion into the blood stream58. With a highly concentrated cytoplasm that possesses a large number of mitochondria (1000–2000/cell), peroxisomes (400–700/cell), lysosomes (~250/cell), Golgi complexes (~50/cell), aggregates of rough and smooth endoplasmic reticulum (~15% of cell volume)58,77,78, hepatocytes are seemingly the most metabolically active cells in the entire body. Accordingly, they have very high oxygen consumption rates (~2.5–5 × 10−5 nmol/cell/min79) when compared to most other cell types.
Together with drug metabolism, the synthesis and secretion of proteins — especially albumin — and the excretion of urea are crucial functions, as well as distinct markers of hepatocyte function and health. Although non-parenchymal cells support and contribute to xenobiotic metabolism80–82, the phase I and II metabolism of exogenous and endogenous compounds are carried out predominantly by hepatocytes70. The effect of phase I reactions, predominantly via the cytochrome P450 (CYP 450) enzyme family located on the smooth endoplasmic reticulum57 is the modification of compounds through oxidation, reduction, and hydroxylation for either excretion or further modification in phase II, or other types of, metabolism54. The subsequent phase II modifications are mostly carried out by cytosolic enzymes termed “transferases”, which conjugate the phase I intermediates with charged species such as glycine, sulphate, glucoronate, or glutathione57. The overall effect of these two phases is the increased water solubility and acidity and decreased toxicity of most parent compounds54. This high metabolic activity of hepatocytes also makes them the prime target for the toxic effects of the compounds that they metabolize. A common route for injury is the toxicity of intermediate metabolites produced by phase I metabolism. We briefly review drug induced liver injuries in section 2.4.
2.2.2 Non-parenchymal cells
Liver sinusoidal endothelial cells (LSECs)
LSECs are sessile cells83, 6–11 μm in cross-section69. They make up 50% of all sinusoidal cells59–76 and constitute 15–20% of the liver by number84 and ~3% by volume69. LSECs can be identified by the expression of SE-185 and CD-3174 surface markers, as well as by their uptake of formaldehyde-treated serum albumin86,87. They form a tubular structure that lines the liver sinusoidal wall with the primary function of transvascular exchange, i.e. filtration, between the blood stream and the tissue59. This exchange is enabled via fenestrations that are clustered into sieve plates67,88,89 with 10–50 fenestrae in each plate84. These fenestrae, specific to the liver endothelium90, are 100–170 nm in diameter, but are dynamic structures whose size responds to the luminal blood pressure, vasoactive substances, drugs, and toxins88,91,92. LSECs show a high capacity of receptor-mediated endocytosis for different molecules, which forms the basis of their scavenger functions89,93,94. Accordingly, together with the Kupffer cells, they form the reticuloendothelial system72,82, which is responsible for scavenging circulating macromolecules and microorganisms from the systemic circulation95. LSECs are also assumed to take part in the regulation of blood flow84 along with the stellate cells through complex signaling between the two cell types96,97.
LSECs are an early target for several toxicants59 because they are directly exposed to blood flow from the portal circulation. In the specific case of acetaminophen/paracetamol, they become swollen and lose their ability to endocytose as early as 30 minutes after administration59,98. They are implicated in many different injury mechanisms, including neutrophil-induced liver injury99 and hepatic fibrosis100. Importantly, they also contribute to both phase I and phase II metabolism, although their contribution on a per cell basis is one to two orders of magnitude lower than that of hepatocytes80,81. A number of in-vitro experiments have now demonstrated that the presence of LSECs has a significant positive effect on the retention of hepatocyte phenotype and metabolic activities85,101,102 and also leads to more representative models of the drug response of hepatic tissues. For further information about LSECs, we refer the readers to the following references72,88,103–105.
Kupffer cells
Kupffer cells, named after Karl W. von Kupffer106,107, are the resident macrophages in the liver, accounting for more than 80% of all macrophages in the body71 and ~15% of the liver cells84. They are 10–13 μm in diameter105, irregularly shaped56, and mobile cells83,107 that adhere to the luminal surface of the sinusoidal wall. They can be identified by their expression of ED-1 and ED-2 surface antigens57,108,109. Phagocytosis and endocytosis of toxicants, particulates, and endotoxins such as lipo-polysaccharides (LPS) are important functions of the Kupffer cells. An equally important function is their secretion of mediators, such as cytokines, prostanoids, oxygen radicals, and proteases71,84, which provide local and long distance cellular signaling cues. While some of these secretions are beneficial for liver regeneration110,111 and host defense71, others may be involved in liver injury56,71. The activation and involvement of Kupffer cells have been implicated in a) diseases including neoplasia70, non-alcoholic fatty liver disease71,112,113, and immunological diseases; b) damage such as ischemia-reperfusion114 and cold-preservation injury84,115; and c) liver toxicity such as acetaminophen99, copper, and iron toxicity116, as well as immune-mediated adverse drug reactions70.
Thus, Kupffer cells play an important role in the acute and chronic responses of the liver to toxic compounds70. In the context of xenobiotic metabolism, three different pathways of interaction between Kupffer cells and hepatocytes have been proposed117: 1) xenobiotics induce Kupffer cell stimulation in a similar manner to LPS stimulation, which in turn limits several hepatic functions; 2) the intermediates resulting from hepatocyte xenobiotic metabolism induce Kupffer cell activation; and 3) xenobiotics induce primary lesions in hepatocytes that are converted into cytotoxic lesions by contact with factors induced by constitutive levels of LPS. Supporting these hypotheses, Milosevic and co-workers117 showed that Kupffer cells co-cultured with hepatocytes show distinctly different nitric oxide (NO) and TNF-α release after stimulation with LPS. Recent unpublished work (Life Technologies118) suggests that the phase I metabolism of hepatocytes can be greatly reduced with addition of pro-inflammatory factors LPS or IL-2, but only when they are co-cultured with Kupffer cells. These studies, along with many others70, point to the prominent interactions of Kupffer cells with hepatocytes and other cells in response to drugs and other challenges. For further information on Kupffer cells and their involvement in hepatic metabolism, we refer the readers to excellent reviews by McCuskey59, Roberts70, Bilzer71, and Laskin72.
Hepatic stellate (Ito) cells
Hepatic stellate cells (HSCs), also called vitamin A-storing cells, lipocytes, interstitial cells, fat-storing cells or Ito cells66, are distinct, star-shaped106,119,120 cells specific to the liver. They make up ~1.4% of the liver volume, and there is about 1 stellate cell for every 8 hepatocytes69. The stellate cells can be identified by CRBP-1121, glial fibrillary acidic protein (GFAP) staining122, or the original gold-chloride method106,120, among others. They are the primary storage site for vitamin A57,97,122 and fat droplets56, and actively participate in controlling the microvascular tone57,70, and thus the blood flow96,97.
A very active interaction established through both cell-cell contact and intracellular signaling between HSCs and LSECs exists and contributes to liver homeostasis66. For example, the HSCs secrete VEGF, which maintains LSEC phenotype123, and the LSECs produce and release NO in return123. While NO keeps the HSCs quiescent123 in a healthy liver, a lack of NO in a diseased state may lead to the activation of HSCs, resulting in excessive ECM secretion and subsequent fibrosis123. Similar to Kupffer cells, HSCs play an important role in modulating drug induced liver injury and hepatocarcinogenesis via the release of growth factors, inflammatory cytokines, and reactive oxygen species upon activation by exogenous insults70,124. For further and more detailed information on the involvement of stellate cells in liver metabolism and injury, we refer readers to excellent reviews by Winau97, Senoo et al.66, and Friedman125.
2.3 Zonation
The liver displays a functionally important gradient of nutrients, hormones, and especially oxygen concentration from the portal triad, the periportal zone (PP), to the central vein, the perivenous zone (PV). The oxygen tension in the PP zone is 60–65 mmHg and in the PV zone is 30–35 mm Hg126–128. These gradients, along with differences in ECM composition, structure, and soluble factors from the PP to PV zones74 manifest as differences in cellular metabolism, including that of xenobiotics and secreted molecules, as well as changes in the morphology and number and phenotypic characteristics of all cell types along the PP-PV axis128–130 (see Fig. 2). This specialization of liver cells is referred to as “zonation”. The acinus structure is generally divided into three different zones from PP (Zone 1) to PV (Zone 3)131–133, which display heterogeneous cell functions.
Figure 2.

Summary of zonation in a liver acinus. Gradients in oxygen, nutrients, hormonal and other factors lead to morphological as well as functional changes from the portal triad to the central vein.
While we discuss the zonation of hepatocytes below, one should note that non-parenchymal cells also display important zonal differences. Specifically, the number of LSECs increases from Zone 1 to Zone 3, while the number of Kupffer cells decreases along the same axis134. Functionally, LSECs display a lower density of fenestrae with larger diameters (>200 nm) in Zone 1 and higher density with smaller pore diameters (<150 nm) in Zone 3135. Kupffer cells, on the other hand, show higher phagocytic activity in Zone 1 compared to Zone 3.
2.3.1 Hepatocyte zonation
Hepatocytes display a distinct heterogeneity of morphology, enzymatic activities, and functional capacity along the three zones of the liver acinus. In contrast to Zone 1 hepatocytes, which are small, Zone 3 hepatocytes are larger, terminally differentiated, and display a much higher number of polyploidy136. While Zone 1 hepatocytes are efficient at oxidative metabolism, fatty acid oxidation, gluconeogenesis, bile acid extraction, ammonia detoxification, and urea and glutathione conjugation, Zone 3 hepatocytes are efficient at glycolysis, liponeogenesis, and CYP 450 biotransformation133,137–139. Zone 2 hepatocytes exhibit intermediate function. Zonal distributions have been observed in both gene expression and activities of important enzymes for a variety of hepatic functions.
Within each lobule, the xenobiotic metabolism is not evenly distributed, as the majority of drug metabolism occurs in PV hepatocytes140. This is especially true for phase I metabolic activity, but elevated levels for some phase II detoxification activities can also be observed in the PP zone. One explanation for this distribution is that by producing most of the phase I metabolic intermediates, which can be highly toxic/reactive and could lead to cell death, in the PV zone141, the liver has evolved to protect itself from organ-wide necrosis142. Supporting this hypothesis, the detoxification of reactive oxygen intermediates by glutathione and glutathione peroxidase, a phase II metabolic activity, is higher in the PP than in the PV zone141,143–145, a setup that protects the liver cells in zones I and II from intermediate-induced damage. In line with these results, hepatocytes in the PV zone are more likely to die from hepatotoxicity than PP hepatocytes140. This metabolic zonation is usually a missing or poorly controlled feature in most in-vitro platforms.
2.4 Liver and drug induced liver injury (DILI)
Because of its central role in drug metabolism, the liver is also one of the main targets for the toxic effects of xenobiotics5,14. There are close to 1000 identified pharmaceutical drugs that result in liver disease, even at a low incidence. Moreover, the liver is directly exposed to 25% of the cardiac output, which can contain high concentrations of intravenously-administered drugs that have not been cleared from the circulation, increasing the risk of toxicity in liver cells.
Pharmaceutical drugs and other xenobiotics are often poorly soluble in aqueous solutions, and thus have to be transformed to a more hydrophilic form to be more readily used and eliminated146. This is achieved through multi-phase metabolic process, as described in section 2.2. An equally important step in drug transformation by the liver is the transport of drugs into the cells, without which the cellular enzymatic metabolism cannot take place. While some drugs enter passively through the cell membrane, others require an active mode of transport147. This is accomplished by “transporter proteins” in the cell plasma membrane that facilitate the transport of chemical substances into and out of cells. Based on the direction of transport, transporters are classified into two broad categories: a) uptake and b) efflux drug transporters. Based on their ontology and sequence, the transporters form two superfamilies named the a) solute linked carrier (SLC) superfamily and b) the adenosine triphosphate (ATP) binding cassette (ABC) superfamily148,149. Because the activity of drug transporters has direct implications for all the subsequent enzymatic activities that take place, the characterization and an improved understanding of these transporters is crucial to all the pharmacokinetic and pharmacodynamics studies, in addition to all the work examining the phase I and II drug metabolizing enzymes.
While most intermediates and end products are harmless thanks to liver metabolism, a non-negligible fraction of intermediates can cause injury to the cells of the liver, as well as to other organ systems. As a result, drug-induced-liver injury (DILI) is the leading (~50%150) cause of acute liver failures151–154, as well as the most important cause of late stage drug failures in clinical trials154,155. In fact, from 1992–2002, the percent of clinical trials that failed because of hepatotoxicity at phase I, II, and III were 43, 23, and 35%, respectively. Moreover, about ~20–30% of all drug withdrawals from the US and EU markets from 1975–2005 was due to hepatotoxicity, and most “black box warnings” on common drugs refer to hepatotoxicity more than any other effect.
Drugs are broadly classified into two groups based on their hepatotoxicity: a) intrinsic hepatotoxins and b) idiosyncratic hepatotoxins155,156. Intrinsic hepatotoxins induce a predictable and common toxic response with respect to dose in a majority of subjects. On the other hand, idiosyncratic hepatotoxicity, which constitutes a sizeable fraction155,157 of drugs, is hard to predict and is not always dose-dependent, but instead depends on many factors such as inter-individual genetic differences, metabolic state, inflammation, or other disease states. This makes idiosyncratic toxicity very difficult to study and almost impossible to detect using animal testing and current analysis tools. Nevertheless, both groups most likely metabolize these toxins into large quantities of highly reactive and toxic metabolites15, which cannot be quickly cleared.
One well-known example of dose-dependent hepatotoxicity is the thoroughly studied case of paracetamol/acetaminophen152,158–161. Conventional wisdom states that Paracetamol is metabolized by the CYP2E1 enzyme to form the intermediate metabolite NAPQI, which is followed by a cascade of disruptive and fatal events if a threshold amount is exceeded. These events range from disturbance of cellular homeostasis and mitochondrial dysfunction to the activation of cell death promoting pathways and the release of drug-modified macromolecules and/or danger signals that initiate an innate and/or adaptive immune response151,154. Recent work indicates that the dose response of drug toxicity can be modulated by LPS exposure162,163, which suggests that inflammatory or diseased states can significantly alter the body’s metabolic response to drugs164,165.
As evidenced from the discussion above, the liver has a highly complex set of responses that may involves hepatocytes and the other non-parenchymal cell types. Furthermore, the pathways that lead to toxicity are not necessarily clear and, except for a few toxins like Paracetamol, have not been well studied. Accordingly, a new generation of in-vitro technologies that attempt to more faithfully mimic the liver ecosystem will be critical for the better understanding required to help solve some of the problems facing the pharmaceutical industry.
3. IN-VITRO PLATFORMS FOR MIMICKING THE LIVER PHYSIOLOGY AND DRUG RESPONSE: A HISTORICAL PERSPECTIVE
A successful in-vitro liver platform is expected to replicate the major liver-specific functions over a prolonged period (>28 days) to allow for both acute and chronic studies in normal and diseased liver pathology. This is quite an undertaking and requires a concerted effort from a large and diverse research effort. Past efforts to mimic the liver physiological and toxicological response in-vitro span decades of work among thousands of researchers. The difficulty and complexity of this task along with the still vast amount of unknowns in the field, resulted in a wide variety of approaches ranging from artificial electrochemical experiments166–168 and use of microsomes169–172 all the way up-to using whole organs173,174. The field is a dynamic one where each advance is adopted slowly and incorporated in the toolbox; this adoption depends on a delicate balance between ease of use, and the longevity and accuracy provided. In the interest of brevity, we only review 1) static suspension and plated culture liver cells and 2) perfused macroscopic and microscopic culture systems. A summary of the platforms we discuss with some of their key success criteria are provided in Table 2.
Table 2.
Comparison table of historical in-vitro platforms.
| Platform name | Description and notes | Cell types PC, NPC NPC types |
Longevity/viability (days/% live) |
Albumin/urea (stability and % control) |
Enzymatic activity (enzymes and % efficacy) |
Drug screening sensi- tivity and specificity |
Figure [references] |
|---|---|---|---|---|---|---|---|
| Perfused Platforms | |||||||
| Micro Hollow Fiber Reactor | Macroscale 3D perfusion-liver bioreactor with microscopic hollow fiber arrangement | Primary human hepatocytes | 23 days, but no direct viability measurement at the end of the study | Comparable albumin, urea, and glucose production in 800 mL, 8 mL, and 2 mL bioreactors | Stable CYP1A, CYP2C9, and CYP3A activity in 2 mL bioreactor for up to 23 days. Comparable (LDH) and (AST) release in 800 mL, 8 mL, and 2 mL bioreactors |
No data |
Fig. 4a [260] |
| Dielectrophoretic 3D liver assembly device | Dielectrophoretic assembly of hepatocytes and endothelial cells | Human hepatocytes and human liver endothelial cells | No data | No data | No data | No data |
Fig. 4b [281] |
| μCCA (micro cell culture analog) cornell group | 3D hydrogel culture in connected microfluidic chambers. Multiple incarnations |
Human hepatocarcinoma cell line HepG2/C3A | 3 days, >90% viability | No data | No data | No data |
Fig. 4c [303, 305] |
| Pearl liver sinusoid system (Millipore, formerly from CellAsic Inc.) | Microfluidic perfusion with microfabricated artificial endothelial barrier |
|
|
|
|
|
Fig. 4d |
| 3D-μFCCS (3D microfluidic cell culture system) Later renamed to 3D HepaToX Chip |
3D microfluidic channel-based cell culture system | Co-culture of rat hepatocytes and rat BMSCs and co-culture of human hepatocarcinoma cell line (HepG2) and a human breast cancer cell line (MCF7) | 3 days, no direct viability measurement | Albumin production for 3 days. Comparable albumin in 3D-μFCCS and 2D monolayer cultures | UDP glucuronyltransferase (UGT) activity measurement for 3 days based on 4-methylumbelliferyl glucuronide (4-MUG) formation. Hepatocytes in the 3D-μFCCS maintain UGT activity at significantly higher level than in 2D monolayer cultures | No data | Fig. 4e |
| Micro-pillar Array Liver Sinusoid Device | Microfluidic, no ECM, micro-pillar array | Cryopreserved human hepatocytes | 14 days constant viability | No data | No data | No data |
Fig. 4e [282] |
| LiverChip from MIT and ZYOXEL Inc. | 3D perfused microreactor culture system later translated to perfused multiwell plate Multiple incarnations |
Rat hepatocytes and spheroids Co-culture of rat hepatocytes and LSEC Co-culture of rat hepatocytes, LSEC, stellate, and Kupffer cells 4. Hepatocyte spheroids 5. Rat hepatocytes, LSEC, stellate, and Kupffer cells |
|
|
|
|
Fig. 4i |
| HμREL®-Flow Liver Culture Device | Microfluidic microscale cell culture analogue (CCA) system (HμREL® chip)15,16 |
|
|
|
|
|
Fig. 4j |
| Membrane Based Perifusion Bioreactor | Perifusion of multiple cell types using a membrane | Hepatocytes and epithelial cell line RL-ET-14 | 14 days, LDH leakage, no direct viability measurement data | No data | Higher EROD activity and inducibility when perfused. Even higher when a co-culture is established. Activity is stable only when both are used | Tested several drugs for cytotoxicity and metabolism. No large scale screening |
[257] |
| Oxygen-permeable Membrane Liver Bioreactor | Macroscopic device 25 μm membrane | Cryopreserved human hepatocytes | 33 days; no direct viability measurement | Stable urea (33 days), stable albumin (22 days), protein secretion | No direct enzymatic activity measurement | Tested effects of diclofenac and interval IL-6 stimulations |
Fig. 3d [266] |
| Hemoshear Liver Platform | Macroscopic transwell sandwich culture based perfusion device | Rat hepatocytes | 14 days, no direct viability measurement | More albumin and urea production in perfused culture for 14 days compared to static cultures | CYP1A1, CYP1A2, CYP2B1, CYP3A2 expression levels higher in perfused cultures compared with non-flow cultures on day 7 | Tested the effects of 3-methyl cholanthrene and dexamethasone in perfused and non-flow cultures on day 7 |
Fig. 3e [269] |
| Modular Bioreactor System, McMB | Macroscopic perfusion chambers in series and parallel | Human hepatocytes | 7–21 days, no direct viability data, shows morphologically healthy cells on day 14 | No data | Shows that detoxification genes are downregulated in static culture conditions but are well maintained in phase I, CYP, enzymes are highly upregulated under flow conditions | No data |
Fig. 3f [261–265] |
| Static Platforms | |||||||
| Hepregen Hepatopac™ | Micropatterned static culture | Rat/human hepatocytes with 3T3-J2 mouse fibroblasts | > 30 days, various methods | Thorough characterization of albumin, urea secretion | Thorough characterization of phase I, II and II activities in different publications | Large scale drug screening (~45 drugs) towards specificity and sensitivity21 |
Fig. 3a [223–225, 231, 234, 243–248] |
| Regenemed Static Scaffold Total Liver Culture | Transwel + nylon scaffolding co-culture | Rat and human hepatocytes and NPC fractions | ~77 days | Stable albumin, urea, fibrinogen and transferrin secretion over the culture period | Characterization of basal and induced activity of CYP3A4, CYP1A1 and CYP2C9 | 7 drug screening. Demonstrated species differences as well as effect of inflammation of drug response |
Fig. 3b [124] |
| InSphero Insight Liver Spheroid Culture System | Hanging drop/spheroid culture co-culture | Rat/human hepatocytes, Kupffer or LSECs | ~28 days, ATP measurement | No data | Characterization of some phase I and phase II enzymes at gene expression and activity level (workshop, website and few publications) | Some information on specific drug toxicities using the model is available. LPS stimulation of the co-culture spheroids and improved response has been shown |
Fig. 3c [162, 249, 250] |
| Hμrel Human (Hμrel Static) Liver Culture Model | Coplanar mixed co-culture | Canine hepatocytes and a mixture of non-parenchymal cells compared to primary human hepatocytes and HepG2 cell line | 31 days | No data | Characterization of basal enzymatic activity | 50 drug screening towards specificity and sensitivity | [251] |
3.1 Static culture of the liver cells
In this section we discuss 1) systems that only incorporate hepatocytes and 2) systems that feature secondary cells, including the non-parenchymal cells of the liver as well as others such as murine fibroblasts or endothelial cell lines; we conclude with a brief look at commercially available static platforms.
3.1.1 Hepatocyte only static culture platforms
Hepatocytes in culture have provided a first-order approximation of an in-vitro liver, for over 40 years. Fresh or cryopreserved primary cells (typically rat or human), hepatoma cell lines (HepG2, Mz-Hep-1, BC2, HepaRG, etc.), and progenitor cell-derived cells (embryonic or induced pluripotent stem cells) each recapitulate certain aspects of in-vivo liver physiology with individual strengths and weaknesses. Here, we only discuss systems that use primary cells, since enzymatic expression and activity are typically much lower in cell lines compared to primary cells175, and only a few toxicology studies using progenitor-derived cells have been published. We refer the readers to recent reviews74,176,177 for detailed information on cell line and explant culture.
Time scale and system complexity
Extending in-vitro culture duration, while maintaining an in-vivo-like phenotype is challenging; hepatocytes often lose function over time when isolated from their native environment30,31,178,179. Nevertheless, culture methods — that both overcome these challenges and provide a convenient work flow for a various applications — have been developed with time scales ranging from several hours to 6 weeks. Methods that provide an appropriate utilization of topology, ECM components, cell-cell contact and soluble factors to mimic the in-vivo liver environment, retain cell phenotype and liver-specific function for a longer time period. Isolated hepatocytes in suspension are widely used for drug clearance22,23 and toxicity24–26 studies. Unfortunately, hepatocyte viability in suspension decreases significantly after ~4 hours180,181 and the method is limited to very short-term studies. Moreover, many cell characteristics lost in the isolated state182,183, including polarity, junctions, bile production, and zonation, cannot be re-established in suspension culture.
Hepatocytes are adherent cells and depend on anchorage to a suitable substrate for the maintenance of differentiated function27. Accordingly, adherent monolayer culture of hepatocytes — on plastic culture dishes28 or ECM coated surfaces (especially collagen type I)29 — is able to slightly extend the culture duration (~1–3 days). Gene expression profiling studies183 indicate significant changes in hepatocyte phase I P450 and phase II metabolism, glucose metabolism, cytoskeleton and ECM, and cell cycle for monolayer cultures over 72 hours. Despite a reduction in expression of biotransformation genes, selected P450 enzyme activities remain inducible184, allowing for short-term use in toxicity studies185.
Spheroid culture methods, usually conducted in suspension, work by inhibiting hepatocyte attachment to vessel walls thereby enforcing cellular aggregation and formation of floating spheroids. Various methods exist for spheroid formation, including mechanical agitation by rotary shaker186 or spinner flask187, hanging drop162,188 or non-adherent surface chemistry189,190. In addition to providing anchorage via cell-cell contact, the 3D organization in spheroids also seems to result in cellular polarity and to some extent the retention of ECM that hepatocytes themselves secrete. Spheroid culture typically maintains many of the liver-specific functions25,177,191, such as albumin, urea, transferrin, and bile secretion, as well as certain phase I and II biotransformation activity for a few weeks162. Despite this encouraging picture, several limitations exist in spheroid culture, including difficulty in imaging, a distribution of nutrients, wastes, and test compounds across the aggregates, and difficulty in scaling the system down for microfluidic applications.
Another advance in long-term function came via the sandwich culture or overlay method30,31,178,179 developed in our labs over two decades ago. Sandwiching hepatocytes between two layers of ECM, typically collagen or Matrigel™ basement membrane matrix, leads to development of stable hepatocyte polarity30 and hepatocyte “plate” structures similar to the in-vivo liver anatomy30. Such polarity, including basal surfaces induced by ECM layers and apical surfaces by cell-to-cell contact, leads to bile canalicular network development192 and bile secretion that resembles in-vivo secretion193. Ease of microscopic imaging is also a benefit of this planar technique.
Matrix sandwich and matrix immobilization methods for hepatocyte culture have significantly increased the viable culture time period, up to 6–8 weeks and enabled stable albumin, urea, transferrin, fibrinogen, and bile salt secretions30,31,178. Moreover, biotransformation activities and induction of many phase I CYP isozymes and phase II enzymes are adequately maintained over at least 2 weeks194. Although the sandwich initially uses one or a few ECM components, hepatocytes themselves secrete ECM and alter the local microenvironment over time195; thus, an important role of the sandwich structure is to act as a scaffold and retain the secreted ECM components196. The success of the sandwich method demonstrates that not only composition but also the topography of the ECM can play an important role in guiding organization and expression of cytoskeletal proteins, cellular polarity and maintaining phenotypic stability69,195,197.
Although several culture configurations have been developed to preserve hepatocyte function and morphology for varying times, translating those methods to microfluidic dimensions has proven difficult. Encapsulation techniques that employ a wide variety of biomaterials have also been used to create more in-vivo like 3D microenvironments for cultured hepatocytes. These biomaterials include alginate198, hyaluronic acid esters199, collagen200,201, and methylated collagen202, and have been able to maintain the liver-specific functions for varying lengths of time. Such encapsulation with biomaterials has been translated successfully in microfluidic devices203 which we also highlight in section 3.3.2, indicating the potential of this approach in efforts towards miniaturization. A promising method, in this context, for creating a thin extracellular matrix is layer-by-layer deposition of polyelectrolyte multilayers204. In this technique, alternating layers of cationic and anionic polymers are deposited via electrostatic attraction205,206 on top of hepatocytes to mimic the space of Disse. While many potential polymers could be utilized for good results in terms of hepatocyte morphology, function, co-culture with LSECs, and CYP 1A1/2 activity have been reported for up to 12 days using chitosan and hyaluronic acid85,207. We have recently been able to translate this approach to a microfluidic device for hepatocyte culture using charge modified collagen strands creating a ECM barrier that is on the order of ~100 nm and have also shown long-term stability of important hepatic functions208.
Hepatocyte stimulation: Soluble factors
Basal medium and supplementary additives for culturing hepatocytes are not standardized and many different formulations are used; the discrepancies in culture configuration and media formulation often make comparisons between different studies and laboratories difficult. Typical basal formulations include DMEM, DMEM/Ham’s F12, and William’s E and these are commonly supplemented with serum, insulin, EGF, corticosteroids (hydrocortisone or dexamethasone), and glucagon to maintain phenotype. While serum is necessary to improve attachment and some hepatic functions, it also causes a range of deleterious effects on morphology and polarity148. In the absence of serum, formulations providing complete amino acids, including proline209, are necessary to support albumin and collagen synthesis.
Many different additional soluble factors, including cytokines and non-physiologic compounds, have been reported to have various beneficial effects on hepatocyte functions. Exogenous cytokine stimulation is a powerful, though potentially non-physiologic, signal to cells. TGF-β1 typically induces apoptosis210 and fibrosis, whereas VEGF can help maintain hepatocyte endothelial cell co-cultures211,212. Addition of up to 2% dimethylsulfoxide (DMSO) to media helps maintain albumin and plasma protein production, morphology, and cellular junctions in hepatocytes in monolayer213, though it may decrease certain CYP P450 activities214. Other compounds reported to improve aspects of hepatocyte function in culture10 include isonicotinamide215, metyrapone216, nafenopin217, and transferrin218.
More complex media formulations have typically maintained hepatocyte function better than basal formulations74,219, though reduction in concentration of additives is likely possible in culture configurations that maintain ECM contacts and cell-cell interactions69. Several attempts have been made to use a factorial design and isolate the effects of specific hormone or cytokine additives on hepatocyte function; in one such example, Zupke et al. demonstrated that glucagon supplementation increases glucose and urea synthesis220. Translation of media requirements from macro-scale tissue culture applications to microfluidic devices, which may have constant or intermittent perfusion, remains unresolved.
3.1.2 Static co-culture systems
Non-parenchymal cells support and contribute to drug metabolism; they also play a crucial role in modulating intrinsic as well as idiosyncratic liver injuries via the release of growth factors, inflammatory mediators as well as reactive intermediates70,71,124. Moreover, the overwhelming evidence demonstrate that these “secondary” cell types enhance hepatocyte function, and prolong the retention of their phenotypes in long-term in-vitro studies9,221–227. With this insight, many systems have been devised to co-culture hepatocytes not only with cells of the liver but also cells from other tissues. Below we review these efforts, which shed light on the question of how we can better recapitulate the liver function by multiple cell culture in-vitro.
2D co-culture approaches
One of the first examples of 2D (coplanar) hepatic co-culture methods was established by Guillizo and co-workers221,222; they demonstrated a significant improvement in both viability and hepatic function in hepatocytes and liver epithelial cells, monolayer co-culture system compared to control group of hepatocyte monolayers. Following this work, it has been shown that by randomly seeding hepatocytes with non-parenchymal cells of liver228,229 or cells of non-hepatic origin, such as fibroblasts230, leads to maintenance of differentiated hepatic function for several weeks through a number of mechanisms including cell-cell contact, and secreted factors such as growth factors and extracellular matrix (ECM) components9,231.
In contrast to random co-cultures, application of microfabrication and patterning approaches has facilitated systematic investigation of the role of homotypic, and heterotypic cell-cell contact on the maintenance of hepatocyte function. By patterning rat hepatocytes and murine 3T3-J2 fibroblasts, our group established that the heterotypic cell-cell contact between hepatocytes and fibroblasts223 as well as homotypic fibroblast interaction224 contributed to enhanced synthetic function of hepatocytes in a co-culture system. More recently, the micropatterning approach was extended to culturing primary human hepatocytes in a multi-well format for drug toxicity screening225 and hepatitis C virus (HCV) infection232. Khetani et al.225 demonstrated the utility of hepatocyte/3T3-J2 fibroblast co-cultures by assessing phase I/II xenobiotic metabolism, bile canalicular transport, secretion of the liver specific products, and susceptibility to hepatotoxins. For HCV infection studies, Ploss et al.232 demonstrated sustained replication of virus for several weeks in human hepatocytes, although the infection was limited to 1–3% of hepatocytes. In addition to fibroblasts, micropatterning approaches have also been applied to patterning of hepatocytes with other cell types such as Kupffer cells with a concomitant increase in the synthetic function233. A comparison between the effects of cell-cell contact and secreted factors was conducted by Hui et al.234 via micro-machined silicon substrates with moving parts which enabled both spatial and temporal control over cell placement. The authors demonstrated the subtle result that maintenance of hepatocyte phenotype by fibroblasts required direct contact for a few hours followed by sustained soluble signals with a spacing between two cell types that is less than 400 μm234.
3D co-culture approaches
While coplanar seeding of cells provided invaluable information about the interactive environment of the liver, recent studies have focused on creating 3D layered structures of hepatic cells in order to supposedly better mimic the liver sinusoid. Ito et al.235 observed enhanced albumin function in co-cultures of hepatocyte and endothelial cells, where layering was achieved by labeling endothelial cells with magnetite cationic liposomes and placing them on top of hepatocytes using a magnet. Another approach relied on growing endothelial cells as a separate sheet and then placing the sheet on top of hepatocytes for creating layered structures236,237. Using this approach, Kim et al.237 were able to maintain hepatocyte phenotype for 4 weeks with well-developed bile canaliculi networks.
A novel 3D co-culture approach was presented by Rajagopalan and co-workers85,204 where they utilized polyelectrolyte layer-by-layer assembly205,206 to layer endothelial cells on a monolayer of hepatocytes. They demonstrated both higher synthetic function and enzymatic activity in co-cultures compared to a control group of pure hepatocytes. In order to include a somewhat crude model of the space of Disse, Katsuta and co-workers238 cultured hepatocytes and endothelial cells on the opposite sides of a micro-porous membrane with stellate cells intercalated in the pores of the membrane. This model was used for investigating the role of stellate cells in mediating intercellular communication between hepatocytes and endothelial cells in the context of endothelial cell morphogenesis. Salerno et al.239 used synthetic biodegradable membranes with human umbilical vein endothelial cells (HUVECs) and primary human hepatocytes to create a layered model, which resulted in enhanced albumin production, urea synthesis, and drug transformation due to heterotypic cell-cell interactions.
Layering can also be achieved using natural ECM rather than synthetic materials. Specifically, Jindal et al.201 utilized collagen as the intervening layer for creating layered co-culture of hepatocytes and endothelial cells. They also established that in the layered structure, surprisingly, proline secreted by endothelial cells contributed to the maintenance of hepatocyte function. The utility of this model was further expanded by integrating a fluorescent reporter clone of endothelial cells and assessing the activation state of endothelial cells under inflammatory conditions240.
Spheroids also provide an alternative for co-cultivating hepatocytes in a 3D configuration. Wong et al.241 exploited a concave micro-well array platform for creating spheroids of hepatocytes and stellate cells. They observed that albumin secretion and drug metabolizing activity was superior in co-cultured spheroids; and also demonstrated that stellate cells played an important role in the formation of stable and uniformly sized spheroidal aggregates. In a similar study, spheroids of hepatocytes and fibroblasts were formed and then cultured in either a bioreactor or a spinner flask242. This study demonstrated significantly improved longevity, albumin production and phase I/II drug metabolizing activity.
Commercial 2D and 3D co-culture platforms
The sandwich method and monolayer culture of pure hepatocytes have been de-facto standards in drug screening and drug discovery studies in the pharmaceutical industry; nevertheless, a few of the aforementioned co-culture approaches have now also reached a commercially viable stage and are available for wider use by academic and industrial establishments. While some of these platforms have been initially cultivated in academic settings, others have been developed directly via commercial efforts. Below we review four of these systems that claim better physiological relevance as well as improved predictive capabilities.
One example of an academic-born technology making its way into the commercial environment is the “Hepatopac” platform developed by Hepregen (Fig. 3a). This platform, which was initially developed in our labs in the late 1990s223,224,231,243,244, was further developed by Khetani and Bhatia at MIT and then by Hepregen225,245–248. This 2D coculture system, as discussed earlier, has two important features: a) use of micropatterning to make rat or human hepatocyte islands of roughly ~100–500 μm and b) use of 3T3-J2 fibroblasts to stabilize and enhance the function of hepatocytes in long-term cultures (> 4 weeks). In its latest iterations, the model has been successfully translated into a multi-well culture plate platform amenable to higher throughput experiments. While the physiological relevance of flbroblasts — of mouse origin — in this system is unclear, recent studies245 on panels of drugs (~40) show improved sensitivity and specificity especially when human hepatocytes were used. The Hepregen platform has been characterized in terms of its enzymatic and functional capabilities and retention of such capabilities.
Figure 3.

Selection of macroscopic or static in-vitro liver models. On top row we share three important platforms which are all designed for static plate like culture while the bottom row show macroscopic perfusion culture systems. (a) Hepregen Hepatopac™ micropatterned co-culture. (b) Regenemed co-culture platform for hepatocyte and NPC co-culture. (c) InSphero Insight platform taking advantage of hanging drop method and concave culture wells. (d) Work of De Bartolo with disk like geometry and membrane oxygenation. (e) Hemoshear platform for hepatocyte culture with ECM for shear protection. (f) Multicompartmental Modular Bioreactor system (MCmB).
Another successful academia industry collaboration, in this realm, is between ETH Zurich and Insphero AG (Schlieren, Switzerland). Their platform (Fig. 3c) features a reincarnation of the hanging drop model combined with a multi-well plate format to reliably produce and culture spherical aggregates (spheroids) of hepatocytes and non-parenchymal cells162. The use of a multi-well plate format allows the easy integration of the abundantly used imaging and automation systems in the industry, thus a seamless integration into current workflows. By tightly controlling the size (~200 μm) of the hepatic aggregates, they have been able to demonstrate high viability and stable function of these spheroids for up to 5 weeks and have conducted several drug toxicity studies with or without inflammatory stimulation162; the system also allows spontaneous zonation within the spheroid. Unpublished data by the company shows significant effect of NPCs on the IC50 values of different compounds highlighting once again the importance of NPCs on drug metabolism. Despite the success and advantages of this platform a notable drawback is the difficulty in imaging a thick tissue construct. Additionally, full characterization and predictive capabilities of this promising platform on a large panel of drugs still remain to be demonstrated. Published249 and unpublished data250 shows high stability of enzymatic expression and activity for long culture periods (~28 days).
Another important static co-culture platform which also originated our labs, and then was transferred to an industrial startup, is the “HμREL-hepatic co-culture” platform from the Hμrel Corporation (North Brunswick, New Jersey). While the company also provides microfluidic “flow” culture products (reviewed in section 3.3.2), their static platforms (i.e. a well-mixed and optimized co-culture of cryopreserved hepatocytes (human, rat, dog and primate origin) with a proprietary stromal cell cohort, has shown longevity (>30 days) and competency in terms of CYP 450 and synthetic functions according to published251 and unpublished data252. Most recently in a large drug panel study (~50) in collaboration with UCB Pharma, the dog co-culture version of this platform has been shown to have high sensitivity (78%) and the highest specificity (73%) among all groups251. Their successful use of cryopreserved dog (canine) hepatocytes also marks an important step towards closing the species gap in drug testing where all species used in animal studies can be compared to in-vitro model results.
The Regenemed (San Diego, CA) platform (Fig. 3b), also features co-culture of human or rat hepatocytes with a full complement of the non-parenchymal liver cells in a 24-well plate format124. It makes use of a removable transwell as well as porous (d ~140 μm) nylon scaffolds to culture a matured NPC fraction on top of hepatocytes that are cultured in the outer well; a cell number ratio of 60% hepatocytes and 40% NPC fraction was deemed optimal. In one report, the platform was demonstrated to be viable and functionally stable for up to 11 weeks as inferred by albumin, transferrin and fibrinogen secretion and urea synthesis124. The feasibility of this platform for drug screening studies was also demonstrated, albeit using a small panel of drugs; a species specific response was observed during the studies and they also noted that specificity of the system is improved when the NPC fraction was included124.
3.2 Flow-based in-vitro liver platforms
Optimal liver function is presumably not only dependent on the coordinated function of the parenchymal and non-parenchymal cells within the hepatic acinus, but also dependent on hepatic blood microcirculation. Aspects of the microcirculation can be simulated in-vitro, via perfusion models, to create a dynamic in-vivo like environment. Through perfusion, nutrient, oxygen, and soluble factors can be replenished in a controlled way so as create a pseudo-steady state of those parameters; similarly secreted factors, biliary secretions, and intermediate or end metabolites can be cleared. This is unlike a static culture where nutrients are consumed in an exponential decay; and end or intermediate products are secreted in an inverse exponential manner; eventually everything is reset to the initial state via media changes. This media cycle is clearly not representative of the physiological state, and also may be stressful for the cells. Spikes in media may be representative of drug injections; however, if the rest of the system is stressed by a lack or overabundance of nutrients and waste, the metabolic response to the drug challenges may be misleading. This problem has long been recognized and researchers have incorporated perfusion in their culture systems to allow for a better in-vivo mimicry.
3.2.1 Macroscopic flow platforms
The early works on macroscopic perfused in-vitro liver systems were primarily motivated by creating a bioartificial liver as an extracorporeal assist device akin to the dialyzers for kidney; however, the bioartificial liver devices went beyond the filtering function by the inclusion of hepatocytes for metabolic functions. Three prominent types of such devices exist 1) flat-plate bioreactors as developed by our group32,34,253,254 and others255,256, 2) hollow fiber37–39 liver assist devices, and 3) macroscopic liver perifusion devices257,258. Although we consider these devices macroscopic, some incorporate microscopic features, for example via SU-8 micropatterning253 in the flat-plates. Some of these platforms were the precursors to today’s microfiuidic devices, and in general, demonstrated enhanced hepatic function over static embodiments32. They also served as the initial proof of concept for critical technologies such as membrane oxygenation32,34,254, and shear reducing microgrooves253,259. While these platforms were initially not intended for drug screening purposes, a recent reincarnation of a scaled down version of the hollow fiber reactor by Zeilenger et al.260 demonstrated such feasibility using human hepatocytes. However, even in their smallest (2 ml) and most successful version of the device, the albumin synthesis and urea excretion declined considerably after a week of perfusion; enzymatic activity, demonstrated via metabolite formation, showed a similar decline.
Other macroscopic devices of note, for their accessibility, longevity and possible commercial success, respectively are: 1) multicompartmental modular bioreactor (MCmB) by Ahluwalia and co-workers261–265, 2) the work of De Bartolo266,267 and that of Gebhardt268, and 3) the Hemoshear platform269. The MCmB261 (Fig. 3f) involves a simple macroscopic low shear environment which uses regular peristaltic pumps, a cylindrical flow chamber on the bottom of which a cell-laden cover slip can be introduced with ease. The MCmB platform was used with co-culture and multi-tissue cultures261,264,265, and enzymatic gene expression was demonstrated for 21 days. The fluidic chambers used in the MCmB platform are commercially available in an easy to use format for any type of perfused cell culture. The work of De Bartolo stands out with respect to longevity of the system, an important requirement for chronic drug response studies. Their platform266 consists of a disk like bioreactor with a 25 μm thick gas permeable membrane for gas exchange (Fig. 3d); they have demonstrated 33 days of stable secretion for culture of cryopreserved human hepatocytes although no direct viability measurement was conducted. They also demonstrated drug clearance studies in addition to IL-6 stimulation of the entire system in intervals. Gebhardt et al.268 showed that addition of RL-ET-14 cells, a rat liver cell line resembling LSECs, to their perifusion system of rat hepatocytes can improve longevity of the system to 14 days in addition to higher stable enzymatic activity and inducibility; this work also demonstrated the potential utility of co-cultured cells for drug clearance as well as drug-drug interactions.
Another platform, the Hemoshear system (originally developed to study endothelial cell hemodynamics267), features a cylindrical macro-perfusion chamber for cells (Fig. 3e) where the hemodynamic environment, i.e. flow, is created in a fashion similar to a cone-and-plate viscometer. The liver version of the platform features a synthetic membrane over the confluent layer of rat hepatocytes in a collagen sandwich configuration; this membrane protects both the cells and the gel structure form the high shear rate (0.6 dynes/cm2) used in their recent study. This recent work267 demonstrates the operation of the system for 14 days with stable secretory function and improved enzymatic activity compared to static cultures; further data with large drug panels are still needed to establish the success of this platform.
3.2.2 Microfluidic platforms
The macroscopic platforms described above provide some evidence that perfusion can improve longevity and function in cultured hepatic systems. An important design consideration when developing tissue analogues is the size of the device and most importantly media volume/height which directly affects the key parameter of “media volume to cell number ratio”. In a typical liver sinusoid (diameter ~5–10 μm), this ratio is about 0.03 nL of blood per hepatocyte270. While achieving this ratio is critical if one desires precise in-vivo mimickry, it is currently not feasible because of: a) technological limitations with regard to fabrication and tissue engineering and b) the lack of appropriate media which must have oxygen carriers in order to deliver sufficient oxygen given the reduced volume. Nevertheless, approaching this ratio is of utmost importance, especially in multicellular tissue constructs and/or multi-organ systems, where one wants to study interactions between two or more cell types through secreted factors and intermediate drug metabolites. Most conventional systems such as traditional culture well plates or current macrofluidic systems262,263,271–273 are at least two orders of magnitude off in their “media volume to cell ratios”, and thus are not suited to capture interactions signaled through the fluid at the physiologically relevant doses of stimulation or drug application. As a rule of thumb, device heights that are 100 μm or smaller provide a much more relevant signaling environment for multicellular systems and can capture interactions among cells that would otherwise be missed in larger systems274. Thus, microfabrication and microfiuidic models appear essential for approaching media volume/cell ratios characteristic of in-vivo values270,275.
Below we discuss microfluidic liver platforms that aim to recapitulate different aspects of the liver taking advantage of technologies that enable precise machining and control of microenvironments276. A brief comparison of these microfluidic platforms along with few macroscopic ones can be found in Table 2 and Figs. 3,4.
Figure 4.

Selection of recent microfluidic in-vitro liver models. Depiction of various microfluidic in-vitro liver technologies. (a) Zeilinger’s work260 with a miniaturized version of hollow fiber perfusion environment. (b) Assembly of hepatocytes and endothelial cells using dioelectrophoresis and their perfusion in the same channel by Schutte et al.281. (c) Micro cell culture analog systeme and multi-tissue interaction by the Cornell group. (d) Sinusoid like perifusion environment with an artificial (microfabricated) endothelial barrier by Lee and co-workers278 that gave rise to the CellASIC Pearl platform. (e) Use of micro-pillars for forced assembly of hepatocytes in works of Toh and Goral.282 (f–h) Various technologies developed in our labs (CEM) featuring various different technologies and demonstrating high throughput capabilities. (i) Zyoxel — MIT culture platform for mono and co-culture of hepatic cells. (j) Hμrel–flow system.
Hepatocyte only microfluidic platforms
A common approach towards liver mimicry in-vitro, via microfabrication, is to create cellular assemblies that resemble hepatic cord/sinusoid like structures. One example is the work by Lee and co-workers277,278 at UC Berkeley that gave rise to a commercial platform via the CellAsic Pearl and Onix systems (EMD Millipore, Billerica, MA). The building block of this platform is a perifusion system where hepatocytes are densely seeded into a narrow, high aspect ratio pocket (Fig. 4d) with a microfabricated artificial barrier featuring fenestrae like structures (2 μm wide); the perifusion is established outside this barrier region thus protecting the cells from mechanical stress. Although the barrier channels are about an order of magnitude larger than endothelial fenestrae, they do create an effective barrier to convective transport which only allows diffusion — a factor that makes the platform amenable to mathematical modeling since the characteristics of the transport barrier are known. Like other commercial platforms this model has also been translated into a culture plate format with 32 individually addressed sinusoid arrays on one plate, and gravitational flow is employed to negate the need for pumps. While the earlier work277,278 on this platform demonstrated high viability of only about a week, recent work indicates hepatocyte culture for over 30 days279.
In a similar fashion, Nakao and co-workers280 cultured primary rat hepatocytes in a 37 μm by 30 μm microfluidic channel, surrounded by an array of 2 μm wide slits. They demonstrated that such narrow and high aspect ratio channels induce hepatocytes to align and form an in-vivo like bile canalicular structure. However, the scalability and long-term viability of the system was not demonstrated. A recent, innovative yet underdeveloped effort in this respect is the creation of sinusoid like structures using dielectrophoretic assembly of primary human hepatocytes and endothelial cells in microfluidic channels281; while the authors demonstrated cord-like structures that can then be perfused in the same channel they were created, they have not yet shown the stability, viability and functionality of these structures.
A recurring theme in the microfluidic work cited so far is either the dilute use280,281 or lack277,278 of ECM proteins which highlights the difficulty of translating some of the successful macroscopic technologies, such as the collagen sandwich method31, to the microfluidic realm. The sandwich configuration relies on hydrogels with typical thicknesses of 100 s of microns that are too bulky to situate easily in enclosed PDMS devices with features as small as ~10 μm and channel heights around ~100 μm. The ECM free approach relies on the premise that the ECM components secreted by hepatocytes and other supporting cells might be able to sustain the long-term hepatic function196; however, the retention of these secreted ECM components is questionable especially in open-loop perfusion systems. One approach that takes this ECM-free approach is the work of Goral et al.282 where they use a dense array of micro-pillars (15 μm tall, pitch is less than a single cell diameter282) both to hold human hepatocytes together and induce a cord like structure. While these investigators demonstrated viability for 14 days, along with formation of 3D bile canalicular structures and healthy expression of connexin 32 and MRP-2, they have not yet investigated broader functions of this system.
Another microfluidic liver platform by Toh and co-workers203,283, dubbed the 3D-μFCCS or the 3D HepaTox Chip, also makes use of a microfabricated endothelial barrier but with 20 μm fenestrae (Fig. 4e). Additionally they use a complex coacervation method using polyelectrolytes to create a 3D matrix for the cultured hepatocytes addressing the need for an initial ECM environment. They demonstrated the versatility of this platform by culturing HepG2, MCF7 and bone marrow stem cells283 initially, and then by using primary rat hepatocytes203,283. The second study203 also demonstrated multiplexing by incorporating a gradient generator for drug dosing purposes. In the same study they also showed higher enzymatic inducibility as well as metabolic activity for up to 72 hours of perfusion culture compared to static plate cultures. Finally, they also conducted a toxicity study for a panel of 5 drug compounds and showed a slightly improved correlation to in-vivo toxicity compared to multi-well plates (R2 = 0.84 vs. R2 = 0.8).
An example of high throughput capability of hepatocyte microfluidic systems comes from our lab which developed a scalable experimental platform that combines microfluidic addressability with quantitative live cell imaging of fluorescent protein transcriptional reporters (the “living cell array”)284–290. The platform uses microvalve arrays to achieve reliable seeding and orthogonal stimulation of multiple fluorescent reporter cell lines while enabling automated time-lapse microscopy to continuously monitor dynamic responses from a 2D matrix of experiments. Using this platform with eight different reporter cells and 8 different stimulating agents, we were able to monitor a remarkable 5000 single time points in a 36-hour period. While this proof of concept work used the H35 hepatoma cell line to create stable GFP reporter cells and was aimed at examining short-term gene expression, the extension to primary hepatocytes for longer periods of time is eminently doable.
Microfluidic co-culture systems
One example of integrating technologies for hepatic tissue engineering and microfabrication comes from our lab where Kane et al.291 combined several previous ideas such as the gas permeable membrane254 as well as a micropatterned co-culture system of hepatocytes with 3T3-J2 fibroblasts224,243 (see Fig. 4g). Cultures were miniaturized and integrated into an 8 × 8 microfluidic well array, similar to the living cell array described above284 (Fig. 4h). Each row or column could be perfused independently with regard to both media and oxygen through separate fluidic manifolds. Stable albumin synthesis and urea excretion was demonstrated for 32 days in this microfluidic array of co-cultures but no enzymatic or toxicity related studies were reported.
A liver platform that has been long in development both academically and in a commercial setting is the Zyoxel platform (see Fig. 4i). This platform stems from early work at MIT292,293 and features cross-flow micro-wells (300 μm wide, 235 μm high), manufactured using silicone or polycarbonate, for perfusion of 3D aggregates of hepatic cells. The cross-flow across hepatocytes is established via a micro-porous membrane at the bottom of the micro-wells. Their early work featured only hepatocytes and demonstrated stable secretory function for up to 15 days. Later incarnations employed co-cocultures with LSECs294,295 and then a full complement of an LSEC enriched parenchymal cell fraction295 for improved stability and mimicry; they also demonstrated spontaneously formed oxygen gradients295 in their scaffolds akin to the in-vivo zonation. These newer incarnations also showed convergence towards a culture plate format complete with pneumatics and sensors, allowing 12 experiments on each plate. Unpublished data on this platform claims longevity of 21 days, and recent publications demonstrate CYP activity296 and drug clearance226 with good in-vitro to in-vivo correlation. While this platform has been around for long time, several issues remain. For example, 1) although the aim of 3D aggregate formation is to create sinusoid like plate structures, the architecture of these aggregates are completely random and reproducibility is relatively poor69,74, 2) the height of the tissue structure interferes with high content methods that rely on optical methods, and 3) the current system appears too complex for widespread adoption124.
Hμrelflow™ (Fig. 4j), a perfusion-driven, microfluidic in-vitro liver platform geared from its inception towards drug and toxicant screening, has been developed by Hμrel Corporation227,297,298. Hμrelflow™’s academic predecessor platform was developed by Michael Shuler and Gregory Baxter of Cornell University (see the discussion on μCCAs). Hμrelflow™ devices are comprised of multiple, fluidically interconnected microscale cell culture compartments which enable simulation of the interaction of test substrates with 2 or more organs, so as to provide enhanced prediction of human physiological response based on PBPK models299,300. Made entirely of a type of plastic selected for its optical clarity and minimized absorption of test substrates301, the platform features flat-bottomed, microscale tissue culture compartments capable of holding adherent cellular materials; the tissue culture compartments are interconnected by microscale channels that enable the device’s culture media to flow in a recirculating pathway throughout the course of the experiment. A recent initiative by the company has specifically focused on integration of small, independently controlled actuation (i.e. pumping) mechanisms into the substrate of each device to reduce the overall culture medium in the device, shortening the circulatory cycle time and enabling Hμrelflow™ to better mimic actual in-vivo recirculation. Maintenance of the platform has been demonstrated up to 6 days with respect to its enzymatic and synthetic function in published work227, and recent unpublished data302 claims over 14 days of stable maintenance. More importantly in the same studies227 clearance data of six drugs showed high in-vitro in-vivo correlation especially in perfused co-culture models of human hepatocytes and stromal cells and the same system also showed a superior metabolite generation compared to all other controls. While more work is needed on this platform to show its retention of enzymatic and functional capacities over longer durations as well as to demonstrate its multi-organ/tissue interaction capabilities, their most recent static co-culture results251 together with their technological advances in micropumping look promising for wider adoption of the system.
With regard to multi-organ interactions in microfluidic systems, with the liver as the primary organ, the field has been advanced by Shuler and co-workers and their micro cell culture analog (μCCA) systems299,303–307. These are, as in the Hurel system, connected microfluidic compartments of different tissue/organ systems with stated physiologically relevant volume ratios as well as in-vivo like residence times for the recirculating media flow. These systems were designed with the notion and premise that they are the microscopic analogues of the in-vivo visceral system and that modeling their response to drug challenges via PBPK approaches can provide important insights and predictions about the real system. These investigators have demonstrated the feasibility of such systems first with a liver-lung299 and a liver-lung-fat tissue model307 using cell lines for all cultures. These early models also demonstrated the clearance of naphtelene and the toxicity of its liver metabolized intermediates on the lung chamber307. More recent examples of the μCCAs demonstrated similar studies for liver-uterus cancer-colon-colon tumor with integrated optical in-situ imaging306, liver-tumor-bone marrow305 and a GI tract model complete with liver, kidney, bone marrow and fat tissues while modeling the intricacy of poorly perfused and well perfused medium flow304. While these μCCAs have been important in establishing the idea of PBPK in the in-vitro microfluidic realm as well as demonstrating multi-organ interactions for toxicity, two obvious drawbacks of this work have been the regular use of cell lines rather than primary cells and the usually short duration (several days) of the studies.
Counterparts of this work in Europe where multiple organ/tissue chips are connected — with the liver as the focal point of the system — has been predominantly developed by Ahluwalia and co-workers and their McMB (Multicompartmental Modular Bioreactor) model261,308,309; and more recently by the body-on-a-chip (BoC) initiative that is led by InSphero AG. While the McMB system consists of fluidically macroscopic modules that are easily connected and disconnected with more common interconnects, the BoC initiative uses a microfluidic translation of the hanging drop spheroid formation and the microfluidic connection of eventual spheroids again in a custom multi-well format310. The McMB model has been commercialized or under the process of commercialization by two companies Kirkstall Ltd. (UK) and IVTech Srl (Italy) where the companies provide off-the-shelf compartments for researchers to develop their own tissue models.
4. A CRITIQUE OF THE HISTORY AND GOING FORWARD: CHALLENGES AND OPPORTUNITIES
As stated earlier one of the major goals of current governmentally supported research initiatives is to develop robust 3D microfluidic tissues that provide improved predictive capabilities of the human response to drugs. While some of platforms claim to meet the arbitrary 28-day duration criterion they fall short in some other category; i.e. they are poorly characterized and/or their physiological relevance is in question or completely unknown. Below we discuss key issues that exist currently as well as ones involve heavy use of microfabricated platforms.
4.1 The need for standardized characterization
In Table 2 we compare various current microfluidic as well as few commercial static culture platforms and provide information about their outputs; there is clearly a significant discrepancy among these platforms in terms of their assessment criteria even amongst the more mature ones. While some groups focus primarily on secretory functions and cell viability as a first indicator of success others march onto drug studies without these more basic validations; few have performed complete characterization of the general health, phenotypic retention and drug response of the platform. This partially stems from the fact that the complete characterization is a long, costly, and challenging task that might not be feasible without adequate support. Nevertheless, in order for the field to move forward as a whole there should be a logical and common checklist to assess the success and guide the evolution of new in-vitro liver models. While the current NIH/DARPA/FDA initiatives pose a challenge of 28 days of viability in perfused organ systems (presumably to match the common practice of 28 animal toxicity studies used by the pharmaceutical industry), the functional and physiological relevance — especially for the liver — warrants deeper discussion. Below we briefly discuss a three-stage characterization strategy and summarize it in Table 3.
Table 3.
Proposed three-stage characterization of in-vitro liver models.
| Stage 1 — Viability and functional stability | ||
| Cell viability | Imaging via complementary fluorescent dyes | Metabolic assays i.e. MTT, Presto Blue |
|
| ||
| Functional assays | Synthetic (albumin, fibrinogen) | Detoxification (urea) |
|
| ||
| Stage 2 — Drug transport and metabolism | ||
| Enzymatic activity, expression |
Phase I CYP 450s (1A2, 3A4, 2C9, 2C19, 2D6, 2B6) activity, inducibility and expression |
Phase II UGT, SULT and others |
|
| ||
| Drug transporter activity | SLC superfamily OATPs NTCP, OCTs | ABC superfamily MRP2, BCRP, MDR1 and MDR2 |
|
| ||
| Stage 3 — Sensitivity and specificity | ||
| Drug panel studies | Efficacious drugs (safe, non-safe) | Non-efficacious drugs (safe, non-safe) |
4.1.1 Stage 1: Cell viability and functional stability
Viability and cell health are the first success indicators of any tissue model. These assessments can be conducted either via fluorescence imaging techniques that rely on a combination of nuclear and cytosolic dyes or via resazurin based transformation assays311,312. Although the functional secretion assays, discussed below, provide some information regarding cell health they provide an incomplete picture where one cannot isolate individual effects. Viability assays should be conducted at short intervals (every day to several days) to demonstrate initial success of any in-vitro liver platform.
Albumin synthesis and urea excretion, are two of the most important markers to deduce the functional capacity of both the liver and its parenchymal cells, the hepatocytes. A consistently high albumin production (~1–5 μg/hr/106 cells) not only makes sure of the synthetic function but is also an indicator of the overall metabolic health of the cells; urea plays a similar role for general metabolic capacity. Recent drug screening studies in micropatterned hepatic co-culture systems show that albumin and urea levels can also be a reasonable but indirect indicator of toxicity245.
4.1.2 Stage 2: Drug metabolism and transport
In addition to the indirect assessment of synthetic and detoxifying capacities, retention of in-vivo like and stable phase I and phase II metabolic activity has possibly become the most important goal. We suggest that every platform should demonstrate stable phase I enzymatic activity, proper inducibility and expression for enzymes from the CYP family; assess phase II conjugative activity, such as glucuronidation (UGT) and sulfation (SULT), at regular intervals; and establish the preliminary toxicological relevance. CYP1A2, CYP3A4, CYP2C9, CYP2C19, CYP2D6, and CYP2B6 have been deemed to be the most import313 phase I enzymes relevant to human drug response; expanded lists can be found in literature150,314,315 and FDA guidance documentations. While full characterization of all enzymatic activity might be cost prohibitive in a purely academic setting, it could very well be established if the platform moves towards heavy commercialization.
A more recent trend in the industry is the push towards better understanding of drug transporter activity in addition to the characterization of the phase I and II enzymatic activity; this is a crucial change since the availability of a drug for metabolism is highly dependent on its kinetics and ability to be transported inside and outside of a cell. Drug transporters play a prominent role in absorption, distribution and elimination of drugs149; thus, their failure or reduced function in-vitro would result in seemingly reduced or impaired metabolism even if the drug metabolism enzymes themselves are intact. Moreover, when a patient takes multiple drugs, they compete for transport pathways resulting in a completely different kinetic and dynamic metabolic picture for both drugs; accordingly an understanding of the way drug transporters handle such interactions has become a crucial piece of the puzzle towards predicting human drug response149. It’s for these reasons that we suggest that a successful in-vitro liver analog should aim to maintain high and stable drug transporter activity; testing for these transporters (Table 3) can be performed by measuring drug uptake and secretion316 as well as by measuring inhibition149. FDA provides general guidance documents for such transporter activity measurements especially in the context of drug-drug interactions.
The four criteria we describe 1) viability, 2) secretory capacity, 3) toxicologically-relevant enzymatic activity, and 4) drug transporter activity should take place in vetting an in-vitro platform for drug based-studies. These seem minimally appropriate before moving on to the next important and final stage, the demonstration of predictive capabilities, in establishing a successful in-vitro liver platform for drug and toxicity testing.
4.1.3. Stage 3: Specificity and sensitivity of in-vitro liver systems
The most important challenge for any platform is to test its’ sensitivity and specificity with a large panel (>100) of diverse drugs of different categories, over long culture terms. These panels should ideally include multiple known drug examples from four categories which are a) safe and efficacious (SE), b) safe but not efficacious (SNE), c) not safe but efficacious (NSE), and d) not safe and not efficacious (NSNE) drugs. Once these panels are established, the platform can be challenged systematically with a range of doses of these drugs; short-term (acute) IC50/LD50 tests as wells as long-term functional marker (albumin, urea) and enzymatic assays can then be performed to assess the dose-response and metabolism of each compound. The next step is then to establish the predictive capabilities of the given platform based on the toxicity correlation between the in-vitro dose response and the in-vivo data available for these known compounds. In this context, two important measures of the success of each platform are a) sensitivity, fraction of correctly predicted positives to all positives in-vivo and b) specificity, the fraction of correctly predicted negatives to all negatives in-vivo where positive or negative is defined in terms of a compounds toxicity. Three examples of such studies with large drug panels (N~45) are demonstrated by Xu et al.317, Khetani et al.245 and Atienzar et al.251
The establishment of a standardized procedure/checklist falls onto the shoulders of the general research community as a whole, funding and overseeing agencies, as well as the end users — the pharmaceutical industry. The FDA, in this regard, provides crucial guidelines which can form the basis of a starting point.
4.2 Zonal recapitulation
As we have discussed above, the liver cells can display a distinct heterogeneity in their morphology, enzymatic activities, and functional capacity along the liver sinusoid38–41. Such heterogeneity in metabolizing and detoxifying capabilities may have significant ramifications in drug testing and screening but this heterogeneity is seldom considered or carefully reproduced in current in-vitro models9. One example that explicitly focuses on recapitulation of zonation is that by Allen and Bhatia318,319 where they use the flat plate bioreactor geometry and active media oxygenation system to induce oxygen gradients across the length of the device. They confirmed the formation of these gradients both through measurements as well as theoretical modeling approaches. They were able to induce a heterogeneous distribution of PEPCK and CYPS similar to in-vivo and demonstrated varying APAP toxicity akin to an in-vivo zonal response. Nevertheless, even this study relies on a passive gradient formation and it is unclear how stable the gradients would be if the system is perturbed by random or deterministic effects.
Most other studies focus on the activity of a single zone which usually ends up being Zone 3, the zone with highest phase I activity and lower phase II detoxification. This means that the generation of intermediate metabolites might be exaggerated while detoxification is underestimated; conversely if Zone 1 conditions are induced, toxicity can be minimized due to high phase II activity. One must consider this to be a potential cause of limited prediction capability of many existing in-vitro liver models that could lead to false negatives and positives and accordingly suggest an active strategy towards recapitulation of zonation as an important avenue of further improvement for in-vitro liver models.
4.3 The need for better detection and instrumentation
The microfluidic realm, which offers good control and precision to recapitulate certain aspects of organ systems in-vitro, comes with its own problems. The micro/nano liter volumes used are both a blessing and a burden in that while these techniques minimize the cost of reagents, it also poses a new problem for the biologists and engineers who routinely handle milliliter volumes both in handling and assaying. Where milliliter volumes can be forgiving, microliter volumes can penalize one for the smallest error.
4.3.1 Instrumentation
Pumping and controlling microliter volumes, and distributing the flow at critical junctions require specialized equipment with high degree of precision. The current solution of pumping via controlled syringes or pressure driven pumps are neither inexpensive nor flexible enough in their operation; moreover they occupy an unwarranted large physical space, hampering the portability and ease of use of these otherwise miniature platforms; commercially available high precision pump systems also fail at the task of handling high throughput systems320. These problems put a high cost of entry and a barrier to wide scale use of microfluidic tissue engineering approaches, currently limiting it to specialized labs. Thus, the field would greatly benefit from new, improved, and integrated technologies that can pump, partition and control the flow of microliter volumes which do not add to the size of the platforms. Some potential solutions and new platforms have been proposed, used, or are under development; these include active miniaturized pumping321,322 and valve solutions, integrated microcontrollers on a tissue culture plate sized platform323 as well as passive pumping solutions279,303,324,325 and resistance based flow splitting307. The accelerated development of these solutions will be important in the quest to minimize the cost and foster large scale adoption of microfluidic drug screening platforms.
4.3.2 Detection
Detection, in the microfluidic realm, is an important problem but also an avenue of great opportunity. On the one hand, most biological assays are optimized and geared towards milliliter volumes of the tissue culture plate and the liters of perfusates from organs or tissue sections which cannot be obtained easily in the microfluidic realm; on the other hand the highly controlled aspect of microfluidics present exciting opportunities for automated and real time detection which can offer new insights and a better mechanistic understanding of metabolic processes. Moreover, microfluidic detection methods, once fully realized, can offer higher sensitivity, minimal invasiveness, and more economical and high throughput/high content assays.
The most popular assays that can be somewhat readily transferred to the microfluidic studies from the macroscopic realm are the optical assays that rely on fluorescent staining. While these are extremely useful end-point studies they generally do not provide continuous real-time data. In this respect, reporter cells284,285,326 provide great promise; such cells natively fluoresce upon stimulation or under certain stress conditions giving specific information about the cell state. Sparse use of such cells with different target signals can provide real time information via optical interrogation of the system. The development of microfluidic small volume analyses such as bead based rolling circle amplification327, and microfluidic ELISAs328,329 and enzymatic assays330,331; and their integration onto the microfluidic tissue culture platforms may bring us closer to near real-time analyses of in-vitro microengineered tissue platforms.
Another avenue of development in the low volume detection comes via the translation of traditional analytical chemistry approaches to the microfluidic world; an additional contribution of such approaches is to free up optical channels that would otherwise be used for a similar analyses. Two examples in this come via two different approaches: a) printed microelectrode arrays for microfluidic integration and322,332 b) droplet based analytical chemistry approaches333–338. We refer the interested readers to a series of reviews by Wikswo and colleagues322,332 in which they discuss the importance of new and improved detection methods in the new microfluidic tissue culture realm.
4.4 The need for better materials
Since its adoption by the Whitesides group for micro-molding techniques102,339 in the 1990s, PDMS has become the staple of microfluidic research as well as other platforms requiring small features. PDMS provides low cost and rapid prototyping capabilities, and upon plasma oxidization, can adhere to itself or other materials without adhesives340. Because PDMS is non-toxic, optically transparent, and oxygen permeable, it seems ideal for culturing and imaging of live cells as in the tissue-on-a-chip models. However, PDMS constructs can suffer from water evaporation, resulting in osmolarity shifts in culture media341, reversal back to a hydrophobic state aft er the initial plasma treatment, and the fact that the material itself displays a small amount of background fluorescence. Another limitation of PDMS, oft en cited as an advantage, is its high gas-permeability, i.e. allowing free diffusion of oxygen and CO2; this makes it hard to control oxygen tension required to induce oxygen-based zonal behavior. Nevertheless, the most significant drawback, for the purposes of drug screening, is the problem of absorption301,342; small hydrophobic molecules, a good fraction of new molecular entities current being developed by the pharmaceutical companies, can easily penetrate and perhaps bind to PDMS.
While strategies to overcome some of these problems have been developed, there still remains a void and an urgent need for PDMS alternatives where absorption is a non-issue. Machined glass and hard plastics are costly alternatives that can fulfill this need; a good strategy might be to consider these for late stage production and not prototypes. The research community has also been working on producing and using alternative polymers such as fluorinated PDMS343, photocurable perfluoroethers (PFPE)344, thermoset polyester345, cyclo-olefin polymer342, the common labware polystyrene (PS)342, and many more; yet, these also oft en come with their own drawbacks and the whole issue becomes one of trade-offs340. Therefore, PDMS is still a suitable material for developing an early platform granted one is aware of its limitations, takes these limitations into account for proper analyses, and eventually replaces PDMS with alternative materials depending on the application. A promising candidate in this regard is the development of novel biocompatible styrenic elastomers such as styrene-ethylene/butylene-styrene (SEBS) which retains the ease of micro-molding similar to PDMS but also provides favorable properties such as low adsorption of hydrophobic molecules like PS346. For more detailed accounts of use of PDMS in biological applications we refer the readers two reviews by Mukhopadhyay340 and Berthier et al.347
4.5 Making sense of the data: Scaling of physical and physiological parameters and predictive models of drug response and toxicity
Translating an in-vivo organ system to an in-vitro screening platform and successfully correlating the output back to the response of the real organ, pose several technical challenges and engineering decisions. While the static culture systems are easier to handle, the recent knowledge base270,275,297 indicates that flow systems have an advantage in not only improving the predictivity of the in-vitro liver systems but also help in retaining hepatic phenotype in long-term experiments. When one settles on using a perfused tissue model, the next question is how to scale the dynamic as well as static parameters so as to create the best mimicry in terms of drug clearance or other outcomes that one is interested in.
For device dimensions most scientists would agree that devices which have a height of 100 μm or less are preferable since they start approaching diameters of the liver sinusoids (on the order of 5–10 um), and thus reduce the signal and/or metabolite dilution problems that many macroscopic platforms face. For dynamic parameters (volumetric flow rate, shear rate, drug dosing and clearance) many integrated approaches such as allometric scaling relying on power law scalings across different mammalian species348–350, integrated physiologically based pharmacokinetic (PBPK) modeling approaches275,351–353 and IVIVE approaches354,355 have been proposed both to guide the parameters as well as work around their limitations when trying to make sense of data.
A very important parameter which controls the adsorption, distribution, metabolism, excretion and toxicity (ADME-TOX) in physiological systems, is the exposure time of the tissue to drugs and other xenobiotics; this is called the “residence time” (~10–20 seconds for the liver356). Accordingly a good first approximation, to mimic in-vivo ADME characteristics in in-vitro fluidic systems is expected by using fluidic residence times similar to the physiological values. Some success in improved PBPK modeling and better predictivity has been claimed299; however, a recent study shows that simply matching residence time can be misleading, for example, in predicting drug clearance275. A concern that arises when focusing on matching residence times is with respect to shear stress; while some liver cells such as the LSECs tolerate shear stress well and do actually need it for proper function, hepatocytes can be injured beyond a shear stress of 0.3 dynes/cm2 33; this hampers the longevity in such systems unless engineered solutions such as microgrooves or indirect perfusion of hepatocytes are used. Perhaps an even more important consideration with residence time matching is the fact that, in-vivo, the parenchymal cells are not directly exposed to flow and actually reside in the perisinusoidal space where the concentrations of nutrients and oxygen are most likely different than the sinusoidal space. We have recently conducted both theoretical and computational analysis of matching drug clearances across different geometries where metabolically active cells are directly exposed to flow. Our preliminary results in very simple theoretical models indicate that matching drug clearance might require matching a combination of Peclet number and the aspect ratio (height vs. length) of the two geometries in question.
In addition to meaningful scaling of dynamic and static parameters, an even more important part in the effort to predict human response to drugs, is the computational and analytical models that can allow us to make sense of the data. While the micro and macro in-vitro platforms aim to act as surrogates for human physiology by providing relevant information on ADME-TOX related parameters, the data they provide cannot be of immediate use except maybe for some cases related to toxicity. For example, studies of clearance of a drug in some static platforms might under-predict the real values by as much as an order of magnitude357; opposite cases of over-prediction is just as likely if the data is taken at face value. Therefore, the field is in need of models with predictive capabilities which connect the dots and bridges the gap between the in-vitro platform realm and the in-vivo reality. In this respect there are several promising approaches discussed in the following reviews353,358–360.
4.6 Towards a more predictive in-vitro microfluidic liver platform
For every in-vitro platform there usually is a trade-off between the cost, complexity, robustness, ease of use of and predictive capabilities. The complexity can arise from the geometry, number of handling steps, intricacy of cell seeding and number of cell types. While the more complex models pose a barrier of entry in terms of their ease of use and cost, they may offer better physiological relevance, longevity, and ultimately higher in-vivo in-vitro correlation of drug response. These are three goals that we are embracing in developing a new 3D microfluidic liver platform where we combine advanced microfabrication and culture of four liver cell types in physiologically relevant ratios.
We posit that in order to best mimic the liver physiological response we need to think from the inside out and identify the smallest representative unit of the liver. The sinusoid (Fig. 1c) is possibly the smallest “functional unit”, and our approach is to linearize this 3D tubular structure into a flat design (Fig. 5), where we have two chambers separated by a PET membrane to simulate the barrier function of an endothelial lining. In one recent publication361, we have described a PDMS on PDMS version of this device and demonstrated long-term stability of function with primary hepatocytes. The flat nature of the design was motivated by the need to incorporate optical readouts using reporter cells as well as conducting regular imaging analyses. In more current embodiments, we have been able to modify our design to a PDMS on glass (Fig. 5) and in preliminary studies we have been able to sustain primary human hepatocytes co-cultured with human cell lines that represents stellate, endothelial and Kupffer cells for 28 days. The unique aspect of this approach is that cells in our device are layered similar to the liver sinusoid: LX-2s (a stellate cell line) are introduced in a pre-gel solution between hepatocytes on glass and the membrane that separates the two chambers; EA.hy926 cells (a LSEC cell line) along with Kupffer cell line U937 cells are seeded on the membrane. This layering resembles the separation between space of Disse and the sinusoidal space. We have also developed methods for primary non-parenchymal cell isolation to replace the cell lines.
Figure 5.
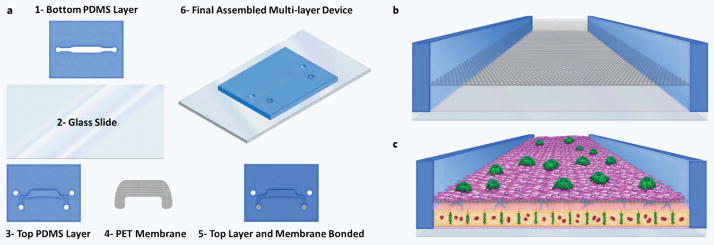
The 3D microfluidic sinusoid analogue developed at our laboratories. (a) Parts and assembly of the current version of our device. Bottom layer (1) is first bonded to the glass slide and then the top section (5) is aligned and bonded to the (1) to create a multi-layer microfluidic device. (b) Cross-sectional view of final assembled device; a PET membrane separate the top and bottom fluidic channels. (c) Final assembled liver sinusoid like tissue in a multi-layer microfluidic device. While human hepatocytes are seeded on an ECM coat on glass slide, stellate cell line is introduced in a pre-gel solution below the membrane. Endothelial cells are introduced on top of the membrane after ECM coating and Kupffer cells are introduced on top of the endothelial lining. The layered structure of this microengineered tissue is similar to the liver sinusoid as depicted in Fig. 1.
The perfusion in our device is conducted on the top layer and further a layer of gel between the hepatocytes and this perfusion provides shear protection for the hepatocytes, akin to the shear protection by endothelium in the liver sinusoid. A guiding principle in our studies is to reduce the media/volume to cell ratio towards that of physiological values; this is driven by the desire to alleviate problems of large fluidic systems which are not well suited to capturing cell-cell or tissue-tissue interactions in connected systems due to dilution of signaling factors. Despite our push towards a more physiological volume/cell ratio, the volume of our device is still relatively large (100 μm in the fluidic top compartment) compared to the volume in a sinusoid with a diameter of 5–10 μm. While further reducing the dimensions would be useful in terms of better capturing interactions this is prohibited by two factors: 1) difficulties in manufacturing and ease of use and 2) more importantly the difficulty of oxygen delivery in such systems without significant advances in artificial oxygen carriers362–364. In terms of drug clearance, as we have discussed above, we have theoretically analyzed this problem to assess if it’s possible to translate clearance values between different geometries such as a device and a sinusoid and found that a combination of channel aspect ratio and Peclet number can be used to guide the flow parameters. As a first approximation we have been using Peclet numbers to primarily guide our flow parameters. Additional computational tools and theoretical analyses will be used in this context to allow us to create a concentration field of nutrients and target drugs that comes as close to the physiological scenario as possible.
Although the initial material used in our platform is PDMS, we are currently testing thermoplastic alternatives that are much better suited for drug screening. We have also developed a new ultrathin collagen coating208 solution for microfluidic applications which circumvents problems that arise when translating hydrogel methodology to the microfluidic realm. We will use this technique to further reduce dimensions in our device and approach more realistic scenarios for interaction between different cell types. While the proposed platform can seem complex, the ultimate aim is to provide both longevity and the best possible representation of the liver physiology resulting in high predictive capability for both acute and chronic drug challenges. As the platform matures and demonstrates its capability, it will be engineered towards more robust handling and ease of use for wider adoption. Further improvements will be made so that it is also more compatible with high-throughput and high-content imaging systems which will be useful tools for drug screening studies.
5. CONCLUSION AND OUTLOOK
It’s evident from this review of in-vitro liver technologies that despite a large body of work conducted by a talented group of scientists and engineers, the field is still somewhat in its infancy in terms of clear standards, procedures and methods for translating in-vitro results to reliable in-vivo predictions. For this very reason funding and direction by government agencies such as in the US and EU are needed to accelerate advances in the field; especially in terms of establishing good practices, standardized challenges, overall characterization of new proposed systems. Despite the excitement currently felt by many in the field, there are critics from both industry and academia who view the current push as headlong and not very thoughtful. Whether it be the “top-down” 10 tissue mandate of DARPA or the “bottom up” tissue integration push of the NIH, critics claim that sub-par systems are being created and connected, whereas more emphasis should be placed on getting individual tissue chips to demonstrate utility and real value first. The pharmaceutical industry views these projects as interesting but sees little possibility of implementation in an industry that wants and needs robust, cost-effective, simple solutions to their problems. The current focus on human cells, although laudatory, may also be misplaced as the bulk of the toxicity data that exists is rat and other animal data and not human data. To some the smarter approach would be to develop all rat tissue models that provide excellent correlations with in-vivo results. Finally, there seems to be a disconnect between some the major problems that exist in drug development and what these systems may provide. It is true that if there is conflicting data among the safety results obtained from different animal species, that a human surrogate might help in deciding whether to pursue further drug development or not. In this case the most predictive, cost-effective system will win out. However, if the problem being considered is the late recognition of toxicity as larger and larger groups of clinical trial phase populations are treated, then these systems may never yield a solution, unless they are developed to represent very large populations of individuals. In theory, thousands of different tissues developed from thousands of IPS cells might be considered a pathway towards a solution, albeit cost-prohibitive in development, and questionable as to its utility until rigorously tested for its predictivity.
On the scientific front, which continue to excite academics and others for their utility in understanding health and disease, many improvements are still needed in terms of tissue engineering materials, and fabrication to engineer more physiologically relevant cellular environments; detection and analytical schemes to improve measurements and better dissect mechanisms of action; theoretical and computational approaches to better bridge the results between in-vitro experiments and in-vivo observations. This means a generation of fantastic opportunities and exciting challenges for scientists and engineers trying address these gaps with innovative ideas and solutions.
Acknowledgments
This work was supported by NIH grants, including a microphysiological systems consortium grant from the NCATS (UH2TR000503), a Ruth L. Kirschstein NRSA Postdoctoral Fellowship from the NIDDK(F32DK098905 for WJM), a K99 — Path to Independence award (K99DK095984 for AB) and a Shriners Fellowship (#84202 for OBU) from the Shriners Hospitals for Children. We thank Robert Freedman of Hμrel Corporation for providing an image of their platform. We thank Dr. Amitabh Bhattacharya (IIT Bombay) for his work on drug clearance scaling studies.
Footnotes
The authors, with the exception of Dr. Martin L. Yarmush, have no professional or financial conflicts, or interest to disclose. Dr. Yarmush serves as a scientific advisor to the Hμrel Corporation.
References
- 1.DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: New estimates of drug development costs. J Health Econ. 2003;22(2):151–185. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 2.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 3.Adams CP, Brantner VV. Spending on new drug development. Health Econ. 2010;19(2):130–141. doi: 10.1002/hec.1454. [DOI] [PubMed] [Google Scholar]
- 4.Adams CP, Brantner VV. Estimating the cost of new drug development: Is it really 802 million dollars? Health Aff (Millwood) 2006;25(2):420–428. doi: 10.1377/hlthaff.25.2.420. [DOI] [PubMed] [Google Scholar]
- 5.Schuster D, Laggner C, Langer T. Why drugs fail — A study on side effects in new chemical entities. Curr Pharm Des. 2005;11(27):3545–3559. doi: 10.2174/138161205774414510. [DOI] [PubMed] [Google Scholar]
- 6.Tollman P, Guy P, Altshuler J, Flanagan A, Steiner M. A Revolution in R&D: How Genomics and Genetics Are Transforming the Biopharmaceutical Industry. Boston Consulting Group; 2001. http://www.bcg.com/documents/file13745.pdf. [Google Scholar]
- 7.Greek R. In: New Insights into Toxicity and Drug Testing. Gowder S, editor. InTech; 2013. pp. 123–152. [Google Scholar]
- 8.Shanks N, Greek R, Greek J. Are animal models predictive for humans? Philos Ethics Humanit Med. 2009;4:2. doi: 10.1186/1747-5341-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guillouzo A. Liver cell models in in vitro toxicology. Env Health Perspect. 1998;106(Suppl 2):511–532. doi: 10.1289/ehp.98106511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wheler C. Replacement, Reduction and Refinement Alternatives: Implementation of the 3 R Alternatives When Using Animals in Science. [presentation], http://www.uregina.ca/research/About_ORS/Workshops/Presentations/Colette_Wheler_3Rs_Presentation.pdf.
- 11.Russell WMS, Burch RL. The Principles of Humane Experimental Technique. Methuen; 1959. [Google Scholar]
- 12.European Commission. Full EU Ban on Animal Testing for Cosmetics Enters into Force. 2013 http://europa.eu/rapid/press-release_IP-13-210_en.htm.
- 13.Hardisty JF, Brix AE. Comparative hepatic toxicity: Prechronic/chronic liver toxicity in rodents. Toxicol Pathol. 2005;33:35–40. doi: 10.1080/01926230590522077. [DOI] [PubMed] [Google Scholar]
- 14.Blomme EA, Yang Y, Waring JF. Use of toxicogenomics to understand mechanisms of drug-induced hepatotoxicity during drug discovery and development. Toxicol Lett. 2009;186:22–31. doi: 10.1016/j.toxlet.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Maddrey WC. Drug-induced hepatotoxicity. J Clin Gastroenterol. 2005;39:S83–S89. doi: 10.1097/01.mcg.0000155548.91524.6e. [DOI] [PubMed] [Google Scholar]
- 16.Wilke RA, Lin DW, Roden DM, Watkins PB, Flockhart D, et al. Identifying genetic risk factors for serious adverse drug reactions: Current progress and challenges. Nat Rev Drug Discov. 2007;6(11):904–916. doi: 10.1038/nrd2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.U.S. FDA. Guidance for Industry Drug-Induced Liver Injury: Premarketing Clinical Evaluation. 2009 http://www.fda.gov/downloads/Drugs/.../Guidances/UCM174090.pdf.
- 18.Zguris JC, Itle LJ, Pishko MV. Microreactor microfluidic systems with human microsomes and hepatocytes for use in metabolite studies. Biomed Microdevices. 2005;7:117–125. doi: 10.1007/s10544-005-1589-9. [DOI] [PubMed] [Google Scholar]
- 19.Emoto C, Murase S, Iwasaki K. Approach to the prediction of the contribution of major cytochrome P450 enzymes to drug metabolism in the early drug-discovery stage. Xenobiotica. 2006;36(8):671–683. doi: 10.1080/00498250600709778. [DOI] [PubMed] [Google Scholar]
- 20.Getek TA, et al. Utility of solution electrochemistry mass spectrometry for investigating the formation and detection of biologically important conjugates of acetaminophen. J Chromatogr. 1989;474(1):245–256. doi: 10.1016/s0021-9673(01)93919-6. [DOI] [PubMed] [Google Scholar]
- 21.Odijk M, et al. Electrochemistry-on-chip for on-line conversions in drug metabolism studies. Biosens Bioelectron. 2010;26(4):1521–1527. doi: 10.1016/j.bios.2010.07.102. [DOI] [PubMed] [Google Scholar]
- 22.Griffin SJ, Houston JB. Comparison of fresh and cryopreserved rat hepatocyte suspensions for the prediction of in vitro intrinsic clearance. Drug Metab Dispos. 2004;32(5):552–558. doi: 10.1124/dmd.32.5.552. [DOI] [PubMed] [Google Scholar]
- 23.Griffin SJ, Houston JB. Prediction of in vitro intrinsic clearance from hepatocytes: Comparison of suspensions and monolayer cultures. Drug Metab Dispos. 2005;33(1):115–120. doi: 10.1124/dmd.33.1.. [DOI] [PubMed] [Google Scholar]
- 24.Moldeus P. Paracetamol metabolism and toxicity in isolated hepatocytes from rat and mouse. Biochem Pharmacol. 1978;27(24):2859–2863. doi: 10.1016/0006-2952(78)90201-0. [DOI] [PubMed] [Google Scholar]
- 25.Vanhaecke T, Rogiers V. Hepatocyte cultures in drug metabolism and toxicological research and testing. Methods Mol Biol. 2006;320:209–227. doi: 10.1385/1-59259-998-2:209. [DOI] [PubMed] [Google Scholar]
- 26.Sokol RJ, et al. Evidence for involvement of oxygen free radicals in bile acid toxicity to isolated rat hepatocytes. Hepatology. 1993;17(5):869–881. [PubMed] [Google Scholar]
- 27.Kataropoulou M, Henderson C, Grant MH. Metabolic studies of hepatocytes cultured on collagen substrata modified to contain glycosaminoglycans. Tissue Eng. 2005;11(7–8):1263–1273. doi: 10.1089/ten.2005.11.1263. [DOI] [PubMed] [Google Scholar]
- 28.Holme JA, Soderlund E, Dybing E. Drug metabolism activities of isolated rat hepatocytes in monolayer culture. Acta Pharmacol Toxicol (Copenh ) 1983;52(5):348–356. doi: 10.1111/j.1600-0773.1983.tb01114.x. [DOI] [PubMed] [Google Scholar]
- 29.Lindblad WJ, et al. Hepatocellular phenotype in vitro is influenced by biophysical features of the collagenous substratum. Hepatology. 1991;13(2):282–288. [PubMed] [Google Scholar]
- 30.Dunn JC, Tompkins RG, Yarmush ML. Long-term in vitro function of adult hepatocytes in a collagen sandwich configuration. Biotechnol Prog. 1991;7:237–245. doi: 10.1021/bp00009a007. [DOI] [PubMed] [Google Scholar]
- 31.Dunn J, Yarmush ML, Koebe HG, Tompkins RG. Hepatocyte function and extracellular matrix geometry: Long-term culture in a sandwich configuration. FASEB J. 1989;3(2):174–177. doi: 10.1096/fasebj.3.2.2914628. [DOI] [PubMed] [Google Scholar]
- 32.Tilles AW, Baskaran H, Roy P, Yarmush ML, Toner M. Effects of oxygenation and flow on the viability and function of rat hepatocytes cocultured in a microchannel flat-plate bioreactor. Biotechnol Bioeng. 2001;73:379–389. doi: 10.1002/bit.1071. [DOI] [PubMed] [Google Scholar]
- 33.Tilles AW, Berthiaume F, Yarmush ML, Tompkins RG, Toner M. Bioengineering of liver assist devices. J Hepatobiliary Pancreat Surg. 2002;9:686–696. doi: 10.1007/s005340200095. [DOI] [PubMed] [Google Scholar]
- 34.Roy P, Baskaran H, Tilles AW, Yarmush ML, Toner M. Analysis of oxygen transport to hepatocytes in a flat-plate microchannel bioreactor. Ann Biomed Eng. 2001;29:947–955. doi: 10.1114/1.1415524. [DOI] [PubMed] [Google Scholar]
- 35.Shinoda M, Tilles AW, Kobayashi N, Wakabayashi G, et al. A bioartificial liver device secreting interleukin-1 receptor antagonist for the treatment of hepatic failure in rats. J Surg Res. 2007;137:130–140. doi: 10.1016/j.jss.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patzer JF., II Oxygen consumption in a hollow fiber bioartificial liver — Revisited. Artif Organs. 2004;28:83–98. doi: 10.1111/j.1525-1594.2004.07150.x. [DOI] [PubMed] [Google Scholar]
- 37.Kelly JH, Sussman NL. The hepatix extracorporeal liver assist device in the treatment of fulminant hepatic failure. ASAIO J. 1994;40(1):83–85. [PubMed] [Google Scholar]
- 38.Sussman NL, Gislason GT, Conlin CA, Kelly JH. The Hepatix extracorporeal liver assist device: Initial clinical experience. Artif Organs. 1994;18(5):390–396. doi: 10.1111/j.1525-1594.1994.tb02221.x. [DOI] [PubMed] [Google Scholar]
- 39.Rozga J, Holzman MD, Ro MS, Griffin DW, Neuzil DF, Giorgio T, Moscioni AD, Demetriou AA. Development of a hybrid bioartificial liver. Ann Surg. 1993;217(5):502–509. doi: 10.1097/00000658-199305010-00010. discussion 509–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van de Bovenkamp M, Groothius GM, Meijer DK, Olinga P. Precision-cut fibrotic rat liver slices as a new model to test the effects of anti-fibrotic drugs in vitro. J Hepatol. 2006;45:696–703. doi: 10.1016/j.jhep.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 41.Olinga P, Schuppan D. Precision-cut liver slices: A tool to model the liver ex vivo. J Hepatol. 2013;58(6):1252–1253. doi: 10.1016/j.jhep.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 42.van de Bovenkamp M, Groothius GM, Draaisma AL, Merema MT, Bezuijen JI, van Glis MJ, Meijer DK, Friedman SL, Olinga P. Precision-cut liver slices as a new model to study toxicity-induced hepatic stellate cell activation in a physiologic milieu. Toxicol Sci. 2005;85:632–638. doi: 10.1093/toxsci/kfi127. [DOI] [PubMed] [Google Scholar]
- 43.Vickers AEM, Saulnier M, Cruz E, Merema MT, Rose K, Bentley P, Olinga P. Organ slice viability extended for pathway characterization: An in vitro model to investigate fibrosis. Toxicol Sci. 2004;82:534–544. doi: 10.1093/toxsci/kfh285. [DOI] [PubMed] [Google Scholar]
- 44.Stieger B. In: Textbook of Hepatology. Rodes J, Benhamou J-P, Blei A, Reichen J, Rizzetto M, editors. Blackwell; 2007. [Google Scholar]
- 45.Lang R, Stern MM, Smith L, Liu Y, et al. Three-dimensional culture of hepatocytes on porcine liver tissue-derived extracellular matrix. Biomaterials. 2011;32(29):7042–7052. doi: 10.1016/j.biomaterials.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 46.Uygun BE, Soto-Gutierrez A, Yaqi H, Izamis ML, et al. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat Med. 2010;16:814–820. doi: 10.1038/nm.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gibb S. Toxicity testing in the 21st century: A vision and a strategy. Reprod Toxicol. 2008;25(1):136–138. doi: 10.1016/j.reprotox.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 48.U.S. National Research Council (Committee on Toxicity Testing and Assessment of Environmental Agents) Toxicity Testing in the 21st Century: A Vision and a Strategy. National Academies Press; 2007. p. xvii.p. 196. [Google Scholar]
- 49.SEURAT-1. Towards the Replacement of In Vivo Repeated Dose Systemic Toxicology Testing. 2013 http://www.seurat-1.eu/
- 50.European Commission CORDIS. Projects & Results Service: The Body-on-a-Chip (BoC) 2014 http://cordis.europa.eu/project/rcn/104027_en.html.
- 51.U.S. NIH. Integrated Microphysiological Systems for Drug Efficacy and Toxicity Testing in Human Health and Disease (UH2/UH3) 2012 http://grants.nih.gov/grants/guide/rfa-files/RFA-RM-11-022.html.
- 52.DARPA. Microphysiological Systems. 2012 http://www.darpa.mil/Our_Work/BTO/Programs/MicrophysiologicalSystems.aspx.
- 53.Guyton AC, Hall JE. Textbook of of Medical Physiology. Saunders; 2006. [Google Scholar]
- 54.Reed S. Essential Physiological Biochemistry — An Organ-based Approach. Wiley; 2009. [Google Scholar]
- 55.Hofmann AF, Hagey LR. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65(16):2461–2483. doi: 10.1007/s00018-008-7568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boyer TD, Wright TL, Manns MP. Zakim and Boyer’s Hepatology: A Textbook of Liver Disease. Elsevier Saunders; 2006. p. xv.p. 1516. [Google Scholar]
- 57.Rodes J, Benhamou J-P, Blei A, Reichen J, Rizzetto M. Textbook of Hepatology. Blackwell; 2007. [Google Scholar]
- 58.Arias IM. In: The Liver: Biology and Pathobiology. Arias IM, Jakoby H, Popper D, Schachter D, Shafritz D, editors. Wiley; 2009. [Google Scholar]
- 59.McCuskey RS. The hepatic microvascular system in health and its response to toxicants. Anat Rec (Hoboken) 2008;291:661–671. doi: 10.1002/ar.20663. [DOI] [PubMed] [Google Scholar]
- 60.Ito Y, McCuskey RS. In: Textbook of Hepatology. Rodes J, Benhamou J-P, Blei A, Reichen J, Rizzetto M, editors. Blackwell; 2007. [Google Scholar]
- 61.Khokha MK, Landini G, Iannaccone PM. Fractal geometry in rat chimeras demonstrates that a repetitive cell division program may generate liver parenchyma. Dev Biol. 1994;165(2):545–555. doi: 10.1006/dbio.1994.1274. [DOI] [PubMed] [Google Scholar]
- 62.Lin DW, Johnson S, Hunt CA. Modeling liver physiology: Combining fractals, imaging and animation. Conf Proc IEEE Eng Med Biol Soc. 2004;5:3120–3123. doi: 10.1109/IEMBS.2004.1403881. [DOI] [PubMed] [Google Scholar]
- 63.Ng YK, Iannaccone PM. Fractal geometry of mosaic pattern demonstrates liver regeneration is a self-similar process. Dev Biol. 1992;151(2):419–430. doi: 10.1016/0012-1606(92)90182-g. [DOI] [PubMed] [Google Scholar]
- 64.Jungermann K, Kietzmann T. Zonation of parenchymal and non-parenchymal metabolism in liver. Annu Rev Nutr. 1996;16:179–203. doi: 10.1146/annurev.nu.16.070196.001143. [DOI] [PubMed] [Google Scholar]
- 65.Kuntz E SpringerLink (Online service) Hepatology, Principles and Practice History: Morphology, Biochemistry, Diagnostics, Clinic, Therapy. Springer-Verlag; 2006. [Google Scholar]
- 66.Senoo H, Yoshikawa K, Morii M, Miura M, Imai K, Mezaki Y. Hepatic stellate cell (vitamin A-storing cell) and its relative — Past, present and future. Cell Biol Int. 2010;34:1247–1272. doi: 10.1042/CBI20100321. [DOI] [PubMed] [Google Scholar]
- 67.Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–163. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- 68.Rappaport AM. The microcirculatory hepatic unit. Microvasc Res. 1973;6:212–228. doi: 10.1016/0026-2862(73)90021-6. [DOI] [PubMed] [Google Scholar]
- 69.Nahmias Y, Berthiaume F, Yarmush ML. Integration of technologies for hepatic tissue engineering. Adv Biochem Eng Biotechnol. 2008;103:309–329. doi: 10.1007/10_029. [DOI] [PubMed] [Google Scholar]
- 70.Roberts RA, Ganey PE, Ju C, Kamendulis LM, Rusyn I, Klaunig JE. Role of the Kupffer cell in mediating hepatic toxicity and carcinogenesis. Toxicol Sci. 2007;96(1):2–15. doi: 10.1093/toxsci/kfl173. [DOI] [PubMed] [Google Scholar]
- 71.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26:1175–1186. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 72.Laskin DL. Sinusoidal lining cells and hepatotoxicity. Toxicol Pathol. 1996;24:112–118. doi: 10.1177/019262339602400115. [DOI] [PubMed] [Google Scholar]
- 73.Zakim D, Boyer TD. Zakim and Boyer’s Hepatology: A Textbook of Liver Disease. Elsevier Saunders; 2003. p. xxiv.p. 1765. [Google Scholar]
- 74.LeCluyse EL, Witek RP, Andersen ME, Powers MJ. Organotypic liver culture models: Meeting current challenges in toxicity testing. Crit Rev Toxicol. 2012;42(6):501–48. doi: 10.3109/10408444.2012.682115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morin O, Normand C. Long-term maintenance of hepatocyte functional activity in co-culture: Requirements for sinusoidal endothelial cells and dexamethasone. J Cell Physiol. 1986;129:103–110. doi: 10.1002/jcp.1041290115. [DOI] [PubMed] [Google Scholar]
- 76.Blouin A, Bolender RP, Weibel ER. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J Cell Biol. 1977;72:441–455. doi: 10.1083/jcb.72.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weibel ER, Stäubli W, Gnägi HR, Hess FA. Correlated morphometric and biochemical studies on the liver cell I. Morphometric model, stereologic methods, and normal morphometric data for rat liver. J Cell Biol. 1969;42(1):68–81. doi: 10.1083/jcb.42.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loud AV. A quantitative stereological description of the ultrastructure of normal rat liver parenchymal cells. J Cell Biol. 1968;37(1):27–46. doi: 10.1083/jcb.37.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Balis UJ, Behnia K, Dwarakanath B, Bhatia SN, Sullivan SJ, Yarmush ML, Toner M. Oxygen consumption characteristics of porcine hepatocytes. Metab Eng. 1999;1(1):49–62. doi: 10.1006/mben.1998.0105. [DOI] [PubMed] [Google Scholar]
- 80.Steinberg P, Lafranconi WM, Wolf CR, Waxman DJ, Oesch F, Friedberg T. Xenobiotic metabolizing enzymes are not restricted to parenchymal cells in rat liver. Mol Pharmacol. 1987;32(4):463–470. [PubMed] [Google Scholar]
- 81.Deleve LD. Dacarbazine toxicity in murine liver cells: A model of hepatic endothelial injury and glutathione defense. J Pharmacol Exp Ther. 1994;268:1261–1270. [PubMed] [Google Scholar]
- 82.Nedredal GI, Elvevold K, Ytrebø LM, et al. Significant contribution of liver nonparenchymal cells to metabolism of ammonia and lactate and cocultivation augments the functions of a bioartificial liver. Am J Physiol Gastrointest Liver Physiol. 2007;293(1):G75–G83. doi: 10.1152/ajpgi.00245.2006. [DOI] [PubMed] [Google Scholar]
- 83.Wisse E, Braet F, Luo D, de Zanger RB, Jans D, Crabbé E, Vermoesen A. Structure and function of sinusoidal lining cells in the liver. Toxicol Pathol. 1996;24:100–111. doi: 10.1177/019262339602400114. [DOI] [PubMed] [Google Scholar]
- 84.Arii S, Imamura M. Physiological role of sinusoidal endothelial cells and Kupffer cells and their implication in the pathogenesis of liver injury. J Hepatobil Pancreat Surg. 2000;7:40–48. doi: 10.1007/s005340050152. [DOI] [PubMed] [Google Scholar]
- 85.Kim Y, Rajagopalan P. 3D hepatic cultures simultaneously maintain primary hepatocyte and liver sinusoidal endothelial cell phenotypes. PLoS One. 2010;5:e15456. doi: 10.1371/journal.pone.0015456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smedsrød B, Pertoft H, Eggertsen G, Sundström C. Functional and morphological characterization of cultures of Kupffer cells and liver endothelial cells prepared by means of density separation in Percoll, and selective substrate adherence. Cell Tissue Res. 1985;241:639–649. doi: 10.1007/BF00214586. [DOI] [PubMed] [Google Scholar]
- 87.Xie G, Wang L, Wang X, Wang L, DeLeve LD. Isolation of periportal midlobular and centrilobular rat liver sinusoidal endothelial cells enables study of zonated drug toxicity. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1204–G1210. doi: 10.1152/ajpgi.00302.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wisse E, de Zanger RB, Charels K, van der Smissen P, McCuskey RS. The liver sieve: Considerations concerning the structure and function of endothelial fenestrae, the sinusoidal wall and the space of Disse. Hepatology. 1985;5(4):683–692. doi: 10.1002/hep.1840050427. [DOI] [PubMed] [Google Scholar]
- 89.Smedsrød B, de Bleser PJ, Braet F, Lovisetti P, Vanderkerken L, Wisse E, Geerts A. Cell biology of liver endothelial and Kupffer cells. Gut. 1994;35(11):1509–1516. doi: 10.1136/gut.35.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.March S, Hui EE, Underhill GH, Khetani S, Bhatia SN. Microenvironmental regulation of the sinusoidal endothelial cell phenotype in vitro. Hepatology. 2009;50:920–928. doi: 10.1002/hep.23085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fraser R, Dobbs BR, Rogers GW. Lipoproteins and the liver sieve: The role of the fenestrated sinusoidal endothelium in lipoprotein metabolism, atherosclerosis, and cirrhosis. Hepatology. 1995;21(3):863–874. [PubMed] [Google Scholar]
- 92.Arias IM. The biology of hepatic endothelial cell fenestrae. Prog Liver Dis. 1990;9:11–26. [PubMed] [Google Scholar]
- 93.Smedsrød B, Johansson S, Pertoft H. Studies in vivo and in vitro on the uptake and degradation of soluble collagen alpha 1(I) chains in rat liver endothelial and Kupffer cells. Biochem J. 1985;228(2):415–424. doi: 10.1042/bj2280415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smedsrød B, Pertoft H, Eriksson S, Fraser JR, Laurent TC. Studies in vitro on the uptake and degradation of sodium hyaluronate in rat liver endothelial cells. Biochem J. 1984;223(3):617–626. doi: 10.1042/bj2230617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seternes T, Sørensen K, Smedsrød B. Scavenger endothelial cells of vertebrates: A nonperipheral leukocyte system for high-capacity elimination of waste macromolecules. Proc Natl Acad Sci USA. 2002;99(11):7594–7597. doi: 10.1073/pnas.102173299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rockey DC. Hepatic blood flow regulation by stellate cells in normal and injured liver. Semin Liver Dis. 2001;21(3):337–349. doi: 10.1055/s-2001-17551. [DOI] [PubMed] [Google Scholar]
- 97.Winau F, Quack C, Darmoise A, Kaufmann SH. Starring stellate cells in liver immunology. Curr Opin Immunol. 2008;20:68–74. doi: 10.1016/j.coi.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 98.Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol. 2010;196:369–405. doi: 10.1007/978-3-642-00663-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65(2):166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 100.Connolly MK, Bedrosian AS, Malhotra A, Henning JR, et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immunogenicity. J Immunol. 2010;185(4):2200–2208. doi: 10.4049/jimmunol.1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kang YB, Rawat S, Cirillo J, Bouchard M, Noh HM. Layered long-term co-culture of hepatocytes and endothelial cells on a transwell membrane: Toward engineering the liver sinusoid. Biofabrication. 2013;5(4):045008. doi: 10.1088/1758-5082/5/4/045008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Duffy DC, McDonald JC, Schueller OJA, Whitesides GM. Rapid prototyping of microfluidic systems in poly(dimethylsiloxane) Anal Chem. 1998;70(23):4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- 103.Elvevold K, Smedsrød B, Martinez I. The liver sinusoidal endothelial cell: A cell type of controversial and confusing identity. Am J Physiol Gastrointest Liver Physiol. 2008;294(2):G391–G400. doi: 10.1152/ajpgi.00167.2007. [DOI] [PubMed] [Google Scholar]
- 104.McCuskey RS, et al. Hepatic microvascular development in relation to the morphogenesis of hepatocellular plates in neonatal rats. Anat Rec A Discov Mol Cell Evol Biol. 2003;275:1019–1030. doi: 10.1002/ar.a.10117. [DOI] [PubMed] [Google Scholar]
- 105.Morin O, Goulet F, Normand C. Liver sinusoidal endothelial cells: Isolation, purification, characterization and interaction with hepatocytes. Revis Biol Cel. 1988;15:1–85. [PubMed] [Google Scholar]
- 106.Kupffer C. Ueber sternzellen der leber [in German] Archiv Mikrosk Anat. 1876;12(1):353–358. [Google Scholar]
- 107.Haubrich WS. Kupffer of Kupffer cells. Gastroenterology. 2004;127(1):16. doi: 10.1053/j.gastro.2004.05.041. [DOI] [PubMed] [Google Scholar]
- 108.Liu H, Cao H, Wu ZY. Isolation of Kupffer cells and their suppressive effects on T lymphocyte growth in rat orthotopic liver transplantation. World J Gastroenterol. 2007;13:3133–3136. doi: 10.3748/wjg.v13.i22.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Valatas V, Xidakis C, Roumpaki H, Kolios G, Kouroumalis EA. Isolation of rat Kupffer cells: A combined methodology for highly purified primary cultures. Cell Biol Int. 2003;27:67–73. doi: 10.1016/s1065-6995(02)00249-4. [DOI] [PubMed] [Google Scholar]
- 110.Selzner N, Selzner M, Odermatt B, Tian Y, Van Rooijen N, Clavien PA. ICAM-1 triggers liver regeneration through leukocyte recruitment and Kupffer cell-dependent release of TNF-alpha/IL-6 in mice. Gastroenterology. 2003;124(3):692–700. doi: 10.1053/gast.2003.50098. [DOI] [PubMed] [Google Scholar]
- 111.Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: Deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci USA. 1997;94(4):1441–1446. doi: 10.1073/pnas.94.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baffy G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J Hepatol. 2009;51:212–223. doi: 10.1016/j.jhep.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Solga SF, Diehl AM. Non-alcoholic fatty liver disease: Lumen–liver interactions and possible role for probiotics. J Hepatol. 2003;38(5):681–687. doi: 10.1016/s0168-8278(03)00097-7. [DOI] [PubMed] [Google Scholar]
- 114.Bilzer M, Gerbes AL. Preservation injury of the liver: Mechanisms and novel therapeutic strategies. J Hepatol. 2000;32(3):508–515. doi: 10.1016/s0168-8278(00)80404-3. [DOI] [PubMed] [Google Scholar]
- 115.Caldwell-Kenkel JC, Currin RT, Tanaka Y, Thurman RG, Lemasters JJ. Kupffer cell activation and endothelial cell damage after storage of rat livers: Effects of reperfusion. Hepatology. 1991;13(1):83–95. [PubMed] [Google Scholar]
- 116.Videla LA, Fernández V, Tapia G, Varela P. Oxidative stress-mediated hepatotoxicity of iron and copper: Role of Kupffer cells. Biometals. 2003;16(1):103–111. doi: 10.1023/a:1020707811707. [DOI] [PubMed] [Google Scholar]
- 117.Milosevic N, Schawalder H, Maier P. Kupffer cell-mediated differential down-regulation of cytochrome P450 metabolism in rat hepatocytes. Eur J Pharmacol. 1999;368(1):75–87. doi: 10.1016/s0014-2999(98)00988-1. [DOI] [PubMed] [Google Scholar]
- 118.Life Technologies. Hepatic Co-Cultures with Hepatocytes and Kupffer Cells. http://www.lifetechnologies.com/us/en/home/life-science/drug-discovery/adme-tox/hepatic-systems/microsomes-s9-fractions-cytosol.html.
- 119.Wake K. “Sternzellen” in the liver: Perisinusoidal cells with special reference to storage of vitamin A. Am J Anat. 1971;132(4):429–462. doi: 10.1002/aja.1001320404. [DOI] [PubMed] [Google Scholar]
- 120.Wake K. Perisinusoidal stellate cells (fat-storing cells, interstitial cells, lipocytes), their related structure in and around the liver sinusoids, and vitamin A-storing cells in extrahepatic organs. Int Rev Cytol. 1980;66:303–353. doi: 10.1016/s0074-7696(08)61977-4. [DOI] [PubMed] [Google Scholar]
- 121.Ghyselinck NB, Båvik C, Sapin V, Mark M, Bonnier D, Hindelang C, et al. Cellular retinol-binding protein I is essential for vitamin A homeostasis. EMBO J. 1999;18:4903–4914. doi: 10.1093/emboj/18.18.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maschmeyer P, Flach M, Winau F. Seven steps to stellate cells. J Vis Exp. 2011;51:2710. doi: 10.3791/2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48(3):920–930. doi: 10.1002/hep.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kostadinova R, Boess F, Applegate D, Suter L, Weiser T, Singer T, Naughton B, Roth A. A long-term three dimensional liver co-culture system for improved prediction of clinically relevant drug-induced hepatotoxicity. Toxicol Appl Pharmacol. 2013;268:1–16. doi: 10.1016/j.taap.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 125.Friedman SL. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jungermann K, Kietzmann T. Oxygen: Modulator of metabolic zonation and disease of the liver. Hepatology. 2000;31(2):255–260. doi: 10.1002/hep.510310201. [DOI] [PubMed] [Google Scholar]
- 127.Jungermann K. Zonation of metabolism and gene expression in liver. Histochem Cell Biol. 1995;103:81–91. doi: 10.1007/BF01454004. [DOI] [PubMed] [Google Scholar]
- 128.Bhatia SN, Toner M, Foy BD, Rotem A, Tompkins RG, Yarmush ML. Zonal liver cell heterogeneity: Effects of oxygen on metabolic functions of hepatocytes. J Cell Eng. 1996;1:125–133. [Google Scholar]
- 129.Traber PG, Chianale J, Gumucio JJ. Physiologic significance and regulation of hepatocellular heterogeneity. Gastroenterology. 1988;95(4):1130–1143. doi: 10.1016/0016-5085(88)90194-1. [DOI] [PubMed] [Google Scholar]
- 130.Wojcik E, Dvorak C, Chianale J, Traber PG, Keren D, Gumucio JJ. Demonstration by in situ hybridization of the zonal modulation of rat liver cytochrome P-450b and P-450e gene expression after phenobarbital. J Clin Invest. 1988;82(2):658–666. doi: 10.1172/JCI113645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rappaport AM. The microcirculatory acinar concept of normal and pathological hepatic structure. Beitr Pathol. 1976;157(3):215–243. doi: 10.1016/s0005-8165(76)80083-2. [DOI] [PubMed] [Google Scholar]
- 132.Davidson AJ, Ellis MJ, Chaudhuri JB. A theoretical approach to zonation in a bioartificial liver. Biotechnol Bioeng. 2012;109:234–243. doi: 10.1002/bit.23279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Burt A, Portmann B, Ferrell L. MacSween’s Pathology of the Liver. Churchill Livingstone; 2006. [Google Scholar]
- 134.Monga SPS. Molecular Pathology of Liver Diseases. Vol. 5. Springer; 2011. p. xxiii.p. 931. 179 illus., 124 illus. [Google Scholar]
- 135.Damania A, Jain E, Kumar A. Advancements in in vitro hepatic models: Application for drug screening and therapeutics. Hepatol Int. 2014;8(1):23–38. doi: 10.1007/s12072-013-9490-8. [DOI] [PubMed] [Google Scholar]
- 136.Celton-Morizur S, Desdouets C. Polyploidization of liver cells. Adv Exp Med Biol. 2010;676:123–135. doi: 10.1007/978-1-4419-6199-0_8. [DOI] [PubMed] [Google Scholar]
- 137.Jungermann K, Katz N. Functional hepatocellular heterogeneity. Hepatology. 1982;2(3):385–395. doi: 10.1002/hep.1840020316. [DOI] [PubMed] [Google Scholar]
- 138.Groothuis GM, Hardonk MJ, Keulemans KP, Nieuwenhuis P, Meijer DK. Autoradiographic and kinetic demonstration of acinar heterogeneity of taurocholate transport. Am J Physiol. 1982;243(6):G455–G462. doi: 10.1152/ajpgi.1982.243.6.G455. [DOI] [PubMed] [Google Scholar]
- 139.Ring JA, Ghabria H, Ching MS, Smallwood RA, Morgan DJ. Fetal hepatic drug elimination. Pharmacol Ther. 1999;84(3):429–445. doi: 10.1016/s0163-7258(99)00046-7. [DOI] [PubMed] [Google Scholar]
- 140.Lindros KO. Zonation of cytochrome P450 expression, drug metabolism and toxicity in liver. Gen Pharmacol. 1997;28(2):191–196. doi: 10.1016/s0306-3623(96)00183-8. [DOI] [PubMed] [Google Scholar]
- 141.Vaananen H. The distribution of cytochrome P-450-mediated drug oxidation and glutathione in periportal and perivenous rat hepatocytes after phenobarbital treatment. J Hepatol. 1986;2(2):174–181. doi: 10.1016/s0168-8278(86)80076-9. [DOI] [PubMed] [Google Scholar]
- 142.Liddle C, Stedman CAM. In: Textbook of Hepatology. Rodes J, Benhamou J-P, Blei A, Reichen J, Rizzetto M, editors. Blackwell; 2007. p. 241. [Google Scholar]
- 143.Smith MT, Loveridge N, Wills ED, Chayen J. The distribution of glutathione in the rat liver lobule. Biochem J. 1979;182(1):103–108. doi: 10.1042/bj1820103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kera Y, Penttila KE, Lindros KO. Glutathione replenishment capacity is lower in isolated perivenous than in periportal hepatocytes. Biochem J. 1988;254(2):411–417. doi: 10.1042/bj2540411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shimizu M, Morita S. Effects of fasting on cadmium toxicity, glutathione metabolism, and metallothionein synthesis in rats. Toxicol Appl Pharmacol. 1990;103(1):28–39. doi: 10.1016/0041-008x(90)90259-w. [DOI] [PubMed] [Google Scholar]
- 146.Gómez-Lechón MJ, Castell JV, Donato MT. Hepatocytes — The choice to investigate drug metabolism and toxicity in man: In vitro variability as a reflection of in vivo. Chem Biol Interact. 2007;168:30–50. doi: 10.1016/j.cbi.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 147.Sugano K, Kansy M, Artursson P, Avdeef A, Bendels S, Di L, Ecker GF, Faller B, Fischer H, Gerebtzoff G, Lennernaes H, Senner F. Coexistence of passive and carrier-mediated processes in drug transport. Nat Rev Drug Discov. 2010;9(8):597–614. doi: 10.1038/nrd3187. [DOI] [PubMed] [Google Scholar]
- 148.Ramboer E, Vanhaecke T, Rogiers V, Vinken M. Primary hepatocyte cultures as prominent in vitro tools to study hepatic drug transporters. Drug Metab Rev. 2013;45(2):196–217. doi: 10.3109/03602532.2012.756010. [DOI] [PubMed] [Google Scholar]
- 149.Zhang L, Strong JM, Qiu W, Lesko LJ, Huang SM. Scientific perspectives on drug transporters and their role in drug interactions. Mol Pharm. 2006;3(1):62–69. doi: 10.1021/mp050095h. [DOI] [PubMed] [Google Scholar]
- 150.Kaplowitz N, DeLeve LD. Drug-Induced Liver Disease. Academic Press; 2013. p. xxiv.p. 746. [10] pages of plates. [Google Scholar]
- 151.Stirnimann G, Kessebohm K, Lauterburg B. Liver injury caused by drugs: An update. Swiss Med Wkly. 2010;140:w13080. doi: 10.4414/smw.2010.13080. [DOI] [PubMed] [Google Scholar]
- 152.Kaplowitz N. Acetaminophen hepatoxicity: What do we know, what don’t we know, and what do we do next? Hepatology. 2004;40(1):23–26. doi: 10.1002/hep.20312. [DOI] [PubMed] [Google Scholar]
- 153.Uetrecht J. Immunoallergic drug-induced liver injury in humans. Semin Liver Dis. 2009;29(4):383–392. doi: 10.1055/s-0029-1240007. [DOI] [PubMed] [Google Scholar]
- 154.Tujios S, Fontana RJ. Mechanisms of drug-induced liver injury: From bedside to bench. Nat Rev Gastroenterol Hepatol. 2011;8:202–211. doi: 10.1038/nrgastro.2011.22. [DOI] [PubMed] [Google Scholar]
- 155.Roth RA, Ganey PE. Intrinsic versus idiosyncratic drug-induced hepatotoxicity — Two villains or one? J Pharmacol Exp Ther. 2010;332:692–697. doi: 10.1124/jpet.109.162651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hewitt NJ, Lechón MJ, Houston JB, Hallifax D, Brown HS, et al. Primary hepatocytes: Current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev. 2007;39(1):159–234. doi: 10.1080/03602530601093489. [DOI] [PubMed] [Google Scholar]
- 157.Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov. 2005;4(6):489–499. doi: 10.1038/nrd1750. [DOI] [PubMed] [Google Scholar]
- 158.Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther. 1973;187(1):211–217. [PubMed] [Google Scholar]
- 159.Jollow DJ, Mitchell JR, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J Pharmacol Exp Ther. 1973;187(1):195–202. [PubMed] [Google Scholar]
- 160.Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther. 1973;187(1):185–194. [PubMed] [Google Scholar]
- 161.Potter WZ, Davis DC, Mitchell JR, Jollow DJ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. 3. Cytochrome P-450-mediated covalent binding in vitro. J Pharmacol Exp Ther. 1973;187(1):203–210. [PubMed] [Google Scholar]
- 162.Messner S, Agarkova I, Moritz W, Kelm JM. Multi-cell type human liver microtissues for hepatotoxicity testing. Arch Toxicol. 2013;87:209–213. doi: 10.1007/s00204-012-0968-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Luyendyk JP, Shores KC, Ganey PE, Roth RA. Bacterial lipopolysaccharide exposure alters aflatoxin B(1) hepatotoxicity: Benchmark dose analysis for markers of liver injury. Toxicol Sci. 2002;68(1):220–225. doi: 10.1093/toxsci/68.1.220. [DOI] [PubMed] [Google Scholar]
- 164.Roth RA, Luyendyk JP, Maddox JF, Ganey PE. Inflammation and drug idiosyncrasy — Is there a connection? J Pharmacol Exp Ther. 2003;307(1):1–8. doi: 10.1124/jpet.102.041624. [DOI] [PubMed] [Google Scholar]
- 165.Shaw PJ, Ganey PE, Roth RA. Idiosyncratic drug-induced liver injury and the role of inflammatory stress with an emphasis on an animal model of trovafloxacin hepatotoxicity. Toxicol Sci. 2010;118(1):7–18. doi: 10.1093/toxsci/kfq168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Alvarez-Lueje A, Bollo S. Stability and drug metabolism assessed by electrochemical methods. Comb Chem High Throughput Screen. 2010;13(8):712–727. doi: 10.2174/138620710791920329. [DOI] [PubMed] [Google Scholar]
- 167.Michel B. From the nucleation of ice crystals in clouds to the formation of frazil ice in rivers. Proc Phys Snow Ice. 1967;1(1):129–136. [Google Scholar]
- 168.Álvarez-Lueje A, Pérez M, Zapata C. In: Topics on Drug Metabolism. Paxton J, editor. InTech; 2012. [Google Scholar]
- 169.Naritomi Y, Terashita S, Kimura S, Suzuki A, Kagayama A, Sugiyama Y. Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab Dispos. 2001;29(10):1316–1324. [PubMed] [Google Scholar]
- 170.Lacarelle B, Marre F, Blanc-Gauthier T, Zhou XJ, Placidi M, Catalin J, Rahmani R. Use of human and animal liver microsomes in drug metabolic studies. Eur J Drug Metab Pharmacokinet. 1991;3:458–465. [PubMed] [Google Scholar]
- 171.Souhaili-el Amri H, Batt AM, Siest G. Comparison of cytochrome P-450 content and activities in liver microsomes of seven animal species, including man. Xenobiotica. 1986;16(4):351–358. doi: 10.3109/00498258609043538. [DOI] [PubMed] [Google Scholar]
- 172.Patterson DS, Roberts BA. The composition of liver microsomes and levels of detoxicating enzymes in 9 animal species. Res Vet Sci. 1970;11(4):399–401. [PubMed] [Google Scholar]
- 173.Gores GJ, Kost LJ, LaRusso NF. The isolated perfused rat liver: Conceptual and practical considerations. Hepatology. 1986;6(3):511–517. doi: 10.1002/hep.1840060331. [DOI] [PubMed] [Google Scholar]
- 174.Sahin S, Karabey Y. Investigation of distribution and elimination of terbutaline sulfate in the perfused rat liver preparation. Eur J Drug Metab Pharmacokinet. 2010;35(1–2):9–14. doi: 10.1007/s13318-010-0002-0. [DOI] [PubMed] [Google Scholar]
- 175.Donato MT, Lahoz A, Castell JV, Gómez-Lechón MJ. Cell lines: A tool for in vitro drug metabolism studies. Curr Drug Metab. 2008;9(1):1–11. doi: 10.2174/138920008783331086. [DOI] [PubMed] [Google Scholar]
- 176.Decaens C, Durand M, Grosse B, Cassio D. Which in vitro models could be best used to study hepatocyte polarity? Biol Cell. 2008;100(7):387–398. doi: 10.1042/BC20070127. [DOI] [PubMed] [Google Scholar]
- 177.Soldatow VY, LeCluyse EL, Griffith LG, Rusyn I. In vitro models for liver toxicity testing. Toxicol Res. 2013;2(1):23–29. doi: 10.1039/C2TX20051A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dunn JCY, Tompkins RG, Yarmush ML. Hepatocytes in collagen sandwich: Evidence for transcriptional and translational regulation. J Cell Biol. 1992;116:1043–1053. doi: 10.1083/jcb.116.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yarmush ML, Toner M, Dunn JCY, Hubel A, Tompkins RG. Hepatic tissue engineering: Development of critical technologies. Ann N Y Acad Sci. 1992;665:238–252. doi: 10.1111/j.1749-6632.1992.tb42588.x. [DOI] [PubMed] [Google Scholar]
- 180.Berry MN, Barritt GJ, Edwards AM. Isolated Hepatocytes: Preparation, Properties and Applications. Vol. 21. Elsevier Science; 2001. [Google Scholar]
- 181.Hengstler JG, Utesch D, Steinberg P, Platt KL, Diener B, Ringel M, et al. Cryopreserved primary hepatocytes as a constantly available in vitro model for the evaluation of human and animal drug metabolism and enzyme induction. Drug Metab Rev. 2000;32(1):81–118. doi: 10.1081/dmr-100100564. [DOI] [PubMed] [Google Scholar]
- 182.Wang H, Gao X, Fukumoto S, Tademoto S, Sato K, Hirai K. Post-isolation inducible nitric oxide synthase gene expression due to collagenase buffer perfusion and characterization of the gene regulation in primary cultured murine hepatocytes. J Biochem. 1998;124(5):892–899. doi: 10.1093/oxfordjournals.jbchem.a022204. [DOI] [PubMed] [Google Scholar]
- 183.Baker TK, Carfagna MA, Gao H, Dow ER, Li Q, Searfoss GH, Ryan TP. Temporal gene expression analysis of monolayer cultured rat hepatocytes. Chem Res Toxicol. 2001;14(9):1218–1231. doi: 10.1021/tx015518a. [DOI] [PubMed] [Google Scholar]
- 184.Sabzevari O, Hatcher M, O’Sullivan M, Kentish P, Gibson G. Comparative induction of cytochrome P4504A in rat hepatocyte culture by the peroxisome proliferators, bifonazole and clofibrate. Xenobiotic. 1995;25(4):395–403. doi: 10.3109/00498259509061860. [DOI] [PubMed] [Google Scholar]
- 185.Guillouzo A, Morel F, Langouët S, Maheo K, Rissel M. Use of hepatocyte cultures for the study of hepatotoxic compounds. J Hepatol. 1997;26(Suppl 2):73–80. doi: 10.1016/s0168-8278(97)80499-0. [DOI] [PubMed] [Google Scholar]
- 186.Li AP, Colburn SM, Beck DJ. A simplified method for the culturing of primary adult rat and human hepatocytes as multicellular spheroids. In Vitro Cell Dev Biol. 1992;28A(9–10):673–677. doi: 10.1007/BF02631045. [DOI] [PubMed] [Google Scholar]
- 187.Wu FJ, Friend JR, Hsiao CC, Zilliox MJ, Ko WJ, Cerra FB, Hu WS. Efficient assembly of rat hepatocyte spheroids for tissue engineering applications. Biotechnol Bioeng. 1996;50(4):404–415. doi: 10.1002/(SICI)1097-0290(19960520)50:4<404::AID-BIT7>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 188.Kelm JM, Timmins NE, Brown CJ, Fussenegger M, Nielsen LK. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol Bioeng. 2003;83(2):173–180. doi: 10.1002/bit.10655. [DOI] [PubMed] [Google Scholar]
- 189.Landry J, Bernier D, Ouellet C, Goyette R, Marceau N. Spheroidal aggregate culture of rat liver cells: Histotypic reorganization, biomatrix deposition, and maintenance of functional activities. J Cell Biol. 1985;101(3):914–923. doi: 10.1083/jcb.101.3.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Du Y, Chia SM, Han R, Chang S, Tang H, Yu H. 3D hepatocyte monolayer on hybrid RGD/galactose substratum. Biomaterials. 2006;27(33):5669–5680. doi: 10.1016/j.biomaterials.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 191.Tong JZ, De Lagausie P, Furlan V, Cresteil T, Bernard O, Alvarez F. Long-term culture of adult rat hepatocyte spheroids. Exp Cell Res. 1992;200(2):326–332. doi: 10.1016/0014-4827(92)90179-c. [DOI] [PubMed] [Google Scholar]
- 192.LeCluyse EL, Audus KL, Hochman JH. Formation of extensive canalicular networks by rat hepatocytes cultured in collagen-sandwich configuration. Am J Physiol. 1994;266(6 Pt 1):C1764–C1774. doi: 10.1152/ajpcell.1994.266.6.C1764. [DOI] [PubMed] [Google Scholar]
- 193.Liu X, Chism JP, LeCluyse EL, Brouwer KR, Brouwer KL. Correlation of biliary excretion in sandwich-cultured rat hepatocytes and in vivo in rats. Drug Metab Dispos. 1999;27(6):637–644. [PubMed] [Google Scholar]
- 194.Kern A, Bader A, Pichlmayr R, Sewing KF. Drug metabolism in hepatocyte sandwich cultures of rats and humans. Biochem Pharmacol. 1997;54(7):761–772. doi: 10.1016/s0006-2952(97)00204-9. [DOI] [PubMed] [Google Scholar]
- 195.Berthiaume F, Moghe PV, Toner M, Yarmush ML. Effect of extracellular matrix topology on cell structure, function, and physiological responsiveness: Hepatocytes cultured in a sandwich configuration. FASEB J. 1996;10(13):1471–1484. doi: 10.1096/fasebj.10.13.8940293. [DOI] [PubMed] [Google Scholar]
- 196.Beken S, Slaus K, De Smet K, Depreter M, Roels F, Vercruysse A, Rogiers V. Effect of extracellular matrix composition on the expression of glutathione s-transferase isoenzymes in organotypical hepatocyte cultures. Toxicol In Vitro. 1999;13(4–5):571–577. doi: 10.1016/s0887-2333(99)00029-6. [DOI] [PubMed] [Google Scholar]
- 197.Semler EJ, Ranucci CS, Moghe PV. Tissue assembly guided via substrate biophysics: Applications to hepatocellular engineering. Adv Biochem Eng Biotechnol. 2006;102:1–46. doi: 10.1007/10_012. [DOI] [PubMed] [Google Scholar]
- 198.Miccheli A, Tomassini A, Capuani G, Di Cocco ME, Sartori E, et al. Energy metabolism and re-establishment of intercellularadhesion complexes of gel entrapped hepatocytes. Cytotechnology. 2000;32(3):219–228. doi: 10.1023/A:1008134005529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Catapano G, De Bartolo L, Vico V, Ambrosio L. Morphology and metabolism of hepatocytes cultured in Petri dishes on films and in non-woven fabrics of hyaluronic acid esters. Biomaterials. 2001;22(7):659–665. doi: 10.1016/s0142-9612(00)00228-3. [DOI] [PubMed] [Google Scholar]
- 200.Gómez Lechón MJ, Jover R, Donato T, Ponsoda X, Rodriguez C, et al. Long term expression of differentiated functions in hepatocytes cultured in three dimensional collagen matrix. J Cell Physiol. 1998;177(4):553–562. doi: 10.1002/(SICI)1097-4652(199812)177:4<553::AID-JCP6>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 201.Jindal R, Nahmias Y, Tilles AW, Berthiaume F, Yarmush ML. Amino acid-mediated heterotypic interaction governs performance of a hepatic tissue model. FASEB J. 2009;23(7):2288–2298. doi: 10.1096/fj.08-114934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Chia SM, Leong KW, Li J, Xu X, Zeng K, Er PN, Gao S, Yu H. Hepatocyte encapsulation for enhanced cellular functions. Tissue Eng. 2000;6(5):481–495. doi: 10.1089/107632700750022134. [DOI] [PubMed] [Google Scholar]
- 203.Toh YC, Lim TC, Tai D, Xiao G, van Noort D, Yu H. A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab Chip. 2009;9(14):2026–2035. doi: 10.1039/b900912d. [DOI] [PubMed] [Google Scholar]
- 204.Rajagopalan P, Shen CJ, Berthiaume F, Tilles AW, Toner M, Yarmush ML. Poly-electrolyte nano-scaffolds for the design of layered cellular architectures. Tissue Eng. 2006;12:1553–1563. doi: 10.1089/ten.2006.12.1553. [DOI] [PubMed] [Google Scholar]
- 205.Boudou T, Crouzier T, Ren K, Blin G, Picart C. Multiple functionalities of polyelectrolyte multilayer films: New biomedical applications. Adv Mater. 2010;22:441–467. doi: 10.1002/adma.200901327. [DOI] [PubMed] [Google Scholar]
- 206.Decher G. Fuzzy nanoassemblies: Toward layered polymeric multicomposites. Science. 1997;277:1232–1237. [Google Scholar]
- 207.Kim Y, Larkin AL, Davis RM, Rajagopalan P. The design of in vitro liver sinusoid mimics using chitosan-hyaluronic acid polyelectrolyte multilayers. Tissue Eng Part A. 2010;16(9):2731–2741. doi: 10.1089/ten.tea.2009.0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.McCarty WJ, Usta OB, Luitje M, Bale SS, Bhushan A, et al. A novel ultrathin collagen nanolayer assembly for 3-D microtissue engineering: Layer-by-layer collagen deposition for long-term stable microfluidic hepatocyte culture. Technology. 2014;2(1):67–74. doi: 10.1142/S2339547814500083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lee J, Morgan JR, Tompkins RG, Yarmush ML. Proline-mediated enhancement of hepatocyte function in a collagen gel sandwich culture configuration. FASEB J. 1993;7(6):586–591. doi: 10.1096/fasebj.7.6.8472895. [DOI] [PubMed] [Google Scholar]
- 210.Oberhammer FA, Pavelka M, Sharma S, Tiefenbacher R, Purchio AF, Bursch W, Schulte-Hermann R. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc Natl Acad Sci USA. 1992;89(12):5408–5412. doi: 10.1073/pnas.89.12.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Cho CH, Berthiaume F, Tilles AW, Yarmush ML. A new technique for primary hepatocyte expansion in vitro. Biotechnol Bioeng. 2008;101(2):345–356. doi: 10.1002/bit.21911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP, Hillan KJ, Ferrara N. Angiogenesis-independent endothelial protection of liver: Role of VEGFR-1. Science. 2003;299(5608):890–893. doi: 10.1126/science.1079562. [DOI] [PubMed] [Google Scholar]
- 213.Isom HC, Secott T, Georgoff I, Woodworth C, Mummaw J. Maintenance of differentiated rat hepatocytes in primary culture. Proc Natl Acad Sci USA. 1985;82(10):3252–3256. doi: 10.1073/pnas.82.10.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Easterbrook J, Lu C, Sakai Y, Li AP. Effects of organic solvents on the activities of cytochrome P450 isoforms, UDP-dependent glucuronyl transferase, and phenol sulfotransferase in human hepatocytes. Drug Metab Dispos. 2001;29(2):141–144. [PubMed] [Google Scholar]
- 215.Inoue C, Yamamoto H, Nakamura T, Ichihara A, Okamoto H. Nicotinamide prolongs survival of primary cultured hepatocytes without involving loss of hepatocyte-specific functions. J Biol Chem. 1989;264(9):4747–4750. [PubMed] [Google Scholar]
- 216.Paine AJ. The maintenance of cytochrome P-450 in rat hepatocyte culture: Some applications of liver cell cultures to the study of drug metabolism, toxicity and the induction of the P-450 system. Chem Biol Interact. 1990;74(1):1–31. doi: 10.1016/0009-2797(90)90055-r. [DOI] [PubMed] [Google Scholar]
- 217.Bayly AC, Roberts RA, Dive C. Suppression of liver cell apoptosis in vitro by the non-genotoxic hepatocarcinogen and peroxisome proliferator nafenopin. J Cell Biol. 1994;125(1):197–203. doi: 10.1083/jcb.125.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Block GD, Locker J, Bowen WC, Petersen BE, Katyal S, Strom SC, et al. Population expansion, clonal growth, and specific differentiation patterns in primary cultures of hepatocytes induced by HGF/SF, EGF and TGF alpha in a chemically defined (HGM) medium. J Cell Biol. 1996;132(6):1133–1149. doi: 10.1083/jcb.132.6.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.LeCluyse EL, Bullock PL, Parkinson A. Strategies for restoration and maintenance of normal hepatic structure and function in long-term cultures of rat hepatocytes. Adv Drug Delivery Rev. 1996;22(1–2):133–186. [Google Scholar]
- 220.Zupke CA, Stefanovich P, Berthiaume F, Yarmush ML. Metabolic effects of stress mediators on cultured hepatocytes. Biotechnol Bioeng. 2000;58(2–3):222–230. [PubMed] [Google Scholar]
- 221.Fraslin JM, Kneip B, Vaulont S, Glaise D, Munnich A, Guguen-Guillouzo C. Dependence of hepatocyte-specific gene expression on cell-cell interactions in primary culture. EMBO J. 1985;4(10):2487–2491. doi: 10.1002/j.1460-2075.1985.tb03960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Guguen-Guillouzo C, Clément B, Baffet G, Beaumont C, Morel-Chany E, Glaise D, Guillouzo A. Maintenance and reversibility of active albumin secretion by adult rat hepatocytes co-cultured with another liver epithelial cell type. Exp Cell Res. 1983;143(1):47–54. doi: 10.1016/0014-4827(83)90107-6. [DOI] [PubMed] [Google Scholar]
- 223.Bhatia SN, Balis UJ, Yarmush ML, Toner M. Probing heterotypic cell interactions: Hepatocyte function in microfabricated co-cultures. J Biomater Sci Polym Ed. 1998;9(11):1137–1160. doi: 10.1163/156856298x00695. [DOI] [PubMed] [Google Scholar]
- 224.Bhatia SN, Balis UJ, Yarmush ML, Toner M. Microfabrication of hepatocyte/fibroblast co-cultures: Role of homotypic cell interactions. Biotechnol Prog. 1998;14(3):378–387. doi: 10.1021/bp980036j. [DOI] [PubMed] [Google Scholar]
- 225.Khetani SR, Bhatia SN. Microscale culture of human liver cells for drug development. Nat Biotechnol. 2008;26:120–126. doi: 10.1038/nbt1361. [DOI] [PubMed] [Google Scholar]
- 226.Dash A, Inman W, Hoffmaster K, Sevidal S, Kelly J, Obach RS, Griffith LG, Tannenbaum SR. Liver tissue engineering in the evaluation of drug safety. Expert Opin Drug Metab Toxicol. 2009;5:1159–1174. doi: 10.1517/17425250903160664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Novik E, Maguire TJ, Chao P, Cheng KC, Yarmush ML. A microfluidic hepatic coculture platform for cell-based drug metabolism studies. Biochem Pharmacol. 2010;79:1036–1044. doi: 10.1016/j.bcp.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Krause P, Saghatolislam F, Koenig S, Unthan-Fechner K, Probst I. Maintaining hepatocyte differentiation in vitro through co-culture with hepatic stellate cells. In Vitro Cell Dev Biol Anim. 2009;45(5–6):205–212. doi: 10.1007/s11626-008-9166-1. [DOI] [PubMed] [Google Scholar]
- 229.Ries K, Krause P, Solsbacher M, Schwartz P, et al. Elevated expression of hormone-regulated rat hepatocyte functions in a new serum-free hepatocyte-stromal cell coculture model. In Vitro Cell Dev Biol Anim. 2000;36(8):502–512. doi: 10.1290/1071-2690(2000)036<0502:EEOHRR>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 230.Kidambi S, Sheng L, Yarmush ML, Toner M, Lee I, Chan C. Patterned co-culture of primary hepatocytes and fibroblasts using polyelectrolyte multilayer templates. Macromol Biosci. 2007;7:344–353. doi: 10.1002/mabi.200600205. [DOI] [PubMed] [Google Scholar]
- 231.Bhatia SN, Balis UJ, Yarmush ML, Toner M. Effect of cell-cell interactions in preservation of cellular phenotype: Cocultivation of hepatocytes and nonparenchymal cells. FASEB J. 1999;13(14):1883–1900. doi: 10.1096/fasebj.13.14.1883. [DOI] [PubMed] [Google Scholar]
- 232.Ploss A, Khetani SR, Jones CT, Syder AJ, et al. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc Natl Acad Sci USA. 2010;107(7):3141–3145. doi: 10.1073/pnas.0915130107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Zinchenko YS, Schrum LW, Clemens M, Coger RN. Hepatocyte and kupffer cells co-cultured on micropatterned surfaces to optimize hepatocyte function. Tissue Eng. 2006;12(4):751–761. doi: 10.1089/ten.2006.12.751. [DOI] [PubMed] [Google Scholar]
- 234.Hui EE, Bhatia SN. Micromechanical control of cell–cell interactions. Proc Natl Acad Sci USA. 2007;104(14):5722–5726. doi: 10.1073/pnas.0608660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ito A, Kamihira M. Tissue engineering using magnetite nanoparticles and magnetic force: Heterotypic layers of cocultured hepatocytes and endothelial cells. Tissue Eng. 2011;10(5–6):833–840. doi: 10.1089/1076327041348301. [DOI] [PubMed] [Google Scholar]
- 236.Harimoto M, Yamato M, Hirose M, Takahashi C, Isoi Y, Kikuchi A, Okano T. Novel approach for achieving double-layered cell sheets co-culture: Overlaying endothelial cell sheets onto monolayer hepatocytes utilizing temperature-responsive culture dishes. J Biomed Mater Res. 2002;62(3):464–470. doi: 10.1002/jbm.10228. [DOI] [PubMed] [Google Scholar]
- 237.Kim K, Ohashi K, Utoh R, Kano K, Okano T. Preserved liver-specific functions of hepatocytes in 3D co-culture with endothelial cell sheets. Biomaterials. 2012;33(5):1406–1413. doi: 10.1016/j.biomaterials.2011.10.084. [DOI] [PubMed] [Google Scholar]
- 238.Kasuya J, Sudo R, Mitaka T, Ikeda M, Tanishita K. Hepatic stellate cell-mediated three-dimensional hepatocyte and endothelial cell triculture model. Tissue Eng Part A. 2011;17(3–4):361–370. doi: 10.1089/ten.TEA.2010.0033. [DOI] [PubMed] [Google Scholar]
- 239.Salerno S, Campana C, Morelli S, Drioli E, De Bartolo L. Human hepatocytes and endothelial cells in organotypic membrane systems. Biomaterials. 2011;32(34):8848–8859. doi: 10.1016/j.biomaterials.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 240.Jindal R, Patel SJ, Yarmush ML. Tissue-engineered model for real-time monitoring of liver inflammation. Tissue Eng Part C Methods. 2010;17(1):113–122. doi: 10.1089/ten.tec.2009.0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Wong SF, No da Y, Choi YY, Kim DS, Chung BG, Lee SH. Concave microwell based size-controllable hepatosphere as a three-dimensional liver tissue model. Biomaterials. 2011;32(32):8087–8096. doi: 10.1016/j.biomaterials.2011.07.028. [DOI] [PubMed] [Google Scholar]
- 242.Leite SB, Teixeira AP, Miranda JP, Tostões RM, Clemente JJ, Sousa MF, Carrondo MJ, Alves PM. Merging bioreactor technology with 3D hepatocyte-fibroblast culturing approaches: Improved in vitro models for toxicological applications. Toxicol In Vitro. 2011;25(4):825–832. doi: 10.1016/j.tiv.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 243.Bhatia SN, Yarmush ML, Toner M. Micropatterning cells in tissue engineering. Methods Mol Med. 1999;18:349–363. doi: 10.1385/0-89603-516-6:349. [DOI] [PubMed] [Google Scholar]
- 244.Bhatia SN, Yarmush ML, Toner M. Controlling cell interactions by micropatterning in co-cultures: Hepatocytes and 3T3 fibroblasts. J Biomed Mater Res. 1997;34(2):189–199. doi: 10.1002/(sici)1097-4636(199702)34:2<189::aid-jbm8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 245.Khetani SR, Kanchagar C, Ukairo O, Krzyzewski S, et al. Use of micropatterned cocultures to detect compounds that cause drug-induced liver injury in humans. Toxicol Sci. 2013;132(1):107–117. doi: 10.1093/toxsci/kfs326. [DOI] [PubMed] [Google Scholar]
- 246.Khetani SR, Bhatia SN. Engineering tissues for in vitro applications. Curr Opin Biotechnol. 2006;17:524–531. doi: 10.1016/j.copbio.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 247.Khetani SR, Szulgit G, Del Rio JA, Barlow C, Bhatia SN. Exploring interactions between rat hepatocytes and nonparenchymal cells using gene expression profiling. Hepatology. 2004;40:545–554. doi: 10.1002/hep.20351. [DOI] [PubMed] [Google Scholar]
- 248.Ukairo O, Kanchagar C, Moore A, Shi J, Gaffney J, Aoyama S, et al. Long-term stability of primary rat hepatocytes in micropatterned cocultures. J Biochem Mol Toxicol. 2013;27(3):204–212. doi: 10.1002/jbt.21469. [DOI] [PubMed] [Google Scholar]
- 249.Kratschmar D, Messner S, Moritz W, Odermatt A. Characterization of a rat multi-cell type 3D-liver microtissue system. J Tissue Sci Eng. 2013;4(2):130. doi: 10.4172/2157-7552.1000130. [DOI] [Google Scholar]
- 250.InSphero AG. InSphero User Group Meeting Boston/Cambridge. 2013 http://www.eventbrite.com/e/insphero-user-group-meeting-bostoncambridge-registration-7518697627.
- 251.Atienzar FA, Novik EI, Gerets HH, Parekh A, Delatour C, et al. Predictivity of dog co-culture model, primary human hepatocytes and HepG2 cells for the detection of hepatotoxic drugs in humans. Toxicol Appl Pharmacol. 2014;275(1):44–61. doi: 10.1016/j.taap.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 252.HμREL Corporation. HμREL® Hepatic Co-cultures. 2014 http://hurelcorp.com/technology/hurel-hepatic-co-cultures.
- 253.Park J, Berthiaume F, Toner M, Yarmush ML, Tilles AW. Microfabricated grooved substrates as platforms for bioartificial liver reactors. Biotechnol Bioeng. 2005;90:632–644. doi: 10.1002/bit.20463. [DOI] [PubMed] [Google Scholar]
- 254.Tilles AW, Balis UJ, Baskaran H, Yarmush ML, Toner M. In: International Symposium on Tissue Engineering for Therapeutic Use. Ikada Y, Ohshima M, editors. Vol. 5. Elsevier; 2000. pp. 59–71. [Google Scholar]
- 255.De Bartolo L, Jarosch-Von Schweder G, Haverich A, Bader A. A novel full-scale flat membrane bioreactor utilizing porcine hepatocytes: Cell viability and tissue-specific functions. Biotechnol Prog. 2000;16:102–108. doi: 10.1021/bp990128o. [DOI] [PubMed] [Google Scholar]
- 256.Shito M. Efficacy of an extracorporeal flat-plate bioartificial liver in treating fulminant hepatic failure. J Surg Res. 2003;111:53–62. doi: 10.1016/s0022-4804(03)00048-9. [DOI] [PubMed] [Google Scholar]
- 257.Gebhardt R, Mecke D. Perifused monolayer cultures of rat hepatocytes as an improved in vitro system for studies on ureogenesis. Exp Cell Res. 1979;124(2):349–359. doi: 10.1016/0014-4827(79)90210-6. [DOI] [PubMed] [Google Scholar]
- 258.Gebhardt R, Mecke D. Permissive effect of dexamethasone on glucagon induction of urea-cycle enzymes in perifused primary monolayer cultures of rat hepatocytes. Eur J Biochem. 1979;97(1):29–35. doi: 10.1111/j.1432-1033.1979.tb13082.x. [DOI] [PubMed] [Google Scholar]
- 259.Horner M, Miller WM, Ottino JM, Papoutsakis ET. Transport in a grooved perfusion flat-bed bioreactor for cell therapy applications. Biotechnol Prog. 1998;14(5):689–698. doi: 10.1021/bp980067e. [DOI] [PubMed] [Google Scholar]
- 260.Zeilinger K, Schreiter T, Darnell M, Söderdahl T, Lübberstedt M, et al. Scaling down of a clinical three-dimensional perfusion multicompartment hollow fiber liver bioreactor developed for extracorporeal liver support to an analytical scale device useful for hepatic pharmacological in vitro studies. Tissue Eng Part C Methods. 2011;17(5):549–556. doi: 10.1089/ten.TEC.2010.0580. [DOI] [PubMed] [Google Scholar]
- 261.Mazzei D, Guzzardi MA, Giusti S, Ahluwalia A. A low shear stress modular bioreactor for connected cell culture under high flow rates. Biotechnol Bioeng. 2010;106:127–137. doi: 10.1002/bit.22671. [DOI] [PubMed] [Google Scholar]
- 262.Vozzi F, Mazzei D, Vinci B, Vozzi G, Sbrana T, et al. A flexible bioreactor system for constructing in vitro tissue and organ models. Biotechnol Bioeng. 2011;108:2129–2140. doi: 10.1002/bit.23164. [DOI] [PubMed] [Google Scholar]
- 263.Vinci B, Duret C, Klieber S, Gerbal-Chaloin S, Sa-Cunhaet A, et al. Modular bioreactor for primary human hepatocyte culture: Medium flow stimulates expression and activity of detoxification genes. Biotechnol J. 2011;6(5):554–564. doi: 10.1002/biot.201000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Guzzardi MA, Domenici C, Ahluwalia A. Metabolic control through hepatocyte and adipose tissue cross-talk in a multicompartmental modular bioreactor. Tissue Eng Part A. 2011;17(11–12):1635–1642. doi: 10.1089/ten.TEA.2010.0541. [DOI] [PubMed] [Google Scholar]
- 265.Guzzardi MA, Vozzi F, Ahluwalia A. Study of the crosstalk between hepatocytes and endothelial cells using a novel multicompartmental bioreactor: A comparison between connected cultures and cocultures. Tissue Eng Part A. 2009;15(11):3635–3644. doi: 10.1089/ten.TEA.2008.0695. [DOI] [PubMed] [Google Scholar]
- 266.De Bartolo L, Salerno S, Morelli S, Giorno L, Rende M, et al. Long-term maintenance of human hepatocytes in oxygen-permeable membrane bioreactor. Biomaterials. 2006;27(27):4794–4803. doi: 10.1016/j.biomaterials.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 267.Hastings NE, Simmers MB, McDonald OG, Wamhoff BR, Blackman BR. Atherosclerosis-prone hemodynamics differentially regulates endothelial and smooth muscle cell phenotypes and promotes pro-inflammatory priming. Am J Physiol Cell Physiol. 2007;293(6):C1824–C1833. doi: 10.1152/ajpcell.00385.2007. [DOI] [PubMed] [Google Scholar]
- 268.Gebhardt R, Wegner H, Alber J. Perifusion of co-cultured hepatocytes: Optimization of studies on drug metabolism and cytotoxicity in vitro. Cell Biol Toxicol. 1996;12(2):57–68. doi: 10.1007/BF00143356. [DOI] [PubMed] [Google Scholar]
- 269.Dash A, Simmers MB, Deering TG, Berry DJ, Feaver RE, et al. Hemodynamic flow improves rat hepatocyte morphology, function and metabolic activity in vitro. Am J Physiol Cell Physiol. 2013;304(11):C1053–C1063. doi: 10.1152/ajpcell.00331.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Maguire T, Usta OB, Yarmush ML. Computational fluid dynamic analysis of a cell-based microfluidic drug screening platform. Nano LIFE. 2010;1:185–194. [Google Scholar]
- 271.Kirkstall, Ltd. Quasi-Vivo® Products. 2015 http://kirkstall.org/index.php/quasi-vivo-products.
- 272.In-vitro Technologies. LiveBox1 (LB1) 2015 http://www.ivtech.it/Products/LiveBox1-%28LB1%29.
- 273.Illa X, Vila S, Yeste J, Peralta C, Gracia-Sancho J, Villa R. A novel modular bioreactor to in vitro study the hepatic sinusoid. PLoS One. 2014;9(11):e111864. doi: 10.1371/journal.pone.0111864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Bale SS, et al. A novel low-volume two-chamber microfabricated platform for evaluating drug metabolism and toxicity [manuscript accepted] Technology. 2015 doi: 10.1142/S2339547815200034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Chen A, Yarmush ML, Maguire T. Physiologically based pharmacokinetic models: Integration of in silico approaches with micro cell culture analogues. Curr Drug Metab. 2012;13:863–880. doi: 10.2174/138920012800840419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Andersson H, van den Berg A. Microfabrication and microfluidics for tissue engineering: State of the art and future opportunities. Lab Chip. 2004;4(2):98–103. doi: 10.1039/b314469k. [DOI] [PubMed] [Google Scholar]
- 277.Zhang MY, Lee PJ, Hung PJ, Johnson T, Lee LP, Mofrad MR. Microfluidic environment for high density hepatocyte culture. Biomed Microdevices. 2008;10:117–121. doi: 10.1007/s10544-007-9116-9. [DOI] [PubMed] [Google Scholar]
- 278.Lee PJ, Hung PJ, Lee LP. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol Bioeng. 2007;97:1340–1346. doi: 10.1002/bit.21360. [DOI] [PubMed] [Google Scholar]
- 279.Lee P, Allen M, Hung P. In: Microfluidic Cell Culture Systems. Bettinger C, Borenstein JT, Tao SL, editors. William Andrew; 2013. pp. 341–355. [Google Scholar]
- 280.Nakao Y, Kimura H, Sakai Y, Fujii T. Bile canaliculi formation by aligning rat primary hepatocytes in a microfluidic device. Biomicrofluidics. 2011;5(2):022212. doi: 10.1063/1.3580753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Schütte J, Hagmeyer B, Holzner F, Kubon M, Werner S, et al. “Artificial micro organs” — A microfluidic device for dielectrophoretic assembly of liver sinusoids. Biomed Microdevices. 2011;13(3):493–501. doi: 10.1007/s10544-011-9517-7. [DOI] [PubMed] [Google Scholar]
- 282.Goral VN, Hsieh YC, Petzold ON, Clark JS, Yuen PK, Faris RA. Perfusion-based microfluidic device for three-dimensional dynamic primary human hepatocyte cell culture in the absence of biological or synthetic matrices or coagulants. Lab Chip. 2010;10:3380–3386. doi: 10.1039/c0lc00135j. [DOI] [PubMed] [Google Scholar]
- 283.Toh YC, Zhang C, Zhang J, Khong YM, Chang S, Samper VD, et al. A novel 3D mammalian cell perfusion-culture system in microfluidic channels. Lab Chip. 2007;7:302–309. doi: 10.1039/b614872g. [DOI] [PubMed] [Google Scholar]
- 284.King KR, Wang S, Irimia D, Jayaraman A, Toner M, Yarmush ML. A high-throughput microfluidic real-time gene expression living cell array. Lab Chip. 2007;7:77–85. doi: 10.1039/b612516f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Thompson DM, King KR, Wieder KJ, Toner M, Yarmush ML, Jayaraman A. Dynamic gene expression profiling using a microfabricated living cell array. Anal Chem. 2004;76(14):4098–4103. doi: 10.1021/ac0354241. [DOI] [PubMed] [Google Scholar]
- 286.Wieder K, King KR, Thompson DM, Zia C, Yarmush ML, Jayaraman A. Optimization of reporter cells for expression profiling in a microfluidic device. Biomed Microdevices. 2005;7(3):213–222. doi: 10.1007/s10544-005-3028-3. [DOI] [PubMed] [Google Scholar]
- 287.Yang E, Foteinou PT, King KR, Yarmush ML, Androulakis IP. A novel non-overlapping bi-clustering algorithm for network generation using living cell array data. Bioinformatics. 2007;23:2306–2313. doi: 10.1093/bioinformatics/btm335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Janorkar A, King KR, Megeed Z, Yarmush ML. Development of an in vitro cell culture model of hepatic steatosis using hepatocyte-derived reporter cells. Biotechnol Bioeng. 2009;102(5):1466–1474. doi: 10.1002/bit.22191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Yang E, Yarmush ML, Androulakis IP. Transcription factor network reconstruction using the living cell array. J Theor Biol. 2009;256:393–407. doi: 10.1016/j.jtbi.2008.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Yarmush ML, King KR. Living-cell microarrays. Annu Rev Biomed Eng. 2009;11:235–257. doi: 10.1146/annurev.bioeng.10.061807.160502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Kane BJ, Zinner MJ, Yarmush ML, Toner M. Liver-specific functional studies in a microfluidic array of primary mammalian hepatocytes. Anal Chem. 2006;78(13):4291–4298. doi: 10.1021/ac051856v. [DOI] [PubMed] [Google Scholar]
- 292.Powers MJ, Domansky K, Kaazempur-Mofrad MR, Kalezi A, Capitano A, et al. A microfabricated array bioreactor for perfused 3D liver culture. Biotechnol Bioeng. 2002;78(3):257–269. doi: 10.1002/bit.10143. [DOI] [PubMed] [Google Scholar]
- 293.Powers MJ, Janigian DM, Wack KE, Baker CS, Beer Stolz D, Griffith LG. Functional behavior of primary rat liver cells in a three-dimensional perfused microarray bioreactor. Tissue Eng. 2002;8(3):499–513. doi: 10.1089/107632702760184745. [DOI] [PubMed] [Google Scholar]
- 294.Hwa AJ, Fry RC, Sivaraman A, So PT, Samson LD, Stolz DB, Griffith LG. Rat liver sinusoidal endothelial cells survive without exogenous VEGF in 3D perfused co-cultures with hepatocytes. FASEB J. 2007;21:2564–2579. doi: 10.1096/fj.06-7473com. [DOI] [PubMed] [Google Scholar]
- 295.Domansky K, Inman W, Serdy J, Dash A, Lim MH, Griffith LG. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip. 2010;10:51–58. doi: 10.1039/b913221j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Sivaraman A, Leach JK, Townsend S, Iida T, Hogan BJ, Stolz DB, et al. A micro-scale in vitro physiological model of the liver: Predictive screens for drug metabolism and enzyme induction. Curr Drug Metab. 2005;6(6):569–591. doi: 10.2174/138920005774832632. [DOI] [PubMed] [Google Scholar]
- 297.Chao P, Maguire TJ, Novik E, Cheng KC, Yarmush ML. Evaluation of a microfluidic based cell culture platform with primary human hepatocytes for the prediction of hepatic clearance in human. Biochem Pharmacol. 2009;78:625–632. doi: 10.1016/j.bcp.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Maguire TJ, Novik E, Chao P, Barminko J, Nahmias Y, Yarmush ML, Cheng KC. Design and application of microfluidic systems for in vitro pharmacokinetic evaluation of drug candidates. Curr Drug Metab. 2009;10:1192–1199. doi: 10.2174/138920009790820093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Sin A, Chin KC, Jamil MF, Kostov Y, Rao G, Shuler ML. The design and fabrication of three-chamber microscale cell culture analog devices with integrated dissolved oxygen sensors. Biotechnol Prog. 2004;20:338–335. doi: 10.1021/bp034077d. [DOI] [PubMed] [Google Scholar]
- 300.Park TH, Shuler ML. Integration of cell culture and microfabrication technology. Biotechnol Prog. 2003;19:243–253. doi: 10.1021/bp020143k. [DOI] [PubMed] [Google Scholar]
- 301.Toepke MW, Beebe DJ. PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip. 2006;6(12):1484–1486. doi: 10.1039/b612140c. [DOI] [PubMed] [Google Scholar]
- 302.HμREL Corporation. HμRELflow™ Microfluidic Platforms. 2014 http://hurelcorp.com/products-services/microfluidics.
- 303.Sung JH, Kam C, Shuler ML. A microfluidic device for a pharmacokinetic-pharmacodynamic (PK-PD) model on a chip. Lab Chip. 2010;10:446–455. doi: 10.1039/b917763a. [DOI] [PubMed] [Google Scholar]
- 304.Mahler GJ, Esch MB, Glahn RP, Shuler ML. Characterization of a gastrointestinal tract microscale cell culture analog used to predict drug toxicity. Biotechnol Bioeng. 2009;104:193–205. doi: 10.1002/bit.22366. [DOI] [PubMed] [Google Scholar]
- 305.Sung JH, Shuler ML. A micro cell culture analog (microCCA) with 3-D hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab Chip. 2009;9:1385–1394. doi: 10.1039/b901377f. [DOI] [PubMed] [Google Scholar]
- 306.Oh TI, Sung JH, Tatosian DA, Shuler ML, Kim D. Real-time fluorescence detection of multiple microscale cell culture analog devices in situ. Cytometry A. 2007;71:857–865. doi: 10.1002/cyto.a.20427. [DOI] [PubMed] [Google Scholar]
- 307.Viravaidya K, Sin A, Shuler ML. Development of a microscale cell culture analog to probe naphthalene toxicity. Biotechnol Prog. 2004;20:316–223. doi: 10.1021/bp0341996. [DOI] [PubMed] [Google Scholar]
- 308.Iori E, Vinci B, Murphy E, Marescotti MC, Avogaro A, Ahluwalia A. Glucose and fatty acid metabolism in a 3-tissue in-vitro model challenged with normo- and hyperglycaemia. PLoS One. 2012;7(4):e34704. doi: 10.1371/journal.pone.0034704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Vinci B, Murphy E, Iori E, Meduri F, Fattori S, Marescotti MC, et al. An in vitro model of glucose and lipid metabolism in a multicompartmental bioreactor. Biotechnol J. 2012;7(1):117–126. doi: 10.1002/biot.201100177. [DOI] [PubMed] [Google Scholar]
- 310.Frey O, Misun PM, Fluri DA, Hengstler JG, Hierlemann A. Reconfigurable microfluidic hanging drop network for multi-tissue interaction and analysis. Nat Commun. 2014;5 doi: 10.1038/ncomms5250. [DOI] [PubMed] [Google Scholar]
- 311.Edd JF, Humphry KJ, Irimia D, Weitz DA, Toner M. Nucleation and solidification in static arrays of monodisperse drops. Lab Chip. 2009;9(13):1859–1865. doi: 10.1039/b821785h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Riss TL, Moravec RA, Niles AL, Benink HA, Worzella TJ, Minor L. In: Assay Guidance Manual. Sittampalam GS, Coussens NP, Nelson H, et al., editors. Eli Lilly & NCATS; 2004. [PubMed] [Google Scholar]
- 313.Huang SM, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, Abraham S, et al. New era in drug interaction evaluation: U.S. Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48(6):662–670. doi: 10.1177/0091270007312153. [DOI] [PubMed] [Google Scholar]
- 314.Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2(6):875–894. doi: 10.1517/17425255.2.6.875. [DOI] [PubMed] [Google Scholar]
- 315.Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 2004;14(1):1–18. doi: 10.1097/00008571-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 316.Szabo M, Veres Z, Baranyai Z, Jakab F, Jemnitz K. Comparison of human hepatoma HepaRG cells with human and rat hepatocytes in uptake transport assays in order to predict a risk of drug induced hepatotoxicity. PLoS One. 2013;8(3):e59432. doi: 10.1371/journal.pone.0059432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Xu JJ, Henstock PV, Dunn MC, Smith AR, Chabot JR, de Graaf D. Cellular imaging predictions of clinical drug-induced liver injury. Toxicol Sci. 2008;105:97–105. doi: 10.1093/toxsci/kfn109. [DOI] [PubMed] [Google Scholar]
- 318.Allen JW, Bhatia SN. Formation of steady-state oxygen gradients in vitro: Application to liver zonation. Biotechnol Bioeng. 2003;82:253–262. doi: 10.1002/bit.10569. [DOI] [PubMed] [Google Scholar]
- 319.Allen JW, Khetani SR, Bhatia SN. In vitro zonation and toxicity in a hepatocyte bioreactor. Toxicol Sci. 2005;84:110–119. doi: 10.1093/toxsci/kfi052. [DOI] [PubMed] [Google Scholar]
- 320.Wu MH, Huang SB, Lee GB. Microfluidic cell culture systems for drug research. Lab Chip. 2010;10(8):939–956. doi: 10.1039/b921695b. [DOI] [PubMed] [Google Scholar]
- 321.Atencia J, Beebe DJ. Steady flow generation in microcirculatory systems. Lab Chip. 2006;6(4):567–574. doi: 10.1039/b514070f. [DOI] [PubMed] [Google Scholar]
- 322.Wikswo J, Block FE, III, Cliffel DE, Goodwin CR, Marasco CC, Markov DA, et al. Engineering challenges for instrumenting and controlling integrated organ-on-chip systems. IEEE Trans Biomed Eng. 2013;60(3):682–690. doi: 10.1109/TBME.2013.2244891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Altschuler SJ, Wu LF. Cellular heterogeneity: Do differences make a difference? Cell. 2010;141(4):559–563. doi: 10.1016/j.cell.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Walker G, Beebe DJ. A passive pumping method for microfluidic devices. Lab Chip. 2002;2(3):131–134. doi: 10.1039/b204381e. [DOI] [PubMed] [Google Scholar]
- 325.Zhu X, Yi Chu L, Chueh BH, Shen M, Hazarika B, Phadke N, Takayama S. Arrays of horizontally-oriented mini-reservoirs generate steady microfluidic flows for continuous perfusion cell culture and gradient generation. Analyst. 2004;129(11):1026–1031. doi: 10.1039/b407623k. [DOI] [PubMed] [Google Scholar]
- 326.Bhushan A, Senutovitch N, Bale SS, McCarty WJ, Hegde M, Jindal R, et al. Towards a three-dimensional microfluidic liver platform for predicting drug efficacy and toxicity in humans. Stem Cell Res Ther. 2013;4(Suppl 1):S16. doi: 10.1186/scrt377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Konry T, Hayman RB, Walt DR. Microsphere-based rolling circle amplification microarray for the detection of DNA and proteins in a single assay. Anal Chem. 2009;81(14):5777–5782. doi: 10.1021/ac900694y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Eyer K, Stratz S, Kuhn P, Küster SK, Dittrich PS. Implementing enzyme-linked immunosorbent assays on a microfluidic chip to quantify intracellular molecules in single cells. Anal Chem. 2013;85(6):3280–3287. doi: 10.1021/ac303628j. [DOI] [PubMed] [Google Scholar]
- 329.Hou F, Zhang Q, Yang J, Li X, Yang X, Wang S, Cheng Z. Development of a microplate reader compatible microfluidic chip for ELISA. Biomed Microdevices. 2012;14(4):729–737. doi: 10.1007/s10544-012-9654-7. [DOI] [PubMed] [Google Scholar]
- 330.Chang C, Sustarich J, Bharadwaj R, Chandrasekaran A, Adams PD, Singh AK. Droplet-based microfluidic platform for heterogeneous enzymatic assays. Lab Chip. 2013;13(9):1817–1822. doi: 10.1039/c3lc41418c. [DOI] [PubMed] [Google Scholar]
- 331.Ross R. Atherosclerosis — An inflammatory disease. N Engl J Med. 2005;340(2):115–126. doi: 10.1056/NEJM199901143400207. http://www.indiana.edu/~k662/articles/athero/athero%20ross.pdf. [DOI] [PubMed] [Google Scholar]
- 332.Velkovsky M, Snider, Cliffel DE, Wiskwo JP. Modeling the measurements of cellular fluxes in microbioreactor devices using thin enzyme electrodes. J Math Chem. 2010;49:251–275. doi: 10.1007/s10910-010-9744-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Chen D, Dua W, Liua Y, Liua W, Kuznetsovb A, et al. The chemistrode: A droplet-based microfluidic device for stimulation and recording with high temporal, spatial, and chemical resolution. Proc Natl Acad Sci USA. 2008;105(44):16843–16848. doi: 10.1073/pnas.0807916105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Konry T, Dominguez-Villar M, Baecher-Allan C, Hafler DA, Yarmush ML. Droplet-based microfluidic platforms for single T cell secretion analysis of IL-10 cytokine. Biosens Bioelectron. 2011;26:2707–2710. doi: 10.1016/j.bios.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Kiss MM, Ortoleva-Donnelly L, Beer NR, Warner J, Bailey CG, et al. High-hroughput quantitative polymerase chain reaction in picoliter droplets. Anal Chem. 2008;80(23):8975–8981. doi: 10.1021/ac801276c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Miller OJ, El Harrakb A, Mangeatb T, Bareta JC, Frenza L, El Debsa B, Mayotet E, et al. High-resolution dose-response screening using droplet-based microfluidics. Proc Natl Acad Sci USA. 2012;109(2):378–383. doi: 10.1073/pnas.1113324109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Guo MT, Rotem A, Heymanab JA, Weitz DA. Droplet microfluidics for high-throughput biological assays. Lab Chip. 2012;12(12):2146–2155. doi: 10.1039/c2lc21147e. [DOI] [PubMed] [Google Scholar]
- 338.Su Y, Zhu Y, Fang Q. A multifunctional microfluidic droplet-array chip for analysis by electrospray ionization mass spectrometry. Lab Chip. 2013;13(10):1876–1882. doi: 10.1039/c3lc00063j. [DOI] [PubMed] [Google Scholar]
- 339.Anderson JR, Chiu DT, Jackman RJ, Cherniavskaya O, McDonald JC, et al. Fabrication of topologically complex three-dimensional microfluidic systems in PDMS by rapid prototyping. Anal Chem. 2000;72(14):3158–3164. doi: 10.1021/ac9912294. [DOI] [PubMed] [Google Scholar]
- 340.Mukhopadhyay R. When PDMS isn’t the best. What are its weaknesses, and which other polymers can researchers add to their toolboxes? Anal Chem. 2007;79(9):3248–3253. doi: 10.1021/ac071903e. [DOI] [PubMed] [Google Scholar]
- 341.Heo YS, Cabrera LM, Song JW, Futai N, Tung YC, Smith GD, Takayama S. Characterization and resolution of evaporation-mediated osmolality shifts that constrain microfluidic cell culture in poly(dimethylsiloxane) devices. Anal Chem. 2007;79(3):1126–1134. doi: 10.1021/ac061990v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Su X, Young EW, Underkofler HA, Kamp TJ, January CT, Beebe DJ. Microfluidic cell culture and its application in high-throughput drug screening: Cardiotoxicity assay for hERG channels. J Biomol Screen. 2011;16(1):101–111. doi: 10.1177/1087057110386218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Thanawala SK, Chaudhury MK. Surface modification of silicone elastomer using perfluorinated ether. Langmuir. 1999;16(3):1256–1260. [Google Scholar]
- 344.Rolland JP, Van Dam RM, Schorzman DA, Quake SR, DeSimone JM. Solvent-resistant photocurable liquid fluoropolymers for microfluidic device fabrication [corrected] J Am Chem Soc. 2004;126(8):2322–2323. doi: 10.1021/ja031657y. [DOI] [PubMed] [Google Scholar]
- 345.Fiorini GS, Lorenz RM, Kuo JS, Chiu DT. Rapid prototyping of thermoset polyester microfluidic devices. Anal Chem. 2004;76(16):4697–4704. doi: 10.1021/ac0498922. [DOI] [PubMed] [Google Scholar]
- 346.Borysiak MD, Bielawski KS, Sniadecki NJ, Jenkel CF, Vogt BD, Posner JD. Simple replica micromolding of biocompatible styrenic elastomers. Lab Chip. 2013;13(14):2773–2784. doi: 10.1039/c3lc50426c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Berthier E, Young EWK, Beebe D. Engineers are from PDMS-land, Biologists are from Polystyrenia. Lab Chip. 2012;12(7):1224–1237. doi: 10.1039/c2lc20982a. [DOI] [PubMed] [Google Scholar]
- 348.Savage VM, Deeds EJ, Fontana W. Sizing up allometric scaling theory. PLoS Comput Biol. 2008;4(9):e1000171. doi: 10.1371/journal.pcbi.1000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws in biology. Science. 1997;276(5309):122–126. doi: 10.1126/science.276.5309.122. [DOI] [PubMed] [Google Scholar]
- 350.White CR, Seymour RS. Allometric scaling of mammalian metabolism. J Exp Biol. 2005;208(Pt. 9):1611–1619. doi: 10.1242/jeb.01501. [DOI] [PubMed] [Google Scholar]
- 351.Baudoin R, Prot JM, Nicolas G, Brocheton J, Brochot C, Legallais C, Benech H, Leclerc E. Evaluation of seven drug metabolisms and clearances by cryopreserved human primary hepatocytes cultivated in microfluidic biochips. Xenobiotica. 2013;43:140–152. doi: 10.3109/00498254.2012.706725. [DOI] [PubMed] [Google Scholar]
- 352.Ghanem A, Shuler ML. Combining cell culture analogue reactor designs and PBPK models to probe mechanisms of naphthalene toxicity. Biotechnol Prog. 2000;16:334–345. doi: 10.1021/bp9901522. [DOI] [PubMed] [Google Scholar]
- 353.Zhao P, Zhang L, Grillo JA, Liu Q, Bullock JM, Moon YJ, et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011;89:259–267. doi: 10.1038/clpt.2010.298. [DOI] [PubMed] [Google Scholar]
- 354.Poulin P, Kenny JR, Hop CE, Haddad S. In vitro-in vivo extrapolation of clearance: Modeling hepatic metabolic clearance of highly bound drugs and comparative assessment with existing calculation methods. J Pharm Sci. 2012;101(2):838–851. doi: 10.1002/jps.22792. [DOI] [PubMed] [Google Scholar]
- 355.Umehara K, Camenisch G. Novel in vitro-in vivo extrapolation (IVIVE) method to predict hepatic organ clearance in rat. Pharm Res. 2012;29(2):603–617. doi: 10.1007/s11095-011-0607-2. [DOI] [PubMed] [Google Scholar]
- 356.Roberts MS, Fraser S, Wagner A, McLeod L. Residence time distributions of solutes in the perfused rat liver using a dispersion model of hepatic elimination: 1. Effect of changes in perfusate flow and albumin concentration on sucrose and taurocholate. J Pharmacokinet Biopharm. 1990;18:209–234. doi: 10.1007/BF01062200. [DOI] [PubMed] [Google Scholar]
- 357.Chiba M, Ishii YY, Sugiyama Y. Prediction of hepatic clearance in human from in vitro data for successful drug development. AAPS J. 2009;11(2):262–276. doi: 10.1208/s12248-009-9103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Golberg A, Linshiz G, Kravets I, Stawski N, Hillson NJ, Yarmush ML, et al. Cloud-enabled microscopy and droplet microfluidic platform for specific detection of Escherichia coli in water. PLoS One. 2014;9(1):e86341. doi: 10.1371/journal.pone.0086341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Roach KL, King KR, Uygun BE, Kohane IS, Yarmush ML, Toner M. High throughput single cell bioinformatics. Biotechnol Prog. 2009;25(6):1772–1779. doi: 10.1002/btpr.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Martin MT, Knudsen TB, Reif DM, Houck KA, Judson RS, Kavlock RJ, Dix DJ. Predictive model of rat reproductive toxicity from ToxCast high throughput screening. Biol Reprod. 2011;85(2):327–339. doi: 10.1095/biolreprod.111.090977. [DOI] [PubMed] [Google Scholar]
- 361.Hegde M, Jindal R, Bhushan A, Bale SS, McCarty WJ, Golberg I, Ustaa OB, Yarmush ML. Dynamic interplay of flow and collagen stabilizes primary hepatocytes culture in a microfluidic platform. Lab Chip. 2014;14(12):2033–2039. doi: 10.1039/c4lc00071d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Castro CI, Briceno JC. Perfluorocarbon-based oxygen carriers: Review of products and trials. Artif Organs. 2010;34(8):622–634. doi: 10.1111/j.1525-1594.2009.00944.x. [DOI] [PubMed] [Google Scholar]
- 363.Henkel-Honke T, Oleck M. Artificial oxygen carriers: A current review. AANA J. 2007;75(3):205–211. [PubMed] [Google Scholar]
- 364.Samaja M, Terraneo L. Impact of hemoglobin concentration and affinity for oxygen on tissue oxygenation: The case of hemoglobin-based oxygen carriers. Artif Organs. 2012;36(2):210–215. doi: 10.1111/j.1525-1594.2011.01296.x. [DOI] [PubMed] [Google Scholar]