

Abstract
MicroRNAs, a class of 22-nucleotide non-coding RNAs, modulate gene expression by associating with the 3’-untranslated regions (3’- UTRs) of messenger RNAs (mRNAs). Although multiple miRNAs are known to be regulated during angiogenesis, their individual roles in blood vessel development are still not fully understood. Herein, we investigate the role of miR-29c in regulating cell cycle and angiogenic phenotype of endothelial cells. The results showed that IGF-1 is highly expressed and down-regulated by miR-29c in human umbilical vein endothelial cells (HUVEC). Consistent with this preliminary finding, introduction of exogenous miR-29c or miR-29c inhibitor alters cell cycle progression, proliferation and tube formation of HUVEC, respectively. Furthermore, by using luciferase reporter assay, we find that the expression of IGF-1, a suppressor transcription factor, is directly regulated by miR-29c through 3’-UTR. In addition, we show that the selective inhibition of PI3K/AKT pathway prior to miR-29c stimulation prevents the expression of angiogenesis suppressor miRNAs that are family and cluster specific. As a conclusion, we find that miR-29c plays a significant role in regulating cell cycle, proliferation and angiogenic properties of HUVECs. This function is likely mediated through IGF-1 proteins at the post-transcriptional level. As a novel molecular target, miR-29c may have a potential value in the treatment of angiogenesis-associated diseases, such as cardiovascular diseases and cancers.
Keywords: miRNA-29c, angiogenesis, endothelial cell, IGF-1
Introduction
MiRNAs (miRNAs) are highly conserved, single-stranded, and non-coding small RNAs, which regulate gene expression at post-transcriptional level by inhibiting protein translation from mRNA or promoting degradation of mRNA [1,2]. Evidence indicates that miRNAs can modulate various aspects of angiogenesis, such as proliferation, migration, and morphogenesis of endothelial cells [3,4]. Much effort has been devoted into the investigation of miRNAs, especially in the area of angiogenesis; however, only limited information has been provided about the angiogenic property of miR-29 family [5]. Although MiR-29c has been found to be highly expressed in endothelium, its functions in endothelium and angiogenesis are still not clarified. Therefore, the role of miR-29c in angiogenesis deserves further investigation. Recently, a clinical study shows that the plasma miR-29c levels of myocardial infarct patients are significantly upregulated during the first 1~5 days [6], which suggests that miR-29c may have a potential value of angiogenesis associated diseases treatment as a novel molecular target [7].
IGF-1, a growth factor that increases neuroprotection, neurogenesis, long-term potentiation, and dendritic growth and complexity [8,9], can also modulate immune functions [10]. For example, IGF-1 reduces inflammatory cytokine responses in the brain and ameliorates lipopolysaccharide (LPS)-induced sickness behaviors that are primarily driven by microglia-dependent production of IL-1b and TNF-α [11]. Moreover, central injection of a viral vector that upregulated IGF-1 in a mouse model of amyotrophic lateral sclerosis reduces microglial secretion of TNF-α and nitric oxide [12,13]. So far there is little information about the angiogenic property of IGF-1, and the relationship between IGF-1 and HUVECs.
In this study, we intend to investigate (1) if miR-29c expression in endothelium is regulated by IGF-1 treatment; and (2) if miR-29c directly targets IGF-1 expression and regulate angiogenesis properties of human endothelial cells. Our results show that miR-29c is an extensively expressed miRNA and highly expressed in HUVECs. Using bioinformatic analysis and luciferase reporter assay, we find that miR-29c acts to target the 3’-UTR of several genes in the IGF-1/PI3K/AKT signal pathway. Over-expression and interference of miR-29c in HUVECs can result in decreased or increased expression of IGF-1 level.
Materials and methods
Cell culture and IGF-1
Human umbilical vein endothelial cells (HUV-ECs) were obtained from Lonza (ALLENDALE, NJ, USA). Cells were cultured in Endothelial Cell Medium (ECM) with 5% fetal bovine serum (FBS), incubated at 37°C, and supplied with 5% CO2 and 95% air. Passages between 3 and 5 were used in all experiments. IGF-1 reagent was purchased from Gibco (Life technologies, Grand Island, NY, USA).
Prediction of miRNAs targeting IGF-1
The miRNA target predicting algorithms miRDB (http://mirdb.org/miRDB/), TargetScan (http://www.targetscan.org/), and PicTar (http://pictar.mdcberlin.de/) were used to predict miRNAs targeting IGF-1 and the binding regions.
Quantitative RT-PCR
The total RNA was extracted by TRIzol (Invitrogen, Carlsbad, CA, USA), using the standard method. The cDNA was synthesized using 1 μg of total RNA and miScript II RT Kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. After reverse transcription of the RNA, cDNA was used as a template in PCR reactions using gene-specific primer pairs (Table 1). Real-time PCR was performed on the ABI 7500 cycler (Applied Biosystems, CA, USA), using miScript SYBR Green PCR Kit (Qiagen, Hilden, Germany). Hsa-RNU6B and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as endogenous controls for miRNAs and mRNAs expression, respectively.
Table 1.
The primers for real-time PCR
| Primer of target | Sequence (5’ to 3’) | Base (bp) |
|---|---|---|
| PI3K forward primer | CAGGTTCCTTCAGTCCTACTCCAGGC | 24 |
| PI3K reverse primer | GCCCAGTCAGCTGATACCATTTAACCG | 26 |
| VEGF forward primer | GCTGCTGTAACGATGAAG | 18 |
| VEGF reverse primer | ATCTGCTGTGCTGTAGGA | 18 |
| AKT forward primer | GGTACCATGGAATACATGCCGAT | 22 |
| AKT reverse primer | GGAAAGCGATGTTACCATTGTGAAG | 25 |
| GAPDH forward primer | GCAAGTTCAACGGCACAG | 18 |
| GAPDH reverse primer | ACGCCAGTAGACTCCACGAC | 20 |
| miR-29c forward | TGACCGATTTCTCCTGGTGTTC | 22 |
| miR-29c reverse | GCGAGCACAGAATTAATACGAC | 22 |
| U6 forward | CTCGCTTCGGCAGCACA | 17 |
| U6 reverse | GCGAGCACAGAATTAATACGAC | 22 |
Transfection and immunocytochemical assay
The miR-29c mimics/inhibitors, obtained from Lifetechnologies (Grand Island, NY, USA), were transfected into HUVECs with the FuGENE 6 transfection reagent (Promega, Madison, USA) at a final concentration of 50/100 nM (HUVECs). The siRictor and siRaptor duplexes were synthesized by GenePharma (Invitrogen, Carlsbad, CA, USA), and a non-silencing siRNA was used as a negative control. The transfection protocol of siRNA was the same as that of miR-29c mimics. Briefly, cells in wells of 96-well plates were grown to 80~90% confluence, and then incubated with small RNA complexes for 6 h before changing the medium.
After transfection, HUVECs were washed with PBS and fixed with 4% paraformaldehyde for 15 min. 0.5% Trixon-100 was used for permeabilization. The cells were blocked in 2% goat serum (diluted in PBS), and then incubated with anti-myosin primary antibodies at 37°C for 1-2 h, followed by fluorescent secondary antibodies at 37°C for 1 h. The nuclei were stained with DAPI (Lifetechnologies) for 10 min. Images were taken using a fluorescence microscope (Olympus, Tokyo, Japan).
Plasmids constructs and luciferase reporter assay
The 3’-UTR of IGF-1 and a mutation sequence were amplified by PCR using the primers with a Bgl II restriction site on each 5’ or 3’ strand. The PCR products were inserted into the Bgl II sites of the pGL3-control vector (Promega, Madison, WI, USA) and identified by DNA sequencing. The wild-type plasmid was created containing the 3’-UTR of IGF-1 with complementary sequence of miR-29c (pGL3- IGF-1 3’-UTR wild), and a mutant plasmid with the mutation sequence without complementary sequence of miR-29c (pGL3- IGF-1 3’-UTR mut). For the luciferase reporter assay, the hUVECs was seeded on 24-well plates and co-transfected using Lipofectamine 2000 (Invitrogen) with 100 ng per well of the resulting luciferase UTR-report vector, 2 ng per well of pRLCMV vector (internal control, Promega), and 20 ng per well of miR-29c precursor molecules or control precursor (Applied Biosystems) following the manufacturer’s instructions. After 24 h, the cells were lysed, and the relative luciferase activity was assessed with the Dual-Luciferase Assay Reporter System (Promega).
Cell proliferation assay
Cell viability was assayed by a colorimetric procedure, using the Cell Counting Kit-8 (Lifetechnologies, NY, USA). The absorbance at 460 nm was determined with a microplate reader. To detect the exact proliferation rates of HUVECs, an EDU (5-ethynyl-20-deoxyuridine) incorporation assay was conducted with a Cell-Light™ EdU In Vitro Imaging Kit (Lifetechnologies, NY, USA). Briefly, cells at 70-80% confluence were treated with 50 nM EDU in ECM medium and incubated for 2 h before fixation in 4% paraformaldehyde. After EDU staining, cell nuclei were stained with Hoechst 33342, and then observed using an inverted fluorescent microscope (Olympus, Tokyo, Japan).
Wound healing assay
The HUVEC wound healing assay was conducted following the method described by [19] with minor modifications. Briefly, HUVECs were first transfected with miR-29c mimics, inhibitors or negative control (NC) RNAs in a 6-well plate, and were allowed to grow overnight until confluent. Next, IGF-1 was added to each well, except for the control one, at a final concentration of 100 ng/ml as a 24 h pre-treatment. The IGF-1 treatment was continued until the end of the test. At the 0 h time point, the cell monolayer in each well was scraped with a sterile 200 μl pipette tip for 3 times to form parallel lines, followed by washing with ECM. The same wound areas were examined and photographed with a Nikon Eclipse TS100 Microscope (Nikon, Japan) at the 0 h and 24 h post-injury time points. The areas of the cells that migrated into the wound fields were measured by Adobe Photoshop software.
In vitro tube network formation assay
For tube network formation assay, each well of the 96-well plates was pre-coated with 50 μl of Matrigel (BD Biosciences, Bedford, MA, USA), and allowed to polymerize for 30 min at 37°C. Then cells were seeded on the Matrigel-coated wells at a density of 2 × 104 cells per well in ECM medium containing 1% FBS at 37°C. Cells started to form tubes at 4 h. Tube formation reached its optimum after 6 h. The tube images were taken at 6-8 h with a digital camera attached to an inverted phase-contrast microscope. The length of the total tube in each well was measured and calculated using the Image software (IPP).
Cell cycle analysis
HUVECs in wells of 6-well plates were transfected with miR-29c mimics, inhibitors or negative control (NC) for 48 h. Then, the cells were harvested for Flow Cytometry analysis. Briefly, cells were fixed in 80% ethanol, suspended in PBS, and incubated with RNase A (0.5 μg/μl) for 30 min at 37°C. Then the cells were stained with propidium iodide on ice for 1 h, and subsequently measured with a FACScan Flow Cytometry (Becton Dickinson, San Jose, USA). The percentage of cells in the G1, S, and G2 phases were analyzed by using the Cell Cycle Analysis Software.
Migration assay
HUVEC migration was determined using a modified two chamber migration assay with a pore size of 8 μm (Transwell chamber, Corning, NY, USA). For migration assay, 1 × 105 cells were seeded in a serum-free medium in the upper chamber. After 12 h incubation at 37°C, cells in the upper chamber were carefully removed with a cotton swab, and the cells which traversed the membrane were fixed in methanol, stained with Giemsa, and photographed in 5 independent × 100 fields for each well.
Western blot
Cells in a 6-well plate were scraped in RIPA lysis buffer with 1 mM PMSF. Proteins (20 μg) were separated on 10% or 12% (for RhoB protein assay) SDS-polyacrylamide gels, and electro-transferred to polyvinylidene difluoride (PVDF) membranes (Lifetechnologies, NY, USA). After a blocking incubation with 5% milk-TBST, the membranes were incubated overnight with primary antibodies, followed by 1 h incubations with a secondary antibody which was conjugated to horseradish peroxidase (1:10000 dilution). After the incubation in an enhanced chemiluminescence reagent (Lifetechnologies, NY, USA), the images were captured by an image reader LAS-4000 system (Olympus, Tokyo, Japan).
Statistical analysis
All experiments were repeated for at least three times. Data were presented as the means ± SEM. Analyses were conducted with a SPSS 19.0 software, using the unpaired Student’s t-test for comparisons of two groups or one-way ANOVA for multiple comparisons. The P values < 0.05 were considered to be statistically significant (Table 1).
Results
IGF-1 treatment down-regulated miR-29c expression
To investigate the effects of treatments with typical IGF-1, the concentrations and time lengths (10, 20 and 40 ng/ml, 12~72 h) on the expression levels of miR-29c were known to correlate with endothelial cell function (Figure 1A). To study the effects of IGF-1 on miR-29c expressions in endothelial cells, we further verified the enhanced expression levels of miR-29c, which were induced by IGF-1. The miR-29c expression was down-regulated significantly by IGF-1 in a time-and-dose-dependent method, and showed expression peaks at 24 h and 40 ng/ml (Figure 1B) (p < 0.05). The data suggested that miR-29c expression can be stimulated by IGF-1 treatment.
A.

HUVECs were incubated with IGF-1 (40 ng/ml) for 72 h. The expression levels of miR-29c was descend significantly after IGF-1 treatment compared to control. (B) HUVECs were incubated with different concentrations of IGF-1 for 24 h, the expression levels of miR-29c was detected by qRT-PCR. Control cells were untreated. (C) Immunocytochemical assay shown the IGF1expression levels after treatment. (D) miR-29c directly down-regulates IGF-1 expression. (E) Predict IGF-1 was potential targets for miR-29c and its validation. Luciference reporter assay was performed to detect the effect of miR-29c and anti-sense miR-29c (F) on the luciference intensity controlled by 3’UTR of IGF-1. Mean ± SD (n = 3), *p < 0.05, **p < 0.01.
IGF-1 is the direct target of miR-29c
Searching the sequence using a TargetScan software, we found that a separate miR-29c-binding seed sequence was conserved through evolution in 3’-UTR of IGF-1 gene. To demonstrate the direct interaction between miR-29c and IGF-1 mRNA, we constructed a luciferase reporter system containing a binding site (IGF-1-3’-UTR-wt) or a mutated site (IGF-1-3’-UTR-mut) located downstream of the pRLCMV luciferase reporter gene. This vector was co-transfected into 293T cells with miR-29c mimics or negative controls. The luciferase activity in the miR-29c group was decreased by 39 % (p < 0.05), compared to negative controls. MiR-29c mimics didn’t affect the luciferase activity in the pGL3-IGF-1-mut vector (Figure 1D). When blocking the expression of miR-29c with miR-29c inhibitor, we found increased luciferase intensities in 293T cells (Figure 1E). On the other hand, the pGL3-IGF-1-mut vector co-transfected with miR-29c inhibitor didn’t show any change of luciferase activity in 293T cells (p < 0.01, respectively, Figure 1F). These results supported the bioinformatics prediction that the 3’-UTR of IGF-1 mRNA could be a target of miR-29c (Figure 1). miR-29c suppresses proliferation and regulation of cell cycle in HCC cells
To detect the roles of miR-29c in HUVECs proliferation, HUVEC were transfected with miR-29c mimics or negative control groups. As shown in Figure 2, over-expression of miR-29c significantly inhibited the growth of HUVECs at about 3 days after infection (p < 0.01), compared to the negative control. The 293T cells showed similar results (p < 0.01). After 5 days, the growth of HUVECs expressing miR-29c was decreased to 45.6% (Figure 2A, 2B). The effect of miR-29c on the proliferation of human endothelial cells was confirmed using an EDU incorporation assay. Similarly, significant differences were observed among cells with different treatments at 24 h (p < 0.01 between miR-29c and inhibitor, Figure 2C and 2D). Then, we examined the cell cycle distribution by FACS after transfection. Compared with the control, the cells transfected with miR-29c showed a significantly higher percentage of cells in G1 phase, and a significantly lower percentage of cells in S phase, which suggested that miR-29c caused G1 arrest (p < 0.05) (Figure 2E and 2F).
Figure 2.

Overexpression of miR-29c reduces HUVECs proliferation in vitro. (A) CCK-8 assays revealed that upregulation of miR-29c reduced cell proliferation of HUVECs and 293T cells (B), compared to negative (NC) transfected cells. (C and D) EDU incorporation assay. In the image overlay, the purple nuclei are EDU stained and indicate proliferating cells, while the blue nuclei are DAPI stained. (E and F) Effect of miR-29c on cell cycle of HUVECs. HUVECs were subjected to Propidium iodide (PI) Flow Cytometry analysis. Overexpression of miR-29c by miR-29c mimic accelerated S to G1 cell cycle transition, while inhibition of miR-29c increase G2 population and S phase population. Data represents Mean ± SE of three independent experiments, *p < 0 .05, **p < 0.01.
MiR-29c promoted tube network formation of HUVECs in vitro
To investigate the influence of miR-29c on the angiogenic properties of endothelial cells, the tube network formations on Matrigel of miR-29c mimics, miR-29c inhibitor or control transfected HUVECs were examined. The results showed that overexpression of miR-29c repressed the tube network formation of HUVECs (Figure 3). Therefore, up-regulated miR-29c in endothelial cells may restrain endothelial cells and inhibit angiogenesis.
Figure 3.
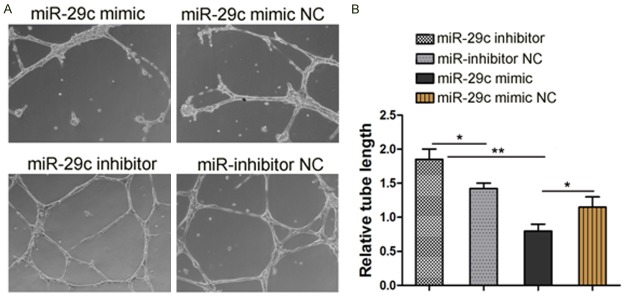
Effect of miR-29c on tube network formation in HUVECs. HUVECs were transfected with miR-29c mimic or miR-29c mimic NC or miR-29c inhibitor/miR-29c inhibitor NC. After 48 hrs, cells in suspension were seeded on wells that precoated with Matrigel. Tube network formations were measured after 6~8 h. A. Representative images of three independent experiments. B. Total tube length was measured with image analysis software and normalized to that of NC group, *p < 0.05, **p < 0.01.
Overexpression miR-29c suppress IGF-1 and PI3K/AKT relevant mRNA expression
We examined the mRNA expression levels of PI3K and AKT, which were key components that related to HUVEC angiogenesis (Figure 4). In the RT-PCR results, a 3.3-fold induction of PI3K mRNA and a 2.4-fold induction of AKT were observed in miR-29c mimics. The IGF-1 and VEGF inductions were significantly reversed by miR-29c mimics and controls (2.23-fold and 1.97-fold, p < 0.05, respectively). The data showed that overexpression miR-29c can down-regulate IGF-1, PI3K/AKT and VEGF mRNA expression.
Figure 4.
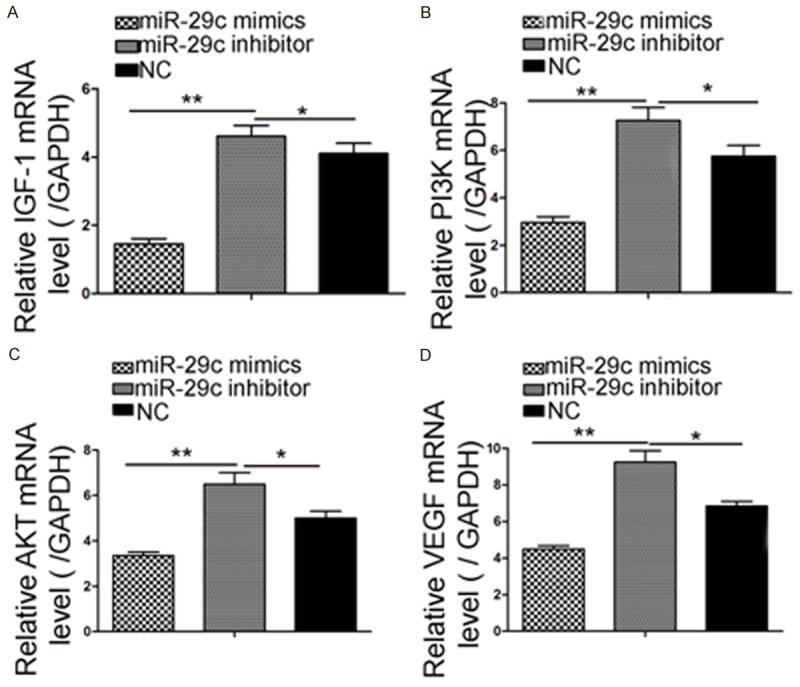
miR-29c overexpression can downregulate IGF-1 mRNA expression. Quantitative RT-PCR results revealed that miR-29c delivery markedly reduced the IGF-1-PI3K/AKT expression. (A) IGF-1, (B) PI3K, (C) AKT, and (D) VEGF mRNA expression. The upregulation of IGF-1 mRNA following miR-29c transfection in HUVECs was analyzed by RT-PCR (*p < 0.05, **p < 0.01).
Effect of miR-29c overexpression suppress HUVECs migration and regulate IGF-1, PI3K/AKT and VEGF pathway activity
We investigated the roles of miR-29c in HUVECs migration. Transwell assay showed that overexpression of miR-29c suppressed cells migration ability of HUVECs by 64.2% (Figure 5A and 5B). Wound healing assay exhibited a similar result (p < 0.05, Figure 5C and 5D). Since VEGF play a crucial role in cell migration and angiogenesis, we then carried out a Western blot and an immunocytochemistry assay to compare the activity of VEGF in miR-29c-overexpressing and control cells. As shown in Figure 5E, VEGF protein expression was dramatically reduced in miR-29c overexpressing cells, compared with the controls (p < 0.01). Then, we used Western blot to examine the PI3K/AKT pathway relevant protein expression in HUVECs. Representative WB showed that up-regulation of miR-29c obviously depressed protein expression of IGF-1, p-PI3K, p-AKT and VEGF expression in HUVECs. (Figure 5G and 5H).
Figure 5.
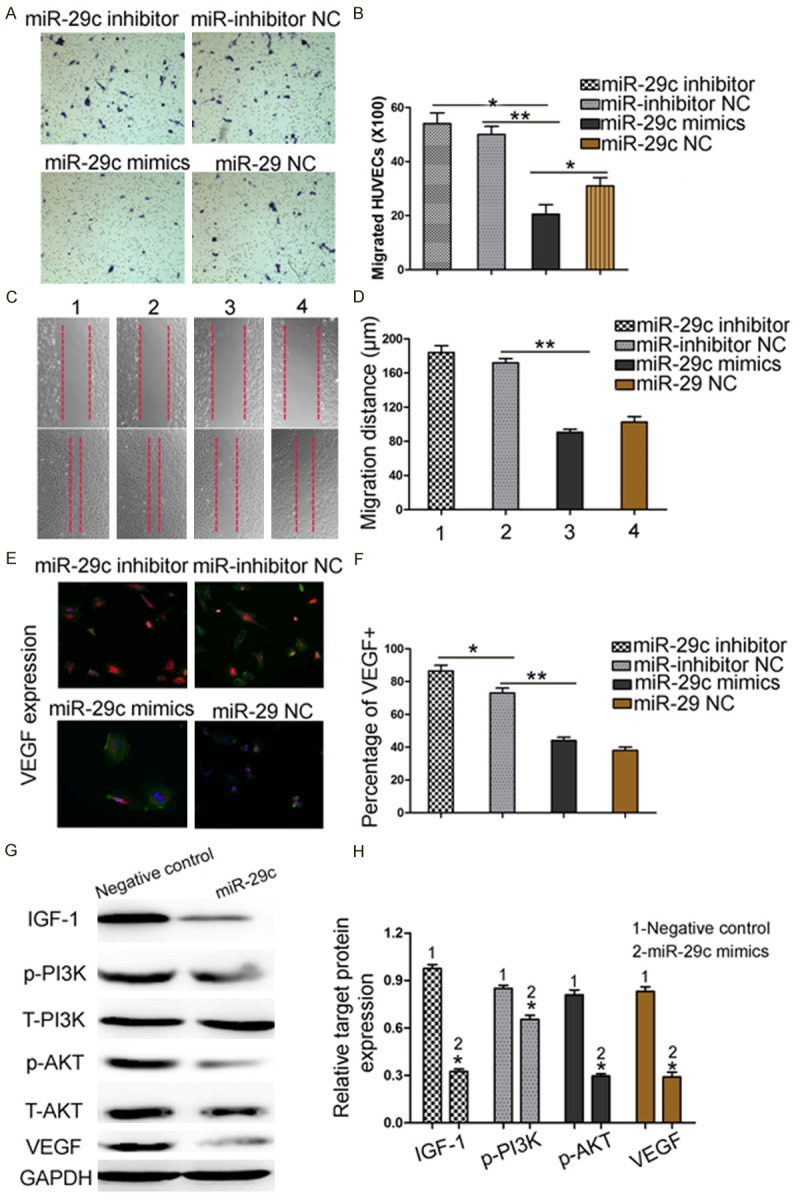
Effect of miR-29c overexpression suppress HUVECs migration and regulate IGF-1 and VEGF protein expression. A, B. Reintroduction of miR-29c depressed cells migration ability by cell migration assay. C, D. The down-regulated expression of miR-29c promoted the migration of HUVECs in a wound-healing assay. E, F. Immunocytochemistry assay to compare the activity of VEGF in miR-29c overexpressing and control cells. G, H. HUVECs that were transfected with miR-29c inhibitors and miR-29c mimics, respectively, were subjected to western blot for IGF-1, p-PI3K, p-AKT and VEGF protein expression. Representative WB showed that up-regulation of miR-29c obviously depressed protein expression of IGF1, p-PI3K, p-AKT and VEGF expression in HUVECs (n = 3 repeats with similar results. *p < 0.05, **p < 0.01).
Discussion
As key pathogenic components of major cardiovascular diseases and tumor microenvironments, miRNAs play important roles in activation of endothelial cells and angiogenesis [14-16]. It is reported that miRNAs are involved in endothelial cell functions [3,15]. In this study, we test the hypothesis that IGF-1 can promote the proliferation and migration of HUVECs, partly through a pathway that involved miRNAs. To the best of our knowledge, this is the first report which identifies the regulation of HUVEC angiogenesis by the miR-29c-IGF-1 pathway. Using microRNA target reporter assay, we show that miR-29c inhibits IGF-1 expression by directly binding to its 3’-UTR and thereby degrading IGF-1 mRNA. By qRT-PCR, we find for the first time that miR-29c is remarkably altered by IGF-1 treatment in a time and dose-dependent manner. Treatment with miR-29c mimics abrogates the IGF-1 augment of HUVEC proliferation and migration, which indicates that miR-29c can mediate the IGF-1-induced augment of endothelial growth and mobility. Endothelium-related miRNAs, such as miR-210, have been reported to be upregulated by hypoxia and involved in modification of angiogenic property of endothelium [17,18]. Our results show that miR-29c expression can be induced by an IGF-1-mimicking reagent in a dose-dependent manner in HUVECs. These data supports our hypothesis that miR-29c may be stimulated by IGF-1 treatment in endothelium.
MicroRNAs can potentially modulate multiple downstream proteins, and the same miRNA may regulate different cells through various mechanisms due to their diverse cell types and microenvironment. Therefore, it is necessary to identify the specific signaling pathway in miR-29c-mediated EC activation [19]. By comparing the differential gene expression profiles between miR-29c overexpressing HUVECs and control HUVECs, we reveal that IGF-1 has a negative correlation with miR-29c. Furthermore, we investigate the effects of alteration of miR-29c expression on cell cycle, proliferation and angiogenic properties in endothelium. The results indicate that G1 to S cell cycle transition of HUVECs can be reduced by miR-29c overexpression. Enforced expression of miR-29c inhibitor in endothelium remarkably promotes cell proliferation and tube network formation on Matrigel. It is reported that miR-29 family noticeable changes in progenitor cell proliferation by expediting G1 to S cell cycle transition [20], and miR-29a expression is upregulated in a metastatic breast cancer cell line [21]. MiR-29a transgenic mice leads to expanded CD5+ B-cell population and development of indolent CLL (chronic lymphocytic leukemia) phenotype [22]. Our results indicate that the functions of miR-29c in endothelial cells are similar to those of hematopoietic progenitor cells. In human osteoblast, miR-29 family increases Wnt signaling by targeting some negative regulators of Wnt signaling [23], which also supports the negative regulation effect of miR-29c on cell proliferation, at least in HUVECs.
The insulin-like growth factor (IGF-1) system includes six binding proteins, three ligands, ex. IGF-1, IGF-II and insulin, and three major receptors, ex. the insulin-like growth factor receptor I (IGF-1R), the insulin receptor (IR) and the insulin-like growth factor receptor 2 (IGF-IIR) [24]. Extensive homology exists between IR and IGF-1R, varying from 45-65% with highly conserved regions within the tyrosine-kinase cassette and substrate binding domain, where homology is upward of 60-80% [25]. Expression of IR and IGF components is widespread throughout the body and is driven by a variety of stimuli under normal physiological conditions including proper nutrition and exercise [26,27]. Insulin resistance is the pathogenic hallmark and strong risk factor for cardiovascular disease, including vascular disease and atherosclerosis [28,29]. Therefore, further exploration of molecular biological mechanisms underlying IGF-1 will be helpful in prevention and treatment of atherosclerosis and its complications [30]. Evidences indicate that miRNA play an important role in atherosclerosis and its related complications [31].
It has been reported that downregulation of miR-145 promotes endothelial lesion formation, whereas elevated miR-155 levels are characteristic of atherosclerotic lesions [32,33]. It has been proved that prolonged growth of endothelial cells in high glucose-containing medium-induced insulin resistance [34-36]. A recent study suggested that miR-29 family played a potent anti-angiogenic role in endothelial cells [5,37]; however, we show that the binding site for miR-29c in IGF-1 3’-UTR is predicted by a bioinformatic algorithm. In addition, luciferase assays show a positive miR-29c-3’-UTR IGF-1 interaction, which indicates that IGF-1 is a direct target of miR-29c.
In short, miR-29c plays a significant role in regulating cell cycle, proliferation and angiogenic properties of HUVECs. This function is likely mediated through IGF-1 protein at the post-transcriptional level. Our results suggest that miR-29c-IGF1-PI3K/AKT-VEGF pathway may act as a potential anti-angiogenic target for treatments of angiogenesis associated diseases.
Conclusions
Our studies have demonstrated that IGF-1 is a novel target of miR-29c. Overexpression of miR-29c decreases angiogenesis of HUVECs cells through the modulation of IGF1-PI3K/AKT pathway. The interplay among miR-29c, IGF1 and the PI3K/AKT-VEGF pathway scratches the surface of the terra incognita (Figure 6). Our results suggest that exogenous overexpression of miR-29c may be considered as a promising strategy for targeted therapies in inhibition of tumor angiogenesis and angiogenesis associated diseases.
Figure 6.
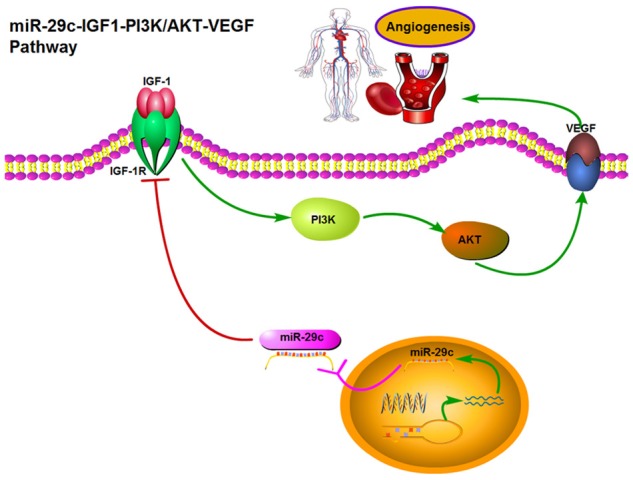
Abridged general view for the interplay among miR-29-PI3K/AKT-VEGF pathway in HUVEC. miR-29c as a angiogenesis suppressor by targeting IGF-1, which decreased the angiogenesis of HUVECs through the modulation of the PI3K/AKT pathway. Overexpression of miR-29c, which suppresses the expression of IGF1, activates the PI3K/AKT-VEGF pathway by increasing the phosphorylation of p-PI3K, p-AKT and VEGF expression, which inhibits cell proliferation and angiogenesis in HUVECs.
Acknowledgements
We thank Prof. Xue Li and Zhou Yu (Wake Forest University Institute for Regenerative Medicine) for the contributed reagents/materials/analysis tools. Project Supported by Program for the Natural Science Foundation of China (81271183, 81000463, 81470772); the Natural Science Foundation of Chongqing (cstc2012jja10053); the Medical Scientific Research Project of Chongqing (20141013, 2013-2-068); Program for Innovation Team Building at Institutions of Higher Education in Chongqing in 2013 and Chongqing Municipal Key Laboratory of Oral Biomedical Engineering of Higher Education.
Disclosure of conflict of interest
No conflicts of interest, financial or otherwise, are declared by the author(s).
References
- 1.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 2.Li W, Liu M, Feng Y, Xu YF, Huang YF, Che JP, Wang GC, Yao XD, Zheng JH. Downregulated miR-646 in clear cell renal carcinoma correlated with tumour metastasis by targeting the nin one binding protein (NOB1) Br J Cancer. 2014;111:1188–1200. doi: 10.1038/bjc.2014.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu YH, Hu TF, Chen YC, Tsai YN, Tsai YH, Cheng CC, Wang HW. The manipulation of miRNA-gene regulatory networks by KSHV induces endothelial cell motility. Blood. 2011;118:2896–2905. doi: 10.1182/blood-2011-01-330589. [DOI] [PubMed] [Google Scholar]
- 4.Pencheva N, Tran H, Buss C, Huh D, Drobnjak M, Busam K, Tavazoie SF. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell. 2012;151:1068–1082. doi: 10.1016/j.cell.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kriegel AJ, Liu Y, Fang Y, Ding X, Liang M. The miR-29 family: genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol Genomics. 2012;44:237–244. doi: 10.1152/physiolgenomics.00141.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lew WY, Bayna E, Dalle Molle E, Contu R, Condorelli G, Tang T. Myocardial Fibrosis Induced by Exposure to Subclinical Lipopolysaccharide Is Associated with Decreased miR-29c and Enhanced NOX2 Expression in Mice. PLoS One. 2014;9:e107556. doi: 10.1371/journal.pone.0107556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Presneau N, Eskandarpour M, Shemais T, Henderson S, Halai D, Tirabosco R, Flanagan AM. MicroRNA profiling of peripheral nerve sheath tumours identifies miR-29c as a tumour suppressor gene involved in tumour progression. Br J Cancer. 2013;108:964–972. doi: 10.1038/bjc.2012.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quesada A, Lee BY, Micevych PE. PI3 kinase/Akt activation mediates estrogen and IGF-1 nigral DA neuronal neuroprotection against a unilateral rat model of Parkinson’s disease. Dev Neurobiol. 2008;68:632–644. doi: 10.1002/dneu.20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez-Martin M, Cifuentes M, Grondona JM, Bermudez-Silva FJ, Arrabal PM, Perez-Figares JM, Jimenez AJ, Garcia-Segura LM, Fernandez-Llebrez P, Fernandez-Llebrez P. Neurogenesis in explants from the walls of the lateral ventricle of adult bovine brain: role of endogenous IGF-1 as a survival factor. Eur J Neurosci. 2003;17:205–211. doi: 10.1046/j.1460-9568.2003.02432.x. [DOI] [PubMed] [Google Scholar]
- 10.Chapes SK, Simske SJ, Forsman AD, Bateman TA, Zimmerman RJ. Effects of space flight and IGF-1 on immune function. Adv Space Res. 1999;23:1955–1964. doi: 10.1016/s0273-1177(99)00456-1. [DOI] [PubMed] [Google Scholar]
- 11.Zhao P, Turdi S, Dong F, Xiao X, Su G, Zhu X, Scott GI, Ren J. Cardiac-specific overexpression of insulin-like growth factor I (IGF-1) rescues lipopolysaccharide-induced cardiac dysfunction and activation of stress signaling in murine cardiomyocytes. Shock. 2009;32:100–107. doi: 10.1097/SHK.0b013e31818ec609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corbo M, Lunetta C, Magni P, Dozio E, Ruscica M, Adobbati L, Silani V. Free insulin-like growth factor (IGF)-1 and IGF-binding proteins-2 and -3 in serum and cerebrospinal fluid of amyotrophic lateral sclerosis patients. Eur J Neurol. 2010;17:398–404. doi: 10.1111/j.1468-1331.2009.02815.x. [DOI] [PubMed] [Google Scholar]
- 13.De Felice B, Guida M, Guida M, Coppola C, De Mieri G, Cotrufo R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene. 2012;508:35–40. doi: 10.1016/j.gene.2012.07.058. [DOI] [PubMed] [Google Scholar]
- 14.Mehta JL, Mercanti F, Stone A, Wang X, Ding Z, Romeo F, Khaidakov M. Gene and miRNA Transcriptional Signatures of Angiotensin II in Endothelial Cells. J Cardiovasc Pharmacol. 2015;65:123–129. doi: 10.1097/FJC.0000000000000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White K, Kane NM, Milligan G, Baker AH. The role of miRNA in stem cell pluripotency and commitment to the vascular endothelial lineage. Microcirculation. 2012;19:196–207. doi: 10.1111/j.1549-8719.2012.00161.x. [DOI] [PubMed] [Google Scholar]
- 16.Roy S, Sen CK. miRNA in wound inflammation and angiogenesis. Microcirculation. 2012;19:224–232. doi: 10.1111/j.1549-8719.2011.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noman MZ, Buart S, Romero P, Ketari S, Janji B, Mari B, Mami-Chouaib F, Chouaib S. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res. 2012;72:4629–4641. doi: 10.1158/0008-5472.CAN-12-1383. [DOI] [PubMed] [Google Scholar]
- 18.Camps C, Buffa FM, Colella S, Moore J, Sotiriou C, Sheldon H, Harris AL, Gleadle JM, Ragoussis J. Hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res. 2008;14:1340–1348. doi: 10.1158/1078-0432.CCR-07-1755. [DOI] [PubMed] [Google Scholar]
- 19.Yin M, Ren X, Zhang X, Luo Y, Wang G, Huang K, Feng S, Bao X, Huang K, He X, Liang P, Wang Z, Tang H, He J, Zhang B. Selective killing of lung cancer cells by miRNA-506 molecule through inhibiting NF-kappaB p65 to evoke reactive oxygen species generation and p53 activation. Oncogene. 2015;34:691–703. doi: 10.1038/onc.2013.597. [DOI] [PubMed] [Google Scholar]
- 20.Wei W, He HB, Zhang WY, Zhang HX, Bai JB, Liu HZ, Cao JH, Chang KC, Li XY, Zhao SH. miR-29 targets Akt3 to reduce proliferation and facilitate differentiation of myoblasts in skeletal muscle development. Cell Death Dis. 2013;4:e668. doi: 10.1038/cddis.2013.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cittelly DM, Finlay-Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P, Jacobsen BM, Sartorius CA, Richer JK. Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene. 2013;32:2555–2564. doi: 10.1038/onc.2012.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pekarsky Y, Croce CM. Is miR-29 an oncogene or tumor suppressor in CLL? Oncotarget. 2010;1:224–227. doi: 10.18632/oncotarget.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapinas K, Kessler CB, Delany AM. miR-29 suppression of osteonectin in osteoblasts: regulation during differentiation and by canonical Wnt signaling. J Cell Biochem. 2009;108:216–224. doi: 10.1002/jcb.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 25.Cao P, Maximov A, Sudhof TC. Activity-dependent IGF-1 exocytosis is controlled by the Ca(2+)-sensor synaptotagmin-10. Cell. 2011;145:300–311. doi: 10.1016/j.cell.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kornprat P, Rehak P, Ruschoff J, Langner C. Expression of IGF-I, IGF-II, and IGF-IR in gallbladder carcinoma. A systematic analysis including primary and corresponding metastatic tumours. J Clin Pathol. 2006;59:202–206. doi: 10.1136/jcp.2005.028480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slaaby R, Schaffer L, Lautrup-Larsen I, Andersen AS, Shaw AC, Mathiasen IS, Brandt J. Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J Biol Chem. 2006;281:25869–25874. doi: 10.1074/jbc.M605189200. [DOI] [PubMed] [Google Scholar]
- 28.Karpe PA, Tikoo K. Heat shock prevents insulin resistance-induced vascular complications by augmenting angiotensin-(1-7) signaling. Diabetes. 2014;63:1124–1139. doi: 10.2337/db13-1267. [DOI] [PubMed] [Google Scholar]
- 29.Vassiliadis E, Barascuk N, Karsdal MA. Atherofibrosis - a unique and common process of the disease pathogenesis of atherosclerosis and fibrosis - lessons for biomarker development. Am J Transl Res. 2013;5:1–14. [PMC free article] [PubMed] [Google Scholar]
- 30.Gatenby VK, Kearney MT. The role of IGF-1 resistance in obesity and type 2 diabetes-mellitus-related insulin resistance and vascular disease. Expert Opin Ther Targets. 2010;14:1333–1342. doi: 10.1517/14728222.2010.528930. [DOI] [PubMed] [Google Scholar]
- 31.Toba H, Cortez D, Lindsey ML, Chilton RJ. Applications of miRNA technology for atherosclerosis. Curr Atheroscler Rep. 2014;16:386. doi: 10.1007/s11883-013-0386-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, Liu Y, Li L, Su B, Yang L, Fan W, Yin Q, Chen L, Cui T, Zhang J, Lu Y, Cheng J, Fu P, Liu F. Involvement of inflammation-related miR-155 and miR-146a in diabetic nephropathy: implications for glomerular endothelial injury. BMC Nephrol. 2014;15:142. doi: 10.1186/1471-2369-15-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang YS, Li SH, Guo J, Mihic A, Wu J, Sun L, Davis K, Weisel RD, Li RK. Role of miR-145 in cardiac myofibroblast differentiation. J Mol Cell Cardiol. 2014;66:94–105. doi: 10.1016/j.yjmcc.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Gursu EM, Ozdemir A, Yalinbas B, Gursu RU, Canbakan M, Guven B, Atasoyu EM, Keskin AT, Elci A, Baru Y. The effect of icodextrin and glucose-containing solutions on insulin resistance in CAPD patients. Clin Nephrol. 2006;66:263–268. doi: 10.5414/cnp66263. [DOI] [PubMed] [Google Scholar]
- 35.Banihani SA, Makahleh SM, El-Akawi Z, Al-Fashtaki RA, Khabour OF, Gharibeh MY, Saadah NA, Al-Hashimi FH, Al-Khasieb NJ. Fresh pomegranate juice ameliorates insulin resistance, enhances beta-cell function, and decreases fasting serum glucose in type 2 diabetic patients. Nutr Res. 2014;34:862–7. doi: 10.1016/j.nutres.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Garg N, Thakur S, McMahan CA, Adamo ML. High fat diet induced insulin resistance and glucose intolerance are gender-specific in IGF-1R heterozygous mice. Biochem Biophys Res Commun. 2011;413:476–480. doi: 10.1016/j.bbrc.2011.08.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan YC, Mei PJ, Chen C, Miao FA, Zhang H, Li ZL. MiR-29c inhibits glioma cell proliferation, migration, invasion and angiogenesis. J Neurooncol. 2013;115:179–188. doi: 10.1007/s11060-013-1223-2. [DOI] [PubMed] [Google Scholar]