

Abstract
Genetic variants surrounding the interferon-λ3 (IFNL3) gene are strongly associated with clearance of hepatitis C virus (HCV). A variant (rs368234815 TT/ΔG) upstream of IFNL3 was recently implicated to control expression of a novel gene termed IFNL4. We conducted genetic analysis of rs368234815 in a chronic HCV patient cohort and molecular studies of IFNL4 in primary human hepatocytes (PHHs). Analysis of PHHs that are heterozygous at rs368234815 revealed that the IFNL4 transcript isoform is rare, accounting for 2% of transcripts arising from IFNL4 loci. Nevertheless, IFNL4 over-expression inhibited replication of multiple Flaviviridae and IFNL4 anti-viral potency required the IFNL receptor. In contrast to IFNL3, IFNL4 was inefficiently secreted and appeared to act in a cell-autonomous manner. Genetic analysis revealed associations of rs368234815 with sustained virological response and pre-treatment viral load. The findings suggest that IFNL4 is an atypical IFNL whose activity may be maladaptive to clearance of HCV infection.
Keywords: Hepatitis C virus, IL28B, Genome-wide association study, Flaviviridae, Sustained virological response
Introduction
Human genetic variation impacts susceptibility to infectious diseases. This is particularly true for hepatitis C virus (HCV), a member of the Flaviviridae and a major world-wide cause of liver disease (Thomas, 2013). Multiple genome-wide association studies have identified polymorphisms near the interferon-λ3 (IFNL3; formerly IL28B) gene that predict efficacy of pegylated IFN-α/ribavirin combination therapy for chronic infection and spontaneous HCV clearance during the acute phase of infection (Ge et al., 2009; Suppiah et al., 2009; Tanaka et al., 2009; Thomas et al., 2009). The most strongly associated single nucleotide polymorphisms (SNPs) found in GWAS studies (known as discovery SNPs) were rs12979860 in European-Americans and African-Americans, and rs8099917 in East Asians. The former is located upstream of IFNL3 and confers >2-fold likelihood of sustained virological response (SVR) between favorable and non-favorable alleles. These SNPs are unlikely to be responsible for improved clearance themselves because they are distally located from known functional elements, such as promoter binding sites or open reading frames. Rather, these variants likely co-segregate with other polymorphisms that exert biological functions related to IFNL expression or activity.
IFNL3 is a member of the type III IFNs, whose genes are clustered together on chromosome 19 (Kotenko et al., 2003; Sheppard et al., 2003). The IFNLs bind a distinct receptor complex that is expressed on epithelial cells (Sommereyns et al., 2008). Receptor binding initiates JAK-STAT signal transduction, leading to expression of interferon-stimulated genes that confer an anti-viral state. Notably, IFNLs inhibit HCV replication in vitro (Marcello et al., 2006; Robek et al., 2005) and IFNL3 genotype has been linked to liver interferon-stimulated gene mRNA expression (Honda et al., 2010; Urban et al., 2010).
Recently, RNA sequencing of primary human hepatocytes (PHH) treated with polyinosinic:cytidylic acid (poly I:C, a mimic of dsRNA) revealed transcriptional activity upstream of the IFNL3 gene (Prokunina-Olsson et al., 2013). Many of the identified transcript isoforms contain putative open reading frames (ORFs). One transcript encoding a 179 amino acid (p179) protein exhibited antiviral activity against a non-infectious HCV replicon when overexpressed and was named IFNL4. Of note, the rs12979860 discovery SNP lies within the first intron of IFNL4 (Fig. 1A). A dinucleotide variant in this region (rs368234815 TT/ΔG) causes a frame shift on these ORFs and may alter translation of IFNL4 mRNA (Prokunina-Olsson et al., 2013). A direct role for IFNL4 in HCV clearance is presently unclear, and whether the IFNL4 protein is endogenously produced in human hepatocytes infected with HCV is uncertain due to lack of Western blot or proteomic data.
Fig. 1.

IFNL4 is anti-viral against infectious Flaviviridae. (A) Relative positions of IFNL3 and IFNL4 mRNAs are indicated along with locations of genetic variants relevant to this study. Proteins produced by IFNL4 transcripts are indicated for the rs368234815 ΔG genotype. (B) Huh7.5 cells transiently transfected with the indicated expression plasmids were infected with HCV, DENV2, or YFV-17D and rates of infection were measured by quantitative immunofluorescence using antibodies against viral antigens. All data are shown as mean±S.D. (C) Representative immunofluorescence images that illustrate levels of virus infection after transfection with the indicated plasmids are shown.
Here, we examined the expression of IFNL4 transcripts in PHHs and human cell lines, characterized IFNL4 secretion and requirement for the IFNL receptor, and performed genetic analysis of rs368234815 in a large well-characterized chronic HCV cohort. While the exact role of IFNL4 in control of HCV infection is still elusive, the molecular and genetic evidence suggest that rs368234815 is likely relevant to viral clearance in patients infected with HCV.
Methods
Genotyping and statistical analysis
DNA from the IDEAL cohort (McHutchison et al., 2009) was used for genotyping. Taqman genotyping design for rs368234815 is: forward primer 5′-TGGGTCCTGTGCACGGTGAT-3′, reverse primer 5′-TCCCTCAGCGCCTTGGCA-3′, and probe 5′-CGCAG (AA/C)GGCCCCCCGG-3′. Taqman genotyping was performed according the protocol suggested by the manufacturer (Life Technologies). Genotypes for rs12979860 were obtained from the Illumina Human 610Quad genotyping array as described previously (Ge et al., 2009). Logistic regression and linear regression were performed in STATA (StataCorp) and R (www.r-project.org). Clinical phenotypes [SVR, rapid virological response (RVR), and pre-treatment viral load] were used as a dependent variable, and genetic polymorphisms (rs12979860 and rs368234815) were used as independent variables as indicated in the table for both logistic and linear regression models. All P values and 95% confidence intervals (CI) reported were calculated from two-tailed distributions. In multiple logistic or linear regression analyses, both rs12979860 and rs368234815 were considered as independent variables to assess independent associations after accounting for the effect of each other.
Cell culture, transfection, and virus infections
Huh7.5 (Blight et al., 2002) and A549 cells were cultured in high glucose DMEM supplemented with 10% heat-inactivated FBS and non-essential amino acids. A2EN cells stably transduced with ISRE-luciferase (Vignola et al., 2010) were kindly provided by Raphael Valdivia (Duke University) and grown in keratinocyte serum-free medium (Invitrogen) supplemented with 30 μg/mL recombinant EGF, 0.1 ng/mL bovine pituitary extract and 0.4 mM CaCl2. For evaluation of IFNL antiviral potency and STAT1 phosphorylation, Huh7.5 cells were transfected with the indicated CMV promoter-driven expression plasmids in 24-well plates using 0.8 μg DNA and 2 μl Lipofectamine 2000 per well unless otherwise indicated. The next day, cells were re-fed media and infected with either HCV JFH1 (Kato et al., 2001), DENV-2 New Guinea C, or YFV 17D. HCV infections were performed at a multiplicity of infection (MOI) of 0.1 whereas flavivirus infections were carried out using an MOI of 1. At 24 (flaviviruses) or 72 (HCV) hours after infection, cells were fixed in cold methanol and stained with either 4G2 anti-envelope (flaviviruses) or C7-50 anti-core (HCV) antibodies as described (Kato et al., 2006). Rates of infection were determined using a Cellomics ArrayScan high content imager. Infectious HCV in cell supernatants were determined by TCID50 assay (Lindenbach, 2009). All reported infection experiments were performed at least three separate times and each experiment had three replicates per condition. Representative experiments are shown in Figs. 1–4.
Fig. 4.
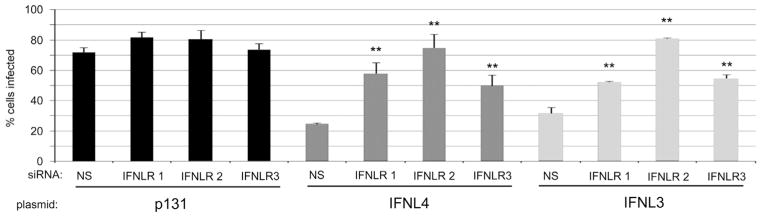
IFNL4 anti-viral activity requires the IFNL receptor. Non-silencing (NS) and three individual IFNLR1 siRNAs were introduced into Huh7.5 cells followed by transfection with the indicated expression plasmids. Subsequently, cells were infected with YFV-17D to evaluate cellular antiviral phenotype. All data show mean values±S.D. Asterisks (**) indicate P values of ≤0.005 as calculated by unpaired Student’s t-test.
For evaluation of IFNL activity in cell supernatants, Huh7.5 were transfected as described above and re-fed with supplemented DMEM 4 h after transfection. 48 h later, cell supernatants were harvested, clarified by centrifugation, and used to treat naïve cells. Subsequently, cell lysates were isolated 6 h after treatment for STAT1 Western blotting, or infected with YFV 24 h after treatment and rates of virus infection determined 24 h post-infection as described above.
For knockdown of IFNLR1, siRNA duplexes were obtained from Qiagen and used to transfect Huh7.5 cells with RNAiMAX (Invitrogen) at a final concentration of 30 nM. Cells were re-fed with media 24 h later and transfected with IFNL expression plasmids as described above. Cells were then infected with YFV 24 h after plasmids transfection and rates of infection determined as described above.
PHH experiments and RT-PCR
Cryopreserved (lot #: Hu8122) or freshly isolated PHHs (lot #: Hu8171) were obtained from Life Technologies. Cells in 24-well plates were maintained in William’s E medium supplemented with the Maintenance Supplement Pack, Serum-free (Life Technologies) 37 °C overnight for recovery. The next day, cells were transfected with 5 μg of polyI:C (Sigma) and 6.4 μl Lipofectamine 2000 per well as described previously (Park et al., 2012). Total RNA was extracted by RNeasy mini column (Qiagen) and treated with on-column DNase to eliminate genomic DNA carry-over. Reverse transcription with random primers (Life Technologies) was subsequently performed to obtain cDNA. Forward primer 5′-GTCCT GTGCA CGGTG ATC-3′ and reverse primer 5′-GATAA CTGGC AATAA ATTTA AACCG GG-3′ were used to generate pooled PCR products from cDNA transcribed from the IFNL4 genomic region. pCR®-Blunt II-TOPO® was used for blunt end insertion of PCR products. Bacterial colonies were randomly picked for sequencing to determine isoform and genotype.
For studies of endogenous IFNL4 mRNAs in A549 cells, confluent monolayers in 24-well dishes were transfected with 1.6 mg polyI:C and 4 μl of lipofectamine 2000 per well. Four hours post-transfection, total RNA was isolated with Trizol reagent and then subjected to reverse transcription using a gene specific reverse primer (5′-TCTTT GATAA CTGGC AATAA-3′). PCR was subsequently performed with this and a forward primer (5′-GTCCT GTGCA CGGTG ATC-3′.
Western blotting, cloning, and RT-qPCR
Cells were lysed using RIPA buffer and subjected to SDS-PAGE and Western blotting. Antibodies to total STAT1 and phospho-STAT1(Tyr 701) were obtained from Cell Signaling Technology. Antibody to polypyrimidine tract-binding protein was kindly provided by M.A. Garcia-Blanco (Duke University). For analysis of cell culture supernatants by Western blot, proteins were concentrated by precipitation with trichloroacetic acid and resuspended in sample buffer prior to fractionation by SDS-PAGE.
IFNL4 and p131 cDNA expression constructs encoding Halo tags were kindly provided by L. Prokunina-Olsson (NCI). For generation of IFNL3 expression constructs, the IFNL3 ORF (AY184374.1) encoding a C-terminal Flag tag was synthesized (Integrated DNA Technologies) and inserted into pcDNA5/FRT/TO (Invitrogen) using KpnI and NotI restriction sites. A comparable IFNL4 expression construct was similarly established (Prokunina-Olsson et al., 2013). Knock down of IFNLR1 mRNA expression was determined RT-qPCR. Briefly, Huh7.5 cells transfected in triplicate with siRNA duplexes (see above) were harvested 48 h post-transfection for RNA isolation (RNeasy, Qiagen) and cDNA synthesis using the High Capacity cDNA synthesis kit (Applied Biosystems). Subsequently, qPCR was performed using Power SYBR green master mix (Applied Biosystems) to measure IFNLR1 transcript levels using forward primer 5′-GGATC TGAAG TATGA GGTGG C-3′ and reverse primer 5′-GTAGA TGGTT CTGGC ACTGA G-3′. Levels of β-2-microglobulin mRNA were analyzed for calibration using forward primer 5′-TGTCT GGGTT TCATC CATCC GACA-3′ and reverse primer 5′-TCACA CGGCA GGCAT ACTCA TCTT-3′.
Results
IFNL4 exhibits broad antiviral activity
The dinucleotide variant rs368234815 (TT/ΔG) near the discovery SNP (rs12979860) has been proposed to influence HCV clearance through the production of IFNL4 in subjects carrying the ΔG allele (Fig. 1A). To expand on these findings, we investigated the effects of IFNL4 over-expression on replication of several infectious Flaviviridae species in Huh7.5 human hepatoma cells (Blight et al., 2002): HCV (JFH-1 strain), dengue virus serotype 2 (DENV2; New Guinea C strain), and the yellow fever virus vaccine (YFV; 17D strain). Transfection of vector plasmid, a construct encoding an inactive isoform of IFNL4 (p131; Fig. 1A), and an IFNL3 expression plasmid served as controls. Replication of each virus was similarly inhibited by expression of IFNL3 and IFNL4 compared to the vector alone and to inactive p131 as measured by immunofluorescence staining for viral antigen followed by quantitative imaging (Fig. 1B and C). The effects of IFNL3 or IFNL4 plasmids were similar over a range of concentrations (Supplementary Fig. 1A), suggesting that the two IFNLs have similar potencies when expressed transiently. Measurements of infectious HCV in supernatants of transfected cells revealed an approximate 10-fold reduction in infectious particles due to either IFNL3 or IFNL4 over expression (Fig. 2A). In addition, STAT1 phosphorylation levels were similar in lysates from cells transfected with either IFNL expression construct (Fig. 2B). These observations confirm the antiviral potency of IFNL4 when exogenously expressed by plasmid transfection.
Fig. 2.

IFNL4 inhibits production of infectious HCV and induces STAT1 phosphorylation after over-expression. (A) Huh7.5 cells were transfected with the indicated plasmids as in Fig. 1 and infected with HCV. Supernanants were collected at 72 h post-infection and titered by TCID50 assay. (B) Western blot analysis of STAT1, STAT1-PO4 and polypyrimidine tract-binding protein (PTB) is shown from cells transfected with the indicated plasmids for 18 h. Quantification of STAT1- PO4 signals normalized to PTB is shown below the STAT1- PO4 blot.
IFNL4 anti-viral activity is cell autonomous
IFNs are typically secreted molecules and trigger paracrine and autocrine signal transduction pathways leading to expression of interferon-stimulated genes (ISGs) that produce an anti-viral phenotype (Schoggins and Rice 2011; Schneider et al., 2014). Recombinant IFNL4 was previously described to lack biological activity for unknown reasons (Prokunina-Olsson et al., 2013). To test whether IFNL4 is functionally secreted from cells, Huh7.5 cells were transfected with expression plasmids as described in Fig. 1 and supernatants were harvested 48 h post-transfection. Subsequently, naïve Huh7.5 cells were treated with supernatants and then infected with YFV to assay for anti-viral effects. The IFNL3-containing supernatant clearly antagonized YFV infection, as expected. In contrast, IFNL4 supernatant lacked detectable antiviral activity (Fig. 3A). We also utilized a stable cell line harboring firefly luciferase under control of an interferon-sensitive response element (ISRE) to analyze supernatants from transfected cells. Whereas supernatant from cells transfected with the IFNL3 plasmid triggered signal transduction to the ISRE promoter at the lowest dose utilized, the supernatants from cells that express IFNL4 did not produce a response (Fig. 3B). Similarly, IFNL3-containing supernatant strongly induced STAT1 phosphorylation in treated cells compared to supernatant from IFNL4-transfected cells (Fig. 3C).
Fig. 3.

IFNL4 is not functionally secreted. (A) Huh7.5 cells were transfected with the indicated plasmids and supernatants harvested 48 h p.t. Naïve cells were infected with YFV-17D 24 h after treatment with the respective supernatants and levels of virus infection determined 24 h later by quantitative immunofluorescence. (B) Supernatants generated after plasmid transfection were used to treat A2EN cells that stably harbor a luciferase transgene under control of an ISRE element. (C) Conditioned media from cells expressing the indicated proteins were used to treat naïve cells for 4 h and the same factors were analyzed by Western blot as in Fig. 2. Quantification of STAT1- PO4 signals normalized to PTB is shown below the STAT1- PO4 blot.
We further analyzed IFNL4 secretion by Western blot analysis. Huh7.5 cells were transfected with plasmids encoding either flag-tagged IFNL4, halo-tagged IFNL4, or halo-tagged p131. After 48 h, equivalent fractions of cell lysates and supernatants were subjected to SDS-PAGE and Western blotting using IFNL4 antibody. Each protein was abundantly expressed in cell lysates and migrated as multiple bands of different apparent molecular weight (Supplementary Fig. 2), possibly reflecting post-translational glycosylation (Hamming et al., 2013). In contrast, each protein was barely detectable in supernatant samples, confirming that IFNL4 and p131 are inefficiently secreted. Together, these data suggest that IFNL4 is an atypical anti-viral factor that does not require secretion for activity in vitro.
The antiviral activity of IFNL4 requires IFNLR1 expression
To be classified as a type III interferon, IFNL4 should functionally require IFNLR1, the class II cytokine receptor that, along with IL10Rβ, transduces signals for all λ-interferons (Kotenko et al., 2003). To determine whether IFNL4 activity requires IFNLR1, we inhibited expression of IFNLR1 in Huh7.5 cells using three independent siRNAs and then expressed p131, IFNL3, or IFNL4 proteins in these cells. Knockdown of IFNLR1 mRNA levels was verified by RT-qPCR due to lack of reliable antibodies for the IFNLR1 protein (Supplementary Fig. 1B). Cells were then infected with YFV to assess anti-viral phenotypes. Transfection of cells with each of the three siRNAs negated the antiviral effects of both IFNL3 and IFNL4 (Fig. 4). These results indicate that IFNLR1expression is required for IFNL4 biological activity.
Endogenous IFNL4 mRNA isoforms are expressed in lung epithelial carcinoma cells
No known immortalized human cell lines are known to express IFNL4. We stimulated multiple cell lines (A549, Huh7, HepG2 and Huh7.5) by transfection with poly (I:C) and performed RT-PCR with pan-amplifying IFNL4 primers. Only A549 carcinoma cells expressed IFNL4 transcripts, each of which contained a novel intron located upstream of the start codon. Specifically, three transcripts were identified, the most abundant of which corresponded to mRNA isoforms predicted to express p107 and p131 proteins in the context of the rs368234815 ΔG genotype (Fig. 5). These mRNA isoforms were also the most abundant transcripts identified in PHHs (see below). The third transcript contained novel splice sites and does not correspond to previously reported IFNL4 mRNAs. These observations suggest that A549 cells may be a tractable model for studies of IFNL4 alternative pre-mRNA splicing and transcriptional regulation.
Fig. 5.
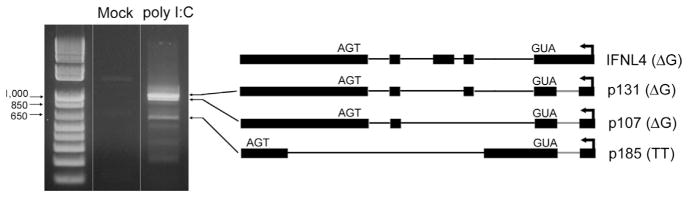
Expression of IFNL4 mRNA isoforms in A549 cells. Cells were either mock-treated or transfected with poly (I:C) and total RNA isolated 4 h post-transfection for RT-PCR detection of IFNL4. Identified transcripts and corresponding predicted proteins based on rs368234815 genotype are depicted on the right. The novel intron found in A549 cells is highlighted in red.
IFNL4 is weakly expressed in primary human hepatocytes
IFNL4 is predicted to be synthesized only by the rs368234815 ΔG allele through alternative splicing (Fig. 1A). We evaluated the relative frequency of IFNL4 mRNA transcripts in primary human hepatocytes (PHHs) from two rs368234815 heterozygous donors stimulated by transfection of poly I:C. We sequenced individual clones derived from RT-PCR amplification using primers designed to hybridize to all known IFNL4 mRNA isoforms. Sanger sequence analysis of 105 individual clones revealed similar ratios of mRNA isoforms between the two donors (Table S1A and B). Approximately 72% of transcripts were derived from the beneficial rs368234815 TT allele (Table 1). This was surprising, as most of these transcripts are predicted targets of nonsense mediated decay (NMD) due to the presence of an in-frame stop codon located upstream of exon junctions. Out of 46 and 59 clones analyzed for each PHH donor, respectively, only 2 were identified in a single donor that encoded functional IFNL4. This suggests that, in the context of rs368234815 heterozygosity, a large majority (~98%) of spliced mRNA transcripts arising from the IFNL4 locus do not produce functional IFNL4 protein.
Table 1.
IFNL4 mRNA isoforms expressed in stimulated PHHs from two donors.
| Transcript | Genotype | Amino acids | Count | Subject to NMD | Frequency (%) |
|---|---|---|---|---|---|
| p131 | TT | 75 | 19 | Yes | 18.1 |
| ΔG | 131 | 12 | No | 11.4 | |
| p179 | TT | 123 | 24 | Yes | 22.9 |
| ΔG | 179 | 2 | No | 1.9 | |
| p170 | TT | 124 | 10 | No | 9.5 |
| ΔG | 170 | 1 | No | 1.0 | |
| p107 | TT | 51 | 11 | Yes | 10.5 |
| ΔG | 107 | 9 | No | 8.6 | |
| Others | TT | – | 12 | – | 11.4 |
| ΔG | – | 5 | – | 4.8 | |
| Total | TT | 76 | 72 | ||
| ΔG | 29 | 28 | |||
| Sum | 105 | 100.0 |
rs368234815 is highly correlated with the discovery SNP (rs12979860) and exhibits stronger statistical association with pre-treatment viral load than rs12979860
To further investigate the association of IFNL4 with clinical phenotypes, we genotyped and analyzed the rs368234815 and rs12979860 variants in the IDEAL Cohort (McHutchison et al., 2009), a large well-characterized sample of patients chronically-infected with HCV (n=794 European-American; n=171 African-American). Linkage disequilibrium (LD) analysis revealed that rs368234815 is strongly associated with rs12979860 in European-American patients (r2=0.977), with somewhat lower LD in African-American patients (r2=0.713). We performed association tests of both rs12979860 and rs368234815 with patient phenotypes (SVR, RVR, and pre-treatment viral load) using a logistic regression or linear regression model to evaluate the correlation of each polymorphism with clinical features. The rs12979860 CC genotype was previously associated with a 2 fold higher chance of achieving SVR compared to that of combined non-CC genotypes (CT and TT), and the CT genotype only showed slightly higher SVR than the TT homozygous patients (Ge et al., 2009), suggesting a mixture between additive and recessive genetic models. Therefore, we performed regression analyses with both recessive and additive models separately (Table 2 and Supplementary Table 2). In the recessive model (rs12979860 CC versus non-CC, and rs368234815 TT/TT versus non-TT/TT), the associations of rs12979860 and rs368234815 with SVR were highly significant in both populations (Table 2). However, because genetic variants surrounding IFNL3 and IFNL4 are in high linkage disequilibrium (inherited together as a linkage disequilibrium block), most polymorphisms in this region exhibit strong association with these clinical phenotypes. In order to question whether rs368234815 shows independent association when controlled for the discovery SNP (rs12979860), which would support a causal role for rs368234815, we performed multiple logistic regression analysis. After correcting for the effect of rs12979860, the P value for rs368234815 remained significant in both populations for SVR (P=0.039 in European-American and 0.042 in African-American), suggesting stronger association of rs368234815 with SVR than the discovery SNP (Table 2). However, the P values of both populations were driven by a small number of patients that have disparate genotype between rs12979860 and rs368234815 and are also of borderline significance (cutoff=0.05). In the additive model, rs368234815 no longer showed statistical significance after correcting for rs12979860 (Supplementary Table 2). Therefore, the genetic evidence for whether rs368234815 is directly causal with respect to SVR is still insufficient and merits further investigation.
Table 2.
Association of candidate causal variants with clinical phenotypes by a recessive genetic model.
| European-American (N=794) LD (r2)=0.977 |
Logistic regression
|
Linear regression | ||||
|---|---|---|---|---|---|---|
| SVR
|
RVR
|
Viral load
|
||||
| OR (95% CI)* | P | OR (95% CI)* | P | coefficient (95% CI) | P | |
| rs12979860 (C/C vs non-C/C) | 7.10 (5.04–10.01) | 5.11 × 10−29 | 7.88 (4.88–12.71) | 2.91 × 10−17 | 14.69 (7.82–21.55) | 2.97 × 10−5 |
| rs368234815 (TT/TT vs non-TT/TT) | 7.42 (5.24–10.50) | 1.31 × 10−29 | 7.53 (4.69–12.07) | 5.76 × 10−17 | 15.06 (8.19–21.92) | 1.92 × 10−5 |
| Multiple logistic regression
|
Multiple linear regression
|
|||||
| rs12979860(correcting for rs368234815) | 0.65 (0.06–6.47) | 0.71 | 10.21 (0.97–106.92) | 0.053 | −9.07 (−56.25–38.1) | 0.706 |
| rs368234815(correcting for rs12979860) | 11.44 (1.13–115.46) | 0.039 | 0.77 (0.075–7.89) | 0.825 | 24.04 (−23.19–71.27) | 0.318 |
|
African-American (N=171)** LD (r2)=0.713 |
Logistic regression
|
Linear regression | ||||
|
SVR
|
RVR
|
Viral load
|
||||
| OR (95% CI)* | P | OR (95% CI)* | P | coefficient (95% CI) | P | |
| rs12979860 (CC vs non-CC) | 5.70 (2.36–13.74) | 1.08 × 10−4 | 6.19 (1.17–32.73) | 0.032 | 28.80 (12.07–45.51) | 0.001 |
| rs368234815 (TT/TT vs non-TT/TT) | 8.04 (3.10–20.81) | 1.79 × 10−5 | 3.47 (0.60–20.28) | 0.167 | 37.53 (20.26–54.81) | 2.99 × 10−5 |
| Multiple logistic regression
|
Multiple linear regression
|
|||||
| rs12979860 (correcting for rs368234815) | 1.34 (0.24–7.44) | 0.74 | 11.02 (0.98–124.06) | 0.052 | −3.32 (−33.27–26.63) | 0.827 |
| rs368234815 (correcting for rs12979860) | 6.23 (1.06–36.48) | 0.042 | 0.45 (0.03–5.97) | 0.548 | 40.46 (8.93–71.98) | 0.012 |
LD is pairwise linkage disequilibrium between rs12979860 and rs368234815.
Odds ratio (95% confidence interval).
In African-Americans, N=157 in RVR.
We previously observed that viral load is significantly correlated with rs12979860 genotype such that patients with the C allele have higher pre-treatment viral load (~1.5-fold; P=1.21 × 10−10) (Ge et al., 2009). Since rs368234815 has lower LD (r2=0.713) with the discovery SNP in African-Americans compared with Caucasians (r2=0.977), there is a higher frequency of disparate genotypes and therefore greater resolution to identify independent effects of rs368234815 and rs12979860. We found that in African-American patients, both rs12979860 and rs368234815 are associated with pre-treatment viral load (P=1.00 × 10−3 for rs12979860 and 2.99 × 10−5 for rs368234815). After correcting for the discovery SNP (rs12979860) in the multiple linear regression analysis, rs368234815 showed independent association with pre-treatment viral load (P=0.012; Table 2). The independent association of rs368234815 with pre-treatment viral load was also evident in multiple linear regression analysis using an additive genetic model (P=0.042 after correcting for rs12979860; Supplementary Table 2). Taken together, these observations suggest that IFNL4 expression may influence pre-treatment viral load in HCV-infected patients.
Discussion
Multiple genome-wide association studies have identified polymorphisms near the IFNL3 gene that predict HCV clearance, but the functional genetic variant(s) contributing to this effect phenotype have been elusive (Ge et al., 2009; Suppiah et al., 2009; Tanaka et al., 2009; Thomas et al., 2009). Here, we analyzed the genetic associations and functional effects of rs368234815. Our in vitro assays clearly indicated that IFNL4 exhibits antiviral potency against multiple members of the Flaviviridae when over-expressed by plasmid transfection in cultured human hepatoma cells. IFNL4 potency also requires the cognate IFNLR1 that is utilized by other IFNL cytokines, and is therefore accurately classified as a member of the IFNL family. An important feature of IFNL4 appears to be its relatively inefficient secretion, suggesting that autocrine signaling through binding to IFNLR1 may occur in the context of endosomes. In agreement with our observations, Hamming et al. (2013) recently reported that over-expressed IFNL4 is not efficiently secreted and requires IFNLR1 for anti-viral activity. Together, these findings establish that IFNL4 is a newly identified type III IFN that is functional if expressed to sufficient levels.
We observed that in rs368234815 heterozygous PHHs, IFNL4 mRNAs transcribed from the rs368234815 ΔG allele are not abundantly expressed, compared to TT allele mRNAs which are predicted to be degraded by NMD. This observation was unexpected, and suggests that endogenous IFNL4 mRNA isoforms may be poorly translated, because NMD requires cytoplasmic translation (Chang et al., 2007), and/or that genetic variation surrounding IFNL4 differentially regulates the transcription of IFNL4 isoforms based on genotype. Moreover, the observation that only ~2% of mRNA transcripts arising from IFNL4 loci in heterozygous primary cells encode the active IFNL4 protein may explain the difficulty of detecting endogenous IFNL4 by traditional Western blot procedures (data not shown). IFNL4 mRNA isoforms that would produce inactive proteins (p107 and p131) were relative abundant in the analyzed PHH cultures and we observed a similar trend in cultured A549 cells where only transcripts excluding the IFNL4-specific exon were detectable. These observations suggest that alternative splicing of IFNL4 pre-mRNA could significantly regulate production of IFNL4 protein and subsequent anti-viral responses. It will be of interest to investigate whether common genetic variants may influence IFNL4 splicing or transcription.
Our findings and those of other groups present an apparent paradox: IFNL4 exhibits clear anti-viral activity in vitro yet its potential for expression is correlated with poor outcome of HCV infection. While this is seemingly incongruous, we suggest that two features of IFNL4 may contribute to its potential maladaptive role in HCV control. First, IFNL4 does not appear to be strongly expressed in primary cells. Second, IFNL4 is not efficiently secreted in vitro and is predicted to act in an autocrine manner in the liver. Based on these observations, it is possible that HCV infection triggers weak hepatic IFNL4 expression that is ineffective at halting productive infection, yet induces a state of refractoriness to secreted IFNL3 and IFN-α. In agreement with this scenario, multiple studies have documented elevated basal ISG expression in livers of patients who respond poorly to IFN-based therapy (Urban et al., 2010; Dill et al., 2011; Kurosaki et al., 2011; Sarasin-Filipowicz et al., 2008; Sheahan et al., 2014) and it is possible that this is driven by expression of IFNL4 in patients with rs368234815 ΔG.
McFarland et al. recently reported that IFNL3 mRNA may be targeted by muscle-specific miRNAs whose expression is somehow triggered by HCV replication. Moreover, a 3′ untranslated region SNP (rs4803217 C/T; Fig. 1A) appears to alter miRNA-mRNA seed base-pairing leading to a small (~30%) increase in IFNL3 expression for the rs4803217 C allele due to escape from miRNA regulation (McFarland et al., 2014). The actual contribution of rs4803217 to HCV clearance is not clear and this variant has not yet been genotyped for association with SVR in a cohort of HCV patients. However, rs368234815 ΔG and rs4803217 T are in high LD (r2=0.966 in European-Americans, 0.701 in African-Americans). Thus, individuals with HCV-induced IFNL4 expression would be predicted to express less IFNL3 in hepatocytes due to post-transcriptional regulation by rs4803217, potentially exacerbating the maladaptive effects of IFNL4 expression.
The association between rs368234815 and pre-treatment viral load in the IDEAL cohort is consistent with previously reported findings that rs368234815 is significantly better correlated with normalized viral load decrease during treatment than the rs12979860 discovery SNP (Prokunina-Olsson et al., 2013). These findings are also consistent with a recent report describing a positive correlation between liver IFNL4 mRNA and HCV RNA levels (Amanzada et al., 2013). However, although the IDEAL cohort is one of the largest and best characterized HCV cohorts available, there is presently not enough statistical evidence for stronger association of rs368234815 with SVR than the discovery SNP in this cohort. Despite this, rs368234815 has been reported to be significantly more predictive of SVR than rs12979860 in a Swiss cohort of chronically-infected patients (Bibert et al., 2013). However, in the Swiss cohort, the reported LD between rs12979860 and rs368234815 is r2=0.91, which is lower than the LD reported in this study (r2=0.977). This discrepancy may be explained by differences in population structure between the two cohorts, although both cohorts are composed of self-reported Caucasians. The extremely high LD in the Caucasian population in the IDEAL cohort may also limit the statistical power to distinguish the independent association of the two variants.
In summary, we explored rs368234815 as a candidate causal variant underlying the GWAS association for HCV clearance using diverse experimental approaches. However, a definitive causal role for rs368234815 in determining HCV clearance (SVR) is still uncertain and merits further investigation, including demonstration of endogenous IFNL4 protein by Western blot and/or proteomic methods.
Supplementary Material
Acknowledgments
We thank Merck Research Laboratories for allowing the use of patient specimens and data from the IDEAL trial. We also thank Ludmila Prokunina-Olsson (NCI) for IFNL4 plasmid constructs and polyclonal antibodies. SB acknowledges support of Mariano Garcia-Blanco (Duke University). We thank Bryan Cullen and Stacia Phillips (both Duke University) for critical reading of the manuscript. SB was supported by a Grant from the NIH (DK082613).
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.virol.2014.12.020.
References
- Amanzada A, Kopp W, Spengler U, Ramadori G, Mihm S. Interferon-lambda4 (IFNL4) transcript expression in human liver tissue samples. PLoS One. 2013;8:e84026. doi: 10.1371/journal.pone.0084026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibert S, Roger T, Calandra T, Bochud M, Cerny A, Semmo N, Duong FH, et al. IL28B expression depends on a novel TT/-G polymorphism which improves HCV clearance prediction. J Exp Med. 2013;210:1109–1116. doi: 10.1084/jem.20130012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- Dill MT, Duong FH, Vogt JE, Bibert S, Bochud PY, Terracciano L, Papassotiropoulos A, et al. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2011;140:1021–1031. doi: 10.1053/j.gastro.2010.11.039. [DOI] [PubMed] [Google Scholar]
- Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- Honda M, Sakai A, Yamashita T, Nakamoto Y, Mizukoshi E, Sakai Y, Yamashita T, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology. 2010;139:499–509. doi: 10.1053/j.gastro.2010.04.049. [DOI] [PubMed] [Google Scholar]
- Hamming OJ, Terczynska-Dyla E, Vieyres G, Dijkman R, Jorgensen SE, Akhtar H, Siupka P, et al. Interferon lambda 4 signals via the IFNlambda receptor to regulate antiviral activity against HCV and coronaviruses. EMBO J. 2013;32:3055–3065. doi: 10.1038/emboj.2013.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- Kato T, Furusaka A, Miyamoto M, Date T, Yasui K, Hiramoto J, Nagayama K, et al. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J Med Virol. 2001;64:334–339. doi: 10.1002/jmv.1055. [DOI] [PubMed] [Google Scholar]
- Kato T, Date T, Murayama A, Morikawa K, Akazawa D, Wakita T. Cell culture and infection system for hepatitis C virus. Nat Protoc. 2006;1:2334–2339. doi: 10.1038/nprot.2006.395. [DOI] [PubMed] [Google Scholar]
- Kurosaki M, Tanaka Y, Nishida N, Sakamoto N, Enomoto N, Honda M, Sugiyama M, et al. Pre-treatment prediction of response to pegylated-interferon plus ribavirin for chronic hepatitis C using genetic polymorphism in IL28B and viral factors. J Hepatol. 2011;54:439–448. doi: 10.1016/j.jhep.2010.07.037. [DOI] [PubMed] [Google Scholar]
- Lindenbach BD. Measuring HCV infectivity produced in cell culture and in vivo. Methods Mol Biol. 2009;510:329–336. doi: 10.1007/978-1-59745-394-3_24. [DOI] [PubMed] [Google Scholar]
- Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, Rice CM. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, McCone J, Nyberg LM, et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med. 2009;361:580–593. doi: 10.1056/NEJMoa0808010. [DOI] [PubMed] [Google Scholar]
- McFarland AP, Horner SM, Jarret A, Joslyn RC, Bindewald E, Shapiro BA, Delker DA, et al. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol. 2014;15:72–79. doi: 10.1038/ni.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, Hergott D, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45:164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Serti E, Eke O, Muchmore B, Prokunina-Olsson L, Capone S, Folgori A, et al. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology. 2012;56:2060–2070. doi: 10.1002/hep.25897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robek MD, Boyd BS, Chisari FV. Lambda interferon inhibits hepatitis B and C virus replication. J Virol. 2005;79:3851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, Bassendine M, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, Heim MH. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci USA. 2008;105:7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan T, Imanaka N, Marukian S, Dorner M, Liu P, Ploss A, Rice CM. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe. 2014;15:190–202. doi: 10.1016/j.chom.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DL. Global control of hepatitis C: where challenge meets opportunity. Nat Med. 2013;19:850–858. doi: 10.1038/nm.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, Nakagawa M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, Kidd J, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban TJ, Thompson AJ, Bradrick SS, Fellay J, Schuppan D, Cronin KD, Hong L, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology. 2010;52:1888–1896. doi: 10.1002/hep.23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignola MJ, Kashatus DF, Taylor GA, Counter CM, Valdivia RH. cPLA2 regulates the expression of type I interferons and intracellular immunity to Chlamydia trachomatis. J Biol Chem. 2010;285:21625–21635. doi: 10.1074/jbc.M110.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.