

Abstract
Previous studies have established that Foxk1 (forkhead box k1) plays an important role in skeletal muscle regeneration. Foxk1 regulates the cell-cycle progression of myogenic progenitors by repressing the cell-cycle inhibitor gene p21. However, the underlying mechanism is not well understood. In the present study, we report the identification of Sds3 (suppressor of defective silencing 3) as an adaptor protein that recruits the Sin3 [SWI (switch)-independent 3]–HDAC (histone deacetylase) repression complex and binds Foxk1. Using GST (glutathione transferase) pull-down assays, we defined the interaction between the Foxk1 FHA (forkhead-associated domain) domain and phospho-Thr49 in Sds3. We demonstrated that the transcriptional repression of Foxk1 is dependent on the Sin3–Sds3 repression complex, and knockdown of Sds3 results in cell-cycle arrest. We further identified the protein kinase CK2 as the protein kinase for Sds3 Thr49 and demonstrated that the protein kinase activity of CK2 is required for proper cell-cycle progression. Analysis of CK2 mutant mice reveals perturbation of skeletal muscle regeneration due to the dysregulation of cell-cycle kinetics. Overall, these studies define a CK2–Sds3–Foxk1 cascade that modulates gene expression and regulates skeletal muscle regeneration.
Keywords: CK2, FHA domain, Foxk1, myogenic progenitors, phosphorylation, suppressor of defective silencing 3 (Sds3)
INTRODUCTION
Forkhead proteins are characterized by a conserved DBD (DNA-binding domain), known as the FH (forkhead) domain or WHD (winged-helix domain) [1,2]. Forkhead proteins play a variety of roles during embryonic development, aging and disease [3–5]. Although the forkhead family members have decreased conservation outside the WHD, several motifs have been shown to have important functional roles. These motifs include the leucine zipper motif, the SIM domain and the FHA (forkhead-associated) domain, which increase the complexity with regards to the functional roles and regulation of the forkhead proteins [6,7]. The FHA domain was originally identified as a conserved domain in a subset of forkhead proteins, and Foxk1 (forkhead box k1) was the prototypic protein [8]. There are four forkhead proteins containing both the FH and FHA domains in yeast (Fkh1, Fkh2, Flh1 and Flh2), but only two members in the human and mouse genomes (Foxk1 and Foxk2) [6]. The FHA domain is present in over 2000 proteins from prokaryotes to eukaryotes, including protein kinases, phosphatases, kinesins, transcription factors, ubiquitin ligases, RNA-binding proteins and others [9]. The FHA domain has been identified as a phosphopeptide-binding motif that functions to recruit interacting proteins and regulate cell-cycle kinetics [10]. Structural studies using X-ray crystallography and NMR techniques have demonstrated that the FHA domain, which contains approximately 100 amino acids, folds in a β-sandwich structure consisting of a three-stranded and a four-stranded anti-parallel β-sheet and several loops and turns linking the β-strands [11]. The β-strands maintain the structure, and the loops and turns contact with the phosphothreonine (pT) residue of the interacting protein. The pT + 3 position residue is also involved in the interaction with the loops and turns, which is referred to as the ‘pT + 3’ rule [11]. Recent studies have revealed the specificity between the FHA domain and interacting phosphothreonine-containing proteins, which is mediated by the van der Waals interaction between the conserved pocket of the FHA domain and the γ-methyl group of threonine, but not serine, residues [12]. Importantly, although Foxk1 is the prototypic FHA-domain-containing protein, the interacting phosphothreonine-containing proteins have not been defined.
Histone deacetylation is an important mechanism for transcriptional repression during development, disease and regeneration [13,14]. Two major HDAC1 (histone deacetylase 1)-containing repression complexes have been well characterized: NuRD (nucleosome remodelling and histone deacetylation) and Sin3 [SWI (switch)-independent 3) complexes [15]. The Sin3 core complex is conserved from yeast to mammals and consists of nine members: Sin3a, Sin3b, HDAC1, HDAC2, Rbbp (retinoblastoma-binding protein) 4, Rbbp7, Sap (Sin3-associated protein) 18, Sap30 and Sds3 (suppressor of defective silencing 3) [16,17]. In the core complex, Sin3a and/or Sin3b are the master scaffold proteins, HDAC1/2 are the histone deacetylation enzymes, Rbbp4/7 recruits the Sin3–HDAC complex to the nucleosome through the interaction with histones H4 and H2A, and Sap18/30 provide additional stabilization to the core complex and may extend the interaction with non-constitutive proteins associated with the Sin3 core complex [16,17]. Genetic studies using conditional knockout mouse models have revealed that the Sin3 proteins are essential for muscle development [18]. Previous studies have demonstrated that Sds3 is important for complex integrity, and studies in yeast have shown that Sds3 mutants modulate the Sin3 transcriptional repression of gene expression [19]. Moreover, overlapping phenotypes of the Sds3-null cells and the Sin3a-null cells further demonstrate the role of the Sin3–Sds3 repression complex in the regulation of embryogenesis, cellular proliferation and apoptosis [20].
The protein kinase CK2 is one of the major serine/threonine protein kinases that is highly conserved from yeast to mammals [21–23]. The tetrameric CK2 holoenzyme is composed of two catalytic subunits, CK2α (gene Csnk2a1 or CK2a1) and/or CK2α′ (gene Csnk2a2 or CK2a2), and two CK2β regulatory subunits (gene Csnk2b or CK2b) [23]. Over 300 substrates have been identified since the discovery of the first CK2 substrate, glycogen synthase [24]. The diversity of the substrates highlights the broad role of CK2 in cellular regulation. Among these roles, cell survival and apoptosis have been investigated extensively [25,26]. CK2 activity is essential for cell viability and suppression of apoptosis through the regulation of the RNA polymerase, Wnt signalling, PI3K (phosphoinositide 3-kinase)/Akt signalling and NF-κB (nuclear factor κB) pathways [27]. The functional role of CK2 in tumorigenesis, transcriptional regulation and cellular proliferation has been well documented [25–28]. For example, CK2 is a positive regulator of the Wnt/β-catenin pathway and modulates cell survival and cell proliferation by regulating multiple components in this pathway [29,30].
The functional role of CK2 during development has been defined through global deletion of CK2 in mice. Homozygous deletion of CK2α results in embryonic lethality due to cardiac defects [31]. The CK2α′ knockout is viable, and initial studies indicate that the mutant male is infertile and has a defect in spermatogenesis [32]. The CK2β-null embryo is non-viable and does not survive early embryogenesis [E (embryonic day) 6.0] owing to a cell-autonomous defect [33,34]. The CK2β-null phenotype was surprising because it is distinct from the CK2α or CK2α′ null phenotypes and suggests additional functional roles for CK2β.
In the present study, we have dissected the mechanism underlying Foxk1 transcriptional repression in MPCs (myogenic progenitor cells). Our work has defined a Foxk1 FHA domain–Sds3 Thr49–CK2 network that regulates p21 gene expression. Furthermore, we have demonstrated that CK2 has an important functional role in the cell-cycle progression of MPCs and muscle regeneration. Collectively, these studies enhance our understanding of the regulation of the MPC population and muscle regeneration by deciphering an important Foxk1-mediated regulatory network.
EXPERIMENTAL
DNA and RNA manipulation
Sin3a and Sin3b expression plasmids were provided by Dr Ronald DePinho (University of Texas MD Anderson Cancer Center, Houston TX, U.S.A.). The other plasmids and their mutants were generated using PCR and verified by DNA sequencing. RNA extraction, RT (reverse transcription) and PCR analyses were performed as described previously [35].
Tissue culture and transcriptional assays
C2C12 myoblasts were cultured in 35-mm-diameter dishes containing DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10 % (v/v) fetal bovine serum and 100 units/ml penicillin/100 μg/ml streptomycin. Approximately 105 cells were transfected with 4 μl of Lipofectamine™ (Invitrogen) and assayed for both luciferase and β-galactosidase activity. Luciferase assays were performed using the Promega Luciferase Assay System following the manufacturer’s instructions. All fold changes in luciferase activity were normalized to β-galactosidase activity and to the vector alone as described previously [36]. All of the transfection experiments were performed in triplicate and replicated three times.
Western blot, co-immunoprecipitation, in vitro translation and GST (glutathione transferase) pull-down assays
Western blot and co-immunoprecipitation assays were performed using standard protocols as described using the following antisera: anti-HA (haemagglutinin) (Santa Cruz Biotechnology and Roche), anti-Myc (Santa Cruz Biotechnology), and anti-phosphothreonine (Cell Signaling Technology) [37]. In vitro protein expression was performed using TNT Quick systems (Promega) according to the manufacturer’s instructions. GST pull-down assays used Escherichia coli BL21 expressing GST-fusion proteins, which were extracted with B-PER Bacterial Protein Extraction Reagent (Pierce Biochemicals) and then purified with glutathione–Sepharose CL-4B (GE Healthcare). GST-fusion proteins bound to Sepharose beads were incubated with 35S-labelled protein product and the BL21 cell extract. The pull-down complex was washed (four times) and resuspended in the sample loading buffer, analysed using a 4–20 % polyacrylamide gel and imaged with a Typhoon PhosphorImager as described previously [36]. To analyse the phosphorylation of the in vitro-translated Sds3, HA–Sds3 or its mutant proteins, they were immunoprecipitated using anti-HA serum (rabbit polyclonal, Santa Cruz Biotechnology), and detected with anti-phosphothreonine serum (mouse monoclonal, Cell Signaling Technology) using Western blotting as described previously [38]. In the phosphatase treatment, the in vitro-translated protein was incubated with CIP (calf intestinal alkaline phosphatase) (New England Biolabs) prior to the GST pull-down assays.
In vitro protein phosphorylation assay
GST–Sds3 (25–71) wild-type and mutant proteins were expressed in E. coli and purified using a glutathione column (GE Healthcare). Purified proteins were incubated with CK2 (New England Biolabs) in a reaction buffer (20 mM Tris/HCl, pH 7.5, 50 mM KCl and 10 mM MgCl2) supplied with 0.2 mM ATP at 30 °C for 10 min. The reaction was terminated by adding 2× sample buffer and loaded on to the SDS/PAGE gel. Threonine phosphorylation was detected using a phosphothreonine antibody (Cell Signaling Technology).
siRNA (small interfering RNA) and cell-cycle analysis
All of the siRNA oligonucleotides and the RISC (RNA-induced silencing complex)-free controls in the present studies were purchased from Dharmacon. The identification of siRNA candidate(s), gene expression and cell-cycle analysis were performed as reported previously [37]. In the transcriptional assays using siRNA treatment, C2C12 myoblasts were transfected with siRNA oligonucleotides for 24 h, then transfected with the expression plasmids and harvested for luciferase reporter expression after an additional 24 h period. All of the siRNA experiments were performed in duplicate and replicated three times.
Protein sequence analysis and statistics
The online program ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) was used to analyse protein sequence conservation. The protein phosphorylation site was analysed using NetPhosK 1.0 (http://www.cbs.dtu.dk/services/NetPhosK/). Student’s t tests were performed to identify significant differences (P < 0.05) between control and experimental samples. Data are presented as means ± S.E.M.
Animal care, CTX (cardiotoxin)-induced muscle regeneration and histology
All of the mice used in these studies were maintained, crossed, genotyped, injected and killed in accordance with an approved Institutional Animal Care and Use Committee protocol at the University of Minnesota. CTX (Calbiochem)-induced muscle injury/regeneration in the adult mouse is an established reliable model to study muscle regeneration [39]. CTX (100 μl of a 10 μM solution) was delivered intramuscularly into the gastrocnemius muscle of the adult age-matched male mice, and the mice were killed at defined time periods: control (uninjured), 1 week and 2 weeks post-injury (n = 3 at each time period). Mice were anaesthetized and perfusion-fixed with 4 % (w/v) paraformaldehyde. The gastrocnemius muscles were harvested, paraffin-embedded, sectioned and stained with haematoxylin and eosin to assess skeletal muscle architecture and myofibre size. The histology of the stained tissues was imaged using a Zeiss Axio Imager M1 microscope equipped with an AxioCam HRc camera, and processed with AxioVision 4.6 software. The muscle XSA (cross-sectional area) was determined in the gastrocnemius muscles using AxioVision 4.6.
RESULTS
The Foxk1 FHA domain recruits the transcriptional repression complex
We have demonstrated previously that Foxk1 promotes MPC proliferation, although the mechanism is unclear [40,41]. We have recently reported that Sin3 proteins interact with Foxk1 and regulate MPCs [42]. To define the transcriptional activity of Foxk1, we used the Gal4–UAS (upstream activating sequence) reporter system. We observed that full-length Foxk1 is a transcriptional repressor. Only the FHA domain (amino acids 81–290) has the transcriptional repressive activity, whereas the N-terminal (amino acids 1–40 and amino acids 40–80) and C-terminal (amino acids 368–719) domains do not (Figures 1A and 1B). The DBD (WHD) has subtle repression activity due to the interference in the interaction between the Gal4 DBD and the UAS motif (results not shown). Previous studies have demonstrated that the FHA domain interacts with phosphothreonine proteins through its conserved residues [11]. To test the hypothesis that the Foxk1 FHA domain recruits the phosphothreonine-containing repressor, we mutated the conserved amino acids within the FHA domain (Figure 1C, upper panel). All four mutations (G112D, S131A, H134L or N152S) attenuated Foxk1 transcriptional repression activity (Figure 1C, lower panel). These studies support the notion that Foxk1 plays an important role in transcriptional repression through its FHA domain in a phosphorylation-dependent mechanism.
Figure 1. Foxk1 recruits the Sin3 repression complex via its FHA domain.
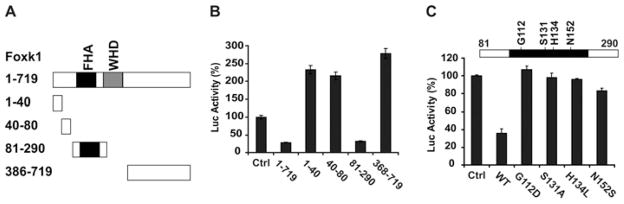
(A) Schematic summary of the Foxk1 deletional constructs used in the transcriptional assays. (B) Full-length Foxk1 or specific deletion mutations fused to the Gal4 DBD are co-transfected into C2C12 cells with the Gal4–UAS–luciferase reporter. Note that full-length Foxk1 represses the reporter. Among the Foxk1 deletional constructs, only the FHA domain (amino acids 81–290) represses the reporter to a similar level as the full-length Foxk1. The Gal4 DBD vector was utilized as the control (Ctrl). (C) The transcriptional activity of the FHA domain was further analysed by mutagenesis using the Gal4–UAS reporter system. Each mutation of the conserved amino acids within the FHA domain attenuates the repression activity. Results are means ± S.E.M. WT, wild-type.
Foxk1 FHA domain binds to Sds3 directly
Previous studies have demonstrated that Foxk1 recruits the Sin3b repression complex [43]. We tested the hypothesis that the Foxk1 FHA domain represses transcriptional activity through the interaction with the Sin3b complex. As shown in Supplementary Figure S1A (at http://www.BiochemJ.org/bj/446/bj4460349add.htm), the interaction between the Foxk1 FHA domain and Sin3b(293), the short isoform of Sin3b protein, was verified using co-IP (co-immunoprecipitation) assays. These results suggest that the Foxk1 FHA domain may function as a trancriptional repression domain through its interaction with the member in the Sin3 repression complex. To further examine the protein–protein interaction between the Foxk1 FHA domain and Sin3b(293), we performed a GST pull-down assay using the 35S-labelled Sin3b(293) protein. We could not detect any signal in the GST-FHA pull-down above that in the control GST pull-down, which indicates that the protein interaction between the Foxk1 FHA domain and Sin3b(293) is indirect (Supplemental Figures S1B and S1C). These results support the notion that there are additional protein(s) serving as an adaptor(s) between the Foxk1 FHA domain and Sin3b. Therefore we examined the ability of each of the nine members in the Sin3a–HDAC repression complex to interact with Foxk1 directly [17]. Using the GST pull-down assay, we observed that only one member of the complex, Sds3, binds to the Foxk1 FHA domain in a direct fashion (Figure 2A). As we previously observed that the transcriptional repression activity of the Foxk1 FHA domain is attenuated by specific mutations in the FHA domain, we examined the effect of the mutations in the FHA domain on the interaction between Sds3 and the FHA domain. As shown in Figure 2(B), specific mutations in the FHA domain completely abolished Sds3 binding to Foxk1. Furthermore, we verified the protein–protein interaction between Sds3 and Foxk1 in C2C12 cells using the co-IP assay (Figure 2C).
Figure 2. The Foxk1 FHA domain binds to Sds3 directly.
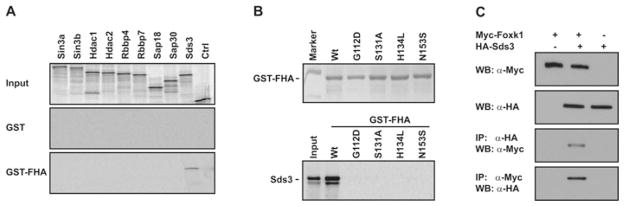
(A) Each member of the Sin3–HDAC repression complex was subcloned into the pCS3 + MT vector, translated in vitro (top panel) and incubated with the GST–FHA domain (bottom panel) using a GST pull-down assay. Only Sds3 interacts with the FHA domain directly. None of the other members of the complex binds to the FHA domain directly. Ctrl, translated protein from the pCS3 + MT vector. (B) The purified GST–FHA domain and its mutations were analysed by SDS/PAGE and then stained with Coomassie Blue (upper panel). Using a GST pull-down assay, the interaction between Sds3 and the mutated FHA proteins was completely abolished (lower panel). (C) Foxk1 binds to the Sds3 identified using the co-IP assay. Myc-tagged Foxk1 and HA-tagged Sds3 were overexpressed in C2C12 myoblasts. Myc–Foxk1 was co-immunoprecipitated with HA–Sds3 in the co-IP assays, and the reciprocal was also true. WB, Western blot; Wt, wild-type.
The Foxk1 FHA domain binds to phospho-Thr49 of Sds3
Having established that the FHA domain of Foxk1 interacts with Sds3, we further defined the Sds3 domain that interacts with Foxk1. Using the GST pull-down assay, we identified a short region (amino acids 25–71) of Sds3 that binds to the FHA domain of Foxk1 (Figures 3A and 3B). As previous studies have demonstrated that the FHA domain binds phosphothreonine proteins [11], we identified two threonine residues, Thr49 and Thr55, in this region (amino acids 25–71). Mutagenesis assays revealed that the T49A/T55A double mutation or T49A single mutation disrupted the Sds3–Foxk1 FHA interaction, but T55A alone had no effect on the interaction (Figure 3C). To examine the Thr49 phosphorylation status of the in vitro-translated Sds3 protein, we performed the immunoprecipitation experiment followed by Western blot analysis. The Sds3 protein was synthesized successfully in both wild-type and mutated constructs (Figure 3D). The phosphothreonine signal was only detectable in the wild-type Sds3, but not in the T49A mutant Sds3 (Figure 3D). These data support the notion that the Thr49 is the major phosphothreonine site in the in vitro-translated Sds3 protein. To analyse the effect of Thr49 phosphorylation on the protein interaction, the in vitro-translated Sds3 protein was treated with CIP prior to the GST pull-down assays. As shown in Figures 3(E) and 3(F), the protein interaction between the Foxk1 FHA domain and Sds3 was attenuated with CIP phosphatase treatment. In addition, we performed immunoprecipitation and Western blot analysis to examine the Thr49 phosphorylation of Sds3 in vivo. As shown in Figure 3(G), the phosphothreonine signal was only detectable in the wild-type Sds3, but not in the T49A mutant Sds3. In summary, we have demonstrated that Thr49 is the major phosphorylated threonine residue in the Sds3 protein in vitro and in vivo, and that the Foxk1 FHA domain binds to the phospho-Thr49 residue of Sds3.
Figure 3. The interaction between Foxk1 FHA domain and Sds3 is mediated by phospho-Thr49 in the Sds3 protein.

(A) Schematic representation of the interaction between the FHA domain and Sds3 deletions. (B) Sds3 deletions were engineered into the pCS3-MT vector and translated in vitro (upper panel). GST pull-down assays were used to map the FHA domain interaction region in Sds3. The amino acids 25–71 region of the Sds3 protein interacts with the Foxk1 FHA domain (lower panel). (C) The threonine residues in Sds3 (25–71) were mutated to alanine individually or in combination (T49A, T55A or T49A/T55A). Mutations of both Thr49 and Thr55 or Thr49 alone disrupt the interaction between Sds3 and the Foxk1 FHA domain, but the Thr55 mutation maintains its association with the Foxk1 FHA domain. (D) The in vitro-translated wild-type Sds3 (WT) is phosphorylated at the threonine residue, but not the mutated Sds3 (T49A), as detected by anti-phosphothreonine serum. (E) In vitro-translated Sds3 is treated with CIP prior to the GST pull-down assay. The protein interaction between Foxk1 FHA domain and Sds3 is attenuated upon phosphatase treatment (Ctrl, non-treated sample). (F) Quantification of the GST pull-down efficiency in (E). The pull-down is the ratio of the signal in the pull-down divided by the signal in the input (*P = 0.02). Results are means ± S.E.M. (G) Thr49 is the major phosphothreonine residue in the Sds3 protein in vivo. HA–Sds3 (WT) or mutated HA–Sds3 (T49A) was transfected into C2C12 cells. HA–Sds3 proteins (WT and T49A mutant) were immunoprecipitated by anti-HA serum, analysed using SDS/PAGE and probed with anti-phosphothreonine serum or anti-HA serum (loading control). Note the phosphothreonine signal in the wild-type Sds3 protein, but no detectable signal was observed in the mutant Sds3 (T49A). Ctrl, non-treated sample; WB, Western blot.
Knockdown of Sds3 results in cell-cycle arrest and decreases Foxk1 activity
Previous reports have shown that Sds3 is a core member of the Sin3–HDAC repression complex and that Sds3 is required early during embryogenesis [20]. To examine the function of Sds3 using the knockdown technique, we initially screened the Sds3 siRNA candidates and identified two siRNA oligonucleotides (#a and #b), which could efficiently knock down endogenous Sds3 in C2C12 cells (Figure 4A). To examine the role of Sds3 in the cell-cycle progression of C2C12 cells, we analysed the cell-cycle profile following Sds3 siRNA treatment, and observed cell-cycle arrest at G0/G1 following siRNA treatment (Figure 4B, #a and #b), which was quantified further (Figure 4C). Consistent with the cell-cycle perturbation, the Cdk (cyclin-dependent kinase) inhibitor genes (p21 and p27) were up-regulated following siRNA treatment (Figure 4D). Interestingly, we have also observed the up-regulation of Foxk1 (Figure 4D). Furthermore, we evaluated the effect of Gal4-DBD–Foxk1 activity following Sds3 knockdown using transcriptional assays. As shown in Figure 4(E), the transcriptional repression of Gal4-DBD–Foxk1 with the RISC-free treatment was approximately 4-fold compared with the Gal4 DBD vector control. The repression was reduced to approximately 2-fold following Sds3 knockdown with siRNA #a or #b.
Figure 4. Knockdown of Sds3 results in cell-cycle arrest in myogenic progenitors.

(A) Two siRNA oligonucleotides were used to knock down endogenous Sds3 efficiently (#a and #b) using qRT-PCR (Ctrl, RISC-free oligonucleotides). (B) Knockdown of Sds3 resulted in G0/G1 cell-cycle arrest as revealed by FACS analysis. (C) Quantification of the cell-cycle phases in (B); *P = 0.003. (D) Gene expression was analysed following Sds3 knockdown using qRT-PCR. Consistent with the cell-cycle arrest, the Cdk inhibitor genes p21 and p27 were up-regulated. (E) Effect of Gal4-DBD–Foxk1 activity upon Sds3 knockdown. The transcriptional repression of Gal4-DBD–Foxk1 was approximately 4-fold compared with the control (Gal4 DBD vector) upon the RISC-free treatment using the Gal4–UAS reporter system. This repression was attenuated to approximately 2-fold following Sds3 knockdown (*P = 0.01; **P = 0.03). Results in (A) and (C–E) are means ± S.E.M.
Sds3 Thr49 is phosphorylated by CK2
To identify the protein kinase(s) that phosphorylates Sds3 Thr49, we analysed the flanking protein sequence of Thr49 in Sds3. This region (amino acids 45–62) is evolutionarily conserved from zebrafish to human (Figure 5A). Using the phosphorylation-site prediction program NetPhosK, we identified CK2 as the best candidate to phosphorylate Thr49 and Thr55 Sds3 residues (Figure 5B). Previous studies have established that CK2 favours serine/threonine residues flanked by acidic amino acids, specifically at the + 1 or + 3 postition [24]. Using GST pull-down assays, we verified their direct interaction (Supplementary Figure S2 at http://www.BiochemJ.org/bj/446/bj4460349add.htm). Next, we examined the in vitro phosphorylation of Sds3 by CK2. Sds3 (25–71) was fused to the GST tag and purified as the substrate (Figure 5C). The in vitro phosphorylation assay revealed that the Thr49 residue was the major CK2 phosphorylation threonine residue. Compared with the phosphothreonine in the wild-type control, it was dramatically reduced in the T49A mutant, mildly reduced in the T55A mutant and undetectable in the double mutant (T49A/T55A) (Figure 5D).
Figure 5. Sds3 Thr49 is the substrate for CK2.
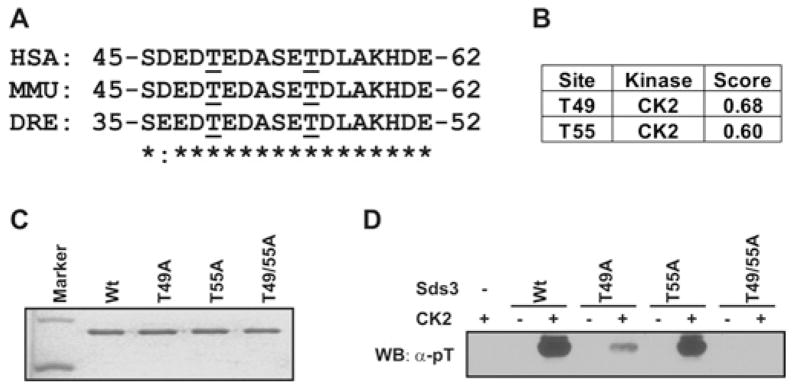
(A) The amino acids of Sds3 (amino acids 45–62) are highly conserved between species, as shown using the alignment program ClustalW2. *, identical amino acids; :, similar amino acids; underlining, threonine residues. DRE, Danio rerio; HSA, Homo sapiens; MMU, Mus musculus. (B) The protein kinases that phosphorylate the threonine residues in Sds3 (amino acids 25–71) were analysed with the program NetPhosK. CK2 was identified as the candidate kinase with the highest score. Both Thr49 and Thr55 were identified as potential phosphorylation sites by CK2. (C) Purified GST–Sds3 (amino acids 25–71) wild-type (Wt) and mutants were analysed using SDS/PAGE and stained with Coomassie Blue. (D) Purified wild-type or mutated Sds3 proteins were incubated with CK2 in vitro and then probed with an anti-phosphothreonine (pT) serum. The threonine phosphorylation was markedly reduced with the Thr49 mutation (T49A), but only marginally reduced with the Thr55 mutation (T55A) and completely abolished in the Thr49/Thr55 double mutant (T49A/T55A). WB, Western blot.
CK2 activity is required for cell-cycle progression in myogenic progenitors
Protein kinase CK2 plays a broad regulatory role during embryogenesis, cardiogenesis and tumorigenesis [31–33]. To investigate their roles in myogenesis, we first examined the gene expression profile of CK2 genes during C2C12 differentiation. As shown in Figure 6(A), CK2b gene expression was down-regulated with cell differentiation, whereas the expressions of CK2a1 and CK2a2 remained relatively stable. As the β subunit is the constitutive member of the CK2 holoenzyme, we knocked down the CK2b gene to examine the effect of CK2 activity on the myogenic progenitors. We identified four siRNA candidates, all of which knocked down CK2b gene expression efficiently (Figure 6B). We observed the cell-cycle perturbation following all of the siRNA treatments (Figure 6C and results not shown), which was further quantified in Figure 6(D). In addition, we also detected the up-regulation of p21 and p27 gene expression (Figure 6E). As shown in Figure 5, we demonstrated that the protein kinase CK2 can phosphorylate the Sds3 Thr49 residue in vitro. To examine the in vivo phosphorylation of Sds3 by CK2, we used the CK2b siRNA to down-regulate CK2 protein activity. We observed that the threonine phosphorylation in Sds3 was significantly reduced (Figures 6F and 6G). Furthermore, we observed the attenuation of the interaction between Sds3 and Foxk1 with CK2b siRNA treatment (Figure 6H). These results support the hypothesis that CK2 is a major protein kinase for the threonine phosphorylation of Sds3 in myogenic progenitors, and CK2 plays an important role in the protein interaction between Foxk1 and Sds3.
Figure 6. CK2 activity is required for proper cell-cycle progression in C2C12 cells.

(A) CK2 gene expression profile during myogenic differentiation. The expression of CK2a1 and CK2a2 is relatively stable during the cell differentiation, whereas CK2b expression is down-regulated steadily. DM, differentiation medium; GM, growth medium. (B) Four siRNA oligonucleotides have been identified and used to knock down the CK2b gene efficiently, which was verified using qRT-PCR [control (Ctrl), RISC-free oligonucleotides]. (C) Knockdown of CK2b results in G0/G1 cell-cycle arrest. A representative sample is shown following siRNA treatment #2. (D) Quantification of the cell-cycle phases in (C) (*P = 0.004). (E) Gene expression profile following CK2b gene knockdown. The Cdk inhibitor genes p21 and p27 were up-regulated following CK2b siRNA treatment. (F) Threonine phosphorylation in the Sds3 protein was reduced following CK2b knockdown. C2C12 cells were treated with CK2b siRNA or RISC-free control (Ctrl), and then transfected with an HA–Sds3 expression plasmid. HA–Sds3 was immunoprecipitated (IP) with anti-HA serum, analysed using SDS/PAGE, and then probed with anti-phosphothreonine (pT) serum (upper panel) or anti-HA serum for loading control (lower panel). (G) Quantification of the ratio of the signal of phosphothreonine to that of the total protein in (F). Knockdown of CK2b resulted in the decease of the signal ratio from 10 % (Ctrl) to 2.8 % (siRNA) (*P = 0.004). (H) Knockdown of CK2b perturbs the protein–protein interaction between Foxk1 and Sds3. The expression of Myc–Foxk1 and HA–Sds3 was detected using an anti-Myc serum and an anti-HA serum respectively. The protein–protein interaction of Foxk1 and Sds3 was analysed by immunoprecipitation with anti-HA serum and Western blot (WB) analysis with anti-Myc serum. Results in (A, B, D, E and G) are means ± S.E.M.
The CK2a2 knockout mice have perturbed skeletal muscle regeneration
Our previous studies have demonstrated the functional role of Foxk1 as a regulator of MPCs during muscle regeneration [40]. The present studies support the hypothesis that CK2 plays an important role in regulating Foxk1 activity in MPCs. The CK2 holoenzyme is composed of two α subunits and two β subunits. Moreover, the α subunits could be CK2α or CK2α′ [23]. Both CK2a1- and CK2b-null mice are embryonic lethal, whereas CK2a2-null mice are viable [31–33]. To evaluate the functional role of CK2α′ during skeletal muscle regeneration, we performed CTX injury with adult male CK2α′ -null and wild-type control mice. As shown in Figure 7(A), the skeletal muscle cellular architecture in the CK2α′ -null mouse was indistinguishable from the wild-type control in the unperturbed state. We observed perturbed muscle regeneration in the CK2α′ -null mice compared with the wild-type controls following CTX-induced muscle injury (Figure 7A). As shown in Figure 7(A), the regenerating myofibre is smaller in the CK2α′-null mice compared with that in the wild-type control 1 week or 2 weeks after CTX injury. The muscle regeneration was further quantified using the cross-sectional area of the regenerating myofibre as an indicator (Figure 7B). To define the molecular regulation program of the muscle in the CK2a2-null mice, we examined gene expression using qRT-PCR (quantitative RT–PCR). As shown in Figure 7(C), we detected an increased expression of the cyclin-dependent kinase inhibitor p21 in the CK2α′ knockout mice. These results further support our hypothesis that muscle regeneration is regulated by CK2α′.
Figure 7. Adult skeletal muscle regeneration is perturbed in CK2a2 mutant mice.

(A) The skeletal muscle architecture and myofibres in CK2a2 mutant mice are indistinguishable from those in the wild-type control mice (uninjured). However, the regenerating myofibres in CK2a2-null mice are smaller than those in the wild-type control 1 week (CTX-1w) or 2 weeks (CTX-2w) after CTX injury (scale bar, 100 μm). (B) Quantification of the myofibre XSA in panel (A). The XSA of myofibres in the wild-type mice (Uninjured) is defined as the baseline 100 % (*P = 0.02; **P = 0.03). (C) qRT-PCR was used to analyse the gene expression in CK2a2 mutant skeletal muscle. The Cdk inhibitor gene p21 was significantly up-regulated, whereas other cell-cycle inhibitor genes, such as Ccng2, Gadd45a, p27, p57 and Rb1, were unchanged compared with wild-type controls (*P = 0.02).
DISCUSSION
Skeletal muscle has a remarkable capacity to restore its cellular architecture upon injury or disease owing to a resident MPC population [44,45]. Although MPCs have a tremendous proliferative capacity, the molecular regulation of this cell population is incompletely defined [46,47]. We previously demonstrated a functional role for Foxk1 during muscle regeneration [40,41]. In the present study, we have further defined the Foxk1 regulatory network in the MPC population and have made three new discoveries that advance our mechanistic understanding of muscle regeneration in the adult mouse.
First, we have identified the direct protein–protein interaction between the Foxk1 FHA domain and the phospho- Thr49 residue in Sds3. Previous studies have implicated a functional role for FHA domain binding proteins and cell-cycle regulation. For example, the FH-FHA protein, Fkh2, regulates both the SWI5 and CLB2 gene expression by interacting with Mcm1 (minichromosome maintenance 1) [48,49]. Another example includes the activation of the Rad53–Dun1 cascade [11,50]. In this latter example, the yeast Rad53 protein harbours one kinase domain, two FHA domains and two SCD domains. Following an appropriate stimulus, Rad53 becomes phosphorylated and forms a dimer through the FHA1 domain where dimerization promotes kinase activation through an activation-loop autophosphorylation. Rad53 recruits its substrate, Dun1, through the interaction between the phospho-SCD1 of Rad53 and the FHA domain of Dun1. Dun1 then becomes activated upon phosphorylation by Rad53. We recently reported that Foxk1 interacts with Sin3 through the SID (SRC1-interaction domain) domain of Foxk1 [42]. This study suggested that the SID domain of Foxk1 may enhance or stabilize the protein–protein interaction between Foxk1 and the Sin3–Sds3 repression complex. In the present study, we demonstrate that Foxk1 harbours an FHA domain that is responsible for the transcriptional repression using the Gal4–UAS reporter system. Additionally, we demonstrated that the Foxk1 FHA domain directly interacts with the Sds3 Thr49 residue. The present study provides an important example of the regulation of cell-cycle kinetics by the FH-FHA protein and Sds3.
Similarly, we have narrowed down the FHA domain-binding motif in Sds3, and we further identified the Thr49 residue that interacts with the Foxk1 FHA domain. We have also demonstrated that the interaction is dependent on the phosphorylation status of Thr49. The FHA-domain-binding consensus motif has been characterized as pT-X-X-D/I/L (the ‘pT + 3 rule’; where X is any amino acid). However, the residues of the + 3 positions are not well conserved between the FHA-domain-interacting proteins [24]. The results of the present study have demonstrated that the Foxk1 FHA domain binds to the Thr49 (T-E-D-A) motif, but not the Thr55 (T-D-L-A) motif. The residues at the + 3 positions are alanine in both cases, which indicates that additional residues may also influence the interaction between the Foxk1 FHA domain and phosphothreonine-containing proteins.
Our second discovery identified CK2 as the kinase that phosphorylates the Sds3 Thr49 residue. Sds3 has been previously shown to be phosphorylated by Cdk5, which augments the Sds3 transcriptional repression activity [51]. In the present study, CK2 was predicted to be the best candidate kinase using a phosphorylation-site prediction program. The results of the present study support the hypothesis that CK2 is a major protein kinase that phosphorylates the threonine residue of Sds3. The CK2 complex interacts with a broad spectrum of substrates via the CK2 catalytic subunits (CK2α and CK2α′) and regulatory subunit (CK2β). Using GST pull-down assays, we verified the interaction between CK2 α/β subunits and Sds3. The CK2 phosphorylation site has been investigated extensively and the substrate minimal consensus has been defined as S/T-X-X-D/E. In the present study, we demonstrated that Thr49 is the major threonine residue in the Sds3 N-terminal region to be phosphorylated by CK2 in vitro and in vivo, although Thr55 could also be phosphorylated by CK2, but to a lesser degree in vitro. There are ten negatively charged residues (including potential serine/threonine residues) flanking Thr49. The negatively charged residues flanking Thr55 are fewer in number (six residues), and there are also positively charged residues in the + 4 (lysine) and + 5 (histidine) positions of Thr55, which are relatively rare in the CK2 phosphorylation motif [24]. We hypothesize that the enrichment of acidic amino acids flanking Thr49 provides a more favourable environment than that of Thr55 for phosphorylation by CK2, which may explain the difference in the phosphorylation assay. Another observation is that there are two serine residues (Ser45 and Ser53) in the binding region that are flanked by negatively charged amino acids. Although previous studies have shown that CK2 prefers the serine residue over the threonine residue, we focused on the threonine residue as it is well established that the FHA domain binds phosphothreonine-containing proteins exclusively. Future studies will examine the phosphorylation of the serine residues by CK2 and the subsequent effect on Sds3 function.
Our third discovery revealed the functional role of CK2 in the cell-cycle progression of myogenic progenitors and muscle regeneration. Although CK2 is believed to be ubiquitous, recent studies indicate that CK2 expression and activity are regulated by co-factors or stimulation [52,53]. In the present study, we observed that the CK2b gene was down-regulated during C2C12 differentiation in contrast with CK2a1 and CK2a2 expression. This dynamic expression pattern suggested that CK2 activity is important for cell-cycle progression. Further studies using siRNA techniques supported this hypothesis, as the down-regulation of CK2b results in cell-cycle arrest and up-regulation of Cdk inhibitor genes. The initial analysis of the CK2α′-null mice revealed increased apoptosis during the later stages of spermatogenesis in the male, which could not be compensated or rescued by CK2α [32]. In the present study, we report another specific functional role for CK2α′ during muscle regeneration, which is not rescued by CK2α. Early studies in yeast have also indicated a requirement for CK2 activity in cell-cycle progression [25]. The regulatory role of CK2 in cell-cycle progression has been further elucidated through the identification of the protein interaction between CK2 and cell-cycle proteins. For example, CK2 interacts with and phosphorylates the cell-cycle inhibitors p21 and p27, resulting in decreased activity and increased cellular proliferation. Consistant with the observation in the Foxk1-null mice, we have also observed the up-regulation of cell-cycle inhibitor p21 in CK2α′-null mice compared with wild-type control in the present study. Taken together, we provide new lines of evidence for the functional role of CK2a2 in the regulation of cell-cycle progression in myogenic progenitors.
In summary, we report a novel signalling cascade in MPCs that govern skeletal muscle regeneration that includes CK2, Sin3, Sds3, Foxk1 and p21. The results of the present study begin to assemble networks that modulate both MPCs and muscle regeneration and provide a platform for future studies using chemical genetics or upstream regulators to promote muscle regeneration in disease and aging skeletal muscle.
Supplementary Material
Acknowledgments
We acknowledge the support of Jennifer L. Fricton, Kathy M. Bowlin and Alicia M. Wallis for assistance with the histological analyses and the animal care.
FUNDING
Funding support was obtained from the National Institutes of Health (National Institute of Arthritis and Musculoskeletal and Skin) [grant numbers 5R01AR047850 and 5R01AR055906 (to D.J.G.)].
Abbreviations used
- Cdk
cyclin-dependent kinase
- CIP
calf intestinal alkaline phosphatase
- co-IP
co-immunoprecipitation
- CTX
cardiotoxin
- DBD
DNA-binding domain
- FH
forkhead
- FHA
forkhead-associated
- Foxk
forkhead box k
- GST
glutathione transferase
- HA
haemagglutinin
- HDAC
histone deacetylase
- MPC
myogenic progenitor cell
- qRT-PCR
quantitative RT–PCR
- Rbbp
retinoblastoma-binding protein
- RISC
RNA-induced silencing complex
- RT
reverse transcription
- Sap
Sin3-associated protein
- Sds3
suppressor of defective silencing 3
- SID
SRC1-interaction domain
- siRNA
small interfering RNA
- SWI
switch
- Sin3
SWI-independent 3
- UAS
upstream activating sequence
- WHD
winged-helix domain
- XSA
cross-sectional area
Footnotes
AUTHOR CONTRIBUTION
Xiaozhong Shi contributed to the concept and design, collection and assembly of data, data analysis and interpretation, and manuscript writing. David Seldin provided the CK2a2 knockout mouse model, contributed to data analysis and wrote the paper. Daniel Garry contributed to the conception and design, and data analysis and interpretation, wrote the paper and provided financial support of the research.
References
- 1.Shimeld SM, Degnan B, Luke GN. Evolutionary genomics of the Fox genes: origin of gene families and the ancestry of gene clusters. Genomics. 2010;95:256–260. doi: 10.1016/j.ygeno.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Wang M, Wang Q, Zhao H, Zhang X, Pan Y. Evolutionary selection pressure of forkhead domain and functional divergence. Gene. 2009;432:19–25. doi: 10.1016/j.gene.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 3.Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007;7:847–859. doi: 10.1038/nrc2223. [DOI] [PubMed] [Google Scholar]
- 4.Partridge L, Bruning JC. Forkhead transcription factors and ageing. Oncogene. 2008;27:2351–2363. doi: 10.1038/onc.2008.28. [DOI] [PubMed] [Google Scholar]
- 5.Hannenhalli S, Kaestner KH. The evolution of Fox genes and their role in development and disease. Nat Rev Genet. 2009;10:233–240. doi: 10.1038/nrg2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol. 2002;250:1–23. doi: 10.1006/dbio.2002.0780. [DOI] [PubMed] [Google Scholar]
- 7.Wijchers PJ, Burbach JP, Smidt MP. In control of biology: of mice, men and Foxes. Biochem J. 2006;397:233–246. doi: 10.1042/BJ20060387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofmann K, Bucher P. The FHA domain: a putative nuclear signalling domain found in protein kinases and transcription factors. Trends Biochem Sci. 1995;20:347–349. doi: 10.1016/s0968-0004(00)89072-6. [DOI] [PubMed] [Google Scholar]
- 9.Durocher D, Jackson SP. The FHA domain. FEBS Lett. 2002;513:58–66. doi: 10.1016/s0014-5793(01)03294-x. [DOI] [PubMed] [Google Scholar]
- 10.Sun Z, Hsiao J, Fay DS, Stern DF. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science. 1998;281:272–274. doi: 10.1126/science.281.5374.272. [DOI] [PubMed] [Google Scholar]
- 11.Mahajan A, Yuan C, Lee H, Chen ES, Wu PY, Tsai MD. Structure and function of the phosphothreonine-specific FHA domain. Sci Signaling. 2008;1:re12. doi: 10.1126/scisignal.151re12. [DOI] [PubMed] [Google Scholar]
- 12.Pennell S, Westcott S, Ortiz-Lombardia M, Patel D, Li J, Nott TJ, Mohammed D, Buxton RS, Yaffe MB, Verma C, Smerdon SJ. Structural and functional analysis of phosphothreonine-dependent FHA domain interactions. Structure. 2010;18:1587–1595. doi: 10.1016/j.str.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 13.Cunliffe VT. Eloquent silence: developmental functions of Class I histone deacetylases. Curr Opin Genet Dev. 2008;18:404–410. doi: 10.1016/j.gde.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayakawa T, Nakayama J. Physiological roles of class I HDAC complex and histone demethylase. J Biomed Biotechnol. 2011;2011:129383. doi: 10.1155/2011/129383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDonel P, Costello I, Hendrich B. Keeping things quiet: roles of NuRD and Sin3 co-repressor complexes during mammalian development. Int J Biochem Cell Biol. 2009;41:108–116. doi: 10.1016/j.biocel.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silverstein RA, Ekwall K. Sin3: a flexible regulator of global gene expression and genome stability. Curr Genet. 2005;47:1–17. doi: 10.1007/s00294-004-0541-5. [DOI] [PubMed] [Google Scholar]
- 17.Grzenda A, Lomberk G, Zhang JS, Urrutia R. Sin3: master scaffold and transcriptional corepressor. Biochim Biophys Acta. 2009;1789:443–450. doi: 10.1016/j.bbagrm.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Oevelen C, Bowman C, Pellegrino J, Asp P, Cheng J, Parisi F, Micsinai M, Kluger Y, Chu A, Blais A, et al. The mammalian Sin3 proteins are required for muscle development and sarcomere specification. Mol Cell Biol. 2010;30:5686–5697. doi: 10.1128/MCB.00975-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vannier D, Balderes D, Shore D. Evidence that the transcriptional regulators SIN3 and RPD3, and a novel gene (SDS3) with similar functions, are involved in transcriptional silencing in S. cerevisiae. Genetics. 1996;144:1343–1353. doi: 10.1093/genetics/144.4.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.David G, Turner GM, Yao Y, Protopopov A, DePinho RA. mSin3-associated protein, mSds3, is essential for pericentric heterochromatin formation and chromosome segregation in mammalian cells. Genes Dev. 2003;17:2396–2405. doi: 10.1101/gad.1109403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed K, Gerber DA, Cochet C. Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol. 2002;12:226–230. doi: 10.1016/s0962-8924(02)02279-1. [DOI] [PubMed] [Google Scholar]
- 22.Pinna LA. Protein kinase CK2: a challenge to canons. J Cell Sci. 2002;115:3873–3878. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- 23.Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 25.St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease. From birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66:1817–1829. doi: 10.1007/s00018-009-9150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinna LA, Allende JE. Protein kinase CK2 in health and disease: protein kinase CK2: an ugly duckling in the kinome pond. Cell Mol Life Sci. 2009;66:1795–1799. doi: 10.1007/s00018-009-9148-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruzzene M, Pinna LA. Addiction to protein kinase CK2: a common denominator of diverse cancer cells? Biochim Biophys Acta. 2010;1804:499–504. doi: 10.1016/j.bbapap.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 28.Duncan JS, Litchfield DW. Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim Biophys Acta. 2008;1784:33–47. doi: 10.1016/j.bbapap.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 29.Song DH, Dominguez I, Mizuno J, Kaut M, Mohr SC, Seldin DC. CK2 phosphorylation of the armadillo repeat region of β-catenin potentiates Wnt signaling. J Biol Chem. 2003;278:24018–24025. doi: 10.1074/jbc.M212260200. [DOI] [PubMed] [Google Scholar]
- 30.Seldin DC, Landesman-Bollag E, Farago M, Currier N, Lou D, Dominguez I. CK2 as a positive regulator of Wnt signalling and tumourigenesis. Mol Cell Biochem. 2005;274:63–67. doi: 10.1007/s11010-005-3078-0. [DOI] [PubMed] [Google Scholar]
- 31.Lou DY, Dominguez I, Toselli P, Landesman-Bollag E, O’Brien C, Seldin DC. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol Cell Biol. 2008;28:131–139. doi: 10.1128/MCB.01119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu X, Toselli PA, Russell LD, Seldin DC. Globozoospermia in mice lacking the casein kinase II alpha’ catalytic subunit. Nat Genet. 1999;23:118–121. doi: 10.1038/12729. [DOI] [PubMed] [Google Scholar]
- 33.Buchou T, Vernet M, Blond O, Jensen HH, Pointu H, Olsen BB, Cochet C, Issinger OG, Boldyreff B. Disruption of the regulatory β subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol Cell Biol. 2003;23:908–915. doi: 10.1128/MCB.23.3.908-915.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blond O, Jensen HH, Buchou T, Cochet C, Issinger OG, Boldyreff B. Knocking out the regulatory β subunit of protein kinase CK2 in mice: gene dosage effects in ES cells and embryos. Mol Cell Biochem. 2005;274:31–37. doi: 10.1007/s11010-005-3117-x. [DOI] [PubMed] [Google Scholar]
- 35.Shi X, Garry DJ. Myogenic regulatory factors transactivate the Tceal7 gene and modulate muscle differentiation. Biochem J. 2010;428:213–221. doi: 10.1042/BJ20091906. [DOI] [PubMed] [Google Scholar]
- 36.Alexander MS, Shi X, Voelker KA, Grange RW, Garcia JA, Hammer RE, Garry DJ. Foxj3 transcriptionally activates Mef2c and regulates adult skeletal muscle fiber type identity. Dev Biol. 2010;337:396–404. doi: 10.1016/j.ydbio.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi X, Bowlin KM, Garry DJ. Fhl2 interacts with Foxk1 and corepresses Foxo4 activity in myogenic progenitors. Stem Cells. 2010;28:462–469. doi: 10.1002/stem.274. [DOI] [PubMed] [Google Scholar]
- 38.Kroll J, Shi X, Caprioli A, Liu HH, Waskow C, Lin KM, Miyazaki T, Rodewald HR, Sato TN. The BTB-kelch protein KLHL6 is involved in B-lymphocyte antigen receptor signaling and germinal center formation. Mol Cell Biol. 2005;25:8531–8540. doi: 10.1128/MCB.25.19.8531-8540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goetsch SC, Hawke TJ, Gallardo TD, Richardson JA, Garry DJ. Transcriptional profiling and regulation of the extracellular matrix during muscle regeneration. Physiol Genomics. 2003;14:261–271. doi: 10.1152/physiolgenomics.00056.2003. [DOI] [PubMed] [Google Scholar]
- 40.Garry DJ, Meeson A, Elterman J, Zhao Y, Yang P, Bassel-Duby R, Williams RS. Myogenic stem cell function is impaired in mice lacking the forkhead/winged helix protein MNF. Proc Natl Acad Sci USA. 2000;97:5416–5421. doi: 10.1073/pnas.100501197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hawke TJ, Jiang N, Garry DJ. Absence of p21CIP rescues myogenic progenitor cell proliferative and regenerative capacity in Foxk1 null mice. J Biol Chem. 2003;278:4015–4020. doi: 10.1074/jbc.M209200200. [DOI] [PubMed] [Google Scholar]
- 42.Shi X, Garry DJ. Sin3 interacts with Foxk1 and regulates myogenic progenitors. Mol Cell Biochem. 2012;366:251–258. doi: 10.1007/s11010-012-1302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Q, Kong Y, Rothermel B, Garry DJ, Bassel-Duby R, Williams RS. The winged-helix/forkhead protein myocyte nuclear factor β (MNF-β) forms a co-repressor complex with mammalian sin3B. Biochem J. 2000;345:335–343. [PMC free article] [PubMed] [Google Scholar]
- 44.Biressi S, Rando TA. Heterogeneity in the muscle satellite cell population. Semin Cell Dev Biol. 2010;21:845–854. doi: 10.1016/j.semcdb.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ten Broek RW, Grefte S, Von den Hoff JW. Regulatory factors and cell populations involved in skeletal muscle regeneration. J Cell Physiol. 2010;224:7–16. doi: 10.1002/jcp.22127. [DOI] [PubMed] [Google Scholar]
- 46.Shi X, Garry DJ. Muscle stem cells in development, regeneration, and disease. Genes Dev. 2006;20:1692–1708. doi: 10.1101/gad.1419406. [DOI] [PubMed] [Google Scholar]
- 47.Kuang S, Rudnicki MA. The emerging biology of satellite cells and their therapeutic potential. Trends Mol Med. 2008;14:82–91. doi: 10.1016/j.molmed.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 48.Kumar R, Reynolds DM, Shevchenko A, Goldstone SD, Dalton S. Forkhead transcription factors, Fkh1p and Fkh2p, collaborate with Mcm1p to control transcription required for M-phase. Curr Biol. 2000;10:896–906. doi: 10.1016/s0960-9822(00)00618-7. [DOI] [PubMed] [Google Scholar]
- 49.Koranda M, Schleiffer A, Endler L, Ammerer G. Forkhead-like transcription factors recruit Ndd1 to the chromatin of G2/M-specific promoters. Nature. 2000;406:94–98. doi: 10.1038/35017589. [DOI] [PubMed] [Google Scholar]
- 50.Branzei D, Foiani M. The Rad53 signal transduction pathway: replication fork stabilization, DNA repair, and adaptation. Exp Cell Res. 2006;312:2654–2659. doi: 10.1016/j.yexcr.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 51.Li Z, David G, Hung KW, DePinho RA, Fu AK, Ip NY. Cdk5/p35 phosphorylates mSds3 and regulates mSds3-mediated repression of transcription. J Biol Chem. 2004;279:54438–54444. doi: 10.1074/jbc.M411002200. [DOI] [PubMed] [Google Scholar]
- 52.Olsten ME, Litchfield DW. Order or chaos? An evaluation of the regulation of protein kinase CK2. Biochem Cell Biol. 2004;82:681–693. doi: 10.1139/o04-116. [DOI] [PubMed] [Google Scholar]
- 53.Montenarh M. Cellular regulators of protein kinase CK2. Cell Tissue Res. 2010;342:139–146. doi: 10.1007/s00441-010-1068-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.