

Abstract
Deletion of the Ikaros (Ikzf1) DNA-binding domain generates dominant-negative isoforms that interfere with Ikaros family activity and correlate with poor prognosis in human precursor B cell acute lymphoblastic leukemias (B-ALL). Here, we show that conditional inactivation of the Ikaros DNA binding domain in early pre-B cells arrests their differentiation at a stage where integrin-dependent niche adhesion augments mitogen-activated protein kinase signaling, proliferation, and self-renewal, and attenuates pre-B cell receptor signaling and differentiation. Transplantation of polyclonal Ikzf1 mutant pre-B cells results in long-latency oligoclonal pre-B-ALL, demonstrating that loss of Ikaros contributes to multistep B-leukemogenesis. These results explain how normal pre-B cells transit from a highly proliferative and stromal-dependent to a stromal-independent phase where differentiation is enabled, providing potential therapeutic strategies for IKZF1 mutant B-ALL.
B cell differentiation is characterized by stage-specific expression of cell surface markers and recombination of the immunoglobulin heavy chain (IgH) and light chain (IgL) genes. These events are responsible for the generation of a large pool of immature B cells from which selection based on antigen receptor specificity takes place1,2. Productive rearrangements at the Igh locus allow pairing of the expressed IgM with the surrogate light chains (SLC), VpreB and λ5, and the proximal signaling molecules Igα and Igβ to form a pre-B Cell Receptor (pre-BCR) signaling complex. Subsequent engagement of the protein tyrosine kinases (PTKs) Lyn, Fyn, Blk and Syk activates signaling cascades supporting pre-B cell proliferative expansion and differentiation3. Loss-of-function mutations in the pre-BCR signaling complex or in associated PTKs cause arrest at an early B cell precursor stage4-10. The pre-BCR, working in concert with the growth-promoting IL-7 cytokine receptor (IL-7R), activates the PI3K-Akt and Mitogen-Activated protein kinases (MAPK) Erk1 and Erk2, thereby providing pre-B cell survival and proliferation11-14.
Pre-BCR signaling also induces differentiation through a distinct set of signaling effectors such as Btk, Slp65 (Blnk) and PLCγ2 (refs. 15-17). These inhibit the PI3K pathway while activating Ca2+ signaling and a network of transcription factors responsible for cell cycle withdrawal and immunoglobulin light chain (IgL) gene rearrangement18-20. Although the importance of pre-BCR signaling in proliferation and differentiation is well established, how the transition between these two disparate phases occurs remains unclear. Loss in IL-7R signaling as well as quantitative and qualitative changes in pre-BCR signaling have been proposed as possible mechanisms underlying this pre-B cell switch.
Human precursor B cell acute lymphoblastic leukemias (B-ALL) frequently display a pre-B cell phenotype, suggesting that a block at the pre-B cell proliferative stage may contribute to leukemogenesis21. Genome-wide studies in human leukemias have identified loss-of-function mutations in genes encoding regulators of B cell differentiation such as PAX5, TCF3, EBF1, and IKZF1 (IKAROS gene) in ~40% of samples from patients with precursor B-ALL22. Notably, IKZF1 mutations, including deletions in the Ikaros DNA-binding domain, were singled out as genetic lesions associated with B-ALL with poor prognosis23-27. Ikaros is required to induce transcription of lymphoid-specific genes in multi-potent progenitors, and its loss leads to developmental arrest prior to B cell lineage specification28,29. Ikaros, together with its family member Aiolos, which is induced after B cell lineage specification30, have been implicated in promoting pre-BCR-mediated differentiation by repressing expression of the SLC of the pre-BCR complex31.
Here, we provide new insight into how pre-B cells switch from proliferation to differentiation, a process that is vulnerable to leukemic transformation. We describe a stromal-adherent self-renewing phase in pre-B cell differentiation that expresses the pre-BCR signaling complex and shows strong activation of the Erk1 and Erk2 and PI3K-Akt proliferation and survival pathways, but which has no Ca2+ signaling potential, normally required for differentiation. Loss in pre-B cell stromal adhesion correlates with attenuation of proliferation, and an increase in the differentiation-inducing components of the pre-BCR signaling complex and the potential for Ca2+ signaling. Importantly, the transition of pre-B cells from a stromal-adherent proliferative to a non-adherent differentiation phase is dependent on Ikaros. Loss of Ikaros augments stromal adhesion in an integrin-dependent manner, locking pre-B cells in a highly proliferative and self-renewing phase from which BALL can arise. Importantly, the survival and proliferation of Ikaros-deficient pre-B cells is strictly dependent on the cooperation between integrin and growth factor receptor signaling, suggesting a new avenue for treatment of IKZF1 mutant, poor-prognosis B-ALL.
Results
The Ikaros family is required for pre-B cell differentiation
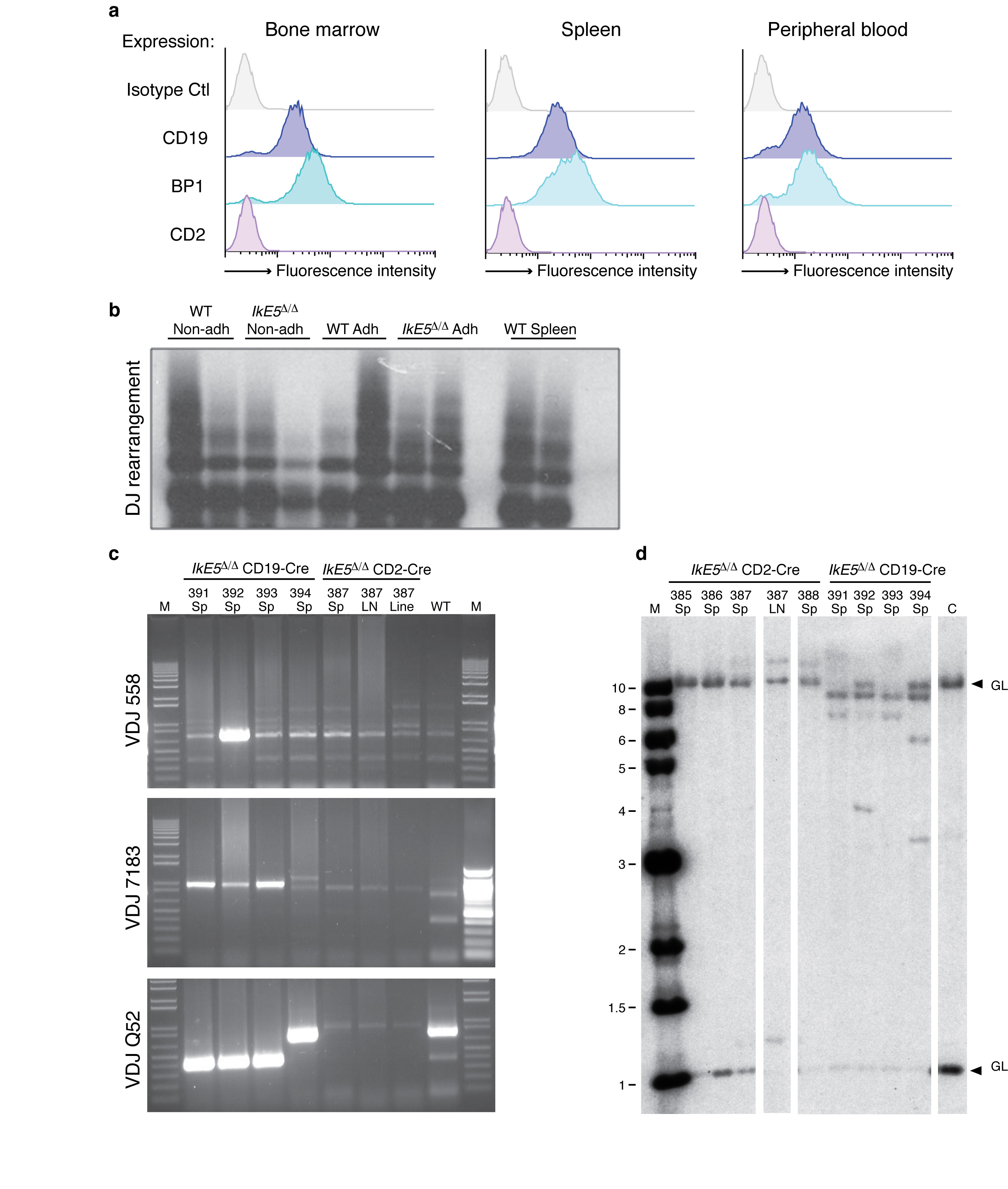
To determine the role of the Ikaros family during B cell differentiation, exon 5 of the Ikzf1 gene (defined hereafter as IkE5), encoding two Ikaros DNA binding zinc fingers, was floxed (IkE5fl/fl; Fig. 1a) and deleted from either the common lymphoid progenitor or the downstream definitive pro-B cell precursor using CD2-Cre or CD19-Cre transgenes, respectively (Supplementary Fig. 1a). Deletion of IkE5 generates Ikaros protein isoforms that lack DNA binding activity and are structurally similar to those encountered in human B-ALL (Ik6)24 (Fig. 1b, pre-B IkE5Δ/Δ. These mutant Ikaros isoforms act in a dominant-negative fashion by dimerizing with co-expressed family members, including Aiolos, and interfering with their DNA binding activity30,32. We confirmed the dominant-negative phenotype by combining the Ikzf3 (Aiolos gene) homozygous null and the Ikzf1 heterozygous null mutations (Ikzf3−/−Ikzf1+/−). Deletion of IkE5 or the combined Ikzf3−/− Ikzf1+/− mutations caused a similar block and expansion of large pre-B cells (CD19+CD43+BP1+; Fig. 1c, d and Supplementary Fig. 1b, c). These normally represent a minor population but were now found in numbers that were similar to those of all bone marrow (BM) B cells in wild type (WT) mice (Fig. 1d). As in WT, the majority of mutant large pre-B cells were in cell cycle (Fig. 1e). The few immature B (CD19+IgM+) cells detected in the IkE5fl/fl CD2-Cre mice (Fig. 1d) had not deleted IkE5fl/fl (Supplementary Fig. 1d), indicating that transition from the large to the small pre-B cell is absolutely dependent on the DNA binding activities of Ikaros gene family members expressed at this stage of differentiation.
Figure 1. Pre-B cell differentiation is dependent on the Ikaros gene family.

a, Strategy to generate a conditional Ikzf1 dominant-negative allele. Non-coding (black) and coding (white) exons, with exon 5 flanked by loxP sites (black arrowheads) for deletion are shown at the Ikzf1 locus. Stars mark zinc fingers involved in DNA binding (E4-E6) or protein dimerization (E8). b, Immunoblot analysis of Ikaros isoforms (Ik-1 and Ik-2) in WT and IkE5Δ/Δ pre-B cells. Shift in size indicates exon 5 deletion. c, Flow cytometric analysis of wild-type (WT) and IkE5fl/fl CD2-Cre bone marrow (BM) cells. Expression of stage-specific markers (as in Supplementary Fig. 1a) identify large pre-B cells (CD19+CD43+BP1+), small pre-B cells (CD19+CD2+IgM−), and immature B cells (CD19+IgM+) in the BM. d, Absolute number of cells/(femur + tibia)×2 in various B cell subsets in WT and IkE5Δ/Δ BM are shown as a graph of means ± standard deviation (s.d.). Asterisks indicate a statistically significant change between WT and mutant B cell subsets (n=10 for WT and mutant; *P < 0.01, **P < 0.0001, two-tailed Student's t-test). e, Representative cell cycle analysis of ex-vivo isolated large pre-B cells from WT and IkE5fl/fl CD2-Cre mice. Gates show relative number of cells in G0/G1 and S/G2/M phase. f, Igh and Igk rearrangements in Ikaros-deficient pre-B cells. Diagram of Igh and Igk loci depicting proximal and distal V, D and J clusters tested for recombination with primers and probes used for detection. Recombination products were amplified by PCR with decreasing amounts of pre-B cell DNA (depicted as black triangles) and with amplification of Ikzf1 non-deleted genomic fragment as loading control. g, Igk recombination fails to rescue the IkE5Δ/Δ large pre-B cell block. Analysis as described in Fig. 1c with intracellular staining for Igκ chain performed on IkE5Δ/Δ and IkE5Δ/Δ: D23 large pre-B cells (CD19+CD43+BP1+).
A hallmark of B cell differentiation is the successful recombination of the Igh locus, a prerequisite for transition to the pre-B cell stage. Both D-J and V-DJ proximal and distal recombination events at the Igh locus were detected at similar frequencies in WT and IkE5Δ/Δ pre-B cells (Fig. 1f and data not shown). However, the low-level Igk rearrangements detected in WT were not seen in mutant pre-B cells (Fig. 1f), indicating either inability to undergo light chain recombination or a block in differentiation prior to Igk recombination. Consistent with recombination only at the Igh locus, the majority of mutant pre-B cells expressed intracellular IgM but not Igκ (Supplementary Fig. 2).
Since Igk recombination is required for B cell maturation, we attempted to rescue the pre-B cell arrest by crossing the IkE5Δ/Δ mice to the D23 transgenic line that expresses a pre-rearranged Igκ chain33. IkE5Δ/Δ D23 pre-B cells were unable to differentiate past the large pre-B (CD19+CD43+ BP1+) stage (Fig. 1g and Supplementary Fig. 2), although both IgM and Igκ chains were expressed intracellularly. This indicates that lack of Igk recombination was not the cause of the maturation defect in Ikaros-deficient pre-B cells. Hence, the transition from large to small pre-B cell is regulated by the Ikaros gene family through a mechanism that is independent of recombination at the IgH or IgL gene loci.
Growth of IkE5Δ/Δ pre-B cells requires adhesion to stroma
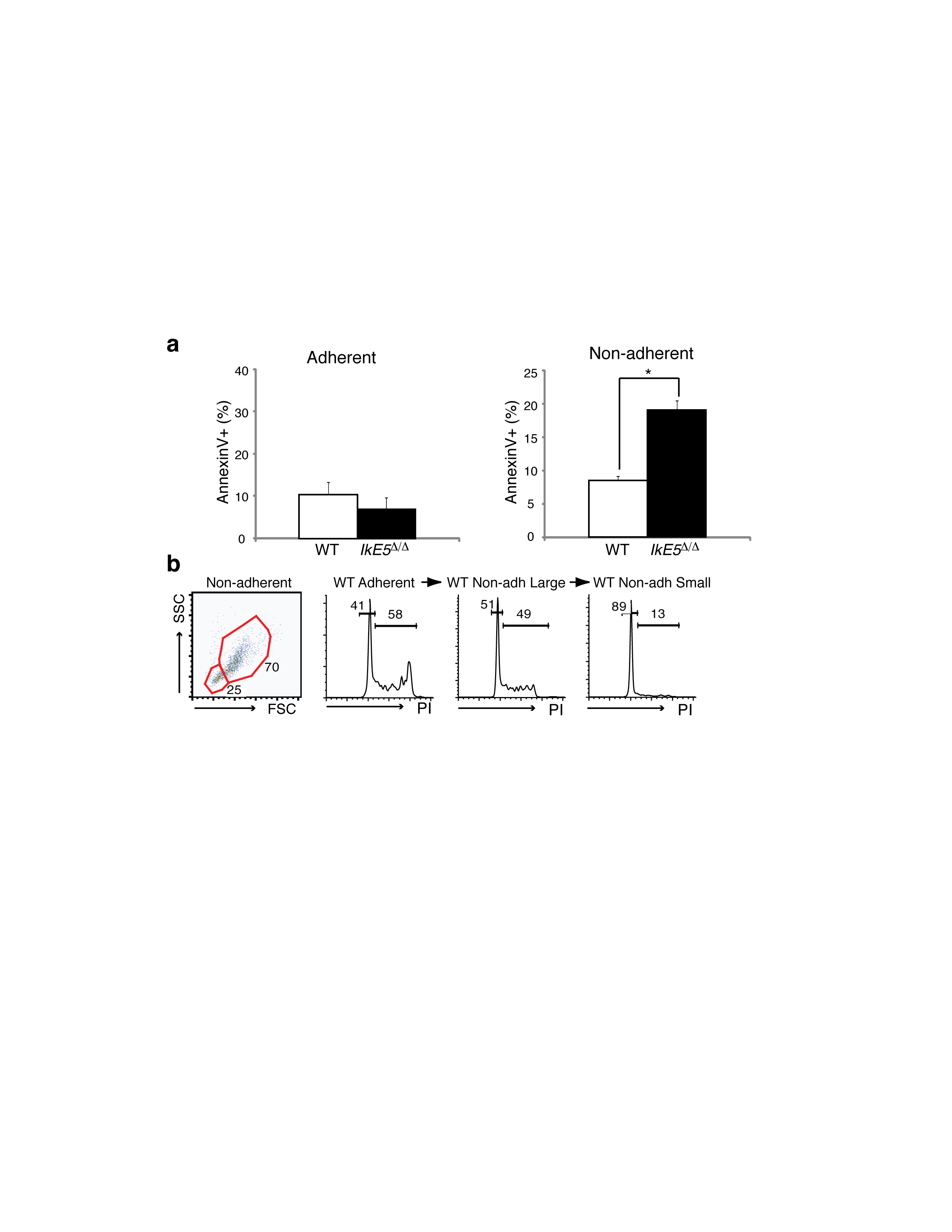
The developmental defect in IkE5Δ/Δ large pre-B cells was further evaluated in in vitro cultures11,34. Under differentiation-inducing conditions (i.e. seven days of stromal-free culture in low concentrations of serum and IL-7), the majority of WT large pre-B cells exited the cell cycle and differentiated into small pre-B (CD19+CD2+IgM−) and immature B (CD19+IgM+CD2+) cells, whereas mutant large pre-B cells (CD19+CD43+BP1+) remained undifferentiated (Fig. 2a). An increase in the concentration of IL-7 promoted the proliferative expansion of WT large pre-B cells but had little effect on their mutant counterparts. In the absence of stroma, survival of IkE5Δ/Δ pre-B cells was greatly compromised compared to WT pre-B cells even in the presence of high concentrations of IL-7, with high levels of apoptosis detected from early time points of culture (Fig. 2b, left panel, and Fig. 2c).
Figure 2. Ikaros-deficient pre-B cells grow only on stroma.

a, Flow cytometric analysis of sorted large pre-B cells (CD19+CD43+BP1+) cultured for 7 days stromal-free with limiting serum and IL-7. Differentiation of WT and IkE5Δ/Δ large pre-B cells is monitored by stage-specific markers. Arrows indicate the direction of pre-B cell differentiation as depicted in Supplementary Fig. 1a. b, Growth of WT and IkE5Δ/Δ large pre-B cells in high, low, and no (5, 0.05, and 0 ng/ml, respectively) IL-7 concentrations under stromal-free conditions (left) or with OP9 BM stroma (right). The mean absolute number of cells obtained in stromal-free (n=5) and stromal-containing (n=4) cultures with replicates for each experiment is shown in a line graph ± s.d. Asterisks denote significant differences between WT and mutant cells (*P < 0.05, **P < 0.01, two-tailed Student's t-test). c, Mean percentage ± s.d. of apoptotic (AnnexinV+) WT and IkE5Δ/Δ large pre-B cells in stromal-free cultures as in Fig. 2b, left panel. d, Cell cycle stage distribution (mean percentage ± s.d. of cells in S+G2+M) of WT and IkE5Δ/Δ large pre-B stromal cultures as in Fig. 2b, right panel. Asterisks in c and d denote significant differences between WT and mutant cells (*P < 0.05, **P < 0.01, two-tailed Student's t-test). e, Cell cycle kinetics of WT and IkE5Δ/Δ large pre-B cells grown on stroma as measured by BrdU pulse-chase. The mean fluorescence intensity (MFI) of BrdU staining is shown at 45 min of pulse and after 48 h of chase.
Although pre-B cell precursors can proliferate and differentiate in the absence of stromal contact, they can only self-renew and undergo greater expansion on stroma34-36. Since IkE5Δ/Δ large pre-B cells proliferated and expanded in vivo in the BM, we tested whether they could grow in vitro on OP9 BM-derived stroma. Under these conditions, IkE5Δ/Δ large pre-B cells grew better than WT especially under limiting concentrations of IL-7 (Fig. 2b, right panel), with ~2 to 10-fold more cells in cycle (Fig. 2d). IkE5Δ/Δ pre-B cells also displayed an increase in cell cycle kinetics compared to WT. Labeling with BrdU showed increased incorporation by IkE5Δ/Δ pre-B cells during pulse and a faster decline during chase (Fig. 2e), indicating shorter cell cycle transitions compared to WT pre-B cells.IkE5Δ/Δ pre-B cells are therefore dependent on stroma for survival and growth, with enhanced proliferation and more rapid cell cycling relative to WT pre-B cells.
Ikaros loss arrests pre-B cells in an adherent phase
A striking morphological difference was apparent between IkE5Δ/Δ and WT large pre-B cell OP9 stromal cultures. The majority of WT pre-B cells were round, light-refracting cells loosely attached to stroma, but the majority of mutant cells had a dark, flat morphology and appeared incorporated into the stromal layer (Fig. 3a, b). Dark, stromal-adherent pre-B cells were also present in WT cultures, but at a much lower frequency (Fig. 3a, b). The few IkE5Δ/Δ non-adherent cells displayed increased apoptosis (Supplementary Fig. 3a), indicating that in the absence of stromal contact, their survival was greatly compromised.
Figure 3. A stromal-dependent self-renewing phase in pre-B cell differentiation is greatly augmented by loss of Ikaros.

a, An adherent phase in pre-B cell differentiation as revealed in stromal cultures of WT and IkE5Δ/Δ large pre-B cells grown in the presence of IL-7 (5 ng/ml). Areas with adherent cells were marked with rectangles (left) and digitally magnified (right). Dotted circle marks the nucleus of OP9 stromal cells used as a stromal reference (scale bar, 30 μm). b, Ratio of adherent to non-adherent cells in WT and IkE5Δ/Δ pre-B cultures at day 2 (D2) and day 3 (D3) with 5 and 0.05 ng/ml of IL-7. The mean ratio is presented ± s.d. Asterisks denote significant differences between WT and mutant pre-B cells at each culture time point (***P < 0.0001, **P < 0.01, *P < 0.05, two-tailed Student's t-test). c, Comparative expression analysis of pre-B cell differentiation genes in adherent and non-adherent pre-B cells. Hierarchical clustering of normalized gene expression values across different conditions is shown. d, Flow cytometric analysis of adherent and non-adherent cells from WT and IkE5Δ/Δ large pre-B cell stromal cultures for CD25 and intracellular Igκ and Igκ. The percentages of positive cells relative to isotype control (grey curve) are indicated. e, Rates of propagation of WT adherent and non-adherent pre-B cell fractions grown with 5 ng/ml of IL-7. The mean number of cells generated by 5 × 104 adherent (dark blue) or non-adherent (light blue) WT pre-B cells after replating on OP9 stroma for 3 days of culture is shown in the top panel. The mean number of adherent and non-adherent subsets recovered from plating either WT adherent or non-adherent pre-B cell stromal cultures is shown in the bottom panel. Error bars indicate s.d. Asterisks indicate a statistically significant difference in the growth (top panel) of WT adherent and non-adherent B cells (*P< 0.05, **P < 0.01, two-tailed Student's t-test). f, Limiting dilution colony forming assay was performed as described previously29. The mean frequency of colony forming cells was calculated based on Poisson distribution and is presented in a line graph ± s.e. g, Re-association of WT and IkE5Δ/Δ pre-B cells after replating on stroma. The mean percentage ± s.d. of stromal-adherent cells, measured 3 hrs after replating is shown. Study was performed with two independent WT and mutant pre-B cell cultures (closed and open symbols), each assayed in ten grids/well. Binding to stroma was calculated per twenty grids and averaged for each cell type (*P < 0.0001, two-tailed Student's t-test).
A progenitor-progeny relationship between adherent and non-adherent pre-B cells was next established in WT cultures. Comparison of transcriptional profiles revealed that small pre-B cell markers (e.g. Igκ, Rag1, Rag2, Irf4, Cd2, and Cd25) expressed at low levels in adherent pre-B cells were induced in the non-adherent fraction, whereas cell cycle-promoting genes, such as Ccnd2, Egr1, Pcna, Igfbp4 and Myc, displayed the opposite expression pattern (Fig. 3c). The overall gene expression of mutant adherent pre-B cells was similar to that of their WT adherent counterparts, although a further reduction in small pre-B cell markers was seen in the mutant cells. The differential expression of small pre-B cell markers, such as CD25 and intracellular Igκ, between WT adherent and non-adherent pre-B cells, was also detected by flow cytometry. Intracellular IgM, a pan pre-B cell marker, was similarly expressed in both WT pre-B cell subsets (Fig. 3d). In the mutant cultures, adherent pre-B cells expressed IgM but no Igκ or CD25, protein consistent with the ex vivo analysis of IkE5Δ/Δ pre-B cells (Fig. 1f, g).
The cell cycle properties of WT adherent and non-adherent pre-B cells were evaluated. Whereas most WT adherent pre-B cells were in cycle, non-adherent WT pre-B cells consisted of large cycling and smaller non-cycling cells at a ratio that decreased with time in culture (Supplementary Fig. 3b and data not shown). WT adherent pre-B cells could be serially passaged on stroma and gave rise to both adherent and non-adherent cells, whereas WT non-adherent pre-B cells gave rise to mostly non-adherent cells with limited proliferative expansion (Fig. 3e).
Given the self-renewing potential of adherent pre-B cells, we compared the clonogenic properties of WT and mutant adherent pre-B cells in a limiting dilution colony-forming assay on stroma (Fig. 3f). Even in the absence of IL-7, the colony-forming potential of IkE5Δ/Δ pre-B cells was high (~20%) and orders of magnitude greater than WT. Although addition of IL-7 had little effect on the ability of IkE5Δ/Δ adherent pre-B cells to form colonies on stroma, it did increase their size by increasing proliferation (Fig. 2d and data not shown). Evaluation of the ability of WT and IkE5Δ/Δ adherent pre-B cells to re-associate with stroma revealed another important difference. Within 3 hrs of replating, 68% of IkE5Δ/Δ pre-B cells rapidly re-bound to stroma, whereas only 15% of WT adherent cells did so even after overnight incubation (Fig. 3g).
Together, these studies provide insight into pre-B cell differentiation by describing the transition from a stromal-adherent to a non-adherent phase. Stromal-adherent pre-B cells express pre-BCR are highly proliferative and have limited self-renewing potential. They are highly dependent on Ikaros for transition to a non-adherent phase where they exit the cell cycle, lose self-renewal capacity, and acquire expression of genes supporting B cell maturation. Loss of Ikaros augments stromal adhesion, self-renewal, and proliferation, pathways that most likely antagonize activation of the pre-B cell differentiation program.
Ikaros loss augments stromal-dependent proliferation
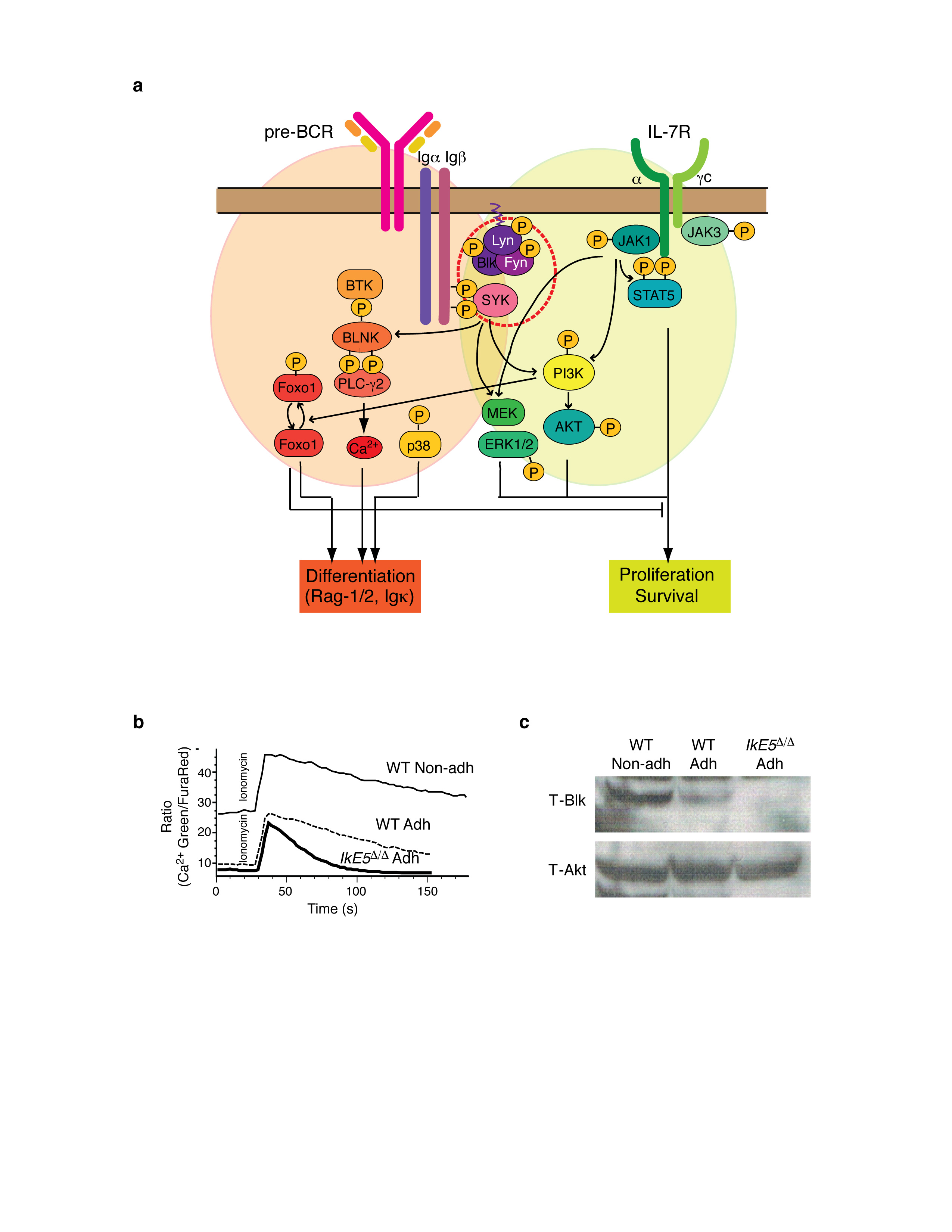
The survival and proliferative expansion of pre-B cells are supported by a combination of pre-BCR and IL-7R signaling that activates the PI3K-Akt and Erk1-2 MAPK pathways (Supplementary Fig 4a and Fig. 4a, b). Both PI3K-Akt and Erk1-2 were active in WT adherent but not in non-adherent pre-B cells (Fig. 4a), which are in the process of exiting the cell cycle (Supplementary Fig. 3b) and upregulating expression of small pre-B cell markers (Fig. 3c, d). Activation of Akt was similar in IkE5Δ/Δ adherent pre-B cells compared to WT, but activation of Erk1 and Erk2 was greatly increased (Fig. 4a). Consistent with a higher Erk1-2 MAPK activity, an increase in Cyclin D2 (Fig. 4a) and cell cycle (Fig. 2d, e) was observed in IkE5Δ/Δ compared to WT large pre-B cells.
Figure 4. Signaling pathways in WT and Ikaros-deficient pre-B cells.

a-b, Immunoblot analysis of proliferation and survival (a) and differentiation (b) signaling pathways activated by IL-7R and pre-BCR is shown. Representative expression and activity of pre-BCR-affiliated PTKs and downstream differentiation-inducing signaling effectors, as described in Supplementary Fig. 4a, are shown from two WT and three IkE5Δ/Δ independent stromal cultures of primary cells after limited in vitro propagation. β-tubulin, T-Btk or T-p38 serve as loading controls for WT and IkE5Δ/Δ pre-B cells and non-adherent WT pre-B cells. c, Intracellular Ca2+ levels (Fura Red, left panel) at steady state and Ca2+ flux (Green/Fura Red, right panel) measured after anti-IgM-stimulation of WT and IkE5Δ/Δ adherent and non-adherent pre-B cells. Fura Red staining and MFI shown on the left site inversely correlates with Ca2+ levels. Data are representative of two independent WT and mutant pre-B cell cultures.
Pre-BCR signaling also supports differentiation to the small pre-B cell stage by activating PLCγ and Ca2+ signaling (Supplementary Fig. 4a). These signaling events are required to switch cells from a proliferative to a quiescent state by inducing transcriptional responses that rely in part on the transcription factor Foxo1 (refs. 18,20). Notably, the baseline level of intracellular Ca2+ was low in both WT and IkE5Δ/Δ adherent pre-B cells but elevated in WT non-adherent pre-B cells, which were the only cells capable of fluxing Ca2+ either upon pre-BCR engagement or ionomycin treatment (Fig. 4c and Supplementary Fig. 4b). Upstream and downstream effectors of Ca2+ signaling such as Blnk and Foxo1 proteins were expressed at low amounts in both WT and IkE5Δ/Δ adherent pre-B cells and were greatly induced in WT non-adherent cells, consistent with small pre-B cell differentiation (Fig. 4b). The low amounts of Foxo1 expressed in adherent pre-B cells were phosphorylated, correlating with active PI3K-Akt in these cells. As previously reported for small pre-B cell differentiation20, p38 MAPK activity was induced from WT adherent to non-adherent pre-B cells but was nearly undetectable in IkE5Δ/Δ adherent pre-B cells (Fig. 4b).
Both the proliferation- and differentiation-inducing arms of pre-BCR signaling are dependent on activation of Fyn, Lyn, Blk and Syk. These PTKs were expressed at similar protein amounts in WT adherent and non-adherent pre-B cells (Fig. 4a). In IkE5Δ/Δ compared to WT adherent pre-B cells, the protein amounts of these key proximal components of pre-BCR signaling (Fyn, Syk, Blk) were greatly reduced, while the amounts of activated (phosphorylated) Lyn were also diminished (Fig. 4b and Supplemental Fig. 4c). The reduced protein expression or activation of these PTKs was unexpected, as it predicts not only a defect in differentiation but also in proliferation of IkE5Δ/Δ pre-B cells, contrary to what we observed both in vivo and in vitro (Figs. 1-2).
IL-7R signaling was examined as a possible mechanism of compensation for the loss in pre-BCR signaling. Phosphorylation of Stat5 (p-Stat5), a measure of IL-7R signaling, was comparable in WT and IkE5Δ/Δ adherent pre-B cells (Fig. 4a). Furthermore and in contrast to WT pre-B cells, IL-7R signaling was unable to support the growth of mutant pre-B cells under stromal-free conditions, and the mutant pre-B cells were only partly dependent on IL-7 for growth on stroma (Fig. 2b and Fig. 3f). Thus, receptors other than IL-7R and pre-BCR must be responsible for activation of survival and proliferation signaling pathways in IkE5Δ/Δ large pre-B cells. Engagement of such receptors is likely to be mediated by interaction of the mutant pre-B cells with stroma.
Increased integrin signaling in IkE5Δ/Δ pre-B cells
A comparative genome-wide transcriptional analysis of primary and cultured WT and IkE5Δ/Δ pre-B cells was performed to reveal potential pathways that might support the aberrant adhesion and growth properties of the mutant population. A signature of genes was deduced that was differentially expressed between both freshly isolated IkE5Δ/Δ and WT large pre-B cells and between IkE5Δ/Δ and WT adherent pre-B cells cultured in vitro (Fig. 5a, b). Up-regulated genes in IkE5Δ/Δ pre-B cells were highly enriched in pathways involved in focal adhesion and remodeling of the actin cytoskeleton (Fig. 5a). Integrins (e.g. Itga1, Itga5, Itgb1) as well as other structural and signaling components of focal adhesions (e.g. Ptk2, Vcl, Actn1, Cttn, Dock1, Rogdi) were shared by many of these pathways (Fig. 5b). The increase in integrin expression was validated at the protein level in both primary and cultured cells. Furthermore, expression of the active isoform of integrin β1 (detected with an activation-specific anti-integrin β1 antibody and elevated levels of phosphorylated focal adhesion kinase (p-FAK), a key downstream effector of integrin signaling, indicated that not only expression but also activation of integrin signaling were elevated in IkE5Δ/Δ pre-B cells (Fig. 5c-f). Although not as pronounced as in IkE5Δ/Δ pre-B cells, significantly higher amounts of FAK and p-FAK were also observed in WT adherent relative to non-adherent pre-B cells, indicating that integrin signaling is also active in WT adherent pre-B cells (Fig. 5d-f).
Figure 5. Increase in integrin signaling mediates adhesion of IkE5Δ/Δ pre-B cells to a stromal niche.

a, Pathway analysis of genes upregulated in IkE5Δ/Δ relative to WT large pre-B cells. Analysis was performed with a signature of upregulated genes shared by ex vivo mutant pre-B cells prior to and after limited stromal expansion. Pathways enriched for integrins and integrin signaling effectors are highlighted in red. b, Upregulated expression of components of the integrin-actin cytoskeleton pathway in primary and cultured WT and IkE5Δ/Δ pre-B cells as defined in Figs. 1 and 3. Hierarchical clustering of normalized gene expression values across different conditions is shown. c, Cell surface expression of integrins α5, β6, and activated β1 in ex vivo sorted and in vitro cultures of large pre-B cells. MFI for integrin staining is provided. d-f, Increase in FAK activation measured by flow cytometry, immunoblot and confocal microscopy. d, Flow cytometric analysis of p-FAK expression in ex-vivo and in vitro cultured large pre-B cells. MFI for p-FAK is indicated. e, Confocal immunofluorescence microscopy detection of activated p-FAK (red channel), GFP-expressing OP9 stroma (green channel), and nuclei (DAPI, blue channel). Scale bar, 25 μm. f, Immunoblot analysis of total FAK and activated p-FAK, with Btk as a loading control as in Fig. 4a. g, Adhesion of WT and IkE5Δ/Δ adherent pre-B cells to fibronectin-coated plates (left panel) in the presence of the fibronectin-derived RGD peptide or the RGE mutant variant (right panel). Asterisks denote significant differences in adhesion between mutant pre-B cells in the presence or absence of RGD or RGE peptides (n=3; *P < 0.05, two-tailed Student's t-test). h, Chemotaxis of WT (circle) and IkE5Δ/Δ (square) pre-B cells measured in a transwell migration assay in the presence of SDF1. The mean percentage of cells recovered at the bottom of the well in two independent studies is shown.
Further evidence for integrin-mediated adhesion was provided by measuring binding of adherent pre-B cells to integrin ligands in vitro. Notably, the frequency by which IkE5Δ/Δ (~80%) or WT (~20%) pre-B cells bound to fibronectin (Fig. 5g) was similar to that with which they bound to stroma (Fig. 3g). A fibronectin peptide (RGD) that binds to integrin α4β1significantly inhibited the binding of both WT and IkE5Δ/Δ pre-B cells, implicating VLA-4 as one of the integrins participating in pre-B cell adhesion (Fig. 5g).
Integrin signaling is involved in pre-B cell chemotaxis in response to CXCL12 (stromal cell-derived factor 1; SDF1)-CXCR4 receptor interactions37. We tested whether elevated integrin signaling in IkE5Δ/Δ pre-B cells affected their chemotactic properties. In sharp contrast to wild-type adherent pre-B cells, their mutant counterparts were unable to migrate in a transwell assay in response to SDF1 (Fig. 5h), indicating that increased integrin signaling reduced chemokine-mediated chemotaxis of the mutant pre-B cells. Consistent with these in vitro data, circulating pre-B cells were not detected in IkE5fl/fl CD19-Cre mice although they were readily seen in WT mice (Supplementary Fig. 5a). Thus, the increase in integrin signaling manifested upon loss of Ikaros in pre-B cells is likely responsible for their stable adhesion to stroma, survival, and proliferative expansion.
IkE5Δ/Δ pre-B cell survival is dependent on FAK activity
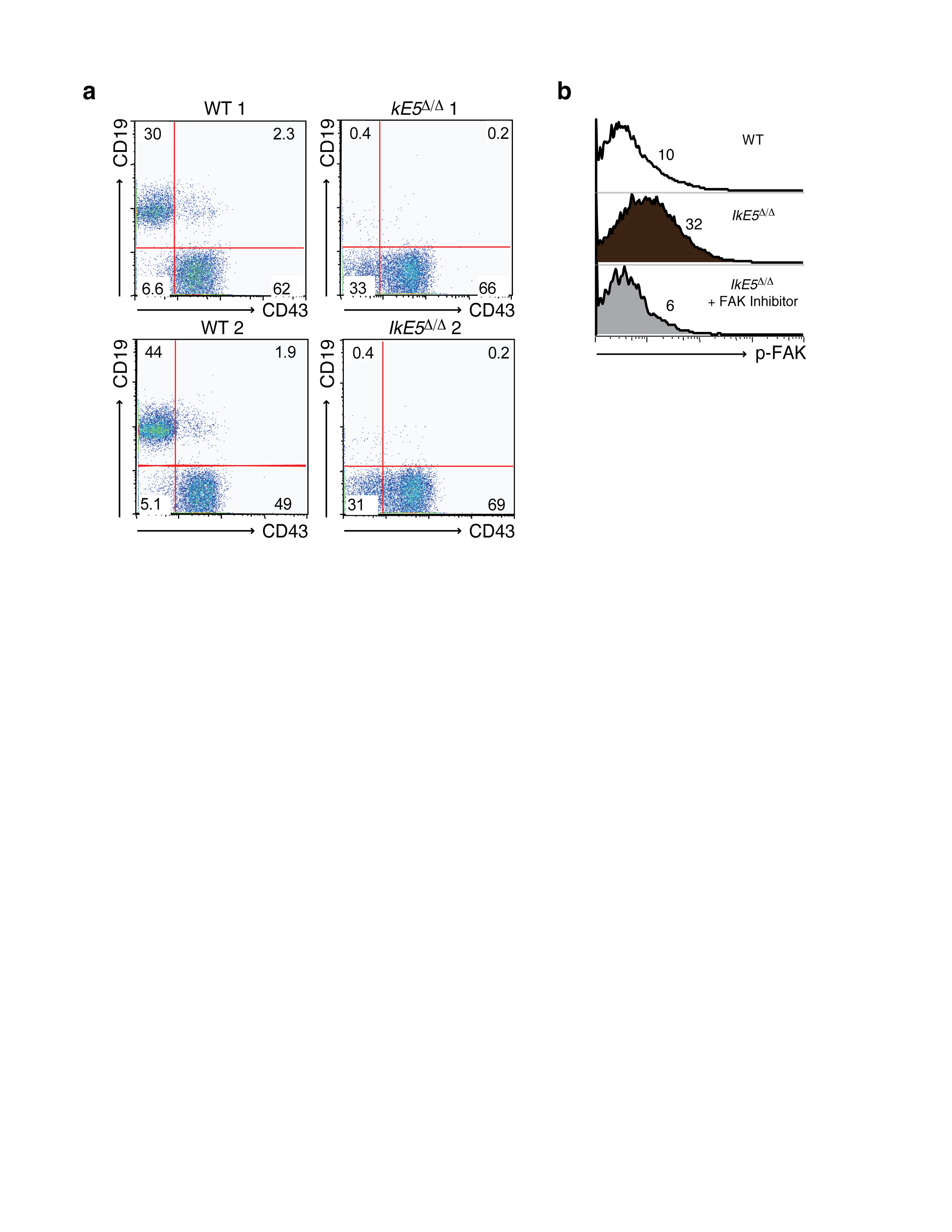
The role of integrin signaling in supporting stromal adhesion and survival of IkE5Δ/Δ pre-B cells was validated by treatment with a small molecule inhibitor (PF-431396) that blocks the kinase activity of FAK (Ptk2) and the related kinase Ptk2b (ref. 38), which together serve as major signaling effectors of the integrin pathway. FAK-Ptk2b inhibitor treatment greatly reduced stromal adhesion not only in IkE5Δ/Δ but also in WT pre-B cells (Fig. 6a). However, the loss in adhesion preceded an increase in apoptosis only in IkE5Δ/Δ pre-B cell and not in WT pre-B cells (Fig. 6b).
Figure 6. FAK inhibition interferes with survival of IkE5Δ/Δ pre-B cells.

a-b. In vitro effects of FAK inhibition on pre-B cell stromal adhesion and survival. The mean percentage ± s.d. of adherent cells (left) and % inhibition of adhesion ± s.d. (right), are shown in (a). The percentage of viable adherent and non-adherent cells recovered in the presence of FAK inhibitor is shown in (b). The data in (a) are from two independent cultures with replicate testing (n=4). For Annexin staining described in (b) replicates were pooled. c-d, In vivo effect of FAK inhibition on IkE5Δ/Δlarge pre-B cells. c, The mean number ± s.d. of pro-B-large pre-B cells (CD19+CD43+) per leg (femur + tibia) of WT (n=2) and IkE5Δ/Δ CD19-Cre (n=3) mice is shown after 3-5 doses of FAK inhibitor (WT, n=3; IkE5Δ/Δ CD19-Cre, n=6) or vehicle control (WT, n=2; IkE5Δ/Δ CD19-Cre, n=3). The effect of FAK inhibitor treatment on total BM B cells (CD19+) in WT mice is also shown. d, Percent of apoptotic cells (mean ± s.d.) of BM cells from panel c. Asterisks in a, c, and d denote significant changes in adhesion, cellularity or survival of WT and mutant large pre-B cells in the presence of the FAK inhibitor vs. control (*P < 0.05, **P < 0.01, ***P <0.001, two-tailed Student's t-test).
The dependence of IkE5Δ/Δ pre-B cells on integrin signaling was also tested in vivo. IkE5fl/fl CD19-Cre mice and WT littermates were given 3-5 doses of an orally bioavailable FAK-Ptk2b inhibitor (PF-562271) or vehicle control and the number of BM pre-B/B cells and apoptotic index was quantified shortly afterwards (Fig. 6c, d). IkE5Δ/Δ large pre-B cells constituted the great majority of BM B cells in vehicle-treated IkE5fl/fl CD19-Cre mice and showed rapid reduction following FAK inhibitor treatment (Fig. 6c). This decrease correlated with an increase in apoptosis that was specific for large pre-B cells and correlated with a specific reduction in activated FAK (p-FAK) in IkE5Δ/Δ large pre-B cells (Fig. 6d and Supplementary Fig. 5b). FAK inhibitor treatment had little effect on the cellularity of WT BM B cells that were mainly comprised of small pre-B and immature B cells. Given the small number of WT large pre-B cells present in the WT BM, it was difficult to discern the effect of the FAK inhibitor treatment on the WT large pre-B cell population.
Taken together, these studies indicate that increased integrin signaling mediated by FAK is responsible for increased stromal adhesion, survival, and accumulation of Ikaros-mutant large pre-B cells under both in vitro and in vivo settings.
Integrin and growth factor signaling cooperate in pre-B cells
We then tested whether integrin-mediated adhesion was sufficient to support stromal-dependent survival and proliferation of IkE5Δ/Δ pre-B cells. The majority of IkE5Δ/Δ pre-B cells plated on fibronectin and collagen died after overnight incubation, indicating that integrin signaling alone could not support their survival (Fig. 7a). In sharp contrast, the majority of WT pre-B cells survived under these conditions.
Figure 7. Cooperation between integrin and growth factor signaling supports survival and proliferation of IkE5Δ/Δ pre-B cells.

a, Effect of integrin and cytokine signaling on WT and IkE5Δ/Δ pre-B cell survival. Mean percent recovery ± s.d. of WT (left) and IkE5Δ/Δ (right) adherent pre-B cells after overnight incubation on plates coated with integrin ligands (fibronectin and collagen, FN+Col) or BSA, in the absence (None) or presence of cytokines (IL-7, SCF, or Both). Asterisks denote significant differences in the number of mutant pre-B cells recovered in the presence of cytokines with or without integrin ligand binding. The data shown is from two independent cultures with replicate testing in each (n=4; *P < 0.01, two-tailed Student's t-test). b, Effect of integrin and cytokine signaling on survival of IkE5Δ/Δ pre-B cells. The mean number ± s.d. of plate-bound and -unbound WT and IkE5Δ/Δ pre-B cells recovered after overnight incubation in plates coated with integrin ligands (FN+Col) in the presence of cytokines (IL-7, SCF, or Both) or without cytokines (None). The mean percent ± s.d. of viable cells in the bound and unbound fractions is shown on the right. Asterisks denote significant changes in number or survival of mutant pre-B cells under the different conditions (n=3; *P < 0.05, **P < 0.01, ***P <0.001, ****P <0.0001 two-tailed Student's t-test). c, Effect of integrin and cytokine signaling on proliferation of IkE5Δ/Δ pre-B cells. The mean percent ± s.d. of cycling cells (S+G2+M) in the bound and unbound fractions of IkE5Δ/Δ pre-B cells as described in Fig. 6b is shown. Asterisks denote significant differences in proliferation of mutant pre-B cells measured when bound or not bound to integrin ligands in the presence of different cytokines (n=3; *P < 0.05, two-tailed Student's t-test).
Stromal niches provide adhesion and growth factor support. Growth factors such as the c-Kit ligand (Stem Cell Factor; SCF) and IL-7 are required for the growth of both early hematopoietic progenitors and lymphoid precursors39,40. In the absence of integrin ligand binding, IL-7 and/or SCF had little or no effect on the survival of IkE5Δ/Δ pre-B cells. However, the combination of integrin engagement and IL-7 or SCF greatly increased IkE5Δ/Δ pre-B cell survival (Fig. 7a, b), and had a smaller but still significant stimulatory effect on the proliferation of IkE5Δ/Δ pre-B cells (Fig. 7c). Thus, augmentation of integrin signaling in IkE5Δ/Δ compared to WT pre-B cells is not only important for maintaining cells in proximity to a stromal niche, but also for cooperating with growth factor signaling to support survival and proliferation, acting in lieu of pre-BCR signaling (Supplementary Fig. 6).
High leukemogenic potential of IkE5Δ/Δ pre-B cells
The rapid development of precursor T-lymphoid neoplasms in IkE5Δ/Δ mice (data not shown and ref. 41) precludes the assessment of B-lymphoid leukemogenesis in these mutant mice. The leukemogenic potential of IkE5Δ/Δ pre-B cells was therefore evaluated by transplantation of this population into immunodeficient NOD-SCID-Il2rg−/− (NSG) recipient mice. Following transplantation with IkE5Δ/Δ pre-B cells isolated from either Cd19-Cre or Cd2-Cre donors, recipient NSG mice uniformly exhibited circulating immature CD19+BP1+CD2− B-lymphoid cells within 7 weeks (data not shown), and developed signs of disseminated leukemia/lymphoma by 3-4 months post-transplant, with weight loss, hyperventilation, and hepatosplenomegaly (mean spleen weight 668 ± 188 mg), whereas recipients of WT pre-B cells remained healthy (Fig. 8a). The disease in recipients of IkE5Δ/Δ Cd19-Cre pre-B cells was somewhat more aggressive than in IkE5Δ/Δ Cd2-Cre recipients (Fig. 8a; median survival 107d vs. 143d; P = 0.0021, Mantel-Cox test). At necropsy, recipients of IkE5Δ/Δ Cd19-Cre pre-B cells had pancytopenia with severe anemia (blood hemoglobin 4.8 ± 0.7 g/dL) that likely contributed to morbidity or death, while IkE5Δ/Δ Cd2-Cre recipients tended to develop hind-limb paralysis and malignant pleural effusions.
Figure 8. Leukemogenic potential of IkE5Δ/Δ pre-B cells.

a, Kaplan-Meier survival curve of NSG mice transplanted with WT or IkE5Δ/Δ pre-B cells. The survival of both cohorts of recipients of IkE5Δ/Δ pre-B cells was significantly shorter than recipients of WT pre-B cells (P = 0.013, Mantel-Cox tests). b, Histopathology of precursor B-cell acute lymphoblastic leukemia/lymphoma derived from IkE5Δ/Δ pre-B cells. (i–iii): Hematoxylin & eosin-stained sections of spleen (i), liver (ii), and BM (iii) from a premorbid NSG recipient (sacrificed day 63 post-transplant) of IkE5Δ/Δ pre-B cells from a CD19-Cre donor. Note the extensive infiltration of all organs with large cells with moderate cytoplasm and prominent nucleoli, and frequent mitotic figures (arrows). Scale bars, 50 μm. (iv) Wright-Giemsa stain of cytospin of BM from this recipient (scale bar, 20 μm). Note predominant population of large lymphoblasts with immature nuclei and basophilic cytoplasm (arrows). c, Integrin expression is elevated in both IkE5Δ/Δ pre-leukemic and leukemic pre-B cells. Percentage of WT, IkE5Δ/Δ pre-leukemic and leukemic pre-B cells expressing integrins α5 (CD49e), α6 (CD49f) and β1 (CD29). d, FAK activation (pFAK) measured by flow cytometry in the presence and absence of FAK inhibitor in WT and mutant pre-B cells. e, FAK inhibition interferes with stromal adhesion of IkE5Δ/Δ preleukemic and leukemic pre-B cells. Inhibitor-treated, closed symbols; vehicle-treated, open symbols. (n=2 each). f, FAK inhibition induces cell death in IkE5Δ/Δ pre-leukemic and leukemic pre-B cells (n=4; *P <10-6, **P <10−7 two-tailed Student's t-test).
Histopathological analysis showed extensive invasion of spleen and liver and involvement of bone marrow with large lymphoid cells of high mitotic index (Fig. 8b). Phenotypic analysis of the malignant IkE5Δ/Δ tumor cells revealed that they were similar to the initially transplanted population with regards to both large pre-B cell surface antigen expression (CD19+CD43+BP1+CD2−; Supplementary Fig. 7a), expression of adhesion molecules (Fig. 8c), and adherence to stroma (see below). However, in contrast to the polyclonal nature of the transplanted IkE5Δ/Δ pre-B cell population (Fig. 1g and Supplementary Fig. 7b), the IkE5Δ/Δ leukemic cells were oligoclonal by Igh gene rearrangement (Supplementary Fig. 7c, d).
Given that the stromal-adhesion phenotype was maintained in IkE5Δ/Δ leukemic pre-B cells, we examined the status of integrin signaling and whether these cells were sensitive to FAK inhibition in vitro. Both integrin expression and FAK activation were elevated in the leukemic cells (Fig. 8c, d). Similar to the pre-leukemic mutant pre-B cells, IkE5Δ/Δ leukemic pre-B cells were highly sensitive to FAK inhibition undergoing both loss of adhesion and dramatic increase in apoptosis (Fig. 8e, f) that correlated with suppression of p-FAK (Fig. 8d). Thus, the arrest at the adherent large pre-B cell stage mediated by loss of Ikaros predisposes this population for transformation to a leukemic state, which however appears to be sensitive to inhibition of adhesion-based signaling pathways supported by FAK, thereby opening a new avenue for therapy of poor prognosis B cell precursor leukemias in humans.
Discussion
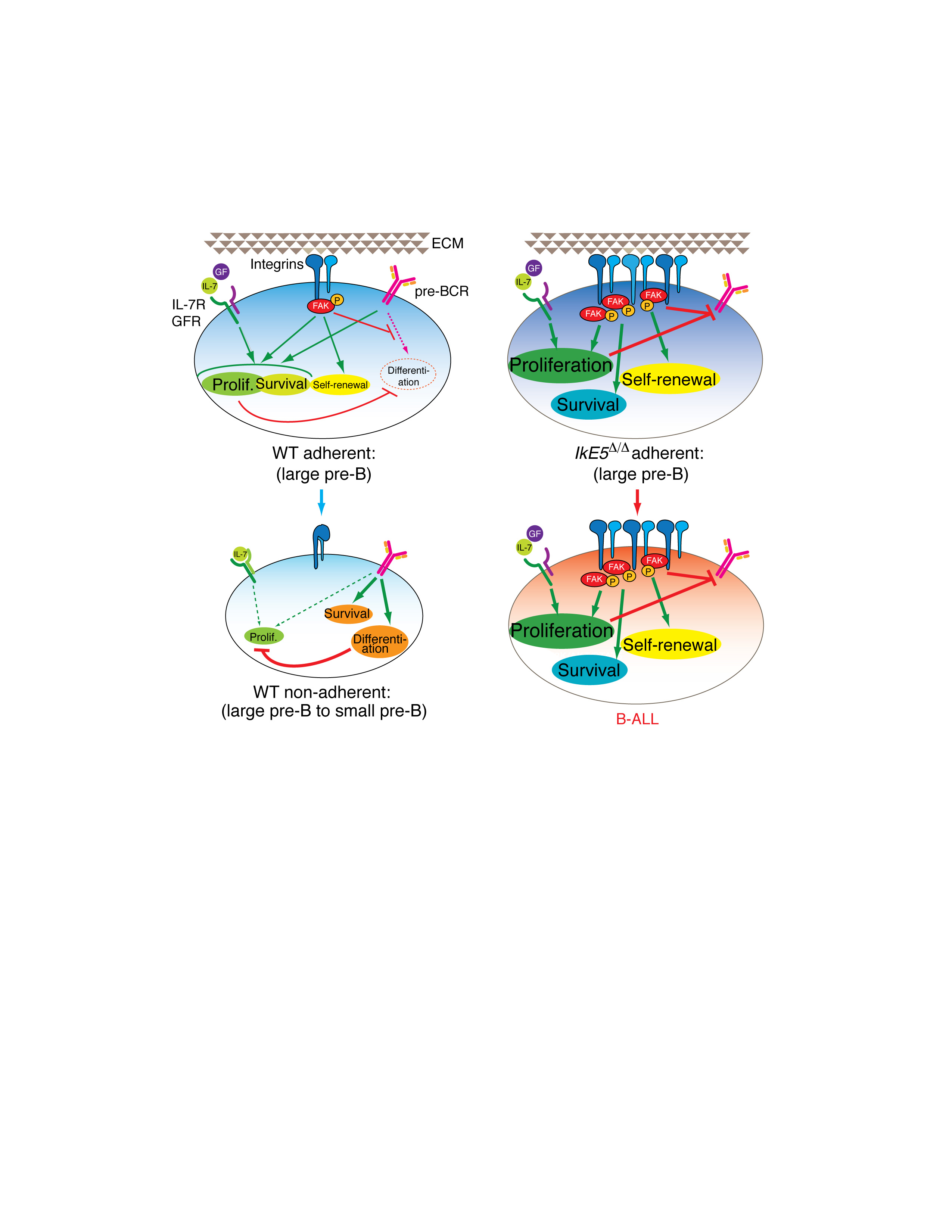
Our studies define a key step in pre-B cell differentiation that is characterized by adherence to bone marrow stroma, self-renewal and proliferative expansion36. Normal pre-B cells transit rapidly through this stromal adherent phase and enter into a non-adherent phase in which self-renewal is lost, proliferation is diminished and differentiation into an Igκ-expressing cell is induced. Loss of Ikaros activity arrests pre-B cells in the adherent, self-renewing, pro-proliferative phase and promotes their transformation to a malignant state.
Ikaros-deficient large pre-B cells show increased expression of structural and signaling components of the focal adhesion and actin cytoskeleton pathways at both the transcription and protein level. Integrins (Itgb1, Itgb3, Itga9, Itga5), vinculin (Vcl), α-actinin (Actn1), myosins (Myo1b, Myl12b), FAK (Ptk2), and the Rac activating GEFRho guanine exchange factors (Arhgef12, Arhgef5), Rho GTPase activating protein 5 (Arhgap5), and Dedicator of cytokinesis (Dock1) were such examples. Extracellular matrix (ECM) components such as laminin (Lamb1), secreted phospho protein (Spp1) and matrix metallopeptidase 14 (Mmp14) were also induced in mutant pre-B cells. These transcriptional changes underscore a strong integrin-signaling environment that is demarcated by high levels of activated FAK, and a stable adhesion and re-adhesion phenotype to both stroma and integrin ligands. WT pre-B cells also demonstrate integrin-dependent adhesion, however, this is transient as the majority of adherent cells rapidly switch to a non-adherent phase and fail to re-adhere upon replating.
Integrins, engaged by the ECM, serve as signaling centers that control actin filament polymerization required for the formation and maturation of focal adhesions42,43. Recent studies suggest that actin also plays a role in organizing adhesion sites and the actinintegrin linkage composition can determine adhesion stability44,45. Cells that lack highly bundled actin structures, such as lymphocytes, have less prominent adhesions46. The actin cross-linker α-actinin, required for the formation of actin filaments47, is upregulated in Ikaros deficient pre-B cells. Actin polymerization and disassembly are regulated by the opposing activities of the Rho and Rac small GTPases48. A potential increase in the Rac activating GEF Dock1 in Ikaros-deficient pre-B cells may contribute to the establishment of an actin environment that is conducive to integrin signaling. Given the low levels of Ca2+ in adherent pre-B cells, recycling of adhesion structures, for example through calpain cleavage of talin, may be ineffective. Consistent with a stable focal adhesion environment, Ikaros-deficient pre-B cells failed to undergo SDF1-mediated chemotaxis in spite of normal or elevated expression of CXCR4 and do not exit the BM microenvironment. In this regard, a recent study reported increased chemotaxis of FAK-deficient pre-B cell precursors from the BM to the periphery49 that is consistent with our observations herein of increased FAK activity in Ikaros-deficient pre-B cells and their inability to migrate from the BM to the periphery.
Notably, in WT pre-B cells, the pro-proliferative and differentiation-inducing arms of pre-BCR signaling are segregated away from one other and into the stromal-adherent and non-adherent phases of pre-B cell differentiation. The presence of IL-7R and pre-BCR together with active Erk1-2 MAPK and PI3K-Akt pathways in WT adherent pre-B cells indicates that receptor signaling is actively contributing to survival and proliferation. As WT pre-B cells detach from stroma, they rapidly turn off Erk1-2 and Akt signaling although expression of pre-BCR and IL-7R persists. An increase in p38 MAPK, Blnk and intracellular Ca2+ is observed, together with a transcriptional induction of pre-B cell differentiation markers. A working model supported by these findings is that in addition to pre-BCR and IL-7R signaling, integrin-mediated adhesion and FAK signaling contribute to the proliferative expansion of early pre-B cells and provide limited self-renewal by keeping them engaged on stroma. Another important effect of integrin signaling is to shut down the differentiation-inducing pathways in pre-B cells either directly or indirectly by promoting proliferation. As adhesion is lost, possibly due to recycling of focal adhesions, the negative effects on differentiation signaling are reduced. Increase in FAK activity has been implicated in the pathogenesis of a variety of cancers by engaging pro-proliferative signaling. Increased integrin and FAK signaling, as in Ikaros-deficient pre-B cells, may be responsible for augmenting self-renewal and proliferation and for further repressing differentiation at this critical developmental stage. A recent study has shown that loss of FAK during B cell differentiation causes a reduction in pre-B and immature B cells49 and is consistent with the above-proposed model of integrin signaling in pre-B cell differentiation.
While integrin-mediated adhesion and activation of the downstream signaling effector FAK are augmented in Ikaros-deficient pre-B cells, all pre-BCR-affiliated PTKs are reduced. As transcription of these PTKs is relatively unperturbed, this effect is probably a consequence of altered protein stability. Feedback mechanisms originating from hyperactive FAK or MAPKs may stimulate degradation of these pre-BCR signaling effectors, thereby limiting the number of pro-proliferative signaling pathways operating in mutant pre-B cells. Loss in proximal pre-BCR signaling does not affect proliferation as this is also supported by other receptor signaling pathways such as growth factor receptors and integrins, but inhibits differentiation, which is solely dependent on the pre-BCR complex.
The switch from the pre-BCR-IL-7R signaling axis in normal pre-B cells to a more ‘progenitor-like’ integrin-growth factor signaling paradigm supports the survival and proliferative expansion of Ikaros-deficient pre-B cells. In fact, mutant pre-B cells cycle more rapidly, possibly due to a niche-mediated increase in MAPK and its downstream targets Cyclin D2 and CDK6. The ability of Ikaros-deficient pre-B cells to respond to different growth factors also highlights a potential to survive in different micro-environments. Aberrant expansion of mutant pre-B cells to non-physiological numbers represents the first step in a leukemic transformation process that evolves rapidly when the Ikaros-deficient pre-B cells are adoptively transferred to an immune-compromised bone marrow environment. However, the fact that the malignant precursor B-ALLs that develop are oligoclonal with respect to Igh gene rearrangement suggests that additional events are necessary for full malignant transformation. Whereas a subset of Igh rearrangements appears to be shared between leukemias arising in different recipients, this further suggests that some steps toward malignant transformation have occurred in the primary mice. Nonetheless, the Ikaros-deficient leukemic pre-B cell clones retain stromal-adherent properties and are still dependent for their survival on FAK activation.
The insights into the mechanisms that support normal pre-B cell differentiation and its aberrant manifestations described herein illuminate potential new strategies for the therapy of B-ALL that are linked to the underlying biology of the pre-B cell. Humans with IKZF1 mutant B-ALL have an inferior prognosis despite intensive treatment, which correlates with persistent residual disease following induction chemotherapy23, 50. Whereas inhibiting FAK causes detachment and death of Ikaros-deficient pre-B cells by depriving them from anchorage-dependent survival, it has little effect on WT pre-B cells. Hence, these and other pathways activated in Ikaros-deficient pre-B cells may provide additional targets for therapeutic intervention in poor-prognosis B-ALL.
Methods
Mice
The IkE5fl/fl mouse line was generated by inserting loxP sites flanking the Ikzf1 exon 5 by a standard gene targeting method. CD2-Cre and CD19-Cre transgenic lines were obtained from Drs. D. Kioussis and K. Rajewsky, respectively. All mice were bred and maintained under pathogen-free conditions in the animal facility at Massachusetts General Hospital, Bldg. 149-8. At the time of analysis, mice were 5–9wk of age. All animal experiments were done according to protocols approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital (Charlestown, MA) and in accordance with the guidelines set forth by the National Institutes of Health.
Antibodies
Antibodies for bone marrow lineage depletion and flow cytometry were purchased from BD Pharmingen, Southern Biotech and eBiosciences. In some cases, hybridoma supernatant containing antibodies against Mac-1,Gr-1, TER119, c-Kit and CD3ε were used. Antibodies and the specific clones used were: CD3 (17A2), CD8α (53-6.7), TCRβ (H57−597), Mac-1 (M1/70), DX5, Gr-1 (RB6-8C5), Ter119 and IgM (R6.60-2 or 11/41), FcγRII/III (2.4G2), CD19 (1D3), c-Kit (ACK2 or 2B8) BP1 (FG35.4), CD43 (S7), CD2 (RM2-5), Ig kappa (187.D), Integrin α5 (HMa5-1), Integrin α6-PE (GoH3) and Integrin β1 (9EG7). For immunoblotting and immunofluorescence, antibodies from Cell signaling technologies raised against the phosphorylated and total protein for Akt (4060/4685), Erk (4377/4695), p38(4511/8690), Stat5 (9351), Lyn (2731/2796), Syk (12358), FAK (3283/8556/3285), Foxo1 (2880/9454), Fyn (4023), Blnk (12168), Btk (8547), and Cyclin D2 (3741) were utilized. Antibodies against total Stat5 (sc-835X), β-tubulin (sc-9104) and Blk (K-23) were purchased from Santa Cruz Biotechnology.
Flow cytometry and cell sorting
. Bone marrow (BM) cells were isolated as previously described51. Briefly, BM cells were harvested from femurs and tibias and subjected to red blood cell (RBC) lysis using ACK buffer (0.15 M ammonium chloride, 10 mM potassium bicarbonate, 0.1 mM EDTA). For large pre-B cell isolation, bone marrow cells were depleted with antibodies against Ter119, Mac-1, Gr-1, IgM, CD3, CD8a, TCRβ, DX5 and positive cells removed with magnetic beads conjugated to goat anti-rat IgG (QIAGEN). Remaining cells after depletion were labeled with various fluorochrome-conjugated mAbs against B-cell markers for phenotypic analysis, and CD19+CD43+BP1+cells were sorted as large-preB cells, for in vitro cultures and RNA-seq analysis and CD19+CD43- CD25+ as small pre-B cells for RNA-seq analysis. The BP1+ fraction of the CD19+CD43+ population expressed lower levels of c-Kit in both WT and IkE5Δ/Δpre-B cells. For analysis of immature IgM+ cells, undepleted BM cells were stained with mAbs against CD19 and IgM and analyzed within the lymphoid population by electronic gating based on size and granularity. For flow cytometry of integrins, cells were stained with either phycoerythrin (PE) conjugates or primary antibody followed by PE-conjugated secondary antibody. Antibody to P-FAK 925 was used for flow cytometry (Cell Signaling, 3284). Flow cytometric analysis was performed using a two-laser FACSCanto™ (BD) or a three-laser MoFlo® (Dako Cytomation). Cell sorting was performed using a three-laser MoFlo®. The resulting files were uploaded to FlowJo (Tree Star) for further analysis.
Intracellular staining
Large Pre B cells were fixed with 2% paraformaldehyde (Electron Microscopy Sciences, PA, USA) in PBS at room temperature for 20 min. After two washes with PBS, the cells were subsequently permeabilized with 0.5% saponinin 2% FCS/PBS for 20 minutes at 4°C. The cells were stained with FITC-conjugated anti-μ or anti-κ antibody for additional 30 min at 4°C, then washed twice in 2% FCS-PBS before analysis on a FACSCanto™ (BD). For intracellular staining for p-FAK, fixed cells were permeablized with 90% methanol for 2 hours and washed. The cells were then incubated with primary antibody for 30 minutes. Cells were washed and incubated with FITC-conjugated secondary antibody for an additional 30 minutes at 4°C washed and analyzed by flow cytometry.
Immunoglobulin gene rearrangement analysis
DNA was isolated from sorted large pre-B cells and analyzed for immunoglobulin heavy and light chain gene rearrangements by PCR approach using primers specific for D-J and V-DJ or V-J rearrangements as described before52, 53. Briefly, For D-JH rearrangement, the DH sense primer was used with a JH3 antisense primer. V- DJ rearrangements were evaluated using a mixture of three different degenerate (at three positions) oligonucleotides homologous to sequences in the conserved framework region 3 (FR3) of the indicated VH gene families and the JH3 antisense primer. For PCR, serial dilutions (1x and 1:3) of the samples were heated to 94°C for 5 min and then subjected to amplification for 35 cycles of 1 min at 94°C, 1 min at 60°C, and 1 min 30 sec at 72°C. After the last cycle, a final extension step at 72°C for 10 min was carried out. PCR products were run on 1% agarose gels, transferred and hybridized with probe upstream of the JH3 primer region. For analysis of the Ig V-J rearrangement, PCR amplification was performed with Vκ sense primer mixture that is degenerate at four positions and Jκ5 antisense oligo. Southern hybridization of PCR-products for V-J rearrangement was performed with probes binding upstream of the Jκ5 region.
Stromal- free cultures
Differentiation in stromal-free cultures was performed as previously described54. Briefly, 2×103 sorted large pre-B cells were plated in opti-MEM media (Gibco) for 4 days (d4) supplemented with 10% FCS, 50 μM 2-ME, 2.4g/L NaHCO3, 100 μg/ml penicillin, 100 μg/ml streptomycin and 5ng/ml of IL-7 (Peprotech). At day 4, cells were washed and re-plated in opti-MEM with 2% FCS and 0.05ng/ml IL-7. After 3 days, cells were harvested, counted and stained for cell surface expression of CD19, BP1, IgM and CD2. For addressing survival and proliferation, large pre-B cells were cultured without stroma for 1-3 days in 0.05-5ng/ml of IL-7. Cells were harvested and analyzed for counts, cell cycle and apoptosis.
Stromal cultures
WT and IkE5Δ/Δsorted large pre-B cells were co-cultured with OP9 stroma in DMEM media (Sigma, D-5671) supplemented with 10% FBS (Sigma, 2442), 50μM, 100 μg/ml penicillin, 100 μg/ml streptomycin, 1X Glutamax (Gibco 35050-062), 10mM HEPES (Gibco, #156-30-80) and 1X Sodium Pyruvate (Gibco, 11360-070) in the presence of indicated amounts of IL-7 as previously described51. Equal number of WT and IkE5Δ/Δ pre-B cells plated in presence of IL-7 were harvested for counts, cell cycle, proliferation and apoptosis at indicated time points. All analysis on cultured large pre-B cells was performed after removal of the OP9–GFP by flow cytometry. Exclusion by electronic gating based on size and granularity was performed.
For calculation of adherent/non-adherent ratios, 5×104 cells were plated on stroma in 0.05-5ng/ml of IL-7 for 1-3 days. The non-adherent cells were harvested followed by PBS wash. The adherent cells were detached with limited trypsinization treatment. Cells from each fraction were counted under a bright-field microscope and ratios were calculated. For re-adhesion assay, equal number of adherent WT and IkE5Δ/Δ cells were allowed to re-attach onto stroma and at 3 hours cells were enumerated as described above.
Limiting dilution analysis
Adherent WT and IkE5Δ/Δadherent large pre-B cells were sorted on stroma in a 96 well plate in step-wise three-fold serial limiting dilution (10 replicates per dilution) from 300 to 1 cells with 0-5ng/ml of IL-7. Colonies were scored visually after 6 days. The mean frequency of colony forming cells was calculated by L-Calc software (Stem Cell Technologies) based on Poisson distribution of the probability of wells scoring positive.
Intracellular calcium and flux measurements
For measurement of intracellular calcium, non-adherent and adherent WT and IkE5Δ/Δ pre-B cells were stained with Fura-red (Life technologies) as per manufacturer's protocol. For calcium flux, cells were harvested into staining buffer that contained 25mM Hepes (pH 7.2), 125 mM NaCl, 5mMKCl, 1mM Na2HPO4, 0.1% glucose and 0.5mM MgCl2, 1 mMCaCl2 and 0.1g BSA just prior to use. Calcium green (Life technologies) and Fura-red were added for 30 minutes at 37°C. Cells were washed twice and re-suspended in staining buffer and placed on ice. Just prior to analysis on FACSCanto™ (BD), the cells were equilibrated to 37°C. Data was acquired for 30 seconds and then pulsed with anti-IgM antibody or ionomycin and acquired for additional indicated time points. Data was analyzed using kinetics platform on FlowJo software (Tree Star).
Apoptosis assay
Cells were stained for apoptosis using Annexin V: Apoptosis detection kit I (BD) according to manufacturer's protocol.
Cell cycle analysis
Cells were harvested at the indicated time points and fixed in 70% cold ethanol overnight at 4°C. Fixed cells were stained with propidium iodide (PI) staining buffer (250 μg/ml RNaseA, 50 μg/ml PI) for 30 min at 37°C and the DNA content was detected by FACS canto. The resulting files were analyzed with FlowJo (Tree Star).
BrdU pulse-chase assay
Cells were labeled for 45min with BrdU washed and then incubated in growth media for up to 48hrs. Cells were harvested at the indicated time points for proliferation analysis using the BrdU flow Kit (BD) per the manufacturer's protocol.
Phase Contrast Microscopy
Phase-contrast pictures of large-preB cells were taken with a Zeiss Axiovert 200M microscope. Prior to microscopy, cells were cultured on OP9-GFP for 24 hours in 5ng/ml of IL-7.
Immuno-blotting
Cells were harvested and whole cell extracts were prepared using RIPA buffer containing 10mM Tris-HCl (pH 8.0), 1mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS and140 mM NaCl. Protease and phosphatase inhibitors (Roche) were added to extraction buffer just prior to use. Equal amounts of protein lysates were separated by SDS-PAGE and transferred to PVDF membranes (Millipore) and probed with indicated antibodies as per manufacturer's protocol.
Immunofluorescence
Cells grown on stroma on Lab-Tek® Chamber Slide (Electron Microscopy Sciences) were fixed with 3% paraformaldehyde for 20 min at room temperature and then permeabilized with phosphate-buffered saline (PBS) plus 0.1% Tween-20 for 45 min. After blocking with PBS containing 2% bovine serum albumin (BSA) for 10 min, the cells were stained with anti-phospho FAK antibody for 45 min, followed by TRITC-conjugated secondary antibody for 30 min. The slides were treated with Vectashield containing DAPI (Vector Laboratories) and mounted. Images were collected using Nikon A1SiR Confocal Microscope and processed using NIS element confocal imaging software.
Adhesion to integrin ligands and in vitro FAK inhibition assay
Adhesion assay were performed in non-TC treated plates (BD). The plates were coated with 10ug/ml fibronectin (FN), collagen (Col.) (Invitrogen) or BSA alone overnight at 4°C. After blocking the plates with 2% BSA for 1 h, equal number of cells were plated and incubated for the indicated time points. At the end of the assay, unbound and bound cells were harvested and enumerated. Percent adhesion was calculated by taking ratio of bound cells over total cells used in assay.
For inhibition assay, 1 μm FAK inhibitor (PF-431396 or PF- 562271, Sigma) or DMSO control was used for treatment and cells were harvested as bound and unbound fractions at 4 or 24 hour post-treatment for calculation of percent adhesion and inhibition of adhesion, apoptosis and cell cycle assays. For peptide mediated blocking of adhesion55, equal number of cells were pretreated for 45 minutes with either 400 μg/ml G-R-G-D-S-P or the control peptide G-R-G-E-S (American Peptide Company) and plated on FN-coated TC dishes. Cells were enumerated for calculation of percent adhesion and percent inhibition.
The effect of growth factors and adhesion was evaluated by plating 2×105adherent pre-B cells on FN and Col. or BSA coated plates in the presence of 5 ng/ml IL-7, 100 ng/ml SCF, both (IL-7 + SCF) or no cytokines for 24 hours. At 24 hours, cells were counted and analyzed for cell cycle and apoptosis. For integrin and growth factor signaling assay, equal number (2×105) cells were plated in FN and Col. or BSA coated plates in presence of 5ng/ml IL-7, 100 ng/ml SCF, both (IL-7 + SCF) or no cytokines for 24 hours. At 24 hours, cells were harvested and counted. Mutant cells were analyzed for cell cycle and apoptosis.
In vivo FAK inhibition assay
WT and IkE5fl/fl CD19cre mice were used for treatment with FAK inhibitor PF-562271 or vehicle (50% DMSO/50%PEG-400). Both cohorts were given a dosage of 25mg/kg/mouse of inhibitor or equal volume of vehicle by oral gavage. Dosage regimens of either 3 or 5 doses were given at ~12 hours apart. At 3 hours after the final dose, mice were sacrificed by CO2 asphyxiation. Bones were flushed and cells were collected and total bone marrow cellularities were estimated. Cells were stained for pre-B cell cell surface markers and PI/Annexin V staining was performed to estimate apoptotic cell frequency.
Transwell migration assay
Transwell plates with (Corning, 3422) were coated with fibronectin (10μg/ml). The wells were washed and blocked with BSA. Serum-free medium with 1% BSA containing the CXCL12 / SDF-1α, (100 ng/ml) was added to the bottom well of the transwell plate. Equal number of cells in 100 μl of serum-free media were added to the upper chambers and incubated for 2 hours at 37°C. At the end of the time point, inserts were removed and migrated cells in bottom wells were counted. Percent migration was calculated by taking ratios of migrated cells over total cells plated in inserts.
RNA-seq, gene expression and pathway analysis
RNA isolated with Trizol was purified using the PureLink RNA mini kit (Ambion). The Truseq RNA sample prep kit was used for construction of cDNA libraries for RNA-sequencing (Illumina). The cDNA libraries were ligated with indexed primers and amplified by 15 cycles of PCR. The amplified libraries were multiplexed and sequenced by the Genome Analyzer at Systems Biology Lab, Harvard University. Read alignment on mouse mm9 assembly was conducted by the BWA algorithm implemented by the DNA Nexus suite. The Deseq algorithm implemented by the R platform was used to determine differential gene expression in freshly sorted WT and IkE5Δ/Δ large pre-B, WT small pre-B as well as in adherent and non-adherent fractions of sorted large pre-B cells after limited propagation on OP9 stroma. Pathway analysis of upregulated genes in IkE5Δ/Δ relative to WT large pre-B was conducted using the Ingenuity software. Heatmaps of normalized tags for gene subsets across WT and IkE5Δ/Δpre-B cell populations were generated with the Avadis software.
Adoptive transfer of purified pre-B cell populations to NSG mice
NOD/SCID/Il2rg−/− (NSG) mice (Jackson Laboratory) were conditioned by 300 cGy gamma irradiation and injected via lateral tail vein with 3× 106 sorted large pre-B (CD19+CD43+BP1+) cells. Diseased mice were characterized by histopathological analysis as previously described56.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This research was supported by an ARRA supplement to 5R01AI42254 and 5R01CA162092 to K.G. N.J. and R.A.V. were supported by NIH grant CA090576. High throughput DNA sequencing and RNA profiling was performed at the Bauer Center for Genomic research, Harvard University, Cambridge. We thank B. Czyzewski for mouse husbandry and Drs. K. White, J.M. Park, B. Morgan, R. Bakshi, E. Alonzo and J. Seavitt for critical review of the manuscript. We thank Drs. R. Bakshi and R. Arya for assistance with statistical analysis and confocal microscopy respectively.
Footnotes
Author Contributions
I.J., T.Y, N.J., X.Q. and J.Z. performed the experiments and edited the manuscript. T.Y. and I.J. made figures; K.G. and R.A.V supervised the research and wrote the manuscript.
The authors declare no competing financial interests.
Accession code
References
- 1.Monroe JG. ITAM-mediated tonic signalling through pre-BCR and BCR complexes. Nat Rev Immunol. 2006;6:283–294. doi: 10.1038/nri1808. [DOI] [PubMed] [Google Scholar]
- 2.Herzog S, Reth M, Jumaa H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat Rev Immunol. 2009;9:195–205. doi: 10.1038/nri2491. [DOI] [PubMed] [Google Scholar]
- 3.Gauld SB, Cambier JC. Src-family kinases in B-cell development and signaling. Oncogene. 2004;23:8001–8006. doi: 10.1038/sj.onc.1208075. [DOI] [PubMed] [Google Scholar]
- 4.Kitamura D, et al. A critical role of lambda 5 protein in B cell development. Cell. 1992;69:823–831. doi: 10.1016/0092-8674(92)90293-l. [DOI] [PubMed] [Google Scholar]
- 5.Gong S, Nussenzweig MC. Regulation of an early developmental checkpoint in the B cell pathway by Ig beta. Science. 1996;272:411–414. doi: 10.1126/science.272.5260.411. [DOI] [PubMed] [Google Scholar]
- 6.Kraus M, et al. Interference with immunoglobulin (Ig)alpha immunoreceptor tyrosine-based activation motif (ITAM) phosphorylation modulates or blocks B cell development, depending on the availability of an Igbeta cytoplasmic tail. J Exp Med. 2001;194:455–469. doi: 10.1084/jem.194.4.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pelanda R, Braun U, Hobeika E, Nussenzweig MC, Reth M. B cell progenitors are arrested in maturation but have intact VDJ recombination in the absence of Ig-alpha and Ig-beta. J Immunol. 2002;169:865–872. doi: 10.4049/jimmunol.169.2.865. [DOI] [PubMed] [Google Scholar]
- 8.Cheng AM, et al. Syk tyrosine kinase required for mouse viability and B-cell development. Nature. 1995;378:303–306. doi: 10.1038/378303a0. [DOI] [PubMed] [Google Scholar]
- 9.Schweighoffer E, Vanes L, Mathiot A, Nakamura T, Tybulewicz VL. Unexpected requirement for ZAP-70 in pre-B cell development and allelic exclusion. Immunity. 2003;18:523–533. doi: 10.1016/s1074-7613(03)00082-7. [DOI] [PubMed] [Google Scholar]
- 10.Saijo K, et al. Essential role of Src-family protein tyrosine kinases in NF-kappaB activation during B cell development. Nat Immunol. 2003;4:274–279. doi: 10.1038/ni893. [DOI] [PubMed] [Google Scholar]
- 11.Marshall AJ, Fleming HE, Wu GE, Paige CJ. Modulation of the IL-7 dose-response threshold during pro-B cell differentiation is dependent on pre-B cell receptor expression. J Immunol. 1998;161:6038–6045. [PubMed] [Google Scholar]
- 12.Fleming HE, Paige CJ. Pre-B cell receptor signaling mediates selective response to IL-7 at the pro-B to pre-B cell transition via an ERK/MAP kinase-dependent pathway. Immunity. 2001;15:521–531. doi: 10.1016/s1074-7613(01)00216-3. [DOI] [PubMed] [Google Scholar]
- 13.Malin S, et al. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat Immunol. 2010;11:171–179. doi: 10.1038/ni.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yasuda T, et al. Erk kinases link pre-B cell receptor signaling to transcriptional events required for early B cell expansion. Immunity. 2008;28:499–508. doi: 10.1016/j.immuni.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 15.Kersseboom R, et al. Bruton's tyrosine kinase cooperates with the B cell linker protein SLP-65 as a tumor suppressor in Pre-B cells. J Exp Med. 2003;198:91–98. doi: 10.1084/jem.20030615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Middendorp S, et al. Tumor suppressor function of Bruton tyrosine kinase is independent of its catalytic activity. Blood. 2005;105:259–265. doi: 10.1182/blood-2004-07-2708. [DOI] [PubMed] [Google Scholar]
- 17.Wen R, et al. Essential role of phospholipase C gamma 2 in early B-cell development and Myc-mediated lymphomagenesis. Mol Cell Biol. 2006;26:9364–9376. doi: 10.1128/MCB.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herzog S, et al. SLP-65 regulates immunoglobulin light chain gene recombination through the PI(3)K-PKB-Foxo pathway. Nat Immunol. 2008;9:623–631. doi: 10.1038/ni.1616. [DOI] [PubMed] [Google Scholar]
- 19.Johnson K, et al. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity. 2008;28:335–345. doi: 10.1016/j.immuni.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 20.Ochiai K, et al. A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat Immunol. 2012;13:300–307. doi: 10.1038/ni.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cobaleda C, Sanchez-Garcia I. B-cell acute lymphoblastic leukaemia: towards understanding its cellular origin. Bioessays. 2009;31:600–609. doi: 10.1002/bies.200800234. [DOI] [PubMed] [Google Scholar]
- 22.Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 23.Mullighan CG, et al. Deletion of IKZF1 and Prognosis in Acute Lymphoblastic Leukemia. N Engl J Med. 2009 doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iacobucci I, et al. Expression of spliced oncogenic Ikaros isoforms in Philadelphia-positive acute lymphoblastic leukemia patients treated with tyrosine kinase inhibitors: implications for a new mechanism of resistance. Blood. 2008;112:3847–3855. doi: 10.1182/blood-2007-09-112631. [DOI] [PubMed] [Google Scholar]
- 25.Iacobucci I, et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell'Adulto Acute Leukemia Working Party (GIMEMA AL WP). Blood. 2009;114:2159–2167. doi: 10.1182/blood-2008-08-173963. [DOI] [PubMed] [Google Scholar]
- 26.Harvey RC, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010 doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Georgopoulos K. Acute Lymphoblastic Leukemia -- On the Wings of IKAROS. N Engl J Med. 2009 doi: 10.1056/NEJMe0809819. [DOI] [PubMed] [Google Scholar]
- 28.Georgopoulos K, et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79:143–156. doi: 10.1016/0092-8674(94)90407-3. [DOI] [PubMed] [Google Scholar]
- 29.Ng SY, Yoshida T, Zhang J, Georgopoulos K. Genome-wide lineage-specific transcriptional networks underscore Ikaros-dependent lymphoid priming in hematopoietic stem cells. Immunity. 2009;30:493–507. doi: 10.1016/j.immuni.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan B, et al. Aiolos, a lymphoid restricted transcription factor that interacts with Ikaros to regulate lymphocyte differentiation. Embo J. 1997;16:2004–2013. doi: 10.1093/emboj/16.8.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson EC, et al. Ikaros DNA-binding proteins as integral components of B cell developmental-stage-specific regulatory circuits. Immunity. 2007;26:335–344. doi: 10.1016/j.immuni.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Sun L, Liu A, Georgopoulos K. Zinc finger-mediated protein interactions modulate Ikaros activity, a molecular control of lymphocyte development. Embo J. 1996;15:5358–5369. [PMC free article] [PubMed] [Google Scholar]
- 33.Pelanda R, Schaal S, Torres RM, Rajewsky K. A prematurely expressed Ig(kappa) transgene, but not V(kappa)J(kappa) gene segment targeted into the Ig(kappa) locus, can rescue B cell development in lambda5-deficient mice. Immunity. 1996;5:229–239. doi: 10.1016/s1074-7613(00)80318-0. [DOI] [PubMed] [Google Scholar]
- 34.Rolink AG, Winkler T, Melchers F, Andersson J. Precursor B cell receptor-dependent B cell proliferation and differentiation does not require the bone marrow or fetal liver environment. J Exp Med. 2000;191:23–32. doi: 10.1084/jem.191.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kierney PC, Dorshkind K. B lymphocyte precursors and myeloid progenitors survive in diffusion chamber cultures but B cell differentiation requires close association with stromal cells. Blood. 1987;70:1418–1424. [PubMed] [Google Scholar]
- 36.Hayashi S, et al. Stepwise progression of B lineage differentiation supported by interleukin 7 and other stromal cell molecules. J Exp Med. 1990;171:1683–1695. doi: 10.1084/jem.171.5.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glodek AM, et al. Focal adhesion kinase is required for CXCL12-induced chemotactic and pro-adhesive responses in hematopoietic precursor cells. Leukemia. 2007;21:1723–1732. doi: 10.1038/sj.leu.2404769. [DOI] [PubMed] [Google Scholar]
- 38.Tse KW, et al. B cell receptor-induced phosphorylation of Pyk2 and focal adhesion kinase involves integrins and the Rap GTPases and is required for B cell spreading. J Biol Chem. 2009;284:22865–22877. doi: 10.1074/jbc.M109.013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rolink A, Streb M, Nishikawa S, Melchers F. The c-kit-encoded tyrosine kinase regulates the proliferation of early pre-B cells. Eur J Immunol. 1991;21:2609–2612. doi: 10.1002/eji.1830211044. [DOI] [PubMed] [Google Scholar]
- 40.Sudo T, et al. Expression and function of the interleukin 7 receptor in murine lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:9125–9129. doi: 10.1073/pnas.90.19.9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winandy S, Wu P, Georgopoulos K. A dominant mutation in the Ikaros gene leads to rapid development of leukemia and lymphoma. Cell. 1995;83:289–299. doi: 10.1016/0092-8674(95)90170-1. [DOI] [PubMed] [Google Scholar]
- 42.Ye F, Kim C, Ginsberg MH. Reconstruction of integrin activation. Blood. 2012;119:26–33. doi: 10.1182/blood-2011-04-292128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wehrle-Haller B. Structure and function of focal adhesions. Curr Opin Cell Biol. 2012;24:116–124. doi: 10.1016/j.ceb.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 44.Galbraith CG, Yamada KM, Galbraith JA. Polymerizing actin fibers position integrins primed to probe for adhesion sites. Science. 2007;315:992–995. doi: 10.1126/science.1137904. [DOI] [PubMed] [Google Scholar]
- 45.Vicente-Manzanares M, Choi CK, Horwitz AR. Integrins in cell migration--the actin connection. J Cell Sci. 2009;122:199–206. doi: 10.1242/jcs.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith A, et al. A talin-dependent LFA-1 focal zone is formed by rapidly migrating T lymphocytes. J Cell Biol. 2005;170:141–151. doi: 10.1083/jcb.200412032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choi CK, et al. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nature cell biology. 2008;10:1039–1050. doi: 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 49.Park SY, et al. Focal adhesion kinase regulates the localization and retention of pro-B cells in bone marrow microenvironments. J Immunol. 2013;190:1094–1102. doi: 10.4049/jimmunol.1202639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waanders E, et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia. 2011;25:254–258. doi: 10.1038/leu.2010.275. [DOI] [PubMed] [Google Scholar]
- 51.Yoshida T, Ng SY, Zuniga-Pflucker JC, Georgopoulos K. Early hematopoietic lineage restrictions directed by Ikaros. Nat Immunol. 2006;7:382–391. doi: 10.1038/ni1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schlissel MS, Corcoran LM, Baltimore D. Virus-transformed pre-B cells show ordered activation but not inactivation of immunoglobulin gene rearrangement and transcription. J Exp Med. 1991;173:711–720. doi: 10.1084/jem.173.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuxa M, et al. Pax5 induces V-to-DJ rearrangements and locus contraction of the immunoglobulin heavy-chain gene. Genes Dev. 2004;18:411–422. doi: 10.1101/gad.291504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Osmond DG, Melchers F, Paige CJ. Pre-B cells in mouse bone marrow: in vitro maturation of peanut agglutinin binding B lymphocyte precursors separated from bone marrow by fluorescence-activated cell sorting. J Immunol. 1984;133:86–90. [PubMed] [Google Scholar]
- 55.Bernardi P, Patel VP, Lodish HF. Lymphoid precursor cells adhere to two different sites on fibronectin. J Cell Biol. 1987;105:489–498. doi: 10.1083/jcb.105.1.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roumiantsev S, de Aos IE, Varticovski L, Ilaria RL, Van Etten RA. The src homology 2 domain of Bcr/Abl is required for efficient induction of chronic myeloid leukemia-like disease in mice but not for lymphoid leukemogenesis or activation of phosphatidylinositol 3-kinase. Blood. 2001;97:4–13. doi: 10.1182/blood.v97.1.4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.