

Abstract
Prodiginines are a family of linear and cyclic oligopyrrole red-pigmented compounds. Herein we describe the in vitro antimalarial activity of 4 natural (IC50 = 1.7-8.0 nM) and 3 sets of synthetic prodiginines against Plasmodium falciparum. Set 1 compounds replaced the terminal non-alkylated pyrrole ring of natural prodiginines and had diminished activity (IC50 >2920 nM). Set 2 and set 3 prodiginines were monosubstituted or disubstituted at either the 3 or 5 position of the right hand terminal pyrrole, respectively. Potent in vitro activity (IC50 = 0.9-16.0 nM) was observed using alkyl or aryl substituents. Metacycloprodiginine and more potent synthetic analogs were evaluated in a P. yoelii murine patent infection using oral administration. Each analog reduced parasitemia by more than 90% after 25 mg/kg/day dosing, and in some cases provided a cure. The most favorable profile was 92% parasite reduction at 5 mg/kg/day, and 100% reduction at 25 mg/kg/day without any evident weight loses or clinical overt toxicity.
Introduction
Malaria remains a major global health threat and an enormous economic burden to disease-endemic nations. Despite increased attention and new commitment to malaria eradication, the disease causes more than a million deaths each year, and drug resistance to each new chemotherapeutic approach has developed, most notably in Plasmodium falciparum, the parasite causing nearly all malaria-related deaths.1 On the heels of the global spread of chloroquine-resistant (CQRa) P. falciparum, resistance has also quickly developed to a variety quinoline analogs, to antifolates, to inhibitors of electron transport, and perhaps most ominously, now to artemisinin.2a-b It is evident that the search for effective novel antimalarial compounds must be comprehensive and must include explorations of chemotypes distinct from the prototypes in clinical use.
Prodiginines are a family of linear and cyclic oligopyrrole red-pigmented compounds with antibacterial,3 anticancer,4 and immunosuppressive activity,5 produced by actinomycetes and other eubacteria. Some prodiginines induce apoptotic effects breaking genomic deoxyribonucleic acid (DNA) strands.6 The prodiginines also shown to have potent in vitro activity against Plasmodium species, at much lower concentrations than seen with mammalian cells.7a-e Despite potent and selective in vitro activity against P. falciparum, the reported efficacy of prodiginines in vivo has either been incomplete, associated with toxicity, or lacking altogether, discouraging their further consideration as antimalarial candidates.
On careful review, however, the previous studies are each limited, either in the identification or variety of compounds tested, by the lack of in vivo correlation studies, or by the route of drug administration in the animals studied. These limitations leave open the question of whether or not there is suitable antimalarial activity among these compounds. Furthermore, previous studies have been limited to naturally occurring prodiginines, leaving open the possibility that some synthetic analogs may have improved in vitro activity or in vivo efficacy or reduced toxicity. We therefore undertook a more comprehensive assessment of the antiplasmodial activity of prodiginines; initially re-assessing the activity of four naturally-occurring prodiginines 1-4 (Figure 1) and subsequently assessing a series of synthetic analogs. Here we report these results, including impressive in vitro potency, SAR, and evidence of in vivo efficacy, including curative efficacy in mice after oral administration.
Figure 1.

Naturally occurring alkyl prodiginines (1-4)
Results and Discussion
Synthesis
The synthesis of various 2-alkyl pyrrole segments 6a-h (Scheme 1) was initiated by converting the readily accessible pyrrole to the corresponding 2-acyl pyrroles 5a-h.8 These acyl pyrroles were then smoothly converted to 6a-h using an excess of NaBH4, in 2-propanol under reflux.9
Scheme 1.
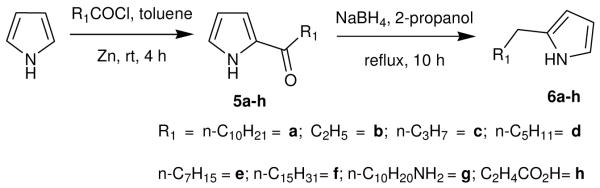
Synthesis of 2-alkylated pyrroles (6a-h)
Synthesis of 3-alkyl pyrrole segments 10a-k (Scheme 2) used the same pyrrole starting material, which treated with phenylsulfonyl chloride in presence of NaOH provided a N-phenylsulfonyl pyrrole (7).10 Use of AlCl3 permitted the regioselective acylation of 7 at the 3 position with the acyl chlorides to obtain the N-phenylsulfonyl-3-acyl pyrroles 8a-k, which were subsequently hydrolyzed under basic (5N NaOH) conditions to give 3-acyl pyrroles 9a-k.11 In the final step 9a-k were reduced to 10a-k using NaBH under reflux.9
Scheme 2.

Synthesis of 3-alkylated pyrroles (10a-k)
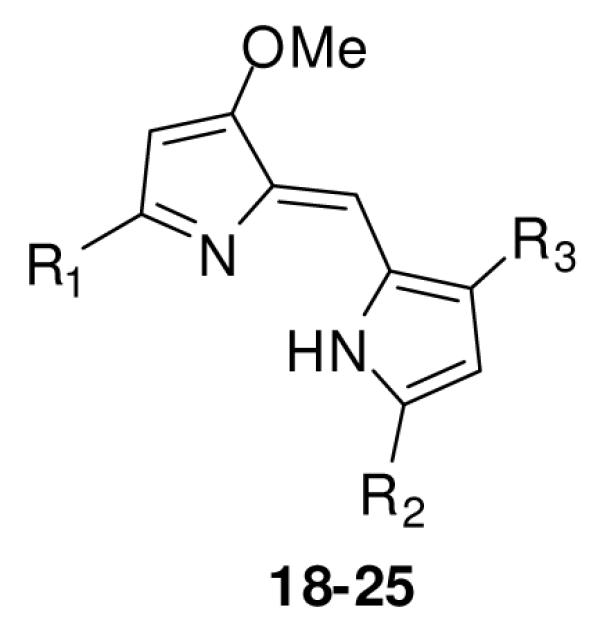
Using literature methodologies, the key intermediate involved in the synthesis of 4-methoxy-2,2′-bipyrrole-5-carboxaldehyde (13) and its analogs 14-17, were prepared via a intermediate 12 from the commercially available 4-methoxy-3-pyrrolin-2-one (11) (Scheme 3).12 Acid-catalyzed condensation of an appropriate alkyl pyrrole segments (2,4-dimethyl pyrrole, 6a-h, and 10a-k) with 13-17 (Scheme 4)13 gave the desired prodiginine analogs 18-25 (Table 1) and 26-43 (Table 2 and 3).
Scheme 3.
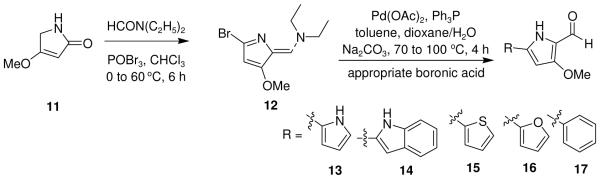
Synthesis of 4-methoxy-2,2′-bipyrrole-5-carbaldehyde (13) and its analogs (14-17).
Scheme 4.

Synthesis of various prodiginine analogs (18-43)
Table 1.
In vitro antimalarial activity of prodiginine analogs where the non alkylated terminal pyrrole (left hand side) has been substituted
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Compd | R1 | R2 | R3 | IC50 (nM) |
Cytotoxicity (nM) |
|
| D6 | Dd2 | |||||
| CQ | - | - | - | 11 | 135 | 16000 |
| 18 |
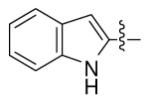
|
CH3 | CH3 | 4250 | 4950 | < 125 |
| 19 |
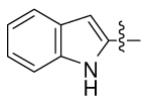
|
n-C11H23 | H | 4060 | 6150 | 3410 |
| 20 |
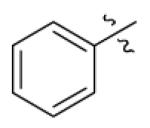
|
n-C11H23 | H | 10470 | 15750 | 3580 |
| 21 |
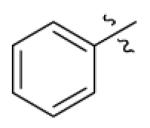
|
CH3 | CH3 | 19410 | 13640 | 7950 |
| 22 |
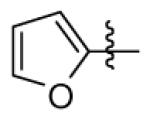
|
n-C11H23 | H | 2920 | 3810 | 980 |
| 23 |
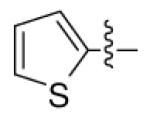
|
CH3 | CH3 | > 25000 | > 25000 | 3530 |
| 24 |
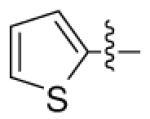
|
n-C11H23 | H | 5940 | 7770 | 2120 |
| 25 |
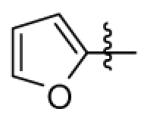
|
CH3 | CH3 | > 25000 | > 25000 | 3890 |
Table 2.
In vitro antimalarial activity of prodiginines containing alkyl substituents at the 3 and 5 positions of the terminal pyrrole ring
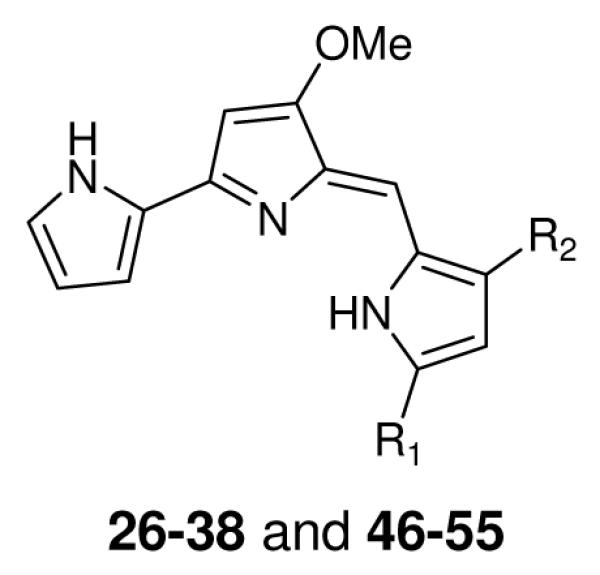
| |||||
|---|---|---|---|---|---|
|
| |||||
| Compd | R1 | R2 | IC50 (nM) |
Cytotoxicity (nM) |
|
| D6 | Dd2 | ||||
| CQ | - | - | 11 | 135 | 16057 |
| 26 | n-C3H7 | H | 2300 | N.D. | N.D. |
| 27 | n-C4H9 | H | 1780 | 1590 | < 125 |
| 28 | n-C6H13 | H | 375 | 450 | < 125 |
| 29 | n-C8H17 | H | 80 | 130 | < 125 |
| 30 | n-C16H33 | H | 300 | 400 | < 125 |
| 31 | n-C11H22NH2 | H | 1700 | N.D. | N.D. |
| 32 | H | (CH2)3COOCH3 | 4500 | N.D | N.D. |
| 33 | H | CH2CH(CH3)2 | 460 | 230 | < 125 |
| 34 | H | n-C4H9 | 80 | 18 | < 125 |
| 35 | H | n-C6H13 | 28 | 7 | < 125 |
| 36 | H | n-C8H17 | 4.6 | 1.8 | < 125 |
| 37 | H | n-C10H21 | 8.0 | 10 | < 125 |
| 38 | H | n-C16H33 | > 25000 | > 25000 | 3628 |
| 46 | CH3 | CH3 | 8900 | 8130 | < 125 |
| 47 | n-C6H13 | n-C3H7 | 4.5 | 4.0 | < 125 |
| 48 | n-C8H17 | n-C3H7 | 2.9 | 2.7 | < 125 |
| 49 | n-C3H7 |
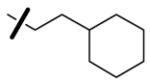
|
1.7 | 1.3 | < 125 |
| 50 | n-C6H13 | n-C6H13 | 1.7 | 1.1 | < 125 |
| 51 | n-C7H15 | n-C6H13 | 2.1 | 1.2 | < 125 |
| 52 | n-C6H13 | n-C8H17 | 4.9 | 2.0 | < 125 |
| 53 | n-C7H15 | n-C8H17 | 6.2 | 2.9 | 3906 |
| 54 | n-C8H17 | n-C8H17 | 92 | 129 | 31250 |
| 55 |
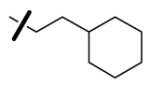
|
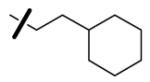
|
5.3 | 3.5 | < 125 |
N.D.: not determined
Table 3.
In vitro antimalarial activity of prodiginines containing aryl substituents at either the 3, or the 3 and 5 positions, of the terminal pyrrole ring
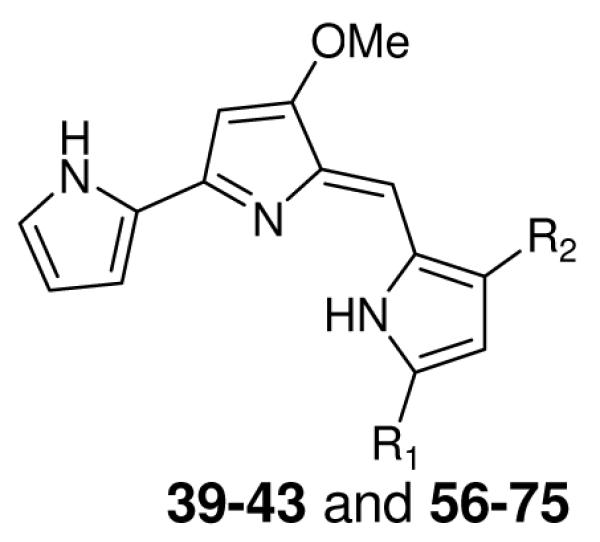
| |||||
|---|---|---|---|---|---|
|
| |||||
| Compd | R1 | R2 | IC50 (nM) |
Cytotoxicity (nM) |
|
| D6 | Dd2 | ||||
| CQ | - | - | 11 | 135 | 16057 |
| 39 | H | C6H5CH2 | 83 | 86 | < 125 |
| 40 | H | 4-OCH3C6H4CH2 | 170 | 156 | < 125 |
| 41 | H | 4-ClC6H4CH2 | 65 | 81 | < 125 |
| 42 | H | 4-BrC6H4CH2 | 90 | 108 | < 125 |
| 43 | H | 2-NaphthylCH2 | 56 | N.D. | N.D. |
| 56 | C2H5 | 4-ClC6H4CH2 | 6.3 | 6.2 | < 125 |
| 57 | n-C3H7 | 4-ClC6H4CH2 | 3.0 | 2.6 | < 125 |
| 58 | n-C6H13 | 4-ClC6H4CH2 | 2.0 | 1.8 | < 125 |
| 59 | n-C7H15 | 4-ClC6H4CH2 | 2.8 | 2.2 | < 125 |
| 60 | n-C8H17 | 4-ClC6H4CH2 | 16.0 | 12.0 | 1105 |
| 61 | 4-ClC6H4CH2 |
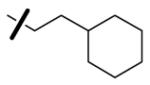
|
3.9 | 2.9 | 1007 |
| 62 | n-C6H13 | 4-FC6H4CH2 | 0.9 | 0.9 | 1462 |
| 63 | n-C8H17 | 4-FC6H4CH2 | 1.3 | 1.2 | < 125 |
| 64 | n-C6H13 | 4-BrC6H4CH2 | 2.9 | 2.8 | < 125 |
| 65 | n-C8H17 | 4-BrC6H4CH2 | 4.0 | 2.9 | < 125 |
| 66 | 4-ClC6H4CH2 | 4-ClC6H4CH2 | 6.1 | 4.8 | < 125 |
| 67 | 4-FC6H4CH2 | 4-FC6H4CH2 | 5.6 | 5.7 | < 125 |
| 68 | 4-BrC6H4CH2 | 4-BrC6H4CH2 | 14.0 | 11.0 | < 125 |
| 69 | 4-FC6H4CH2 | 4-ClC6H4CH2 | 6.1 | 6.1 | < 125 |
| 70 | 4-BrC6H4CH2 | 4-ClC6H4CH2 | 8.3 | 7.7 | < 125 |
| 71 | 4-BrC6H4CH2 | 4-FC6H4CH2 | 5.7 | 5.1 | < 125 |
| 72 | 2,4-Cl2C6H3CH2 | 2,4-Cl2C6H3CH2 | 12.6 | 11.0 | < 125 |
| 73 | 2,6-F2C6H3CH2 | 2,6-F2C6H3CH2 | 14.7 | 18.3 | < 125 |
| 74 | 3-FC6H4CH2 | 3-FC6H4CH2 | 5.1 | 6.7 | < 125 |
| 75 | 2-ClC6H4CH2 | 2-ClC6H4CH2 | 3.6 | 4.9 | < 125 |
N.D.: not determined
A new synthetic strategy, outlined in Scheme 5, was used to synthesize 3,5-disubstituted prodiginine analogs 46-55 (Table 2) and 56-75 (Table 3). The 2,4-disubstituted pyrroles 44 were synthesized from either the corresponding 2 or 3-acyl pyrroles (5 or 9) with their corresponding acyl chlorides and AlCl3.14 Subsequent reduction with NaBH4 gave the desired 2,4-disubstituted pyrroles 45.9 Acid-catalyzed condensation of these 45 with 13 provided the desired 3,5-disubstituted prodiginine analogs 46-75 (Table 2 and 3).13
Scheme 5.

Synthesis of 2,4-disubstitued pyrroles (45), and the corresponding prodiginine analogs (46-75).
Biological Activity
The natural and synthetic prodiginines were assayed for their in vitro antimalarial activity against P. falciparum pansensitive D6 with the chloroquine (CQ) as a reference drug. A direct comparative assessment of the antimalarial activity of natural products, prodigiosin (1), undecylprodiginine (2), metacycloprodiginine (3), and streptorubin B (4) (Figure 1) revealed they all had remarkably potent activity with very low IC50 values (8 nM, 7.7 nM, 1.7 nM, and 7.8 nM, respectively) against P. falciparum strain D6, and slightly more active than CQ (11 nM). The natural prodiginine 3 was consistently observed to be the most potent of the 4 natural products, and demonstrated that changes in the alkyl component of prodiginines affected the in vitro antimalarial activity. Based on these observations a series of new analogs of prodiginines with similar core structure were generated.
In the early stages, a set of analogs 18-25 (Table 1), in which an indole, phenyl, thiophen and furan replaced the terminal non alkylated pyrrole ring (left hand side) of core moiety, were generated. In vitro analysis of the activity of these compounds against P. falciparum pansensitive D6 demonstrated activity (IC50 >2920 nM) significantly diminished relative to the natural prodiginines 1-4 (IC50 = 1.7-8.0 nM). This work suggested that the left hand side pyrrole ring of the prodiginines is important for the potent antimalarial activity. We subsequently tested a hypothesis that the alkyl chain on right hand side pyrrole of the prodiginines might represent an opportunity to make potent and selective antimalarials with the desired “drug-like” properties. Subsequent analogs were synthesized (Scheme 4 and 5) to obtain a SAR for the alkyl groups at the terminal alkylated pyrrole of the prodiginines.
One set of these analogs 26-38 (Table 2), and 39-43 (Table 3) were monosubstituted and contained different alkyl chains or aryl groups at either the 3 or 5 position of the terminal pyrrole ring (right hand side) (Scheme 4). These compounds were screened for their in vitro antimalarial activity P. falciparum strain D6 and the results were shown in Table 2 and 3. Of the monoalkylated analogs, compounds 26, 27, 31, 32, and 38 showed the poorest activity with IC50 values above 1700 nM against D6. Compounds 28, 30, and 33, all exhibited slightly better activity with IC50 values 375 nM, 300 nM, and 460 nM respectively against D6. Analogs, 29, 34, 35, 36 and 37 were the most potent of the mono alkylated prodiginine analogs with IC50 values of 80 nM, 80 nM, 28 nM, 4.6 nM, and 8.0 nM respectively against D6. These observations and the activity of the natural product 2, demonstrates that C6-C11 alkyl chain length at either the 3 or 5 position of right hand side pyrrole ring is required for optimal activity of monoalkylated prodiginines. The presence of either an amine group (see 31; IC50 = 1700 nM (D6) versus 2; IC50 = 7.7 nM (D6)) or methylcarboxylate (see 32; IC50 = 4500 nM (D6) versus 34; IC50 = 80 nM (D6)) at the end of the alkyl chain leads to a decrease in activity. The 3-aryl monosubstituted prodiginine analogs 39-43 (Table 3) exhibited moderate inhibitory activity with IC50 values of 83 nM, 170 nM, 65 nM, 90 nM, and 56 nM respectively against D6. While the presence of a halogen substituent appeared to lead to increase in activity (see 41; IC50 = 65 nM (D6) versus 39; IC50 = 83 nM (D6) and 40; IC50 = 170 nM (D6)), the activity was poorer than that seen for the more potent monoalkylated prodiginines (such as 2, and 35-37).
The final set of prodiginine analogs generated had substituents at both position 3 and 5 of the terminal pyrrole ring (right hand side). The substituents were either two alkyl chains (46-55), an alkyl chain and an aryl group (56-65), or two aryl groups (66-75). These compounds 46-75 (Tables 2 and 3), were screened for in vitro animalarial activity against P. falciparum strain D6. Among the dialkylated analogs 49-51 showed potent in vitro antimalarial activity with IC50 values 1.7 nM, 1.7 nM and 2.1 nM respectively (Table 2) against D6. Other dialkylated prodiginines 47, 48, 52, 53, and 55 also exhibited excellent activity with IC50 values 4.5 nM, 2.9 nM, 4.9 nM, 6.2 nM, and 5.3 nM respectively against D6, and demonstrated that they are more active than the monoalkylated prodiginines (2, and 26-38). The two exceptions were analogs 46 (IC50 = 8900 nM), and 54 (IC50 = 92 nM) which contained a total alkyl chain length of two and sixteen carbon respectively on right hand side pyrrole ring of the core moiety. From both the mono and dialkylated series the data clearly shown that prodiginines containing a combined C8-C15 alkyl chain length had the most potent in vitro antimalarial activity (Figure 2).
Figure 2.

SAR of combined alkyl chain length for mono and dialkylated prodiginines and in vitro antimalarial activity against D6 strain of P. falciparum
The analogs 56-65, which contains one alkyl and one aryl substituents on right hand pyrrole ring were also tested for in vitro antimalarial activity against P. falciparum strain D6. Among these analogs, 62 (IC50 = 0.9 nM and 63 (IC50 = 1.3 nM) (Table 3) were the most potent and comparable to the most potent dialkylated prodiginines 49-51 and the natural prodiginine 3 (IC50 = 1.7 nM). In this series (56-65) all compounds, with the exception of 60 (IC50 = 16.0 nM), were extremely potent (IC50 < 6.3 nM), significantly more than the corresponding analogs 39-43 which have an aryl substituent, but lack the alkyl substituent. The in vitro activity results of chloro (56-61), fluoro (62 and 63) and bromo (64 and 65) substituted analogs of prodiginine indicated that the bromo substituted analogs 64 and 65 were slightly less potent than chloro and fluoro substituted analogs (Table 3).
The final set of analogs 66-75, contains two aryl substituent groups on right hand side pyrrole ring. In this set, most of the compounds (66, 67, 69-71, 74, and 75) exhibited potent activity (IC50 < 8.3 nM). The exceptions were 68 (IC50 = 14.0 nM), which contains a bromine atom on both aryl substituents, and 72 (IC50 = 12.6 nM) and 73 (IC50 = 14.7 nM) in which each aryl group is dihalogenated (Table 3). A comparison of the most potent compounds in this series, with analogs 47-65 revealed that the in vitro activity for prodiginines bearing two aryl substituents is slightly less than for disubsititued prodiginines in which either one or both of the substituents is an alkyl chain. That said, a general pattern of more potent in vitro activity for disubstutued prodignines compared to monosubstituted prodiginines was observed.
The majority of the new synthetic prodiginines were also tested alongside CQ against the multidrug-resistant (MDR) Dd2. In all cases there was no significant change in the activity of each analog between the D6 and Dd2 (Table 1-3), indicating no cross-resistance with CQ. The most potent synthetic prodiginines were active in the 1 nM range against Dd2, compared to 135 nM for CQ.
A number of the synthetic prodiginines were tested against a number of mammalin cell lines and IC50 values in the 500-2000 nM ranges was not observed (data not shown). A general assay with mitogen stimulated murine splenic lymphocytes was used to evaluate all the synthetic prodiginines (Tables 1-3) and to determine if synthetic changes led to reduced toxicity.15 The majority of the synthetic prodiginines exhibited much greater toxicity (IC50 values < 125 nM) in this particular assay. There were prodiginines which had slightly less toxicity (1000-4000 nM), but these were in all cases also associated with markedly reduced activitiy against P. falciparum, and were still more toxic than CQ (16000 nM). Thus using these particular in vitro assays, the antimalarial activity and toxicity for the current set of synthetic prodiginines exhibit comparable SAR.
The in vivo antimalarial efficacy of the natural prodiginine 3 and a selection of the more potent synthetic analogs were evaluated in a P. yoelii murine patent infection. In initial proof-of-principle testing, administration of 25 mg/kg/day 3 by gavage using a caprylic/capric triglyceride vehicle resulted in greater than 99% reduction in parasitemia (0.28% vs 54.1% in controls); at 100 mg/kg/day parasite clearance was complete, and was accompanied by temporary weight loss, and three of four mice were cured. This data provided the first demonstration of oral effectiveness of prodiginines. Equally important, unlike previous reports of parenteral dosing, efficacy after gavage dosing was observed to be without evident toxicity. These finding indicated that antiplasmodial oral efficacy might be achievable and led to similar in vivo testing of some of the more potent synthetic prodiginines. Examples of impressive potency against murine malaria was observed and is briefly summarized here (Table 4). Analogs 48, 49, 57, 58, and 66 each reduced parasitemia by more than 90%, the exceptions were 41 (38%) and 60 (72%), after 5 mg/kg/day dosing, without any evidence of weight loss or toxicity. 41, 48,49, 57, 58, 60 and 66 each reduced parasitemia by ≥ 90%, after 25 mg/kg/day dosing, however, weight loss was observed during the dosing course with a few analogs (Table 4). However, the weight reduction was temporary and for unknown reasons (including dehydration, fasting of tested mice or toxicity at the higher dose). While an exhaustive in vivo analysis of prodiginine analogs was not conducted, the temporary weight loss was primarily associated with analogs with alkyl substituents. The prodiginines 41 and 66 by contrast have no alkyl substituents and have one or two aryl substituents. Of these compounds tested, 66 had the most favorable profile: 92% parasite reduction at 5 mg/kg/day, 100% reduction at 25 mg/kg/day without any evident weight loss or clinical overt toxicity. Although testing consisted only of equidose comparisons between compounds, without full dose ranging or efforts to define curative dose levels, it is worth mentioning that there was one cure each in the 60 and 58 treated groups (25 mg/kg/day).
Table 4.
In vivo antimalarial efficacy of prodiginine analogs in P. yoelii murine infection
|
Compd |
Dose mg/kg/day |
No. of treated mice |
% Group mean weight; Day3 vs Day0 |
% Parasitemia reduction vs controls; Day3 |
|---|---|---|---|---|
| Controls | - | 5 | 100.8 | 0 |
| 41 | 5 | 5 | 101.2 | 38 |
| 41 | 25 | 5 | 105.0 | 90 |
| 48 | 5 | 5 | 100.1 | 90 |
| 48 | 25 | 4 | 92.4 | 100 |
| 49 | 5 | 5 | 101.5 | 97 |
| 49 | 25 | 4 | 84.5 | 100 |
| 57 | 5 | 5 | 102.9 | 94 |
| 57 | 25 | 5 | 87.6 | 100 |
| 58 | 5 | 5 | 100.3 | 99 |
| 58 | 25 | 4 | 96.0 | 100 |
| 60 | 5 | 5 | 100.2 | 72 |
| 60 | 25 | 3 | 93.4 | 100 |
| 66 | 5 | 5 | 107.5 | 92 |
| 66 | 25 | 5 | 102.5 | 100 |
Conclusions
Although natural prodiginines 3,7d and heptylprodigiosin (76) 7e,16 have demonstrated remarkable in-vitro potency against growth of P. falciparum the mode of action is unknown, and a SAR has not been reported. Herein, we have prepared a set of prodiginine analogs via simple and efficient pathways from commercially available inexpensive starting materials and reagents. These standardized protocols, can be used to generate a large number of prodiginines in a grams scale from a single batch. This current work has suggested that the non alkylated terminal pyrrole (left hand side) of the natural prodiginines is required for in vitro activity, but that the substituents at the alkylated terminal pyrole (right hand side) can be varied. Specifically in vitro activity comparable or better (IC50 ≤ 1 nM) than most of the natural prodiginines was observed for analogs with either alkyl or aryl substituents at both the 3 and 5 position of the right hand side pyrrole, In addition these synthetic prodiginines are equally effective against P. falciparum pansensitive D6 and MDR Dd2.
Previous results of in vivo testing, have also cast doubt on the suitability of prodiginines as antimalarial candidates. Against P. berghei in mice, subcutaneous injection of natural prodiginines 1,7a 76,7e 2, 3, butylcycloheptylprodiginine (77), and cyclononylprodiginine (78),7b,16 have each been shown to extend survival, however, either without cure or at doses resulting in more toxic deaths than cures. Natural prodiginine 1 was shown to be completely ineffective for antimalarial activity, even at toxic doses, for oral treatment of Rhesus monkeys infected with P. cynomolgi.7c In the current study it has been demonstrated for the first time that prodiginines can be administered orally and produce marked parasite clearance, including cures in some cases, without evident weight lose and toxicity. Nonetheless, preliminary in vitro assays continue concerns associated with the toxicity of prodiginines, and the quest for analogs with improved antimalarial activity but reduced toxicity continues.
Experimental Section
Purification of natural prodiginines (1-4)
Natural product 1 was purchased from the developmental therapeutics program NCI/NIH. Using minor modifications to literature procedures, 2, 4 were isolated from S. coelicolor M511,17 and 3 was isolated from S. longisporus ruber.13a
General
1H NMR spectra were run on Bruker AMX-400 MHz spectrometer in CDCl3, 13C NMR spectra were recorded at 100 MHz in CDCl3. Chemical shifts are reported as values in ppm relative to CHCl3 (7.26) in CDCl3 and TMS was used as internal standard. HRMS (ESI) were recorded on high-resolution (30,000) thermo LTQ-Orbitrap Discovery hybrid mass spectrometry (San Jose, CA). Reagents were from Aldrich and used as supplied, unless otherwise noted reactions which required the use of anhydrous, inert atmosphere techniques were carried out under an atmosphere of Argon/Nitrogen. Chromatography was executed with silica gel (230-400 mesh) and neutral-alumina using mixtures of ethyl acetate and hexane as eluents. Analytical HPLC analyses were performed on a Supelco Discovery HS C18 column (4.6 × 250 mm) with a linear elution gradient ranging from CH3OH:CH3CN:H2O (40%:10%:50%) to CH3OH (100%) in 0.15% trifluoroacetic acid at a flow rate of 1 mL/min. A purity of ≥95% has been established for all tested compounds, exception of 25, 26 and 33 achieved 93%, 81% and 86% respectively.
Representative procedure for the synthesis of 1-(1H-pyrrol-2-yl)-octan-1-one (5e)
A mixture of pyrrole (2.0 g, 29.8 mmol), octanoyl chloride (7.8 g, 44.7 mmol), and zinc powder (3.88 g, 59.7 mmol) in toluene (50 mL) was stirred at room temperature for 4 h. The reaction mixture was quenched with saturated sodium bicarbonate solution (50 mL), and extracted with ethyl acetate (3 × 30 mL). The combined organic layer was washed with water, dried over anhydrous Na2SO4, and evaporated under reduced pressure. The crude product was chromatographed on silica gel to afford the title compd 5e (3.16 g, 55%). 1H NMR (CDCl3, 400 MHz) δ 9.71 (br s, 1H), 7.02 (m, 1H), 6.91 (m, 1H), 6.27 (m, 1H), 2.75 (t, J = 7.5 Hz, 2H), 1.70 (m, 2H), 1.33 (m, 8H), 0.92 (t, J = 7.1 Hz, 3H).
Representative procedure for the synthesis of 2-octyl-1H-pyrrole (6e)
To a stirred solution of 5e (1.0 g, 5.18 mmol) in 150 mL of 2-propanol (IPA) at 25 °C was added slowly sodium borohydride (NaBH4) (1.34 g, 36.26 mmol), and the reaction mixture was heated at reflux for 10 h. The hot reaction mixture was poured into 150 mL of ice water, and the solution was acidified with 10% aqueous HCl. The suspension was extracted with dichloromethane (3 × 50 mL), the combined organic extracts were washed with water, brine, and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was chromatographed on silica gel to afford the title compd 6e (714 mg, 77%). 1H NMR (CDCl3, 400 MHz) δ 7.79 (br s, 1H), 6.56 (m, 1H), 6.04 (m, 1H), 5.83 (m, 1H), 2.50 (t, J = 7.6 Hz, 2H), 1.53 (m, 2H), 1.28 (m, 10H), 0.79 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 132.9, 116.0, 108.2, 104.8, 31.9, 29.7, 29.5, 29.4, 29.3, 27.8, 22.7, 14.1.
Synthesis of compound 7
Pyrrole (10 g, 149 mmol) was added to a well-agitated suspension of NaOH (17.9 g, 447 mmol) in 100 mL of 1,2-dichloroethane. This mixture was then cooled to 0 °C and stirred for 10 min. Following, in which a solution of phenylsulfonyl chloride (31.5 g, 173 mmol) in 20 mL of 1,2-dichloroethane was added drop wise over a period of 20 min. Thirty minutes after the completion of addition, the reaction was allowed to come to room temperature and left stirring overnight. The reaction was quenched by pouring onto 300 mL of distilled water. The organic layer was separated, and the aqueous layer was extracted with dichloromethane (3 × 100 mL). The combined organic extract was washed with distilled water to neutrality and dried over anhydrous Na2SO4. Removal of the solvent in vacuo gave 7 as white crystalline solid (21.6 g, 70%). This solid was subsequently washed with hot hexane in greater than 65% overall yield.
Representative procedure for the synthesis of (1-benzenesulfonyl-1H-pyrrol-3-yl)-(4-chloro-phenyl)-methanone (8i)
To a stirred suspension of anhydrous AlC13 (3.85 g, 29 mmol) in 50 mL of 1,2-dichloroethane at 25 °C was added slowly octanoyl chloride (5.0 g, 27 mmol). The resulting solution was stirred at 25 °C for 10 min, a solution of 7 (5.0 g, 24 mmol) in 10 mL of 1,2-dichloroethane was added slowly, and the mixture was stirred at 25 °C for 3 h. The reaction was quenched with ice and water, and the product was extracted into dichloromethane (3 × 100 mL). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and evaporated under reduced pressure to afford crude product 8i (8.0 g, 100%) as white solid. The crude product was used for next step without further purification.
Representative procedure for the hydrolysis of 8i
A solution of 8i (8.0 g, 24 mmol) in 100 mL of 1,4-dioxane was stirred with 100 mL of 5N NaOH at 25 °C for 20 h. The organic layer was collected, and the aqueous layer was thoroughly extracted with ethyl acetate. The combined extracts were washed with brine, dried over anhydrous Na2SO4, and evaporated under reduced pressure to give (4-Chloro-phenyl)-(1H-pyrrol-3-yl)-methanone (9i) as a solid material (4.6 g, 100%).
Synthesis of (5-bromo-3-methoxy-pyrrol-2-ylidenemethyl)-diethyl-amine (12)
To a mixture of diethylformamide (2.68 g, 26.5 mmol) and chloroform (10 mL) at 0 °C was added drop wise a solution of phosphorus oxybromide (6.32 g, 22.1 mmol) in chloroform (10 mL). The resulting suspension was stirred at 0 °C for 30 min, and the solvent was removed by rotary evaporation to obtain the Vilsmeier complex as a white solid. After drying in vacuo for 20 min, chloroform (10 mL) was added to the solid and cooled to 0 °C. A solution of 4-methoxy-3-pyrrolin-2-one (11) (1.0 g, 8.8 mmol) in chloroform (20 mL) was added drop wise and the mixture was warmed to room temperature, and then heated at 60 °C for 5 h. The mixture was poured onto ice water (75 mL), and the pH of the aqueous solution was adjusted to pH 7–8 by treatment with 2N NaOH. Ethyl acetate (40 mL) was added to the resulting precipitate and the mixture was filtered over celite to remove the black solid containing phosphorus salts. The two layers were separated and the aqueous layer was extracted with EtOAc (3 × 100 mL). The organic layers were combined, washed with brine (3 × 200 mL), dried over anhydrous Na2SO4, filtered and the solvent was removed by rotary evaporation to furnish the crude bromo enamine 12. The residue was filtered over a pad of silica gel (50 mL) using a 10% EtOAC/hexanes as eluent to obtain the enamine as an oil, which upon drying in vacuo led to a solid (1.64 g, 72%). 1H NMR (CDCl3, 100 MHz) δ 6.90 (s, 1H), 5.50 (s, 1H), 4.05 (q, J = 7.1 Hz, 2H), 3.65 (s, 2H), 3.30 (q, J = 7.1 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H), 1.20 (t, J = 7.1 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ165.3, 138.7, 133.5, 120.8, 96.4, 58.0, 51.2, 44.5, 18.3, 21.2.
Representative procedure for the preparation of compound 13
To a degassed solution of toluene (5 mL) were added Pd(OAc)2 (303 mg, 1.3 mmol) and PPh3 (1.6 g, 6.1 mmol). The mixture immediately turns to bright yellow color and was stirred at 70 °C for 30 min under N2. A solution of 12 (3.5 g, 13.5 mmol) and N-Boc-2-pyrroleboronic acid (3.72 g, 17.6 mmol) in 10% water/dioxane (50 mL) was degassed and purged with N2. The solution was transferred to the suspension of Pd(PPh3)4 in toluene followed by the addition of Na2CO3 (4.31 g, 40.6 mmol). The mixture was stirred for 3 h at 100 °C and then poured onto water (100 mL), the pH of the solution was lowered to pH 7 with 2N HCl. The brown precipitate was recovered by filtration over a fritted disc funnel and washed with water then acetone. The yellow solid was washed with 10 mL of CHCl3 then Et2O (2 × 10 mL). The desired product 13 is obtained as a yellow solid and used without further purification (2.32 g, 90%). 1H NMR (DMSO-d6, 400 MHz) δ 11.40 (br s, 1H), 11.20 (br s, 1H), 9.30 (s, 1H), 6.90 (t, J = 4.4 Hz, 1H), 6.75 (s, 1H), 6.25 (d, J = 4.4 Hz, 1H), 6.10 (d, J = 4.4 Hz, 1H), 3.80 (s, 3H).
Representative procedure for the synthesis of 4′-methoxy-5′-(3-octyl-1H-pyrrol-2-ylmethylene)-1H,5′H-[2,2′]bipyrrolyl (36)
To a stirred suspension of 13 (220 mg, 1.15 mmol) and compound 10d (269 mg, 1.5 mmol) in anhydrous methanol (10 mL) was added methanolic 2N HCl (catalytic amount). The resulting bright colored solution was stirred for 5 h at room temperature. The methanol was removed under reduced pressure; the crude solid was dissolved in ethyl acetate (50 mL), and washed with saturated NaHCO3 solution (2 × 25 mL). The organic layer was dried over anhydrous Na2SO4, the solvent was removed under reduced pressure, and the crude product was chromatographed on neutral alumina as stationary phase and hexane-ethyl acetate as mobile phase to afford the final prodiginine analog 36 (256 mg, 63%). 1H NMR (CDCl3, 400 MHz) δ 6.92 (s, 1H), 6.77 (m, 1H), 6.72 (dd, J = 1.2, 2.4 Hz, 1H), 6.60 (d, J = 2.4 Hz, 1H), 6.21 (dd, J = 0.9, 2.7 Hz, 1H), 6.06 (s, 1H), 5.97 (d, J = 2.5 Hz, 1H), 4.00 (s, 3H), 2.58 (t, J = 7.6 Hz, 2H), 1.57 (m, 2H), 1.28 (m, 10H), 0.90 (t, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 168.5, 159.8, 139.8, 134.6, 128.5, 126.8, 125.3, 122.5, 113.4, 112.6, 110.4, 110.2, 95.1, 58.4, 31.9, 31.4, 29.5 (2C), 29.3, 26.1, 22.7, 14.1; HRMS (ESI) calcd for C22H29N3O (M+H)+: 352.2383, found. 352.2387. Note- Most of these prodiginine analogs were in the form of HCl salt (Addition of ethereal HCl to the pure prodiginine analog).
Representative procedure for the synthesis of 1-(5-octanoyl-1H-pyrrol-3-yl)-octan-1-one (44)
To a stirred suspension of anhydrous AlC13 (1.34 g, 10.1 mmol) in 30 mL of 1,2-dichloroethane at 25 °C was added slowly octanoyl chloride (1.34 g, 8.1 mmol). The resulting solution was stirred at 25 °C for 10 min, a solution of 9i (1.3 g, 6.7 mmol) in 10 mL of 1,2-dichloroethane was added slowly, and the mixture was stirred at 25 °C for 2 h. The reaction was quenched with ice and water, and the product was extracted into dichloromethane (3 × 50 mL). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and evaporated under reduced pressure to afford 44 (2.14 g, 100%) as white solid. The crude product was washed with hot hexane in greater than 96% of overall yield. 1H NMR (CDCl3, 400 MHz) δ 10.26 (br s, 1H), 7.60 (dd, J = 1.5, 1.9 Hz, 1H), 7.32 (dd, J = 0.9, 1.5 Hz, 1H), 2.78 (m, 4H), 1.72 (m, 4H), 1.38-1.25 (m, 16H), 0.89 (m, 6H); 13C NMR (CDCl3, 100 MHz) δ 196.1, 192.2, 132.6, 127.5, 127.2, 115.2, 39.8, 38.1, 31.7 (2C), 29.4, 29.3, 29.1, 29.0, 25.1, 24.7 (2C), 22.6, 14.1 (2C).
Representative procedure for the synthesis of 2,4-dioctyl-1H-pyrrole (45)
To a stirred solution of 44 (1.3 g, 4.0 mmol) in 150 mL of 2-propanol at 25 °C was added slowly sodium borohydride (2.11 g, 57.0 mmol), and the reaction mixture was heated at reflux for 10 h. The hot reaction mixture was poured into 150 mL of ice water, and the solution was acidified with 10% aqueous HCl. The suspension was extracted with dichloromethane (3 × 50 mL). The combined organic extracts were washed with water, brine, and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure to give 45, and the crude product was chromatographed on neutral alumina to afford the title compd 45 (865 mg, 73%). 1H NMR (CDCl3, 400 MHz) δ 7.60 (br s, 1H), 7.40 (s, 1H), 5.76 (s, 1H), 2.53 (t, J = 7.6 Hz, 2H), 2.41 (t, J = 7.6 Hz, 2H), 1.57 (m, 4H), 1.33 (m, 18H), 0.87 (m, 6H); 13C NMR (CDCl3, 100 MHz) δ 132.8, 124.9, 112.8, 105.4, 31.9 (2C), 31.2, 29.7, 29.6, 29.5, 29.4, 29.3, 29.2, 27.9, 27.4, 27.2, 22.7 (2C), 14.1 (2C).
Representative procedure for the synthesis of 4′-methoxy-5′-(5-octyl-3-propyl-1H-pyrrol-2-ylmethylene)-1H,5′H-[2,2′]bipyrrolyl, hydrochloride (48)
To a stirred suspension of 13 (100 mg, 0.52 mmol) and compound 45 (151 mg, 0.68 mmol) in anhydrous methanol (10 mL) was added methanolic 2N HCl (Catalytic amount). The resulting bright colored solution was stirred for 5 h at room temperature. The methanol was removed under reduced pressure; the crude solid was dissolved in ethyl acetate (50 mL), and washed with saturated NaHCO3 solution (2 × 25 mL). The organic layer was dried over anhydrous Na2SO4, the solvent was removed under reduced pressure, and the crude product was chromatographed on neutral alumina as stationary phase and hexane-ethyl acetate as mobile phase to afford the final prodiginine analog 48 (143 mg, 69%). Note-Most of these prodiginine analogs were in the form of HCl salt (Addition of ethereal HCl to the pure prodiginine analog). 1H NMR (CDCl3, 400 MHz) δ 12.79-12.60 (3 br s, 3H), 7.20 (m, 1H), 7.04 (s, 1H), 6.90 (m, 1H), 6.33 (m, 1H), 6.08 (br s, 2H), 4.01 (s, 3H), 2.92 (t, J = 7.7 Hz, 2H), 2.62 (t, J = 7.5 Hz, 2H), 1.75 (m, 2H), 1.64 (m, 2H), 1.41-1.31 (m, 10H), 0.97 (t, J = 7.3 Hz, 3H), 0.87 (t, J = 7.1 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 165.6, 153.4, 147.4, 145.5, 126.7, 124.2, 122.3, 120.1, 116.7, 113.2, 112.7, 111.6, 92.7, 58.7, 31.9, 29.4, 29.3, 29.2, 29.1, 28.4, 28.3, 24.2, 22.7, 14.1, 13.9; HRMS (ESI) calcd for C25H36N3O (M+H)+. 394.2853, found. 394.2853.
In vitro antimalarial activity: P. falciparum growth inhibition
P. falciparum pansensitive D6 and MDR Dd2 (parasites MRA-285, MRA-156, ATCC, Manassas, Virginia) were cultivated under standard conditions, as previously described,18 and in vitro susceptibility to test compounds was determined by measuring growth after 72 hours in the presence of varying concentrations of test compound or CQ (positive control), or no drug; each condition in quadruplicate. Growth inhibition was determined using the SYBR Green I-based fluorescence assay19 with minor modifications as previously described,20 and expressed as the compound concentration inhibiting growth by 50% (IC50).
In vivo antimalarial efficacy: P. yoelii murine patent infection
Female outbred CF-1 mice (Charles River, Wilmington, MA) were injected intravenously via tail vein with 5 × 105 erythrocytes infected with P. yoelii-Kenya (parasite MRA-428, ATCC, Manassas, VA), and groups of five mice were then treated either with test compound or vehicle only, once each day for three days beginning 24 hours after infection. All compounds were administered by gavage in caprylic/capric triglyceride vehicle (Miglyol 812, Sasol, Hamburg, Germany). One day after the final dose, thin blood smears were made from all mice, stained using a modified Giemsa method, and parasitemia ((infected erythrocytes/total erythrocytes) × 100) was determined using microscopy and compared to untreated controls. Mice without detectable parasitemia were observed and monitored by serial examination of blood smears. Infected mice were euthanized; those remaining parasite-free for 28 days were considered cured.
Alamar Blue assay for mammalian cell viability
The general cytotoxic effects of prodiginine analogs on host cells were assessed by a functional assay as described previously15 using murine splenic lymphocytes induced to proliferate and differentiate by concanavalin A. Splenic lymphocytes isolated from C57BL/6J mice were washed twice in RPMI 1640 medium, and resuspended in complete RPMI containing 10% fetal bovine serum (FBS), 50 μg/mL penicillin/streptomycin, 50 μM β-mercaptoethanol, and 1 μg/mL concanavalin A. Cells (100 μL/well) then were seeded into 96-well flat-bottom tissue culture plates containing drug solutions (100 μL) serially diluted with complete culture medium to a final cell density of 2 × 105 per well. The plates were then incubated for 72 h in a humidified atmosphere at 37 °C and 5% CO2. An aliquot of a stock solution of resazurin (Alamar Blue, prepared in 1 × PBS) was then added at 20 μL per well (final concentration, 10 μM), and the plates were returned to the incubator for another 24 h. After this period, the fluorescence in each well was measured in a Gemini EM plate reader with an excitation wavelength at 560 nm and an emission wavelength at 590 nm. IC50 values were determined by non-linear regression analysis of logistic concentration-fluorescence intensity curves (GraphPad Prism software).
Supplementary Material
Acknowledgment
This work was supported by a grant from the National Institutes for Health (GM077147).
Footnotes
Abbreviations. CQR, chloroquine-resistant; DNA, deoxyribonucleic acid; SAR, structure-activity relationship; CQ, chloroquine; IC50; half maximal inhibitory concentration; nM, nanomolar; MDR, multidrug-resistant
Supporting Information Available. Compounds characterization data and spectra (NMR and HRMS) of all the prodiginine analogs associated with this article are available as supplementary data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Greenwood BM, Fidock DA, Kyle DE, Kappe SH, Alonso PL, Collins FH, Duffy PE. Malaria: progress, perils, and prospects for eradication. J. Clin. Invest. 2008;118:1266–1276. doi: 10.1172/JCI33996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Hyde JE. Drug-resistant malaria. Trends Parasitol. 2005;21:494–498. doi: 10.1016/j.pt.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. Artemisinin resistance in Plasmodium falciparum malaria. N Engl. J. Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alihosseini F, Ju KS, Lango J, Hammock BD, Sun G. Antibacterial Colorants: Characterization of Prodiginines and Their Applications on Textile Materials. Biotechnol Prog. 2008;24:742–747. doi: 10.1021/bp070481r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Regourd J, Al-Sheikh Ali A, Thompson A. Synthesis and Anti-Cancer Activity of C-Ring-Functionalized Prodigiosin Analogs. J. Med. Chem. 2007;50:1528–1536. doi: 10.1021/jm061088f. [DOI] [PubMed] [Google Scholar]
- 5.D’Alessio R, Bargiotti A, Carlini O, Colotta F, Ferrari M, Gnocchi P, Isetta A, Mongelli N, Motta P, Rossi A, Rossi M, Tibolla M, Vanotti E. Synthesis and Immunosuppressive Activity of Novel Prodigiosin Derivatives. J. Med. Chem. 2000;43:2557–2565. doi: 10.1021/jm001003p. [DOI] [PubMed] [Google Scholar]
- 6.Ho TF, Ma CJ, Lu CH, Tsai YT, Wei YH, Chang JS, La JK, Cheuh PJ, Yeh CT, Tang PC, Chang JHT, Ko JL, Liu FS, Yen HCE, Chang CC. Undecylprodigiosin selectively induces apoptosis in human breast carcinoma cells independent of p53. Toxicology and Applied Pharmacology. 2007;225:318–328. doi: 10.1016/j.taap.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 7.(a) Castro AJ. Antimalarial activity of prodigiosin. J. Nature. 1967;213:903–904. doi: 10.1038/213903a0. [DOI] [PubMed] [Google Scholar]; (b) Gerber NN. A new prodiginine (prodigiosin-like) pigment from streptomyces. Antimalarial activity of several prodiginines. J. Antibiot. (Tokyo) 1975;28:194–199. doi: 10.7164/antibiotics.28.194. [DOI] [PubMed] [Google Scholar]; (c) Davidson DE, Jr, Johnsen DO, Tanticharoenyos P, Hickman RL, Kinnamon KE. Evaluating new antimalarial drugs against trophozoite induced Plasmodium cynomolgi malaria in rhesus monkeys. Am. J. Trop. Med. Hyg. 1976;25:26–33. doi: 10.4269/ajtmh.1976.25.26. [DOI] [PubMed] [Google Scholar]; (d) Isaka M, Jaturapat A, Kramyu J, Tanticharoen M, Thebtaranonth Y. Potent In Vitro Antimalarial Activity of Metacycloprodigiosin Isolated from Streptomyces spectabilis BCC 4785. Antimicrob. Agents Chemother. 2002;46:1112–1113. doi: 10.1128/AAC.46.4.1112-1113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lazaro JEH, Nitcheu J, Predicala RZ, Mangalindan GC, Nesslany F, Marzin D, Concepcion GP, Diquet B. Heptyl prodigiosin, a bacterial metabolite, is antimalarial in vivo and non-mutagenic in vitro. J. Nat. Toxins. 2002;11:367–377. [PubMed] [Google Scholar]
- 8.Yadav JS, Reddy BVS, Kondaji G, Rao RS, Kumar SP. Zinc-mediated acylation and sulfonation of pyrrole and its derivative. Tetrahedron Lett. 2002;43:8133–8135. [Google Scholar]
- 9.Huggins MT, Lightner DA. Intramolecular hydrogen bonding between remote termini. Tetrahedron. 2001;57:2279–2287. [Google Scholar]
- 10.Zelikin A, Shastri VR, Langer R. Facile Synthesis of 3-Alkylpyrroles. J. Org. Chem. 1999;64:3379–3380. doi: 10.1021/jo9823339. [DOI] [PubMed] [Google Scholar]
- 11.Kakushima M, Hamel P, Frenette R, Rokach J. Regioselective synthesis of acylpyrroles. J. Org. Chem. 1983;48:3214–3219. [Google Scholar]
- 12.Dairi K, Tripathy S, Attardo G, Lavallee J-F. Two-step synthesis of the bipyrrole precursor of prodigiosins. Tetrahedron Lett. 2006;47:2605–2606. [Google Scholar]
- 13.(a) Wasserman HH, Rodgers GC, Keith DD. Metacycloprodigiosin, a tripyrrole pigment from Streptomyces longisporus rubber. J. Am. Chem. Soc. 1969;91:1263–1264. doi: 10.1021/ja01033a065. [DOI] [PubMed] [Google Scholar]; (b) Wasserman HH, Keith DD, Nadelson J. Synthesis of metacycloprodigiosin. J. Am. Chem. Soc. 1969;91:1264–1265. doi: 10.1021/ja01033a066. [DOI] [PubMed] [Google Scholar]; (c) Wasserman HH, Keith DD, Rodgers GC. The structure of metacycloprodigiosin. Tetrahedron. 1976;32:1855–1861. [Google Scholar]; (d) Wasserman HH, Gosselink E, Keith DD, Nadelson J, Sykes RJ. The synthesis of [9](2,4)pyrrolophanes. Tetrahedron. 1976;32:1863–1866. [Google Scholar]; (e) Wasserman HH, Keith DD, Nadelson J. The synthesis of metacycloprodigionsin. Tetrahedron. 1976;32:1867–1871. [Google Scholar]
- 14.See the experimental section for detailed procedures.
- 15.(a) Ahmed SA, Gogal RM, Jr., Walsh JE. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: an alternative to [3H]thymidine incorporation assay. J Immunol Methods. 1994;170:211–224. doi: 10.1016/0022-1759(94)90396-4. [DOI] [PubMed] [Google Scholar]; (b) Zhi-Jun Y, Sriranganathan N, Vaught T, Arastu SK, Ahmed SA. A dye-based lymphocyte proliferation assay that permits multiple immunological analyses: mRNA, cytogenetic, apoptosis, and immunophenotyping studies. J Immunol Methods. 1997;210:25–39. doi: 10.1016/s0022-1759(97)00171-3. [DOI] [PubMed] [Google Scholar]; (c) Kelly JX, Smilkstein MJ, Brun R, Wittlin S, Cooper RA, Lane KD, Janowsky A, Johnson RA, Dodean RA, Winter R, Hinrichs DJ, Riscoe MK. Discovery of dual function acridones as a new antimalarial chemotype. Nature. 2009;459:270–273. doi: 10.1038/nature07937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.See the supporting information for the structures of natural prodiginines 76, 77, and 78.
- 17.Chen K, Rannulu NS, Cai Y, Lane P, Liebl AL, Rees BB, Corre C, Challis GL, Cole RB. Unusual odd-electron fragments from even-electron protonated prodiginine precursors using positive-ion electrospray tandem mass spectrometry. J Am Soc Mass Spectrom. 2008;19:1856–1866. doi: 10.1016/j.jasms.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 18.Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Hinrichs D, Riscoe MK. Antimalarial quinolones: Synthesis, potency, and mechanistic studies. Exp Parasitol. 2008;118:487–497. doi: 10.1016/j.exppara.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burgess SJ, Selzer A, Kelly JX, Smilkstein MJ, Riscoe MK, Peyton DH. A Chloroquine-like Molecule Designed to Reverse Resistance in Plasmodium falciparum. J. Med Chem. 2006;49:5623–5625. doi: 10.1021/jm060399n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.