

Abstract
Abstract
A series of mono-alkylcarboxylic acid derivatives of tetraphenylporphyrin have been prepared. All the porphyrins were completely characterized by use of mass, 1H NMR, UV–visible, and fluorescence spectroscopy. Experimental log P were determined by use of reversed-phase thin-layer chromatography with use of log P Rekker. These porphyrins are potential photosensitizers in photodynamic therapy.
Graphical abstract
 .
.
Keywords: Porphyrins, Sensitizers, Hydrophobicity, Fluorescence spectroscopy
Introduction
Hematoporphyrin (1), the first compound that was tried in photodynamic therapy (PDT), contains, besides the basic porphyrin ring, two carboxyethylene groups. Photofrin, currently the most frequently used porphyrin-type compound in anticancer therapy, was obtained from hematoporphyrin (1) (Fig. 1) [1]. Photofrin molecules contain carboxyl (carboxyethylene) groups which give the compound its hydrophobic and hydrophilic properties. Photofrin preparations are, however, a mixture of several porphyrin compounds. Numerous research teams around the world have been trying to obtain novel compounds with precisely determined structure and controlled biological properties [2–4]. Several of these compounds are derivatives of tetraphenylporphyrin (2) (tetraarylporphyrin, Fig. 1). The aryl fragments in these porphyrins are modified by substituting hydrogen atoms with various groups altering the hydrophobic–hydrophilic character of the whole molecule, most often without changing its photochemical properties.
Fig. 1.
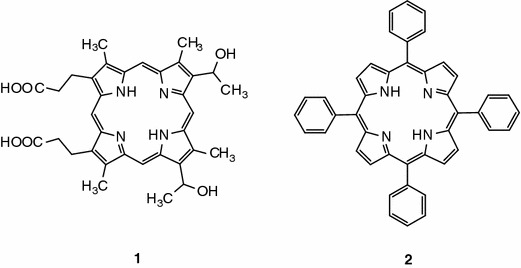
Hematoporphyrin and tetraphenylporphyrin
Simple tetraarylporphyrins are highly soluble in slightly polar solvents, most often halogenated or DMF; these however cannot be used in biological investigations except at the stage of preparing photosensitizer–carrier systems, e.g. liposomes. Most synthetic porphyrins are not very soluble in simple alcohols. Some porphyrins are soluble in water which makes them suitable for basic biological studies. Introduction of strongly polar groups, for example ammonium or sulfone groups, enables dissolution of tetraarylporphyrins in water. Suitably modified porphyrins can be incorporated into lipid membranes, micelles, or liposomes; in this form they are delivered into cells [5–13]. Porphyrin carriers of this type are very promising, because most of the porphyrins obtained synthetically are strongly hydrophobic which makes them useless in investigations of direct PDT or other therapeutic applications. Derivatives with acidic moieties at the ends of alkyl chains have not yet been studied for their applicability in photodynamic therapy. Compounds with alkyl chains of different lengths separating the porphyrin fragment from the carboxyl group were chosen for study. Hydrophobic–hydrophilic character is responsible for a variety of the properties of organic compounds, for example solubility, permeability through semi-permeable membranes (for example cell membranes), and ability to anchor in lipid layers. Most importantly, for PDD and PDT it is responsible for the manner of transport of photosensitizer molecules into the living organism. The easiest way to verify the assumed properties of a compound is by synthesis of the compound.
In search of novel potential photosensitizers we obtained a series of carboxyalkyloxy tetraphenylporphyrin derivatives 4–8 (Fig. 2). Carboxyl groups are attached to one of the phenyl rings of tetraphenylporphyrin via alkyl chains of different lengths. Previously, compounds with n = 1 and n = 3 were obtained as intermediates during the synthesis of more complex tetraphenylporphyrin derivatives. In particular, cholesterol derivatives of carboxymethylporphyrin (n = 1) were synthesized and their gelator properties described [14]. Butyric acid (n = 3) was attached to arylporphyrin via an ether bond in order to obtain a porphyrin with an easily modifiable fragment (in this case a COOH group). Next, it was substituted using either lipophilic guanosine in order to examine the self-assembly of this group [15] or with a tyrosine group when obtaining modified TiO2 clusters in which photoinduced electron transfer occurred [16]. Amide derivatives of this compound were also obtained as potential agents for photodynamic therapy of cancers [17]. Glycosidic porphyrins obtained from the butyric acid derivative of tritolylphenylporphyrin were also designed as potential PDT agents [18].
Fig. 2.

Synthesized compounds
Results and discussion
To the best of our knowledge no derivatives with carboxyl groups at the distal ends of alkyl chains have been investigated for their usefulness in PDT. We thus decided to investigate compounds with alkyl chains of different lengths separating the porphyrin fragment from the carboxyl group. To synthesize alkyl precursors with n = 4, 5, 6, 7, and 11 we used the classic method of obtaining carboxyl derivatives starting from suitable dibromoalkyl derivatives and malonic acid diethyl ester [19, 20]. The yields of the obtained diethyl (bromoalkyl)malonates were not very high but this could be expected, especially for shorter alkyl chains. These compounds quite easily undergo intramolecular cyclization reaction under the reaction conditions [21, 22]. Hydroxyphenyltritolylporphyrin (3) was obtained according to Ref. [23]. Diethyl (ω-bromoalkyl)malonates were coupled to a hydroxyl fragment of porphyrin using NaOH in DMF (Scheme 1). Under the reaction conditions the monocarboxy derivatives were obtained directly; in the presence of NaOH hydrolysis of the ester took place, whereas DMF distillation at an elevated temperature led to simultaneous decarboxylation of the malonic acid derivative. Exceptional for this synthetic route were the reactions of diethyl (3-bromopropyl)malonate leading to a malonic acid derivative and of diethyl (4-bromobutyl)malonate which gave a pentyloxy derivative of tetraphenylporphyrin. The properties of the obtained derivative were compared with those of the similar compound obtained by direct alkylation of the hydroxyphenyl derivative of porphyrin with 1-bromopentane.
Scheme 1.

Porphyrins 4 and 5 were obtained by a different method, direct reaction of a suitable ethyl ester of ω-bromoalkyl acid with hydroxyl derivative of porphyrin (Scheme 2). The esters were hydrolysed to the corresponding carboxylic derivatives.
Scheme 2.

All of the obtained porphyrins 4–8 had characteristic common bands in the visible part of the UV–visible spectrum. They have very strong absorption peaks centred at 422 nm (Soret band) and four other peaks centred at 519, 555, 594, and 650 nm (Q bands). The spectra clearly identify the porphyrinic chromophores of compounds 4–8. The minor differences between the absorption maxima of the porphyrins implied their photosensitizing properties are also comparable. UV–visible data for all the compounds are listed in Table 1.
Table 1.
UV–visible spectra of compounds 4–9, 11, and 12 [λ(log ε)] in dichloromethane solution
| Derivative | Soret/nm | QIV/nm | QIII/nm | QII/nm | QI/nm |
|---|---|---|---|---|---|
| 4 | 422 (5.38) | 517 (3.95) | 554 (3.77) | 592 (3.51) | 650 (3.63) |
| 5 | 422 (5.54) | 518 (4.04) | 555 (3.84) | 592 (3.57) | 651 (3.58) |
| 6 | 422 (5.57) | 519 (4.10) | 555 (3.86) | 594 (3.60) | 650 (3.58) |
| 7 | 422 (5.58) | 519 (4.17) | 555 (3.94) | 594 (3.67) | 650 (3.66) |
| 8 | 422 (5.57) | 519 (4.13) | 555 (3.90) | 594 (3.63) | 650 (3.67) |
| 9 | 422 (5.70) | 519 (4.30) | 554 (4.07) | 593 (3.81) | 649 (3.77) |
| 11 | 422 (5.46) | 518 (4.05) | 555 (4.05) | 594 (3.55) | 650 (3.52) |
| 12 | 422 (5.11) | 517 (3.67) | 554 (3.47) | 594 (3.21) | 651 (3.31) |
Fluorescence spectra of all porphyrins and TPP as a reference are similar in shape, and consist of two bands at 654 and 720 nm. From Table 2 we can see that the quantum yields of all the porphyrins are almost identical with that of the reference (TPP). The photostability of compounds 4–9, 11, and 12 was determined for their chloroform solutions at 25 °C. The results presented in Table 3 show good photostability of the compounds which indicates their potential usefulness for PDT.
Table 2.
Fluorescence emission maxima, wavelengths (λ max), and fluorescence quantum yields (ΦF) for compounds 4–9 and 11 and tetraphenylporphyrin (TPP) as reference; log P Rekker and log P exp for compounds 5–9 and 11
| Photosensitizer | λ max1/nm | λ max2/nm | ΦF | log P Rekker | log P exp |
|---|---|---|---|---|---|
| 4 | 653 | 720 | 0.11 | – | – |
| 5 | 652 | 718 | 0.11 | 9.08 | 8.25 |
| 6 | 654 | 719 | 0.13 | 9.60 | 10.79 |
| 7 | 654 | 721 | 0.11 | 10.12 | 12.05 |
| 8 | 654 | 720 | 0.11 | 12.19 | 18.48 |
| 9 | 654 | 720 | 0.11 | 9.39 | 8.85 |
| 11 | 654 | 720 | 0.11 | 9.55 | 8.92 |
| TPP | 652 | 718 | 0.1 | – | – |
Table 3.
Photostability of compounds 4–9, 11, and 12
| Photosensitizer | 0 | 15 | 30 | 45 | 60 | 75 | 90 |
|---|---|---|---|---|---|---|---|
| 4 | 100 | 99 | 99 | 98 | 97 | 97 | 95 |
| 5 | 100 | 99 | 96 | 96 | 95 | 95 | 95 |
| 6 | 100 | 100 | 99 | 99 | 97 | 96 | 96 |
| 7 | 100 | 100 | 99 | 98 | 98 | 97 | 97 |
| 8 | 100 | 99 | 98 | 99 | 96 | 95 | 95 |
| 9 | 100 | 98 | 98 | 97 | 96 | 96 | 96 |
| 11 | 100 | 99 | 99 | 98 | 97 | 97 | 95 |
| 12 | 100 | 100 | 98 | 97 | 96 | 96 | 96 |
For irradiation, visible light from two 36-W fluorescent lamps was used and the light intensity was adjusted to 16.5 W/m2. Photostability was determined by monitoring the intensity of the Soret band after a defined irradiation time (0–90 min)
Mass spectrometric analysis of all the synthesized porphyrins was performed by use of the ESI–MS technique. The mass spectra (positive mode) of all the porphyrins contained a base peak corresponding to the intact porphyrin—no fragment ions were detected. For porphyrins 6–8 and 10 the most intense ions appeared when sodium cations were present in the solutions.
Lipophilicity is an important molecular descriptor very often correlated with the biological activity of drugs and other compounds. It is characterized, for example, by the logarithm of a partition coefficient (log P). We determined log P values of all the compounds by a TLC method. Table 2 shows values calculated by use of the  method and the experimental log P values. Log P
exp was calculated by use of the equation [24]:
method and the experimental log P values. Log P
exp was calculated by use of the equation [24]:
where 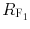 and
and 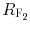 are retardation factors for methanol–chloroform 8:2 and 7:3 (v/v), respectively, as mobile phase. Log P
Rekker was calculated, in compliance with the constant fragmentations method proposed by Rekker, by use of the equation [25, 26]:
are retardation factors for methanol–chloroform 8:2 and 7:3 (v/v), respectively, as mobile phase. Log P
Rekker was calculated, in compliance with the constant fragmentations method proposed by Rekker, by use of the equation [25, 26]:
where f is a constant representing the lipophilicity of a defined structural fragment and a describes how many times a specific fragment is repeated in a molecule. Experimental results show that additional methylene units in the carbon chain result in an increase of the compound’s lipophilicity. We obtained log P exp = 1.49 for tetrakis(3-hydroxyphenyl)porphyrin as reference compound. The literature shake-flask log P value for this compound is 2.02 [27].
Solubility in polar solvents is a serious obstacle for the synthesized compounds. As could be expected, elongation of the alkyl chain led to increased hydrophobicity of the whole molecule. As a result, this often leads to aggregation and precipitation of porphyrins from solutions used for biological investigations. Currently, all these compounds are undergoing biological investigation using liposomal formulations which can substantially reduce the extent of photosensitizer aggregation and can be valuable carriers and delivery systems.
Conclusions
Novel mono-carboxyalkyloxyphenyl derivatives of porphyrin were synthesized and their physicochemical properties were examined. Quantum yields of all the obtained porphyrins are almost identical with that of the reference compound (TPP). Information about the lipophilicity of the examined compounds can prove helpful in further research on their potential medical uses. Their balanced hydrophobic–hydrophilic properties make it possible to place them in systems (e.g. micelles, liposomes) similar to natural biological membranes.
Experimental
All chemical reagents were purchased from Aldrich or Acros and were used without further purification. 1H NMR and 13C NMR spectra were recorded in CDCl3 using a Varian spectrometer (400 MHz) with TMS as reference. UV–visible spectra were recorded in dichloromethane solutions using a Genesys 6 (ThermoSpectronic) spectrophotometer. Fluorescence spectra of the samples were recorded on a Varian Eclipse Cary-fluorescence spectrophotometer. ESI MS spectra were acquired using an LCQ DUO Finnigan Thermoquest or a Varian MS-500 instrument. Porphyrin solutions were directly injected into the ion source by use of a syringe pump.
General procedure for diethyl (ω-bromoalkyl)malonates
Diethyl malonate (0.05 mol), 1,ω-dibromoalkane (0.05 mol), 0.375 g tetrabutylammonium iodide (1 mmol), and 15 cm3 toluene were placed in a flask. A solution of 17.25 g K2CO3 in 15 cm3 water was added. The obtained mixture was stirred and heated to reflux for 20 h. After cooling to room temperature the solid was removed by suction. The organic phase was separated and the aqueous phase was extracted with toluene (3 × 10 cm3). The combined organic layers were washed with water and dried over anhydrous magnesium sulfate. After evaporating the solvent the product was distilled under reduced pressure.
Diethyl (3-bromopropyl)malonate
Yield 16%; b.p.: 140–145 °C (7–8 mbar), lit. 135 °C (4 mbar) [28], 121–123 °C (2 mbar) [19].
Diethyl (4-bromobutyl)malonate
Yield 16%; b.p.: 104–106 °C (3–4 mbar), lit. 133–136 °C (2 mbar) [29].
Diethyl (5-bromopentyl)malonate
Yield 21%; b.p.: 178–180 °C (8 mbar), lit. 153 °C (3 mbar) [30]; 159–161 °C (3 mbar) [31].
Diethyl (6-bromohexyl)malonate
Yield 17%; b.p.: 175–178 °C (9 mbar), lit. 185–187°°C (10 mbar) [32]; 193 °C (12 mbar) [31].
Diethyl (10-bromodecyl)malonate
Yield 20%; b.p.: 222–224 °C (7–8 mbar), lit. 123 °C (0.02 mbar) [20, 33].
General procedure for 5-[4-(carboxyalkyloxy)phenyl]-10,15,20-tritolylporphyrins
5-(4-Hydroxyphenyl)-10,15,20-tritolylporphyrin (0.24 mmol), solid NaOH (0.8 mmol), and 20 cm3 DMF were placed in a round-bottomed flask. The mixture was stirred at room temperature until NaOH was dissolved. Then 2 mmol diethyl (ω-bromoalkyl)malonate were added and the resulting mixture was stirred at room temperature for 24 h. After evaporation of the DMF under reduced pressure 50 cm3 dichloromethane was added to the residue. The resulting solution was washed with water (5 × 50 cm3) and dried over MgSO4. After evaporation of the solvent the crude product was washed with petroleum ether (20 cm3) and chromatographed on silica with dichloromethane.
5-[4-(5-Carboxypentyloxy)phenyl]-10,15,20-tritolylporphyrin (5, C53H46N4O3)
Yield 14%; 1H NMR (400 MHz, CDCl3): δ = 8.86 (m, 8H), 8.10 (d, 2H, J = 8.4 Hz), 8.09 (d, 6H, J = 8.0 Hz), 7.54 (d, 6H, J = 8.0 Hz), 7.25 (d, 2H, J = 8.4 Hz), 4.24 (t, 2H), 2.70 (s, 9H), 2.51 (t, 2H), 2.01 (q, 2H), 1.86 (q, 2H), 1.72 (q, 2H), −2.76 (bs, 2H) ppm; ESI–MS: m/z = 787 (100) [M + H]+.
5-[4-(6-Carboxyhexyloxy)phenyl]-10,15,20-tritolylporphyrin (6, C54H48N4O3)
Yield 57%; 1H NMR (400 MHz, CDCl3): δ = 8.86 (m, 8H), 8.11 (d, 2H, J = 8.4 Hz), 8.10 (d, 6H, J = 8.0 Hz), 7.55 (d, 6H, J = 8.0 Hz), 7.27 (d, 2H, J = 8.4 Hz), 4.27 (t, 2H), 3.55 (t, 2H), 2.71 (s, 9H), 2.10–2.00 (m, 4H), 1.77 (q, 2H), 1.53 (q, 2H), –2.76 (bs, 2H) ppm; ESI–MS: m/z = 823 (100) [M + Na]+.
5-[4-(7-Carboxyheptyloxy)phenyl]-10,15,20-tritolylporphyrin (7, C55H50N4O3)
Yield 14%; 1H NMR (400 MHz, CDCl3): δ = 8.85 (m, 8H), 8.10 (d, 2H, J = 8.4 Hz), 8.09 (d, 6H, J = 8.0 Hz), 7.54 (d, 6H, J = 8.0 Hz), 7.27 (d, 2H, J = 8.4 Hz), 4.24 (t, 2H), 3.50 (t, 2H), 2.70 (s, 9H), 1.99 (q, 4H), 1.66 (q, 4H), 1.52 (m, 2H), −2.76 (bs, 2H) ppm; ESI–MS: m/z = 837 (100) [M + Na]+.
5-[4-(11-Carboxyundecyloxy)phenyl]-10,15,20-tritolylporphyrin (8, C59H58N4O3)
Yield 18%; 1H NMR (400 MHz, CDCl3): δ = 8.85 (m, 8H), 8.10 (d, 2H, J = 8.4 Hz), 8.09 (d, 6H, J = 8.0 Hz), 7.55 (d, 6H, J = 8.0 Hz), 7.27 (d, 2H, J = 8.4 Hz), 4.25 (t, 2H), 3.43 (t, 2H), 2.70 (s, 9H), 2.00 (q, 2H), 1.90 (q, 2H), 1.62 (q, 2H), 1.53–1.20 (m, 12H), −2.76 (bs, 2H) ppm; ESI–MS: m/z = 894 (100) [M + Na]+.
5-[4-[4,4-Bis(ethoxycarbonyl)butyloxy]phenyl]-10,15,20-tritolylporphyrin (9, C57H52N4O5)
Yield 27%; 1H NMR (400 MHz, CDCl3): δ = 8.84 (m, 8H), 8.10 (d, 2H, J = 8.4 Hz), 8.09 (d, 6H, J = 7.6 Hz), 7.54 (d, 6H, J = 7.6 Hz), 7.24 (d, 2H, J = 8.4 Hz), 4.27 (q, 4H), 3.56 (t, 1H), 2.69 (s, 9H), 2.27 (q, 2H), 2.04 (q, 2H), 1.51 (bs, 2H), 1.33 (t, 6H), −2.76 (bs, 2H) ppm; ESI–MS: m/z = 873 (100) [M + H]+.
5-(4-Pentyloxyphenyl)-10,15,20-tritolylporphyrin (10, C52H46N4O)
Yield 14%; 1H NMR (400 MHz, CDCl3): δ = 8.87 (m, 8H), 8.11 (d, 2H, J = 8.4 Hz), 8.10 (d, 6H, J = 8.0 Hz), 7.55 (d, 6H, J = 8.0 Hz), 7.27 (d, 2H, J = 8.4 Hz), 4.23 (t, 2H), 2.71 (s, 9H), 1.99 (q, 2H), 1.62 (q, 2H), 1.53 (q, 2H), 1.05 (t, 3H), −2.73 (bs, 2H) ppm; ESI–MS: m/z = 765 (100) [M + Na]+, 743 (100) [M + H]+; UV–Vis (CH2Cl2): λ max (log ε) = 422 (5.54), 519 (4.13), 555 (3.89), 593 (3.63), 650 (3.60) nm.
General procedure for 5-[4-[ω-(ethoxycarbonyl)alkyloxy]phenyl]-10,15,20-tritolylporphyrins
5-(4-Hydroxyphenyl)-10,15,20-tritolylporphyrin (100 mg, 0.15 mmol), 5 mg NaH (0.2 mmol), and 50 cm3 DMF were placed in a round-bottomed flask and stirred at room temperature for 30 min. Then 1 mmol ethyl ω-bromoalkylcarboxylate was added and the resulting mixture was stirred at 80 °C for 48 h. After evaporation of the DMF under reduced pressure 50 cm3 dichloromethane was added to the residue. The resulting solution was washed with water (3 × 50 cm3) and dried over MgSO4. After evaporation of the solvent the crude product was washed with petroleum ether (20 cm3) and chromatographed on silica with dichloromethane–ethanol 30:1 (v/v).
5-[4-[4-(Ethoxycarbonyl)butyloxy]phenyl]-10,15,20-tritolylporphyrin (11, C54H48N4O3)
Yield 33%; 1H NMR (400 MHz, CDCl3): δ = 8.85 (m, 8H), 8.11 (d, 2H, J = 8.0 Hz), 8.09 (d, 6H, J = 7.6 Hz), 7.55 (d, 6H, J = 7.6 Hz), 7.27 (d, 2H, J = 8.0 Hz), 4.28 (t, 2H), 4.21 (q, 2H), 2.71 (s, 9H), 2.52 (q, 2H), 2.03 (m, 4H), 1.33 (t, 3H), −2.76 (bs, 2H) ppm; 13C NMR (125 MHz, CDCl3): δ = 173.19, 155.75, 139.33, 137.32, 135.68, 134.57, 134.53, 130.89, 127.42, 120.09, 120.05, 113.67, 60.44, 33.36, 33.03, 32.04, 23.55, 21.53, 14.26 ppm; ESI–MS: m/z = 801 (100) [M + H]+.
5-[4-[5-(Ethoxycarbonyl)pentyloxy]phenyl]-10,15,20-tritolylporphyrin (12, C55H50N4O3)
Yield 29%; 1H NMR (400 MHz, CDCl3): δ = 8.85 (m, 8H), 8.11 (d, 2H, J = 8.0 Hz), 8.10 (d, 6H, J = 7.6 Hz), 7.55 (d, 6H, J = 7.6 Hz), 7.27 (d, 2H, J = 8.0 Hz), 4.26 (t, 2H), 4.19 (q, 2H), 2.71 (s, 9H), 2.44 (t, 2H), 2.01 (q, 2H), 1.84 (q, 2H), 1.69 (q, 2H), 1.31 (t, 3H), −2.76 (bs, 2H) ppm; 13C NMR (125 MHz, CDCl3): δ = 173.75, 158.89, 139.36, 137.32, 135.61, 134.54, 129.60, 129.07, 127.43, 120.10, 112.72, 67.98, 60.35, 34.40, 29.23, 25.90, 24.90, 21.55, 14.35 ppm; ESI–MS: m/z = 815 (100) [M + H]+.
General procedure for 5-(4-carboxyalkyloxyphenyl)-10,15,20-tritolylporphyrins from ethyl ester
5-[4-(ω-Ethoxycarbonylalkyloxy)phenyl]-10,15,20-tritolylporphyrin (0.03 g, ~0.04 mmol) dissolved in 10 cm3 dichloromethane was added to a solution of sodium ethoxide (1 g sodium dissolved in 10 cm3 ethanol). One drop of water was added to the mixture. The reaction mixture was stirred for 3 h at room temperature. The solution was then acidified to pH 7 with 1 M HCl, washed with water (3 × 30 cm3), dried over Na2SO4, and concentrated. Purification by column chromatography on silica gel (elution with CH2Cl2–EtOH, 25:1 (v/v) afforded the product.
5-[4-(4-Carboxybutyloxy)phenyl]-10,15,20-tritolylporphyrin (4, C52H44N4O3)
Yield 24%; 1H NMR (400 MHz, CDCl3): δ = 8.85 (d, 8H), 8.09 (d, 8H, J = 8.0 Hz), 7.54 (d, 6H, J = 8.0 Hz), 7.24 (d, 2H, J = 8.0 Hz), 4.26 (t, 2H), 2.76 (s, 9H), 2.59 (q, 2H), 2.03 (q, 2H), 1.62 (m, 2H), −2.76 (bs, 2H) ppm; ESI–MS: m/z = 773 (100) [M + H]+.
5-[4-(5-Carboxypentyloxy)phenyl]-10,15,20-tritolylporphyrin (5) was obtained by this method in 19% yield.
Acknowledgments
Financial support by the Polish State Committee for Scientific Research (grant no. R 05 043 03) is gratefully acknowledged. The work of two authors (Marcin Rojkiewicz and Grzegorz Zięba) was partially supported by a PhD scholarship within the framework of the “University as a Partner of the Economy Based on Science” (UPGOW) project, subsidized by the European Social Fund (EFS) of the European Union.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Schaffer M, Schaffer PM, Hofstetter A, Dühmke E, Jori G. Photochem Photobiol Sci. 2002;1:438. doi: 10.1039/b202849b. [DOI] [PubMed] [Google Scholar]
- 2.Nyman ES, Hynninen PH. J Photochem Photobiol B. 2004;73:1. doi: 10.1016/j.jphotobiol.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Bonnett R. Chem Soc Rev. 1995;24:19. doi: 10.1039/cs9952400019. [DOI] [Google Scholar]
- 4.Sternberg ED, Dolphin D, Brückner C. Tetrahedron. 1998;54:4151. doi: 10.1016/S0040-4020(98)00015-5. [DOI] [Google Scholar]
- 5.Ballut S, Makky A, Loock B, Michael J-P, Maillard P, Rosilio V. Chem Commun. 2009;8:224. doi: 10.1039/b816128c. [DOI] [PubMed] [Google Scholar]
- 6.Ricchelli F. J Photochem Photobiol B. 1995;29:109. doi: 10.1016/1011-1344(95)07155-U. [DOI] [PubMed] [Google Scholar]
- 7.Voszka I, Galántai R, Maillard P, Csik G. J Photochem Photobiol B. 1999;52:92. doi: 10.1016/S1011-1344(99)00107-4. [DOI] [Google Scholar]
- 8.Tsuchida E, Komatsu T, Arai K, Nishide H. Chem Commun. 1993;8:730. [Google Scholar]
- 9.Nagata N, Kugimiya S, Kobuke Y. Chem Commun. 2001;8:689. doi: 10.1039/b009458g. [DOI] [Google Scholar]
- 10.Lavi A, Weitman H, Holmes RT, Smith KM, Ehrenberg B. Biophys J. 2002;82:2101. doi: 10.1016/S0006-3495(02)75557-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Postigo F, Mora M, De Madariaga MA, Nonell S, Sagristá ML. Int J Pharm. 2004;278:239. doi: 10.1016/j.ijpharm.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Voszka I, Szabό Z, Csik G, Maillard P, Grόf P. J Photochem Photobiol B. 2005;79:83. doi: 10.1016/j.jphotobiol.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Dror S, Bronshtein I, Wiehe A, Röder B, Senge MO, Ehrenberg B. J Photochem Photobiol. 2006;82:695. doi: 10.1562/2005-09-01-RA-669. [DOI] [PubMed] [Google Scholar]
- 14.Tian HJ, Inoue K, Yoza K, Ishi-I T, Shinkai S (1998) Chem Lett 871
- 15.Masiero S, Gottarelli G, Pieraccini S (2000) Chem Commun 1995 [DOI] [PubMed]
- 16.Yu J, Chen J, Wang X, Zhang B, Cao Y (2003) Chem Commun 1856 [DOI] [PubMed]
- 17.Sol V, Lamarche F, Enache M, Garcia G, Granet R, Guilloton M, Blais JC, Krausz P. Bioorg Med Chem. 2006;14:1364. doi: 10.1016/j.bmc.2005.09.071. [DOI] [PubMed] [Google Scholar]
- 18.Chaleix V, Sol V, Huang Y-M, Guilloton M, Granet R, Blais JC, Krausz P (2003) Eur J Org Chem 1486
- 19.Ikegami S, Asai T, Tsuneoka K, Matsumara S, Akaboshi S. Tetrahedron. 1974;30:2087. doi: 10.1016/S0040-4020(01)97343-0. [DOI] [Google Scholar]
- 20.Matsubara H, Seto K, Tabuchi H, Imazaki H, Takahashi S. Bull Chem Soc Jpn. 1993;66:578. doi: 10.1246/bcsj.66.578. [DOI] [Google Scholar]
- 21.Cort AD, Mandolini L, Masci B. J Org Chem. 1983;48:3979. doi: 10.1021/jo00170a020. [DOI] [Google Scholar]
- 22.Casadei MA, Galli C, Mandolini L. J Org Chem. 1981;46:3127. doi: 10.1021/jo00328a030. [DOI] [Google Scholar]
- 23.Little RG, Anton JA, Loach PA, Ibers JA. J Heterocycl Chem. 1975;12:343. doi: 10.1002/jhet.5570120226. [DOI] [Google Scholar]
- 24.Niestrój A. J Planar Chromatogr–Mod TLC. 2007;20:483. doi: 10.1556/JPC.20.2007.6.15. [DOI] [Google Scholar]
- 25.Rekker RF, Mannhold R (1992) In: Calculation of drug lipophilicity. VCH, Weinheim
- 26.Mannhold R, Rekker RF. Perspect Drug Discov. 2000;18:1. doi: 10.1023/A:1008782809845. [DOI] [Google Scholar]
- 27.Serra AC, Pineiro M, Rocha Gonsalves AM, Abrantes M, Laranjo M, Santos AC, Botelho MF. J Photochem Photobiol B. 2008;92:59. doi: 10.1016/j.jphotobiol.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 28.van Tamelen EE, Foltz RL. J Am Chem Soc. 1969;91:7372. doi: 10.1021/ja01054a026. [DOI] [Google Scholar]
- 29.Tatarinova VI, Vasiliev AA, Petrosyan VA (1990) Izv Akad Nauk SSSR Ser Khim 2646
- 30.Knipe AC, Stirling CJM (1968) J Chem Soc B 67
- 31.Salmon-Legagneur F, Nevéu C (1956) Bull Soc Chim 1345
- 32.Buchta E, Fischer M. Chem Ber. 1966;99:1509. doi: 10.1002/cber.19660990514. [DOI] [Google Scholar]
- 33.Zieger K, Weber K. Liebigs Ann Chem. 1934;512:164. doi: 10.1002/jlac.19345120114. [DOI] [Google Scholar]