

Abstract
Abstract
Chiral acyclic and macrocyclic amines derived from trans-1,2-diaminocyclohexane in complexes with diethylzinc efficiently catalyze asymmetric hydrosilylation of aryl–alkyl and aryl–aryl ketones with enantiomeric excess of the product up to 86 %. A trianglamine ligand with a cyclic structure or the presence of an additional coordinating group increases the enantioselectivity of the reaction, in comparison with catalysis by a simple acyclic N,N′-dibenzyl-1,2-diaminocyclohexane ligand. In addition, the effect of the asymmetric activation of the catalyst by a variety of alcohols and diols is studied.
Graphical Abstract

Electronic supplementary material
The online version of this article (doi:10.1007/s00706-012-0754-0) contains supplementary material, which is available to authorized users.
Keywords: Homogeneous catalysis, Metal complexes, Ligands, Macrocycles, Asymmetric activation, Zinc
Introduction
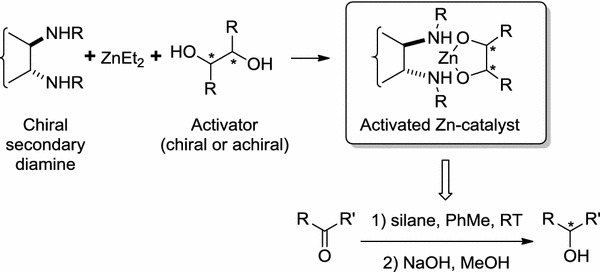
Secondary alcohols are an important class of chiral building blocks in organic synthesis and form structural fragments of numerous biologically active compounds [1]. Major asymmetric catalytic methods for preparation of secondary alcohols rely mainly either on asymmetric hydrogenation of ketones, with use of chiral rhodium complexes as catalysts in both homogeneous and heterogeneous approaches, or on oxazaborolidine-catalyzed asymmetric reduction of ketones with diborane [2–6]. Before the re-invention of the reductive properties of polymethylhydrosiloxane (PMHS), a safe and inexpensive by-product of the silicon industry, hydrosilylation of C=O bonds seemed a less convenient method because of the toxicity and cost of monomeric silanes. Since the discovery of Zn-diamine-catalyzed asymmetric hydrosilylation of prochiral ketones, several other methods for enantioselective reduction of the C=O bonds have been developed [7–23]. They are mainly based on the use of chiral transition metal complexes with P,P-bidentate ligands, P,S-ligands, N-ligands, or N-heterocyclic carbene ligands. Although the enantioselectivity obtained by use of these complexes is >90 %, use of such complexes suffers from high cost and elaborate preparation. In contrast, use of catalytic systems based on zinc has emerged as a promising and advantageous method for metal-catalyzed asymmetric hydrosilylation of prochiral ketones (Scheme 1).
Scheme 1.

According to the original procedure developed by Mimoun et al. [7] the reaction requires chiral 1,2-diamine as ligand for dialkylzinc. Modifications could be made either by judicious choice of chiral ligand, by adding an activator, or by changing the reaction conditions (temperature, solvent). The ligands were usually obtained from commercially available amines, for example 1,2-diamino-1,2-diphenylethane or 1-phenylethylamine. However, enantiomerically pure trans-1,2-diaminocyclohexane (DACH) is the preferred source of a virtually unlimited number of diamine structures. Apart from simple acyclic N,N′-disubstituted derivatives, the ability to make complex structures of triangular, rhombic, or spherical character from suitable substrates is well documented for DACH [24–29]. Another possibility of modifying the structure and properties of macrocyclic DACH derivatives is available by further N-substitution and/or by formation of additional chiral centers at the benzylic positions and introduction of other functional groups in the ligand structure [30, 31]. Chiral macrocyclic tetramine and hexamine macrocycles derived from trans-1,2-diaminocyclohexane in complexes with diethylzinc efficiently catalyze asymmetric hydrosilylation of aryl–alkyl ketones and imines with enantiomeric excess of the product up to 99 % [32, 33].
Introduction of two different chiral centers into the ligand structure may give rise to synergistic effects, leading to and increase in the enantioselectivity of hydrosilylation reaction [34]. The basic concept consists in the idea of “asymmetric activation”, introduced by Mikami and co-workers and subsequently applied to several enantioselective transformations [35–39]. By following this concept, Ushio and Mikami have developed an efficient method for hydrosilylation of ortho-substituted benzophenones by use of chiral [Zn(diamine)(diol)] complexes, with chirality residing in the diamine part of the complex (Scheme 2) [38]. Prochiral benzophenones can be enantioselectively reduced with up to 96 % ee of the product. The effect of an activator on the stereochemistry of the product was negligible, because enantioselectivity was comparable irrespective of whether (R) or (S)-BINOL, or ethylene glycol was used.
Scheme 2.
Recently Bette et al. [39, 40] reported Zn-promoted direct reduction of a variety of ketones to the corresponding alcohols with PMHS in protic solvents. Although the chemical yields of the reactions were quantitative, moderate enantioselectivity (ee up to 55 %) was obtained by use of a variety of enantiopure diamine ligands. Another possible means of improving the efficiency of the catalyst system was by changing the reaction conditions from an aprotic to a protic solvent—a rather uncommon situation in organometallic chemistry.
The main objective of this work was to test several acyclic or cyclic ligands derived from enantiomerically pure trans-1,2-diaminocyclohexane, which are secondary or tertiary diamines, imines, or heteraphanes with additional substituents with potential coordinating character (Fig. 1).
Fig. 1.
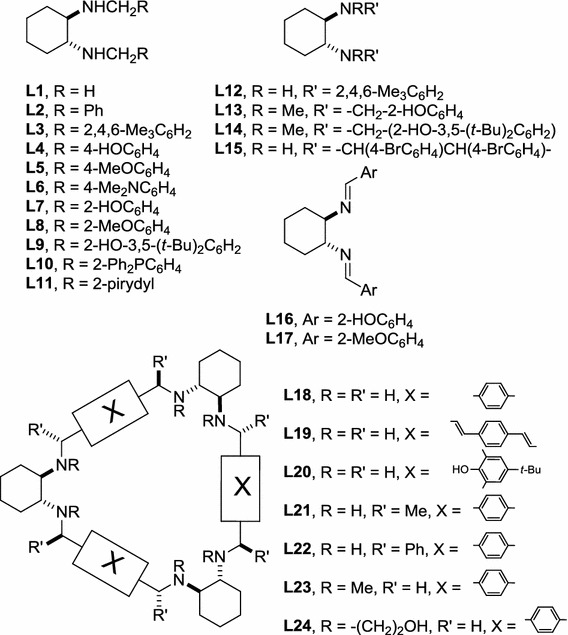
Acyclic and cyclic ligands used in this study
According to Mimoun’s model and our recent results, coordination of the ketone substrate to the ZnR2–amine complex results in release of an alkane (RH) molecule with the formation of Zn···O coordinated species in which one of the nitrogen atoms forms a bond to the carbon atom of the carbonyl group [7, 30]. Complexation of the ketone molecule with C–N bond formation is accompanied by significant changes of the conformation of the ligand around the coordination site. Alternatively, hydrosilylation may take place by direct silane attack on the activated carbonyl group without significant change of structure of the complex [30]. Ligands used in this study may act according to the first or second model.
This study was inspired by the work of Ushio and Mikami [38] and Costa et al. [37], who reported that enantiopure [Zn(diamine)] species could be activated by either enantiopure BINOL or achiral 1,2 or 1,3-diol ligands to form efficient catalysts for asymmetric hydrosilylation of ketones. We were particularly interested in the application of achiral or chiral diols as activators, because the number of available diols is higher than the number of chiral diamines and because the diamines are usually synthesized from the corresponding diols.
We tested several diols (Fig. 2), either chiral or achiral, in combination with appropriate diamines. For chiral diols both enantiomers and racemic mixtures were used, if available.
Fig. 2.
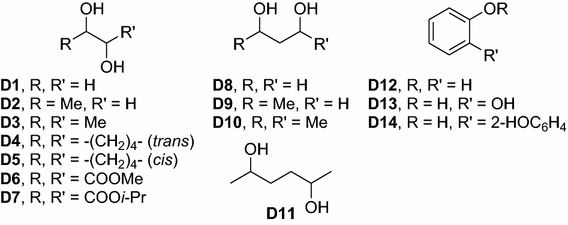
Diols and alcohols used as activators in [Zn(diamine)(diol)]-catalyzed hydrosilylations
The effects of temperature and the structure of the silane were also investigated. As test substrate for the [Zn(diamine)(diol)]-catalyzed reactions we selected 4-methylacetophenone; the general reaction conditions were as shown in Scheme 1, where R = 4-MeC6H4 and R′ = Me.
Results and discussion
Effect of the structure of the amine ligand on the efficiency of the catalytic system
We initially examined the effect of ligand structure on the yield and enantioselectivity of Zn-catalyzed hydrosilylation of 4-methylacetophenone (Fig. 3). The catalyst load in all cases was 3.5 mol %. Diphenylsilane was used as the reducing agent and all reactions were carried out in dry and degassed toluene for 24 h at room temperature.
Fig. 3.

Effect of ligand structure on conversion and enantioselectivity of hydrosilylation of 4-methylacetophenone. Reaction conditions: 3.5 mol % catalyst, 1.2 equiv. silane, reaction time 24 h, toluene, RT; conversions and enantiomeric excesses were determined by HPLC with a Chiralpak IA column, average of two runs
Among 24 ligands tested nine (L4, L5, L9, L12–L14, L16, L22, and L23) did not promote hydrosilylation under these conditions (Fig. 3, entries 4, 5, 9, 12–14, 16, and 22–23), resulting in recovery of the starting material. These non-effective ligands contain either polar or bulky groups in different positions on the benzene ring (L4, L5, L9, L16) or have an additional substituent on the nitrogen atom (L13, L14, L23).
Ligands having additional hydroxy (L4) or methoxy groups (L5) in the para position of the benzene ring were inactive in the hydrosilylation reaction, in contrast with ligand L6, with dimethylamino groups in the para positions (Fig. 3, entry 6). It is interesting that the conformationally labile ligands L7 and L20 with hydroxy groups in the ortho positions which do not form planar complexes with zinc, are, nevertheless, active hydrosilylation catalysts, with conversions of the substrate almost quantitative, although with low enantiomeric excess of the product (Fig. 3, entries 7 and 20).
Catalytically inactive acyclic (L13 and L14) and macrocyclic (L23) ligands are distinguished by additional methyl substituents on the nitrogen atoms. This indicates that the presence of an N–H bond in the ligand structure is essential if the reaction is to proceed. Macrocyclic ligand L24, characterized by the presence of hydroxyethyl substituents on the nitrogen atoms, is an exception. Although it activates the catalyst, the enantioselectivity obtained by use of L24 was low and the best result was obtained when six equivalents of ZnEt2 per one equivalent of the ligand were used (ee 15 %, conversion of the substrate 99 %).
Although salen L16 is catalytically inactive, apparently because of the formation of dimeric species [41] its methoxy derivative L17 promotes the hydrosilylation reaction (Fig. 3, entry 17).
The presence of an N–H hydrogen atom is a prerequisite for catalytic activity; it is, however, not sufficient for catalysis of the hydrosilylation reaction. In secondary diamine L12, an aniline derivative, delocalization of the nitrogen lone pair to the aromatic ring reduces its coordination ability. More importantly, the specific, almost parallel conformation of the aromatic rings in L12 does not allow the zinc atom to be included in the coordination space of the nitrogen atom [42]. This makes the ligand ineffective for catalysis.
In L22 the phenyl groups on the benzylic carbon atoms occupy positions perpendicular to the average plane of the macrocycle [43, 44] and thus strongly stabilize the triangular conformation of L22. Steric crowding and lack of flexibility of the ligand apparently prohibit substrate binding. In contrast with L22, less sterically crowded L21 catalyses the hydrosilylation reaction efficiently (Fig. 3, entry 21).
The enantiomeric excess of the product obtained by use of L15 is the second best for all ligands tested; however, conversion of the substrate does not exceed 5 % (Fig. 3, entry 15). Therefore L15 is not efficient as a ligand.
These results clearly show that both overcrowding and too much space around the coordination site make the efficiency of the catalytic system problematic. Ligands L1 and L2 differ in substitution of nitrogen atoms. Although both ligands are catalytically active in terms of conversion of the substrate (over 99 and 98 %, respectively for L1 and L2; Fig. 3, entries 1 and 2), the enantioselectivity of the reaction with use of N-benzyl derivative L2 is ca twice as high (76 %) as that obtained by use of the simple N-methyl DACH derivative L1 (37 %) and may be further increased if the diamine is part of a regular trianglamine L18 structure. In this case conversion of the substrate is quantitative and the macrocyclic structure of the ligand results in an increase in the enantioselectivity (82 %) of the process (Fig. 3, entry 18).
Efficiency of the catalyst may be further tuned by introduction of additional coordinating groups, although with varying results. Replacement of the phenyl group by a 2-pyridyl group (L11) results in an increase of conversion at the expense of enantioselectivity (52 %), in comparison with L2 (Fig. 3, entry 11). Introduction of a diphenylphosphine group in the ligand L10 structure results in an increase of enantioselectivity (to 72 %; Fig. 3, entry 10), but enantioselectivity is still lower than with use of L2. The best result in terms of product enantioselectivity (86 %) and conversion of the substrate (>99 %) was obtained by use of methoxy-substituted ligand L8 (Fig. 3, entry 8). Direct (Me)O···Zn interactions are excluded in the complex of L8 and dialkylzinc. According to computation at the PBE0/6-311++G(d,p) level [45] the stable conformer L8 ZnMe2 (Fig. 4b), in which a direct O···Zn interaction is possible, is of higher energy (19.7 kJ mol−1) than the conformer with an intramolecular NH···O(Me) hydrogen bond (Fig. 4).
Fig. 4.

Structures of stable conformers L8 ZnMe2 (a) and L8 ZnMe2 (b) calculated at the PBE0/6-311++G(d,p) level of theory (some hydrogen atoms were omitted for clarity; distances are in Å)
Screening of the ligands was conducted in conjunction with screening of the type of silane used as the reducing agent. Selected results are shown in Table 1.
Table 1.
Effect of silane structure on conversion and enantioselectivity of the hydrosilylation of 4-methylacetophenone
| Entry | Silane | Ligand | Conversion/% | ee/% |
|---|---|---|---|---|
| 1 | PMHS | L2 | >99 | 55 |
| 2 | (EtO)3SiH | L8 | >99 | 85 |
| 3 | Ph2SiH2 | L8 | >99 | 86 |
| 4 | PMHS | L11 | >99 | 41 |
| 5 | PMHS | L18 | 99 | 81 |
| 6 | (EtO)3SiH | L18 | >99 | 78 |
| 7 | Et3SiH | L18 | Traces | 7 |
| 8 | PhSiH3 | L18 | >99 | 78 |
| 9 | Me2PhSiH | L18 | 0 | 0 |
3.5 mol % catalyst, 1.2 equiv. silane, reaction time 24 h, toluene, RT; conversion and enantiomeric excess were determined by HPLC with a Chiralpak IA column (average of two runs)
In the test reaction none of the silanes tested (PMHS, Et3SiH, (EtO)3SiH, Me2PhSiH, and PhSiH3) performed better than diphenylsilane in combination with L8. The decrease of enantioselectivity was small when (EtO)3SiH was combined with L8 (drop to 85 %; Table 1, entry 2) or L18 (drop to 81 %; Table 1, entry 4), compared with the combination of Ph2SiH2 and L8. In other cases, however, it was significant. Among silanes tested, two (Et3SiH, Me2PhSiH) did not promote hydrosilylation under the above mentioned conditions. In all cases discussed in this paragraph the product of hydrosilylation had S absolute configuration.
Hydrosilylation of aryl–alkyl or aryl–aryl ketones catalyzed by [Zn(L8)] or [Zn(L18)] complexes
Having established L8 and L18 as the best performing chiral ligands, a variety of alkyl–aryl and aryl–aryl ketones were reduced by using 3.5 mol % catalyst, 1.2 equiv. silane, and toluene as solvent. The most interesting results are summarized in Table 2.
Table 2.
[Zn(Diamine)]-catalyzed hydrosilylation of prochiral ketones
| Entry | Substrate | Silane | Ligand | ee/% [yield/%]a |
|---|---|---|---|---|
| 1 | C6H5COC2H5 | Ph2SiH2 | L8 | 74 (S)b |
| 2 | C6H5COC2H5 | Ph2SiH2 | L18 | 85 (S) |
| 3 | C6H5COC2H5 | PMHS | L18 | 86 [71] (S) |
| 4 | C6H5COC6H11 | Ph2SiH2 | L8 | 74 (S) |
| 5 | C6H5COC6H11 | Ph2SiH2 | L18 | 83 (S) |
| 6 | 4-MeO–C6H4COCH3 | Ph2SiH2 | L8 | 79 [44] (S) |
| 7 | 4-MeO–C6H4COCH3 | Ph2SiH2 | L18 | 43 [60] (S) |
| 8 | 4-F–C6H4COCH3 | PMHS | L18 | 78 (S) |
| 9 | 4-F–C6H4COCH3 | PhSiH3 | L18 | 81 (S) |
| 10 | 4-F–C6H4COCH3 | Ph2SiH2 | L18 | 82 [63] (S) |
| 11 | 4-NC–C6H4COCH3 | Ph2SiH2 | L8 | 80 [26] (S) |
| 12 | 4-NC–C6H4COCH3 | PMHS | L18 | 71 (S) |
| 13 | 4-NC–C6H4COCH3 | PhSiH3 | L18 | 69 (S) |
| 14 | Indanone | Ph2SiH2 | L8 | 32 (S) |
| 15 | Indanone | Ph2SiH2 | L18 | 69 [75] (S) |
| 16 | 1-Tetralone | Ph2SiH2 | L8 | 84 [91] (S) |
| 17 | 1-Tetralone | Ph2SiH2 | L18 | 71 [97] (S) |
| 18 | 2-Tetralone | Ph2SiH2 | L8 | 39 [75] (S) |
| 19 | 2-Tetralone | Ph2SiH2 | L18 | 12 [62] (S) |
| 20 | C6H5COCF3 | Ph2SiH2 | L8 | 15 [43] (R) |
| 21 | C6H5COCF3 | Ph2SiH2 | L18 | 19 [54] (R) |
| 22 | C6H5COCF3 | PMHS | L18 | 11 [50] (R) |
| 23 | 2,4,6-Me3C6H2COCF3 | Ph2SiH2 | L8 | 6 (R) |
| 24 | 2,4,6-Me3C6H2COCF3 | PMHS | L18 | 19 [52] (R) |
| 25 | 4-F–C6H4COCF3 | Ph2SiH2 | L8 | 6 (R) |
| 26 | 4-F–C6H4COCF3 | PMHS | L18 | 9 [34] (R) |
| 27 | 3,5-(CF3)2–C6H3COCH3 | Ph2SiH2 | L8 | 68 [76] (S) |
| 28 | 3,5-(CF3)2–C6H3COCH3 | PMHS | L18 | 16 [64] (S) |
| 29 | 3-Me–C6H4COC6H5 | Ph2SiH2 | L8 | 13 [81] (S) |
| 30 | 3-Me–C6H4COC6H5 | Ph2SiH2 | L18 | 1 (S) |
| 31 | 4-Me–C6H4COC6H5 | Ph2SiH2 | L8 | 7 (S) |
| 32 | 4-Me–C6H4COC6H5 | Ph2SiH2 | L18 | 11 [92] (S) |
| 33 | 2-Cl–C6H4COC6H5 | Ph2SiH2 | L8 | 62 (R) |
| 34 | 2-Cl–C6H4COC6H5 | Ph2SiH2 | L18 | 72 [84] (R) |
| 35 | 4-Cl–C6H4COC6H5 | Ph2SiH2 | L8 | 26 (R) |
| 36 | 4-Cl–C6H4COC6H5 | Ph2SiH2 | L18 | 26 [47] (R) |
3.5 mol % catalyst, 1.2 equiv. silane, reaction time 24 h, toluene, RT; conversion and enantiomeric excess were determined by HPLC with a Chiralpak IA column (average of two runs)
aIsolated yield
bAbsolute configuration
Of all substrates ethyl phenyl ketone gave the best results in terms of enantioselectivity and yield of the reaction (ee up to 86 %; Table 2, entries 2 and 3). The presence of a bulky alkyl group in cyclohexyl phenyl ketone resulted in a small decrease of enantioselectivity (83 %; Table 2, entry 5). For both compounds, use of macrocycle L18 provided better results than the use of acyclic ligand L8.
Enantiomeric purity varied significantly when cyclic ketones, 1-tetralone, 2-tetralone, and 1-indanone, were used as the substrates. The best result was obtained in the reduction of 1-tetralone by diphenylsilane in the presence of ligand L8 (ee 84 %, yield 91 %; Table 2, entry 16). Better yield (97 %) at the expense of enantioselectivity (drop to 71 %) was achieved when zinc complex with L18 was used as the catalyst for reduction of 1-tetralone (Table 2, entry 17). The opposite trend was observed on use of 1-indanone—higher enantiomeric excess (69 %) was obtained by use of L8 (Table 2, entry 15). 2-Tetralone was an exception—because of the low stereodifferentation of both faces of the substrate molecule, enantioselectivity of the reaction did not exceed 40 % when L8 was used as ligand (Table 2, entry 18).
4-Methoxyacetophenone, 4-cyanoacetophenone, and 4-fluoroacetophenone gave the products of hydrosilylation with good ee (79–82 %; Table 2, entries 6–7 and 10–13).
Pharmaceutically important aryl trifluoromethyl ketones were hydrosilylated with low enantioselectivity (ee 19 %; Table 2, entries 21 and 24, and 9 %; Table 2, entry 26). The lack of success was in agreement with previously reported data [30]. On the other hand, trifluoromethyl groups attached to the aryl ring affected reaction enantioselectivity to a small extent only. When 3,5-bis(trifluoromethyl)phenyl methyl ketone was used the enantioselectivity of the reaction (up to 68 %) was over four times more than when phenyl trifluoromethyl ketone was used, and the product was obtained with good chemical yield (76 %; Table 2, entry 27) when L8 was used as a ligand.
Hydrosilylation of substituted benzophenones gave mixed results (Table 2, entries 29–36). The best result (ee 72 %, yield 84 %) was obtained when 2-chlorobenzophenone was reduced chemoselectively by diphenylsilane with use of L18 (Table 2, entry 34).
Asymmetric activation of the [Zn(diamine)] catalyst
Use of an activator in the [Zn(diamine)]-catalyzed hydrosilylation reactions has the potential to increase both enantioselectivity and the reaction rate. In our study we tested several diol–amine combinations including acyclic ligands L1, L2, macrocycle L18, and diol activators D1–D14 (Figs. 1, 2). Note that L2 was used in either the racemic or enantiomerically pure form.
Standard conditions involved generation of the zinc complex by addition of an equimolar amount of diethylzinc in hexane to a solution of the ligand in toluene (or any other solvent) followed by addition of an equimolar amount of the appropriate diol. After 0.5 h the ketone and the silane were added and reaction was performed at room temperature for 24 h and then quenched with methanolic NaOH solution (Scheme 2). Changing of the order of addition of ZnEt2, diamine, activator, silane, and substrate did not affect enantioselectivity or conversion rate.
Although the nature of the silane had only a negligible effect on the conversion rate and enantioselectivity of the reaction, slightly better results were obtained when diphenylsilane was used as the reducing agent.
The effect of activator structure and the reaction conditions on the efficiency of the catalytic system was studied in the test reaction of hydrosilylation of 4-methylacetophenone. The results are summarized in Table 3.
Table 3.
Effect of ligand structure and solvent on the efficiency of [Zn(diamine)(diol)]-catalyzed hydrosilylation of 4-methylacetophenone
| Entry | Solvent | Ligand | Activator | Conversion/% | ee/% |
|---|---|---|---|---|---|
| 1 | Toluene | L1 | D1 | 100 | 41 |
| 2 | Toluene | L2 | D1 | 98 | 75 |
| 3 | Toluene | L2 | D1 a | >99 | 78 |
| 4 | Toluene | L18 | D1 | 99 | 82 |
| 5 | Toluene | L1 | (rac)-D9 | 100 | 40 |
| 6 | Toluene | L18 | (rac)-D9 | 96 | 83 |
| 7 | Toluene | L1 | (R)-D9 | 100 | 41 |
| 8 | Toluene | L18 | (R)-D9 | 97 | 73 |
| 9 | Toluene | L18 | (R)-D9 a | 92 | 73 |
| 10 | Toluene | L1 | (S)-D9 | 100 | 38 |
| 11 | Toluene | L18 | (S)-D9 | >99 | 81 |
| 12 | THF | L18 | D1 | >99 | 81 |
| 13 | CH2Cl2 | L18 | D1 | >99 | 69 |
| 14 | Toluene | L18 | MeOHb | 98 | 79 |
| 15 | Toluene | L18 | MeOHa | >99 | 84 |
| 16 | MeOH | L18 | None | 97 | 61 |
| 17 | Toluene | (rac)-L2 | (R)-D2 | >99 | 4 |
| 18 | Toluene | L2 | (rac)-D2 | 99 | 77 |
| 19 | Toluene | None | D6 | 0 | 0 |
| 20 | Toluene | None | D6 a | 0 | 0 |
| 21 | Toluene | (rac)-L2 | D6 | >99 | 5 |
| 22 | Toluene | L2 | D6 | >99 | 72 |
| 23 | Toluene | None | D7 | 0 | 0 |
| 24 | Toluene | (rac)-L2 | D7 | >99 | 6 |
3.5 mol % catalyst, 1.2 equiv. diphenylsilane, reaction time 24 h, toluene, RT; conversion and enantiomeric excess were determined by HPLC with a Chiralpak IA column (average of two runs)
a2 equiv. activator were used
b1 equiv. MeOH was used
The effect of amine and diol structure on the enantioselectivity of [Zn(diamine)(diol)]-catalyzed hydrosilylation was investigated for two arbitrarily chosen activators D1 and D9. Enantioselectivity obtained for [Zn(diamine)D1]-catalyzed hydrosilylations ranged from 41 (L1) to 82 % (L18) and increased slightly (to 83 %) if 2 equiv. D1 were used for 1 equiv. L18 (Table 3, entries 1–4). The presence of the stereogenic center in D9 did not significantly affect the stereoselectivity of the reaction. Comparison of the results obtained for Zn(L1) and Zn(L18) complexes activated by either (rac), (R), or (S)-D9 shows the advantage of cyclic over acyclic structure of the amine ligand and in each series similar results were obtained when either (rac)-D9 or enantiomerically pure D9 was used as the activator (Table 3, entries 5–11). For [Zn(L18)(D9)]-catalyzed hydrosilylations of 4-methylacetophenone, for example, product enantioselectivity was: 83, 73, and 81 % for (rac), (R), and (S)-D9, respectively. The enantioselectivity of the non-activated [Zn(L18)]-catalyzed reaction was 82 %.
Changing the solvent from toluene to THF or dichloromethane slightly improved conversion in the [Zn(L18)(D1)]-catalyzed hydrosilylation of 4-methylacetophenone, from 98 to 100 %, at the expense of enantioselectivity (drop to 81 or 69 % ee of the product; Table 3, entries 12 and 13). Substituting the diol in the catalyst structure with 2 or 1 equiv. methanol resulted in a 1 and 6 % (respectively for 2 or 1 equiv. MeOH) drop in enantioselectivity of the hydrosilylation reaction performed in toluene (Table 3, entries 14 and 15). The same reaction could be performed in pure methanol (Table 3, entry 16). Although conversion of the substrate was still high, enantiomeric purity of the product decreased substantially (drop to 61 %). In contrast with the reduction of ketones, a similar reaction performed for imines gave better results in protic solutions than in any non-polar or aprotic media [31].
The stereochemical course of the reaction is controlled by the chirality of the diamine part of catalytically active species. Although use of (rac)-L2 with enantiomerically pure diols D2, D6, and D7 preserved high conversion of the substrate, we observed a substantial decrease of enantioselectivity (Table 3, entries 17, 21, and 24). Use of enantiomerically pure L2 with (rac)-D2 as the catalytic system provided the product with 77 % ee (Table 3, entry 18). It should be noted that when used alone, the diol or [Zn(diol)] were not catalytically active species (Table 3, entries 19, 20, and 23).
Full screening of the activators was conducted for [Zn(L18)]-catalyzed hydrosilylation of 4-methylacetophenone. All reaction were carried out in toluene on a 1 mmol scale, using 3.5 mol % L18, equimolar amounts of ZnEt2 and the activator, and 1.2 mmol silane. Conversion and enantiomeric excess were determined by HPLC with a Chiralpak IA column. The results are summarized in Fig. 5.
Fig. 5.

Effect of activator structure on enantioselectivity in [Zn(L18)]-catalyzed hydrosilylation of 4-methylacetophenone. First row represents achiral or racemic activators, second and third rows summarize results obtained for activators of R or S absolute configuration at the stereogenic centers, respectively
The results show that all combinations of L18 and the activator were catalytically active. As was mentioned above, conversion of the substrate was high (over 92 %) and enantiomeric excess of the product was usually ca. 80 %.
More detailed inspection of the results summarized in Fig. 5 shows that the highest enantioselectivity (ee 85 %) in the hydrosilylation of 4-methylacetophenone was achieved with trianglamine ligand L18 and (S)-D2 or (S,S)-D4 as activator, with diphenylsilane as reducing agent (Fig. 5, entries 2 and 4). Replacing (S)-D2 or (S,S)-D4 by the simple diol D1 resulted in a 3 % drop of enantioselectivity, with unchanged conversion (Table 3, entry 4). Replacing the aliphatic alcohol by phenol as activator had no profound effect on either enantioselectivity or conversion of the substrate. The product of hydrosilylation had S absolute configuration irrespective of the amine–alcohol combination used.
Summary
In this work we tested a wide range of DACH-based amine ligands, activators, and reactants for asymmetric hydrosilylation of prochiral ketones. The L8-ZnEt2 and L18-ZnEt2 (1:1) complexes catalyze the hydrosilylation of 4-methylacetophenone by diphenylsilane in toluene solution, with up to 86 % ee of the product. The enantioselectivity is higher than that with use of other acyclic or cyclic ligands. Because both L8 and L18 can be conveniently prepared by a one-pot procedure from the inexpensive tartrate salt of DACH and o-anisole or terephthalaldehyde, the Zn complexes of L8 and L18 are good alternatives to previously reported chiral catalysts.
A small effect of activator structure on enantioselectivity and significant large increases in the conversion and yield of the reaction seem consistent with the mechanism postulated by Bette et al. [39] and by us, supported by computational experiments [46]. According to this proposal the diol moiety does not sterically affect the substrate in the transition state; it does, however, activate the zinc ion acting as catalyst [39, 40].
Experimental
NMR spectra were recorded on Bruker BioSpin 400 (400 MHz) or Varian MR 300 (300 MHz) instruments at 25 °C using CDCl3 as solvent. Chemical shifts are reported in ppm relative to the TMS peak (1H and 13C NMR spectra). Spectral assignments were obtained by analysis of chemical shifts and by comparison with literature data. Mass spectra were recorded on an AMD-402 spectrometer. HPLC analysis was performed at room temperature with an Hitachi LaChrom Elite system equipped with a Chiralpak IA column. Details of HPLC separation of the enantiomers are given in the Supplementary Information (Table SI1). Absolute configurations of the products were determined by comparison of measured optical rotation with the literature data [47–62].
Ligands L1–L3, L5, L7–L18, L21–L24 were prepared by procedures reported in the literature [24–30, 43, 63–71]. Ligands L4, L4, L19, and L20 were obtained from the corresponding imines [28, 65, 72], by use of a procedure reported elsewhere [24]. NMR spectra and melting points for all ligands and products of hydrosilylation reactions were in agreement with literature data (systematic names of L1–L3, L5, L7–L18, L21–L24 and some physical data are given in the Supplementary Information, Table SI2) [24–30, 41, 43, 47–71]. Diols D1–D14 were commercial products.
(R,R)-N,N′-Bis[(4-hydroxyphenyl)methyl]-1,2-cyclohexanediamine (L4, C20H26N2O2)
Yield 86 %; white solid; m.p.: 141–144 °C (ethyl acetate–hexane); 1H NMR (400 MHz, CDCl3): δ = 7.11–7.08 (m, 4H), 6.79–6.72 (m, 4H), 3.79 (d, J = 12.7 Hz, 2H), 3.51 (d, J = 12.8 Hz, 2H), 3.27 (s, 2H), 2.26–2.24 (m, 2H), 2.17–2.13 (m, 2H), 2.06 (s, 2H), 1.75–1.73 (m, 2H), 1.42–1.35 (m, 4H) ppm; 13C NMR (101 MHz, CD3OD): δ = 158.3, 131.1, 129.7, 116.5, 60.5, 50.0, 30.7, 25.8 ppm; IR (KBr):  = 3,368, 3,295, 3,234, 2,940, 1,610, 1,598, 1,516, 1,247, 996, 829 cm−1; HR-EI-MS (m/z): [M]+ calcd for C20H26N2O2 326.4326, found 326.4350.
= 3,368, 3,295, 3,234, 2,940, 1,610, 1,598, 1,516, 1,247, 996, 829 cm−1; HR-EI-MS (m/z): [M]+ calcd for C20H26N2O2 326.4326, found 326.4350.
(R,R)-N,N’-Bis[(4-dimethylaminophenyl)methyl]-1,2-cyclohexanediamine (L6, C24H36N4)
Yield 83 %; white solid; m.p.: 140–146 °C (ethyl acetate–hexane); 1H NMR (300 MHz, CDCl3): δ = 7.12–6.97 (m, 4H), 6.65–6.50 (m, 4H), 3.90–3.86 (m, 2H), 3.85–3.81 (m, 2H), 2.95–2.91 (s, 12H), 2.43–2.39 (m, 2H), 1.92–1.85 (m, 2H), 1.84–1.80 (m, 2H), 1.69–1.65 (m, 2H), 1.38–1.34 (m, 2H), 1.33–1.29 (m, 2H) ppm; 13C NMR (75 MHz, CDCl3): δ = 160.7, 151.7, 129.2, 124.9, 111.5, 73.9, 40.2, 33.4, 24.7 ppm; IR (KBr): 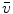 = 3,430, 2,920, 2,852, 2,826, 1,633, 1,610, 1,528, 1,370, 1,181, 807 cm−1; HR-EI-MS (m/z): [M]+ calcd for C24H36N4 380.5694, found 380.5701.
= 3,430, 2,920, 2,852, 2,826, 1,633, 1,610, 1,528, 1,370, 1,181, 807 cm−1; HR-EI-MS (m/z): [M]+ calcd for C24H36N4 380.5694, found 380.5701.
(2R,3R,16R,17R,30R,31R)-1,4,15,18,29,32-Hexaaza-(2,3:16,17:30,31)-tributano-(8,11:22,25:36,39)-trietheno-1,2,3,4,5,14,15,16,17,18,19,28,29,30,31,32,33,42-octadecahydro[42]annulene (L19, C54H72N6)
Yield 84 %; white solid: m.p.: >320 °C (dichloromethane–hexane); 1H NMR (300 MHz, CDCl3): δ = 7.25 (s, 6H), 6.53 (d, J = 15.2 Hz, 6H), 6.22–6.16 (m, 6H), 3.29–3.25 (m, 6H), 3.19–3.15 (m, 6H), 2.55–2.49 (m, 6H), 1.72 (s, 6H), 1.69–1.61 (m, 6H), 1.52–1.45 (m, 6H), 1.44–1.34 (m, 12H) ppm; 13C NMR (126 MHz, CDCl3): δ = 136.3, 130.5, 128.8, 126.4, 126.1, 61.1, 49.1, 49.0, 31.4, 25.0 ppm; IR (KBr): 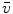 = 3,424, 3,296, 2,928, 2,854, 2,370, 1,757, 1,450, 1,238, 1,120, 1,050, 964, 750 cm−1; HR-EI-MS (m/z): [M]+ calcd for C54H72N6 805.1897, found 805.1905.
= 3,424, 3,296, 2,928, 2,854, 2,370, 1,757, 1,450, 1,238, 1,120, 1,050, 964, 750 cm−1; HR-EI-MS (m/z): [M]+ calcd for C54H72N6 805.1897, found 805.1905.
(2R,3R,11R,12R,20R,21R)-1,4,10,13,19,22-Hexaaza-(2,3:11,12:20,21)-tris(tetramethylene)-7,16,25-trihydroxy-(6,8:15,17:24,26)-tris(2-tert-butyl-1-propen-3-yl)-1,2,3,4,5,9,10,11,12,13,14,18,19,20,21,22,23,27-octadecahydro[27]annulene (L20, C54H84N6O3)
Yield 93 %; glass; 1H NMR (400 MHz, CDCl3): δ = 7.09 (s, 6H), 5.05 (s, 3H), 3.81 (m, 12H), 2.53–2.51 (m, 6H), 1.96 (s, 6H), 1.69–1.61 (m, 6H), 1.53–1.45 (m, 6H), 1.44–1.34 (m, 39H) ppm; 13C NMR (75 MHz, CDCl3): δ = 154.5, 154.0, 140.8, 125.0, 123.9, 60.3, 48.8, 48.2, 33.8, 31.6, 31.4, 30.8, 24.8 ppm; IR (KBr): 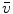 = 3,269, 2,950, 2,858, 1,609, 1,484, 1,362, 1,215 cm−1; HR-EI-MS (m/z): [M]+ calcd for C54H84N6O3 865.6605, found 865.6596.
= 3,269, 2,950, 2,858, 1,609, 1,484, 1,362, 1,215 cm−1; HR-EI-MS (m/z): [M]+ calcd for C54H84N6O3 865.6605, found 865.6596.
General procedure for Zn(diamine)]-catalyzed hydrosilylation of ketones
In a 5-cm3 round-bottomed flask 12.5 mm3 ZnEt2 (1 M in hexane, 0.0125 mmol) and the appropriate chiral ligand (0.0125 mmol) were dissolved in 1.5 cm3 freshly distilled toluene and stirred under an argon atmosphere for 30 min. The corresponding ketone (0.344 mmol), or its solution in 1 cm3 toluene, and the silane (0.413 mmol) were then added to the mixture. The resulting solution was stirred at room temperature for 24 h. NaOH (1 M in MeOH; 1 cm3) was then added with vigorous stirring. The reaction mixture was stirred for an additional hour at room temperature and then the solvents were evaporated. The residue was dissolved in a mixture of 10 cm3 H2O and 1 cm3 10 % HCl and extracted with diethyl ether (3 × 10 cm3). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, H2O, and brine, dried over anhydrous MgSO4, and concentrated under vacuum. The product was purified by column chromatography on silica gel with hexane–EtOAc (10:1) as eluent.
General procedure for Zn(diamine)(diol)]-catalyzed hydrosilylation of 4-methylacetophenone
In a 5-cm3 round-bottomed flask 12.5 mm3 ZnEt2 (1 M in hexane, 0.0125 mmol), the appropriate chiral ligand (0.0125 mmol), and the diol (0.0125 mmol) were dissolved in 1.5 cm3 freshly distilled toluene and stirred under an argon atmosphere for 30 min. 4-Methylacetophenone (0.344 mmol) and silane (0.413 mmol) were then added to the mixture. The resulting solution was stirred at room temperature for 24 h, then 1 cm3 NaOH (1 M in MeOH) was added with vigorous stirring. The reaction mixture was stirred for an additional hour at room temperature and the solvents were then evaporated. The residue was dissolved in a mixture of 10 cm3 H2O and 1 cm3 10 % HCl and extracted with diethyl ether (3 × 10 cm3). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, H2O, and brine, dried over anhydrous MgSO4, and concentrated under vacuum. The product was purified by column chromatography on silica gel with hexane–EtOAc (10:1) as eluent.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We are grateful to the Ministry of Science and Higher Education (Poland) for financial support (grant no. PBZ-KBN-NN204 555 939).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
References
- 1.Blaser HU, Spindler F, Studer M. Appl Catal A Gen. 2001;221:119. doi: 10.1016/S0926-860X(01)00801-8. [DOI] [Google Scholar]
- 2.Ojima I. Catalytic Asymmetric Synthesis. New York: Wiley-VCH; 2000. [Google Scholar]
- 3.Bartók M. Chem Rev. 2010;110:1663. doi: 10.1021/cr9002352. [DOI] [PubMed] [Google Scholar]
- 4.Mallat T, Orglmeister E, Baiker A. Chem Rev. 2007;107:4863. doi: 10.1021/cr0683663. [DOI] [PubMed] [Google Scholar]
- 5.Bartók M. Curr Org Chem. 2006;10:1533. doi: 10.2174/138527206778249612. [DOI] [Google Scholar]
- 6.Osawa T, Harada T, Takayasu O. Curr Org Chem. 2006;10:1513. doi: 10.2174/138527206778249676. [DOI] [Google Scholar]
- 7.Mimoun H, de Saint Laumer JY, Giannini L, Scopelliti R, Floriani C (1999) J Am Chem Soc 121:6158
- 8.Marciniec B. Hydrosilylation: a comprehensive review on recent advances. Berlin: Springer; 2009. [Google Scholar]
- 9.Nishiyama H. Hydrosilylation of carbonyl and imino groups. In: Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive asymmetric catalysis. Berlin: Springer; 1999. p. 267. [Google Scholar]
- 10.Diez-González S, Nolan SP. Org Prep Proced Int. 2007;39:523. doi: 10.1080/00304940709458641. [DOI] [Google Scholar]
- 11.Carpentier J-F, Bette V. Curr Org Chem. 2002;6:913. doi: 10.2174/1385272023373851. [DOI] [Google Scholar]
- 12.Riant O, Mostefaï N, Courmarcel J (2004) Synthesis 2943
- 13.Hayashi T. Acc Chem Res. 2000;33:354. doi: 10.1021/ar990080f. [DOI] [PubMed] [Google Scholar]
- 14.Roy AK. Adv Organomet Chem. 2007;55:1. doi: 10.1016/S0065-3055(07)55001-X. [DOI] [Google Scholar]
- 15.Diez-González S, Nolan SP. Acc Chem Res. 2008;41:349. doi: 10.1021/ar7001655. [DOI] [PubMed] [Google Scholar]
- 16.Brunner H, Becker R, Riepl G. Organometallics. 1984;3:1354. doi: 10.1021/om00087a006. [DOI] [Google Scholar]
- 17.Nishiyama H, Yamaguchi S, Kondo M, Itoh K. J Org Chem. 1992;57:4306. doi: 10.1021/jo00041a049. [DOI] [Google Scholar]
- 18.Evans DA, Michael FE, Tedrow JS, Campos KR. J Am Chem Soc. 2003;125:3534. doi: 10.1021/ja012639o. [DOI] [PubMed] [Google Scholar]
- 19.Gade LH, César V, Bellemin-Laponnaz S. Angew Chem Int Ed. 2004;43:1014. doi: 10.1002/anie.200353133. [DOI] [PubMed] [Google Scholar]
- 20.Song C, Ma C, Ma Y, Feng W, Ma S, Chai Q, Andrus MB. Tetrahedron Lett. 2005;46:3241. doi: 10.1016/j.tetlet.2005.03.026. [DOI] [Google Scholar]
- 21.Shaikh NS, Enthaler S, Junge K, Beller M. Angew Chem Int Ed. 2008;47:2497. doi: 10.1002/anie.200705624. [DOI] [PubMed] [Google Scholar]
- 22.Hosokawa S, Ito J, Nishiyama H. Organometallics. 2010;29:5773. doi: 10.1021/om1009186. [DOI] [Google Scholar]
- 23.Yun J, Buchwald SL. J Am Chem Soc. 1999;121:5640. doi: 10.1021/ja990450v. [DOI] [Google Scholar]
- 24.Gawroński J, Kołbon H, Kwit M, Katrusiak A. J Org Chem. 2000;65:5768. doi: 10.1021/jo000623v. [DOI] [PubMed] [Google Scholar]
- 25.Gawroński J, Gawrońska K, Grajewski J, Kwit M, Plutecka A, Rychlewska U. Chem Eur J. 2006;12:1807. doi: 10.1002/chem.200500887. [DOI] [PubMed] [Google Scholar]
- 26.Kuhnert N, Straßnig C, Lopez-Periago AM. Tetrahedron Asymmetry. 2002;13:123. doi: 10.1016/S0957-4166(02)00065-4. [DOI] [Google Scholar]
- 27.Gawroński J, Kwit M, Grajewski J, Gajewy J, Długokińska A. Tetrahedron Asymmetry. 2007;18:2632. doi: 10.1016/j.tetasy.2007.10.033. [DOI] [Google Scholar]
- 28.Kwit M, Skowronek P, Kołbon H, Gawroński J. Chirality. 2005;17:S93. doi: 10.1002/chir.20119. [DOI] [PubMed] [Google Scholar]
- 29.Kwit M, Gawroński J. Tetrahedron Asymmetry. 2003;14:1303. doi: 10.1016/S0957-4166(03)00130-7. [DOI] [Google Scholar]
- 30.Gérard S, Pressel Y, Riant O. Tetrahedron Asymmetry. 2005;16:1889. doi: 10.1016/j.tetasy.2005.04.025. [DOI] [Google Scholar]
- 31.Bandini M, Melucci M, Piccinelli F, Sinisi R, Tommasi S, Umani-Ronchi A (2007) Chem Commun 4519 [DOI] [PubMed]
- 32.Gajewy J, Kwit M, Gawroński J. Adv Synth Catal. 2009;351:1055. doi: 10.1002/adsc.200800801. [DOI] [Google Scholar]
- 33.Gajewy J, Gawroński J, Kwit M. Org Biomol Chem. 2011;9:3863. doi: 10.1039/c1ob05074e. [DOI] [PubMed] [Google Scholar]
- 34.Mastranzo VM, Quintero L, de Parrodi CA, Juaristi E, Walsh PJ. Tetrahedron. 2004;60:1781. doi: 10.1016/j.tet.2003.12.039. [DOI] [Google Scholar]
- 35.Mikami K, Matsukawa S. Nature. 1997;385:613. doi: 10.1038/385613a0. [DOI] [Google Scholar]
- 36.Ohkuma T, Doucet H, Pham T, Mikami K, Korenaga T, Terada M, Noyori R. J Am Chem Soc. 1998;120:1086. doi: 10.1021/ja972897e. [DOI] [Google Scholar]
- 37.Costa AM, Jimeno C, Gavenonis J, Carroll PJ, Walsh PJ. J Am Chem Soc. 2002;124:6929. doi: 10.1021/ja0166601. [DOI] [PubMed] [Google Scholar]
- 38.Ushio H, Mikami K. Tetrahedron Lett. 2005;46:2903. doi: 10.1016/j.tetlet.2005.02.135. [DOI] [Google Scholar]
- 39.Bette V, Mortreux A, Savoia D, Carpentier J-F. Adv Synth Catal. 2005;347:289. doi: 10.1002/adsc.200404283. [DOI] [Google Scholar]
- 40.Bette V, Mortreux A, Lehmann CW, Carpentier J-F (2003) Chem Commun 332 [PubMed]
- 41.Meyer N, Roesky PW. Z Anorg Allg Chem. 2007;633:2292. doi: 10.1002/zaac.200700188. [DOI] [Google Scholar]
- 42.Kwit M, Gawroński J. Tetrahedron. 2003;59:9323. doi: 10.1016/j.tet.2003.09.090. [DOI] [Google Scholar]
- 43.Savoia D, Gualandi A, Stoeckli-Evans H. Org Biomol Chem. 2010;8:3992. doi: 10.1039/c004873a. [DOI] [PubMed] [Google Scholar]
- 44.Gualandi A, Grilli S, Savoia D, Kwit M, Gawroński J. Org Biomol Chem. 2011;9:3863. doi: 10.1039/c0ob01192d. [DOI] [PubMed] [Google Scholar]
- 45.Robb MA, Cheeseman JR, Montgomery JA, Vreven T Jr, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03 (Revision D.0), Gaussian Inc, Wallingford
- 46.Gajewy J, Gawroński J, Kwit M (2012) Organometallics (submitted)
- 47.Morris DJ, Hayes AM, Wills M. J Org Chem. 2006;71:7035. doi: 10.1021/jo061154l. [DOI] [PubMed] [Google Scholar]
- 48.Utsukihara T, Misumi O, Kato N, Kuroiwa T, Horiuchi CA. Tetrahedron Asymmetry. 2006;17:1179. doi: 10.1016/j.tetasy.2006.04.007. [DOI] [Google Scholar]
- 49.Le Roux E, Malacea R, Manoury E, Poli R, Gonsalvi L, Peruzzinia M. Adv Synth Catal. 2007;349:309. doi: 10.1002/adsc.200600350. [DOI] [Google Scholar]
- 50.Biswas K, Prieto O, Goldsmith PJ, Woodward S. Angew Chem Int Ed. 2005;44:2232. doi: 10.1002/anie.200462569. [DOI] [PubMed] [Google Scholar]
- 51.Shimizu H, Igarashi D, Kuriyama W, Yusa Y, Sayo N, Saito T. Org Lett. 2007;9:1655. doi: 10.1021/ol070289q. [DOI] [PubMed] [Google Scholar]
- 52.Singh RP, Twamley B, Fabry-Asztalos L, Matteson DS, Shreeve JM. J Org Chem. 2000;65:8123. doi: 10.1021/jo005622h. [DOI] [PubMed] [Google Scholar]
- 53.Yong KH, Chong JM. Org Lett. 2002;4:4139. doi: 10.1021/ol026901p. [DOI] [PubMed] [Google Scholar]
- 54.Xu Q, Zhou H, Geng X, Chen P. Tetrahedron. 2009;65:2232. doi: 10.1016/j.tet.2009.01.058. [DOI] [Google Scholar]
- 55.Dearden MJ, Firkin CR, Hermet J-PR, O’Brien P. J Am Chem Soc. 2002;124:11870. doi: 10.1021/ja027774v. [DOI] [PubMed] [Google Scholar]
- 56.Homann MJ, Vail RB, Previte E, Tamarez M, Morgan B, Dodds DR, Zaks A. Tetrahedron. 2004;60:789. doi: 10.1016/j.tet.2003.10.123. [DOI] [Google Scholar]
- 57.Stampfer W, Kosjek B, Faber K, Kroutil W. J Org Chem. 2003;68:402. doi: 10.1021/jo026216w. [DOI] [PubMed] [Google Scholar]
- 58.Wu X, Liu X, Zhao G. Tetrahedron Asymmetry. 2005;16:2299. doi: 10.1016/j.tetasy.2005.06.010. [DOI] [Google Scholar]
- 59.Duan HF, Xie J-H, Shi W-J, Zhang Q, Zhou Q-L. Org Lett. 2006;8:1479. doi: 10.1021/ol060360c. [DOI] [PubMed] [Google Scholar]
- 60.Stanchev S, Rakovska R, Berova N, Snatzke G. Tetrahedron Asymmetry. 1995;6:183. doi: 10.1016/0957-4166(94)00374-K. [DOI] [Google Scholar]
- 61.Ohkuma T, Koizumi M, Yoshida M, Noyori R. Org Lett. 2000;2:1749. doi: 10.1021/ol0000814. [DOI] [PubMed] [Google Scholar]
- 62.Salvi NA, Chattopadhyay S. Tetrahedron. 2001;57:2833. doi: 10.1016/S0040-4020(01)00134-X. [DOI] [Google Scholar]
- 63.Lee EK, Kim SH, Jung BH, Ahn WS, Kim GJ. Tetrahedron Lett. 2003;44:1971. doi: 10.1016/S0040-4039(03)00069-8. [DOI] [Google Scholar]
- 64.Etxebarria J, Degenbeck H, Felten AS, Serres S, Nieto N, Vidal-Ferran A. J Org Chem. 2009;74:8794. doi: 10.1021/jo9015425. [DOI] [PubMed] [Google Scholar]
- 65.Kylmälä T, Kuuloja N, Xu Y, Rissanen K, Franzén R (2008) Eur J Org Chem 4019
- 66.Sun J, Zhu C, Dai Z, Yang M, Pan Y, Hu H. J Org Chem. 2004;69:8500. doi: 10.1021/jo040221d. [DOI] [PubMed] [Google Scholar]
- 67.Gao JX, Yi XD, Xu PP, Tang CL, Wan HL, Ikariya T. J Organomet Chem. 1999;592:290. doi: 10.1016/S0022-328X(99)00565-3. [DOI] [Google Scholar]
- 68.Zhang G, Yashima E, Woggon WD. Adv Synth Catal. 2009;351:1255. doi: 10.1002/adsc.200900069. [DOI] [Google Scholar]
- 69.Aoyama H, Tokunaga M, Kiyosu J, Iwasawa T, Obora Y, Tsuji Y. J Am Chem Soc. 2005;127:10474. doi: 10.1021/ja051750h. [DOI] [PubMed] [Google Scholar]
- 70.Yeori A, Goldberg I, Shuster M, Kol M. J Am Chem Soc. 2006;128:13062. doi: 10.1021/ja0647654. [DOI] [PubMed] [Google Scholar]
- 71.Hesemann P, Moreau JJE, Soto T. Synth Commun. 2003;33:183. doi: 10.1081/SCC-120015698. [DOI] [Google Scholar]
- 72.Sarnicka A, Starynowicz P, Lisowski J. Chem Commun. 2012;48:2237. doi: 10.1039/c2cc16673a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.