

Synopsis
Advances in understanding the biology of melanoma have provided great insights about the mechanisms of chemoresistance and its genetic heterogeneity in parallel with advances in drug design culminating in recent major treatment breakthroughs using small molecules inhibitors in metastatic melanoma (MM). While clinical benefit of targeted therapies has been unquestionable, future advances can only be possible if we better understand the interplay between genetic aberrations and role of other crucial non-genetic changes yet to be identified by such projects as the Cancer Genome Atlas Project (TCGA) in Melanoma. Combination therapies, either among small molecule inhibitors themselves and/or with immunotherapies may be the optimal strategy to prevent development of drug resistance that is inherently linked with such targeted therapies.
Keywords: Melanoma, Next Generation Sequencing, BRAF inhibitors, Ocular Melanoma, Immunotherapies
1. Introduction
1.1. Historic Overview
Similar to those for other cancers, targeted therapies for MM have only been under investigation for a little more than a decade. Before 2010 treatment of MM had achieved minimal progress since the ‘70s when dacarbazine was approved, when a ‘one-size-fits-all’ approach with various chemotherapeutic approaches had been applied to nearly all cancers. In clinical trial after clinical trial, chemotherapies in MM were proved to be largely ineffective compared to dacarbazine1. In fact, the minimal clinical benefit from systemic treatments was so predictable that clinical efficacy benchmarks were built around the statistical design for future phase II clinical trials in MM2. During this frustrating era, few immunotherapies were proved to be promising with durable clinical benefit in a small subset of patients with MM or high risk for relapse melanoma1. More than any other time from the past, treatment of MM is currently being shaped around targeted therapies administered in particular melanoma subgroups, given in precisely defined schedules, alone or in combination with other targeted therapies or various other immunotherapeutic approaches.
1.2 Better Understanding of the Biology of Melanoma was the Driving Force Behind Clinical Development of Targeted Therapies
It is becoming increasingly understood that cancers have distinct aberrations in particular cellular processes, in particular DNA repair pathways, which make them either relatively sensitive3 or refractory4 to systemic chemotherapies. Melanoma has one of the highest mutation frequencies5 and frequently shows elevated expression of DNA repair proteins6. Four important points are remarkable with respect to genetic aberrations in melanoma:
Only a handful of genes are more frequently mutated (Figure 1) or show gene copy number alterations (amplifications or deletions, Figure 2) than others7,8, whereas the clinical importance of most other genetic aberrations is currently unclear.
While most frequently mutated genes bear mutational ‘hotspots’ (‘canonical’ mutations), increasing evidence suggests the presence of non-canonical mutations (Figure 1) that can only be identified using NGS methodologies.
The most frequently mutated genes are components of two principal signaling pathways, the Ras-Raf-MEK-ERK and the PI3K-Akt-mTOR signaling pathway (Figure 3). The activation status of these kinases within each of these pathways are not independent from each other and dynamically adjust to environmental changes, including targeted treatments9.
The most frequently occurring mutations BRAF and RAS proteins are paradoxically not related to sun exposure, nor can they be found in early stages of melanoma, or even premalignant conditions10, and are retained during later stages of melanoma11.
More than one mutation and/or gene copy alteration can coexist within melanoma, which can have important clinical implications12 (Figures 1 and 2).
Response to immunotherapies is independent from mutational status13.
Figure 1.
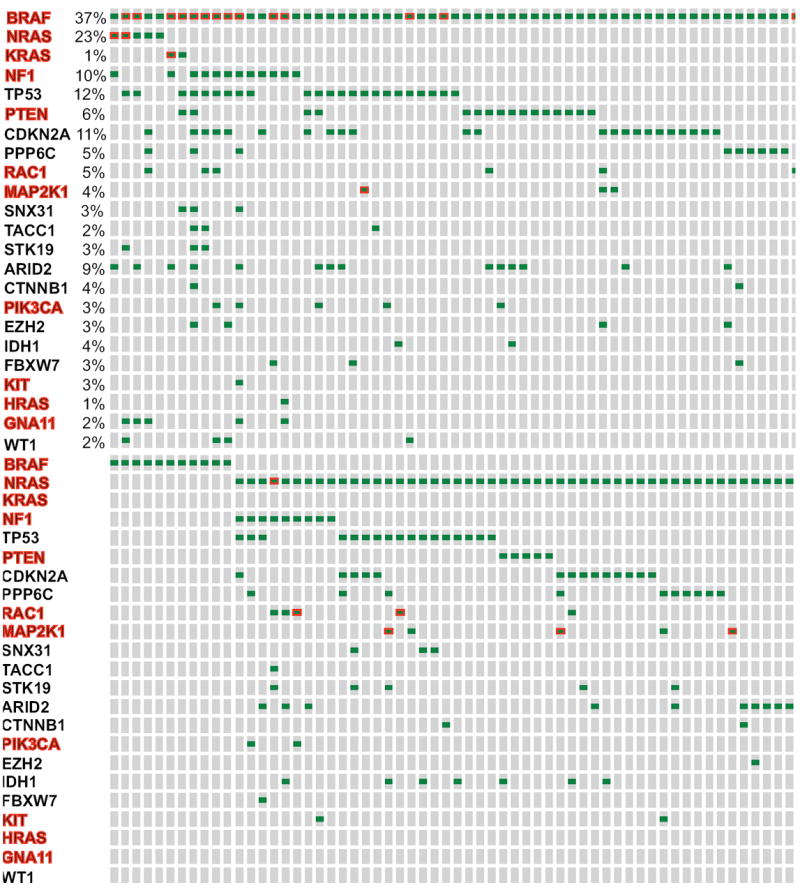
Frequency of somatic mutations that were previously identified in the work presented in the Hodis et al. Cell 2012 and applied in the publicly available cohort of cutaneous melanoma samples that has been collected as part of the Cancer Genome Atlas Project. Only 257 out of the so far (August 17, 2014) analyzed 375 samples that bear mutations (green dots) in any of these genes are shown. NF1, HRAS, and KRAS are also presented to emphasize the 4 emerging subgroups on the basis of BRAF, RAS, NF1 mutations, versus no mutation (triple-wild type group). Mutations highlighted with red border signify non-canonical mutations. Most genes, highlighted in red, are components of the BRAF/MEK/ERK, or PI3K/Akt signaling (see Figure 3 for further details). Analysis was performed using the cBioportal for Cancer Genomics (www.cbioportal.org) in compliance with early publication of results from the website, ad per Cerami et al. Cancer Discov 2012 and Gao et al. Sci Signal. 2013.
Figure 2.
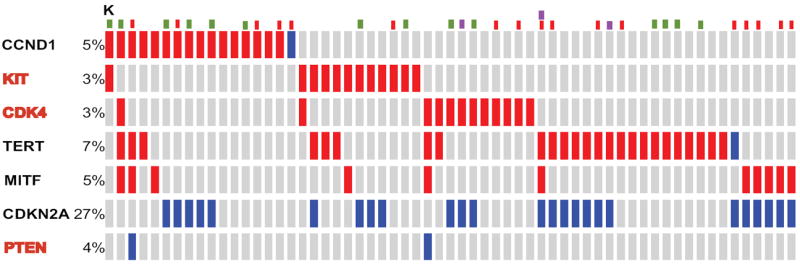
Frequency of gene copy number alterations that were previously identified in the work presented in the Hodis et al. Cell 2012 and applied in the publicly available cohort of cutaneous melanoma samples that has been collected as part of the Cancer Genome Atlas Project. Only 145 out of the so far (Aug 17, 2014) analyzed 375 sample that bear gene amplifications (red bars), or gene deletions (blue bars) in any of these genes are shown. Samples with BRAF (red dots), RAS (green dots), or NF1 (purple dots) are also shown for associations. Analysis was performed using the cBioportal for Cancer Genomics (www.cbioportal.org) in compliance with early publication of results from the website, ad per Cerami et al. Cancer Discov 2012 and Gao et al. Sci Signal. 2013.
Figure 3.
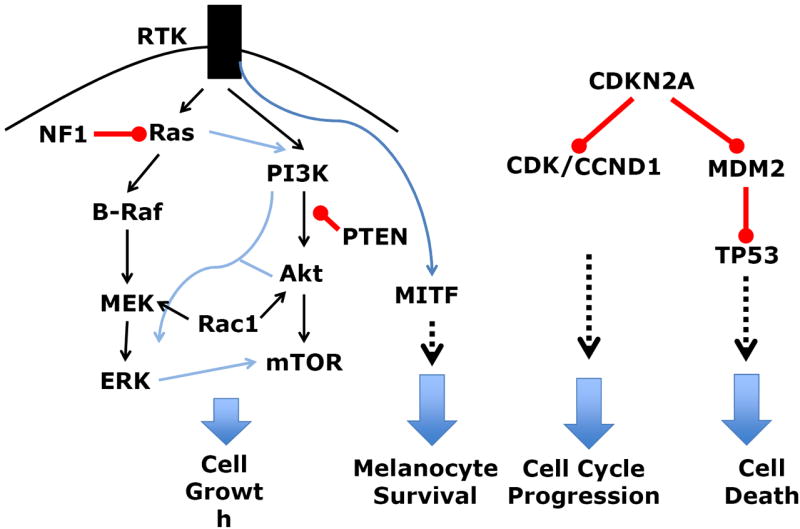
Cellular processes disrupted in melanoma as a result of genetic aberrations (mutations or gene copy number alterations). See Figure 1 and 2 for details regarding the type and frequency of genetic aberrations. Red lines indicate inhibition, blue and blank indicate activation.
In preliminary analyses of mutations of more than 350 cutaneous melanoma specimens as part of the Cancer Genome Atlas (TCGA), cutaneous melanomas can be conventionally classified in 4 different mutational groups (Figure 1)8:
‘Hotspot’ mutations in the BRAFV600 as well as immediately adjacent codons,
‘Hotspot’ mutations of the RAS oncogenes (N-, K-, or H-RAS) with the predominance of those occurring in NRAS (>90%),
Mutations of the neurofibromatosis 1 gene (NF1), an inhibitor of RAS signaling (Figure 4) without any concurrent hotspot mutations in the BRAF and NRAS (approximately 10%),
No mutations in any of the above described genes.
Figure 4.
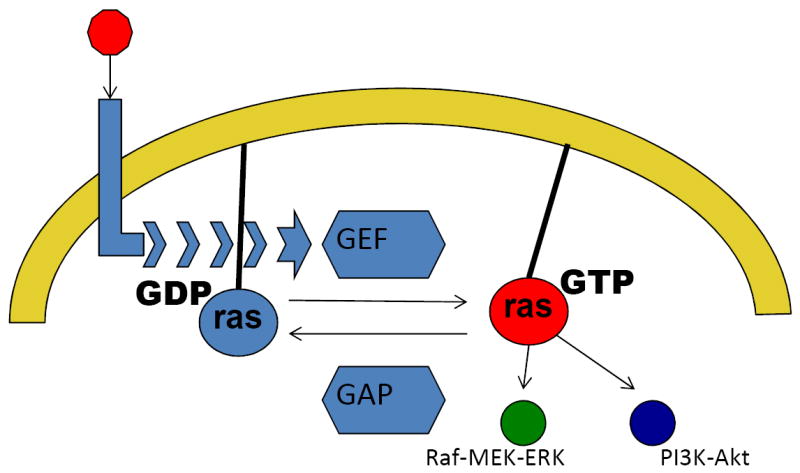
Simplified diagram on the regulation of the RAS superfamilty of small GTPases and the role of NF1. RAS proteins become active versus inactive if bound to GTP and GDP, respectively. Extracellular growth factor signals (red circle) are transmitted through growth factor receptors (light blue) to guanine nucleotide exchange factors (GEF), which activate RAS (red) by exchanging GDP for GTP. In contrast, GTPase activating proteins, including NF1, tend to keep RAS proteins in their inactive state (light blue). Please note the promiscuity of RAS signaling which activate several signaling pathways, including the MAPK as well as the PI3K-Akt pathway.
2. Treating Patients with MM in the Era of Small Molecule Inhibitors
The approach to a patient with MM has dramatically changed since 2010 with the advent of small inhibitor therapies, especially for BRAF-mutant patients. In addition to factors such as patient’s performance status, tumor doubling time, ability to perform metastatectomy, comorbid factors, AJCC staging system guidelines, knowledge about the mutation status for at least BRAF, NRAS and KIT, is becoming the standard of care for prognostic14 and treatment reasons. NGS methodologies with the ability to sequence several hundred cancer-associated genes are increasingly being incorporated in standard therapeutic decisions and have so far revealed that the mutational landscape of melanoma is more complex than was originally thought15.
Metastatectomy remains the best treatment for patients who can become completely free of distant metastatic disease with surgery16. However, controversies exist with respect to the optimal sequencing of systemic therapies for patients with unresectable MM. Retrospective analysis of BRAF-mutant patients who participated in the European Ipilimumab Expanded Access program suggest that the overall survival (OS) of patients with BRAF-mutant melanoma who originally received a single-agent MAPK inhibitor (MAPKi) followed by ipilimumab was inferior to that of patients who received ipilimumab first, followed by MAPKi13. Current guidelines suggest that in the absence of a clinical trial, patients with relatively asymptomatic, slow tumor kinetics, M1a/M1b disease —all presumed surrogate factors of a relatively more functional host immune system— should be initially considered for an immunotherapy followed by targeted therapy, if available, or chemotherapy.
3. Treatment with Single-Agent Targeted Agents
3.1 All Three FDA-approved Targeted Therapies in Metastatic Melanoma are for Patients with BRAF-Mutant Melanoma
As of May 2013 there are three FDA-approved MAPKi for MM: the two BRAF inhibitors (BRAFi), vemurafenib and dabrafenib, and the first-in-class MEK inhibitor (MEKi), trametinib. Their approval was based on randomized phase III trials in which each of the investigational agents was compared against dacarbazine for patients with unresectable BRAFV600E(K)-mutant MM17-19. All important clinical endpoints were in favor of the investigational agents. Five key observations were remarkable:
Responses were seen early during treatment, suggesting direct antitumor effect. However, further investigation showed that BRAFi may actually have effects upon the tumor microenvironment, such as increased influx of effector CD8+ cells infiltrating the tumor20, upregulation of immune checkpoint proteins within the tumor21, and suppression of angiogenic molecules, such as VEGF22. These effects may be either secondary to suppression of downstream actions of BRAF oncogenic signaling within melanoma cells themselves, or secondary to paradoxical activation of BRAF signaling within immune cells23.
Although antitumor responses have been impressive, the majority of responses were partial. Early studies using novel proteomic methods from analysis of the adaptive responses of the kinome in various melanoma cell lines following treatment with single-agent BRAFi or MEKi24 reveal incomplete suppression of the activity of all components of the RAF-MEK1/2-ERK pathway.25
Development of secondary resistance was seen in the majority of patients. Although general mechanisms of resistance will be described below, a surrogate clinical marker for durable responses to BRAFi was the ability to achieve complete antitumor response26,27.
All three treatments are orally administered and overall well tolerated with side effects similar to other previously FDA-approved tyrosine kinase inhibitors. The most interesting class-specific side effect was the small but significantly higher incidence of cutaneous squamous carcinomas, especially when pre-existing skin lesions bear KRAS or HRAS mutations28. Side effects from trametinib include high blood pressure, diarrhea, bleeding, coagulopathies, cardiomyopathy, and ocular toxicities.
All trials were conducted in patients with extracranial MM. Nevertheless, phase II trials using BRAFi in patients with active brain metastases showed considerable intracranial antitumor responses29,30. These results were somewhat unexpected in view of the fact that none of the three agents achieves significant drug levels within the CNS in preclinical studies (31 and references therein). The mechanism for such an antitumor effect within the brain is currently unknown, but it is reasonable to speculate that the blood-brain-barrier is, to a certain extent, compromised allowing for drug to enter the brain, a hypothesis that is currently being clinically tested (ClinicalTrials.gov Identifier NCT01978236).
3.2 Non-FDA-Approved but Promising Therapies for Patients with MM: More MAPK Pathway Inhibitors, Other Pathways, and Effects on Other Melanoma Subtypes
3.2.1 Do other BRAF inhibitors have a role in BRAF-mutant melanoma?
Encorafenib (LGX818) is the third BRAFi in advanced stages of clinical development. In contrast to other BRAFi, it has an extremely long dissociation half-life (30 hours versus 2 and 0.5 hours for dabrafenib and vemurafenib, respectively), a property that makes encorafenib extremely potent. In line with these preclinical data, a phase I trial of single-agent encorafenib in patients with BRAF-mutant MM showed that antitumor responses were also seen among patients who have previously received BRAFi32, suggesting that prolonged inhibition of the target may further improve therapeutic benefit.
3.2.2. Treatment of Patients with NRAS-Mutant Melanoma
NRAS-mutant melanomas have an overall worse prognosis than the more abundant BRAF-mutant melanomas14. This is attributed to the relative ‘promiscuity’ of the RAS family of proteins to signal through multiple signaling pathways, including the MAPK and the PI3K/Akt pathway, as opposed to RAF proteins that are relatively more committed to signal through the MAPK pathway (Figure 3)33. Due to difficulties in developing direct RAS inhibitors, current efforts focus on inhibiting druggable downstream effectors of RAS proteins, such as MEK. A phase II trial of binimetinib (MEK162), a highly specific MEK inhibitor, in patients with NRAS-mutant melanoma showed an approximately 20% partial response34, a promising result that is currently being investigated in a large randomized phase III trial (NCT01763164). Of note, no responses were seen with trametinib35.
3.2.3 Targeting Mutations in Rare Melanomas
cKIT is a type III transmembrane receptor tyrosine kinase that mediates signaling via various pathways and plays an important physiologic role for the development and maintenance of various cells, including melanocytes. A number of genetic aberrations of the cKIT gene have been observed in melanoma from both cutaneous (acral and chronically sun-damaged skin) and mucosal primary, including mutations in particular exons (e.g. 11 and 13) and gene amplifications36. Recently reported trials using various cKIT inhibitors focusing on melanomas that bear cKIT genetic aberrations are proof-of-principle that melanomas driven by constitutively active proto-oncogenes can be successfully targeted by small molecule inhibitor therapies. These trials show that antitumor responses can occur particularly in patients with activating mutations, as opposed to gene amplifications, and can occasionally be durable36.
Over the last few years, significant advances have been made towards understanding the biology of ocular melanoma. Primary ocular melanomas frequently bear activating, mutually exclusive hotspot mutations in two of the guanine nucleotide-binding proteins for Aq and A11 (GNAQ and GNA11)37 which lead to constitutive activation of phospholipase C (PLC) β and MAPK. In addition, approximately half of primary uveal melanomas bear inactivating mutations in the BRCA1 associated protein 1 (BAP1), a genetic event that usually follows that of GNAQ/GNA11, and is associated with propensity for distant metastases38. Early clinical trials suggest that targeting ocular melanomas with MEKi is associated with better clinical benefit compared to standard chemotherapy39, a result that is currently being confirmed in a large phase III trial (NCT01974752). A phase I clinical trial of a novel PLCβ inhibitor, AEB071, in patients with ocular melanoma showed early promising safety and efficacy results40. The role of other targeted treatments against ocular melanoma with BAP1 mutation is currently under investigation (NCT01587352).
4. Therapies Using Combinations of Small Molecule Inhibitors
4.1. Combinations Among Small Molecule Inhibitors
Concurrent suppression of BRAF plus MEK results in more potent and sustained suppression of ERK signaling, presumably due to incomplete suppression of the MAPK pathway by either agent alone25. In fact, treatment of patients with BRAFV600E,K-mutant MM with dabrafenib and trametinib (D+T) led to a higher incidence of complete antitumor responses41. In line with this, the combination of D+T was associated with more prolonged progression-free survival compared to dabrafenib alone in a randomized phase III trial, COMBI-d42. In this trial, D+T was overall well tolerated with lower frequency of keratoacanthomas and squamous cell carcinomas, although significant pyrexia, more frequent dose reductions, interruptions, and permanent discontinuations were observed in the D+T compared to dabrafenib alone arm. Nevertheless, the safety and efficacy data led to the FDA approval of D+T as a frontline therapy for patients with BRAFV600E,K-mutant melanoma. Randomized phase III trials testing the efficacy of concurrent BRAF plus MEK inhibition with other BRAFi+MEKi are underway43,44.
Given the high incidence of NRAS mutations that coexist with CDKN2A locus genetic aberrations7,8 and the strong preclinical rationale to combine MEK plus CDK inhibitors in NRAS-driven genetically engineered mouse models45, a clinical trial of combined MEKi and CDK inhibition using binimetinib plus LEE011 is underway. Preliminary results suggest that this treatment combination induces antitumor responses in approximately 1/3 of the analyzed patients, although dosing schedules in relation to toxicity and tolerability are currently being addressed46.
4.2. Combinations of Small Molecule Inhibitors and Immunotherapies
It is becoming increasingly understood that small molecule inhibitor therapies and immunotherapies have complementary strengths and limitations47 and various preclinical models have suggested a synergistic mechanism of action between these two treatment modalities23,48,49. Current treatment combinations are being tested in BRAF-mutant patients, given the lack of effective targeted therapies in other groups. Scheduling of these treatments —concurrent versus sequential— as well as selection of optimal drug combinations is important not only to minimize toxicity, but also to avoid potential small molecule inhibitor-mediated immunosuppression50, and optimize treatment effect granted by each drug. Despite early concerns that this combination could be associated with considerable toxicity, early results from subsequent ongoing trials suggest that the observed toxicities may not be only drug-class effect, but also related with the particular drug combination tested51. As these and more other trials (NCT01656642 and NCT01988896) mature, and even evolve into large randomized studies in the near future, we will be able to better assess durability of responses, a clinical benefit that is infrequently shared among targeted therapies.
5. Treatment Resistance
Based on the history of targeted therapies in other malignancies, resistance to targeted therapies in melanoma was highly expected. Several excellent reviews have been written for this important topic52. In the case of BRAF-mutant melanoma, the most extensively studied subtype with respect to drug resistance, these mechanisms may or may not involve reactivation of the ERK signaling pathway. In fact, on most occasions BRAF-mutant, MAPKi-resistant melanoma remain ‘addicted’ to MAPK signaling, which explains why the OS of patients who continue to be treated beyond disease progression with MAPKi is significantly longer compared to those who switch to non-MAPKi-based treatments53. Another important aspect is the role of PI3K/Akt signaling in primary as well as secondary drug resistance, either via acquired genetic alteration or secondary activation of the pathway by endogenous (RAS) or environmental factors54,55. So far, approaches to overcome resistance after it is developed have been challenging, and current efforts focus upon prevention of drug resistance. It is anticipated that small molecule inhibitors with superior pharmacodynamic properties (e.g. encorafenib), and/or addition of a third targeted therapy to the BRAF/MEKi backbone may overcome the challenge of turning the drugs’ cell inhibitory effect to an actual cell killing one. While trials testing combinations of three small molecule inhibitors are underway (e.g. NCT02110355), toxicity and tolerability will be a significant concern, in addition to efficacy. This is a particular concern when MAPKi were previously combined with inhibitors of the PI3K/Akt pathway56.
6. Summary/Discussion
While great progress has been achieved towards finding effective treatments for patients with BRAF-mutant melanoma, therapeutic advances are only beginning to occur in RAS-mutant melanoma. The biology of melanomas that bear no oncogenic mutations for BRAF/RAS has only recently begun to be systematically investigated57, but the subgroup that bears NF1 mutations, which is negative inhibitor of RAS signaling58, suggests that an even greater proportion of melanomas than what was originally thought may actually be addicted to RAS or RAF/MEK signaling. Clearly, targeted therapies provide a clinical benefit that is only durable in a small subset of patients. Nevertheless, the remarkable ability of small molecule inhibitors to suppress tumor growth leads to a better functioning immune system dysfunction, which, even if only temporary, may pave the way for better and more durable responses using immunotherapies. The tremendous efficacy of such targeted therapies in distant MM is already being investigated in earlier stages of disease where risk of relapse is high (NCT1682083 and 01667419). It is actually the administration of such treatments at an earlier stage that is expected to significantly change the natural history of disease over the next decade, especially the development of challenging future complications that are inherently difficult to treat (e.g. brain metastases).
Finally, the notion of ‘targeted’ therapies may likely be further expanded in the near future, thanks to projects such as TCGA. As part of this project, clinicopathologically well-annotated, snap-frozen tumors undergo profiling across multiple molecular data platforms for downstream integrative bioinformatics analysis (DNA and RNA sequencing as well as microRNA, methylation and proteomic profiling). This high-order investigation will most likely identify important non-genetic ‘targets’ amenable to therapeutic manipulation8.
Key Points.
Although melanoma bears one of the highest number of mutations per given DNA length among other malignancies, the most abundant mutations primarily affect two major signaling pathways.
Next generation sequencing methodologies (NGS) targeting a panel of cancer-related genes may better capture heterogeneity of melanoma and assist in treatment decisions.
Several genetic aberrations (mutations, copy number aberrations) can coexist within a particular melanoma, which may be of prognostic and therapeutic significance.
Although BRAF mutant melanomas have been the most successful melanoma subset for targeted therapies, progress is ongoing for other melanoma subtypes as well (e.g. RAS-mutant, ocular).
Treatment strategies are more successful in preventing, as opposed to treating, secondary drug resistance; combination treatments among targeted therapies and/or with immunotherapies may be more successful than single-agent approaches.
Footnotes
Stergios Moschos has served as consultant for Genentech, Amgen, and Merck. Dr.Pinnamaneni has no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee C, Collichio F, Ollila D, et al. Historical review of melanoma treatment and outcomes. Clin Dermatol. 2013;31:141–147. doi: 10.1016/j.clindermatol.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 2.Korn EL, Liu PY, Lee SJ, et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J Clin Oncol. 2008;26:527–534. doi: 10.1200/JCO.2007.12.7837. [DOI] [PubMed] [Google Scholar]
- 3.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jewell R, Conway C, Mitra A, et al. Patterns of expression of DNA repair genes and relapse from melanoma. Clin Cancer Res. 2010;16:5211–5221. doi: 10.1158/1078-0432.CCR-10-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watson I. Comprehensive Molecular Characterization of Regional Metastatic Melanoma. Oral presentation at the The Cancer Genome Atlas 3rd Annual Scientific Symposium; Bethesda, MD: National Institutes of Health; 2014. [Google Scholar]
- 9.Johnson GL, Stuhlmiller TJ, Angus SP, et al. Molecular pathways: adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin Cancer Res. 2014;20:2516–2522. doi: 10.1158/1078-0432.CCR-13-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poynter JN, Elder JT, Fullen DR, et al. BRAF and NRAS mutations in melanoma and melanocytic nevi. Melanoma Res. 2006;16:267–273. doi: 10.1097/01.cmr.0000222600.73179.f3. [DOI] [PubMed] [Google Scholar]
- 11.Colombino M, Capone M, Lissia A, et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol. 2012;30:2522–2529. doi: 10.1200/JCO.2011.41.2452. [DOI] [PubMed] [Google Scholar]
- 12.Nathanson KL, Martin AM, Wubbenhorst B, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436) Clin Cancer Res. 2013;19:4868–4878. doi: 10.1158/1078-0432.CCR-13-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ascierto PA, Simeone E, Sileni VC, et al. Clinical experience with ipilimumab 3 mg/kg: real-world efficacy and safety data from an expanded access programme cohort. J Transl Med. 2014;12:116. doi: 10.1186/1479-5876-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jakob JA, Bassett RL, Jr, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118:4014–4023. doi: 10.1002/cncr.26724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeck WR, Parker J, Carson CC, et al. Targeted next generation sequencing identifies clinically actionable mutations in patients with melanoma. Pigment Cell Melanoma Res. 2014;27:653–663. doi: 10.1111/pcmr.12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sosman JA, Moon J, Tuthill RJ, et al. A phase 2 trial of complete resection for stage IV melanoma: results of Southwest Oncology Group Clinical Trial S9430. Cancer. 2011;117:4740–06. doi: 10.1002/cncr.26111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323–332. doi: 10.1016/S1470-2045(14)70012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 19.Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 20.Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18:1386–1394. doi: 10.1158/1078-0432.CCR-11-2479. [DOI] [PubMed] [Google Scholar]
- 21.Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–1231. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khalili JS, Liu S, Rodriguez-Cruz TG, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18:5329–5340. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koya RC, Mok S, Otte N, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012;72:3928–3937. doi: 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duncan JS, Whittle MC, Nakamura K, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Angus SP, Stuhlmiller TJ, Reuther R, et al. Defining the adaptive kinome response to BRAF and MEK inhibition in melanoma (abstr 4761). AACR Annual Meeting; San Diego, CA. 2014. [Google Scholar]
- 26.Kim K, Amaravadi RK, Flaherty KT, et al. Significant long-term survival benefit demonstrated with vemurafenib in ongoing phase I study. Presented in the Society of Melanoma Research Annual Meeting; Hollywood, CA. 2012. [Google Scholar]
- 27.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087–1095. doi: 10.1016/S1470-2045(12)70431-X. [DOI] [PubMed] [Google Scholar]
- 30.Dummer R, Goldinger SM, Turtschi CP, et al. Vemurafenib in patients with BRAF(V600) mutation-positive melanoma with symptomatic brain metastases: final results of an open-label pilot study. Eur J Cancer. 2014;50:611–621. doi: 10.1016/j.ejca.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Vaidhyanathan S, Mittapalli RK, Sarkaria JN, et al. Factors influencing the CNS distribution of a novel MEK-1/2 inhibitor: implications for combination therapy for melanoma brain metastases. Drug Metab Dispos. 2014;42:1292–1300. doi: 10.1124/dmd.114.058339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dummer R, Robert C, Nyakas M, et al. Initial results from a phase I, open-label, dose escalation study of the oral BRAF inhibitor LGX818 in patients with BRAF V600 mutant advanced or metastatic melanoma (abstr 9028). ASCO Annual Meeting; Chicago IL. 2014. [Google Scholar]
- 33.Rodriguez-Viciana P, Sabatier C, McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol. 2004;24:4943–4954. doi: 10.1128/MCB.24.11.4943-4954.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–256. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 35.Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–789. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Curtin JA, Busam K, Pinkel D, et al. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24:4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 37.Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carvajal RD, Sosman JA, Quevedo JF, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311:2397–2405. doi: 10.1001/jama.2014.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piperno-Neumann S, Kapiteijn E, Larkin JMG, et al. Phase I dose-escalation study of the protein kinase C inhibitor AEB071 in patients with metastatic uveal melanoma (abstr 9030). ASCO Annual Meeting; Chicago, IL. 2014. [Google Scholar]
- 41.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long GV, Stroyakovsky DL, Gogas H, et al. COMBI-d: A randomized, double-blinded, Phase III study comparing the combination of dabrafenib and trametinib to dabrafenib and trametinib placebo as first-line therapy in patients with unresectable or metastatic BRAFV600E/K mutation-positive cutaneous melanoma (abstr 9011). ASCO Annual Meeting; Chicago, IL. 2014. [Google Scholar]
- 43.Ribas A, Gonzalez R, Pavlick A, et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: a phase 1b study. Lancet, Oncol. 2014;15:954–965. doi: 10.1016/S1470-2045(14)70301-8. [DOI] [PubMed] [Google Scholar]
- 44.Kefford R, Miller WH, Tan DS-W, et al. Preliminary results from a phase Ib/II, open-label, dose-escalation study of the oral BRAF inhibitor LGX818 in combination with the oral MEK1/2 inhibitor MEK162 in BRAF V600-dependent advanced solid tumors (abstr 9029). ASCO Annual Meeting; Chicago, IL. 2013. [Google Scholar]
- 45.Kwong LN, Costello JC, Liu H, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–1510. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sosman JA, Kittaneh M, Lolkema JK, M P, et al. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: Early encouraging clinical activity (abstr 9009). ASCO (ed); ASCO 2014 Annual Meeting; Chicago, IL. [Google Scholar]
- 47.Ribas A, Hersey P, Middleton MR, et al. New challenges in endpoints for drug development in advanced melanoma. Clin Cancer Res. 2012;18:336–341. doi: 10.1158/1078-0432.CCR-11-2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mok S, Koya RC, Tsui C, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74:153–161. doi: 10.1158/0008-5472.CAN-13-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu C, Peng W, Xu C, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19:393–403. doi: 10.1158/1078-0432.CCR-12-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boni A, Cogdill AP, Dang P, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–5219. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 51.Igor Puzanov, Callahan Margaret K, Linette Gerald P, et al. Phase 1 study of the BRAF inhibitor dabrafenib with or without the MEK inhibitor trametinib in combination with ipilimumab for V600E/K mutation–positive unresectable or metastatic melanoma (abstr 2511). ASCO Annual Meeting; Chicago IL. 2014. [Google Scholar]
- 52.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 53.Chan M, Haydu L, Menzies AM, et al. Clinical characteristics and survival of BRAF-mutant (BRAF+) metastatic melanoma patients treated with BRAF inhibitor dabrafenib or vemurafenib beyond disease progression (abst 9062). ASCO Annual Meeting; Chicago IL. 2014. [Google Scholar]
- 54.Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2014;4:69–79. doi: 10.1158/2159-8290.CD-13-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trunzer K, Pavlick AC, Schuchter L, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31:1767–1774. doi: 10.1200/JCO.2012.44.7888. [DOI] [PubMed] [Google Scholar]
- 56.Algazi AP, Posch C, Ortiz-Urda S, et al. A phase I trial of BKM120 combined with vemurafenib in BRAFV600E/k mutant advanced melanoma (abstr 9101). ASCO Annual Meeting; Chicago IL. 2014. [Google Scholar]
- 57.Mar VJ, Wong SQ, Li J, et al. BRAF/NRAS wild-type melanomas have a high mutation load correlating with histologic and molecular signatures of UV damage. Clin Cancer Res. 2013;19:4589–4598. doi: 10.1158/1078-0432.CCR-13-0398. [DOI] [PubMed] [Google Scholar]
- 58.Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74:2340–2350. doi: 10.1158/0008-5472.CAN-13-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]