

Abstract
Defining human protein interaction networks has become essential to develop an overall, systems-based understanding of the molecular events that sustain cell growth in normal and disease conditions. To characterize protein interaction networks from human cells, we have undertaken the development of a systematic, unbiased technology pipeline that couples experimental and computational approaches. This discovery engine is central to the Human Proteotheque Initiative (HuPI), a multidisciplinary project aimed at building a repertoire of comprehensive maps of human protein interaction networks, the Human Proteotheque. The information contained in the Proteotheque is made publicly available through an interactive web site that can be consulted to visualize some of the fundamental molecular connections formed in human cells and to determine putative functions of previously uncharacterized proteins based on guilt by association. The process governing the evolution of HuPI towards becoming a repository of accurate and complete protein interaction maps is described.
Keywords: protein interaction networks, human proteotheque initiative, transcription machinery, integrative systems biology
Introduction
Human cells function through the action of thousands of proteins that control their growth and differentiation. Most human proteins rarely work alone, but rather, assemble with other proteins into complexes to concertedly exert their function (Spirin and Mirny 2003; Rives and Galitski 2003; Alberts 1998). In addition, functionally related protein complexes forming specific cellular machineries interact together during different biological processes, such as gene transcription, DNA replication and repair, and others. The situation is further complicated by the fact that any given polypeptide could assemble into more than one protein complex, making the network of protein interactions sustaining cell growth and differentiation not only very complex in its organisation, but also in its dynamic of assembly (Han et al. 2004; Li et al. 2004). This unique situation highlights the pivotal role of protein interaction networks in cell function. Consequently, mapping their topology is a key issue in biomedical research, and the development of efficient technologies for doing so is an important challenge to modern research in proteomics and systems biology (Ranish et al. 2003; Link et al. 1999; Coulombe et al. 2004).
If we postulate that any given interaction made by a protein within a cell achieves a specific function, one can envision two main reasons for investing efforts into building accurate, complete protein interaction maps and for making them available to the scientific community. First, protein interaction networks, considered here as the set of interactions a protein makes with other proteins, DNA, and RNA molecules and metabolites, are deemed to represent the fingerprint of the physiological status of a cell and their modulation is predicted to represent the signature of specific disease conditions, including those observed in cancers and viral infections (for example, see Cui et al. 2007). Publicly available protein interaction maps will undoubtedly accelerate the discovery process in biomedical research because they would reveal new, more global (i.e., systemic) molecular descriptions of specific cellular conditions, such as those encountered in disease; these maps may well represent the new generation of biomarkers, being more accurate and specific as they are based on multiple parameters. These maps will also reveal new targets for drug discovery. Second, a large fraction of the proteins encoded by the human genome remain uncharacterized and their precise function, unknown. By identifying protein interaction partners, it becomes possible to determine putative functions of many previously un-characterized proteins (see below for examples in human cells). A number of experimental methods have been developed to identify protein–protein interactions, including the yeast two-hybrid (Y2H) method and the affinity purification – mass spectrometry (MS) approach. These methods have been used to characterize protein interaction networks and, in some cases, for defining protein function based on guilt-by-association criteria in the bacteria, yeast, fly, and worm systems (Giot et al. 2003; Ho et al. 2002; Uetz et al. 2000; Ito et al. 2001; Gavin et al. 2006; Gavin et al. 2002; Krogan et al. 2006; Butland et al. 2005; Li et al. 2004). Recently, the Y2H and the affinity purification – MS approaches have been applied to generate protein interaction maps for a fraction of the human proteome and to determine the function of previously uncharacterized human proteins (Jeronimo et al. 2007; Stelzl et al. 2005; Rual et al. 2005; Ewing et al. 2007). In addition to experimental determination, various computational methods have been used to predict protein interaction networks by using information from publicly available databases, such as BIND, MIPS and HPRD (Xia et al. 2004; Shoemaker and Panchenko 2007; Bader et al. 2003; Mewes et al. 2002; Peri et al. 2003). Consequently, defining protein interaction networks is invaluable for deciphering protein function in health and disease.
The literature contains a myriad of papers reporting on protein interactions (for reviews, see Devos and Russell 2007; Cusick et al. 2005). Efforts to curate and integrate these interactions into public databases have emerged in different projects around the world (Orchard et al. 2007). These projects are important and valuable. However, because of the heterogeneity of the data and the experimental procedures used to derive the various datasets, integration efforts become extremely difficult and their results raise important questions in terms of their completeness and accuracy. This notion of accuracy and completeness is a highly relevant issue that needs to be considered seriously when developing protein interaction maps that are highly valuable and, as far as possible, not misleading. Clearly, the challenge and value of building helpful protein interaction maps is immense.
Building a repertoire of meaningful protein interaction maps: an overview of the Human Proteotheque Initiative (HuPI)
The Human Proteotheque Initiative (HuPI) is a multidisciplinary, ongoing project aimed at generating comprehensive maps of the protein interaction networks that underlie cellular functions in humans (see schematic representation in Fig. 1). The maps of protein interaction networks built in the course of the HuPI project, the HuPI maps, will be deposited in a repertoire, the Human Proteotheque, which will be made publicly available as an atlas describing some fundamental human molecular networks. Because the HuPI project is still in its infancy, the Human Proteotheque is still rudimentary at the time of this writing. To generate valuable tools for scientists interested in basic biological and biomedical research, the HuPI project must conform to three main criteria. First, the data must be both accurate and complete. In other words, both the specificity and the sensitivity of the interaction datasets must be set to maximum values while developing and applying the overall experimental procedure. To achieve this goal, we have elected to develop an experimental pipeline, termed the HuPI discovery engine, in which data acquisition and analysis is performed in a highly systematic manner, favouring automation when possible to prevent bias that is sometimes the consequence of human decisions in experimental setups. The HuPI discovery engine is a constantly evolving pipeline that is developed towards systematic, unbiased operating procedures. Second, the data must be analysed, mined, and integrated in such a way that (i) all the relevant information is extracted and stored and (ii) this information is transferred into interaction maps that are comprehensive. As mentioned above, systematic procedures must be developed to ensure that the data is analyzed in an unbiased manner. For this reason, computational approaches are key to this downstream part of the HuPI discovery engine (Fig. 1). Third, both the protein interaction datasets and the comprehensive maps of their network of connections must be made available to the scientific community through the internet. A web-interfaced database is being developed to help the users find all the information relevant to their research. Graphical tools for the visualization and navigation of the protein interaction maps play a central role in the development of the HuPI database and web site.
Fig. 1.

Overview of the Human Proteotheque Initiative (HuPI). A collection of cell lines each expressing affinity-tagged proteins is used in a systematic, unbiased technology pipeline, termed the HuPI discovery engine, to characterize human protein interaction networks. Computational procedures are used to select high-confidence protein interactions and to build comprehensive maps, i.e., the HuPI-Maps, of high-density interaction networks.
The first generation of the HuPI discovery engine: systematic characterization of the protein–protein interaction network for the human transcription machinery
Over the past few years, we have taken an important step forward in describing protein complexes and their interaction networks in human cells. The developed procedure constitutes the basis of the HuPI discovery engine. Starting with components of the general transcription apparatus, namely RNA polymerase II and its general factors, we have used in vivo pull-down experiments with affinity-tagged polypeptides expressed at physiological levels to purify protein complexes in native conditions and defined their components using MS (Coulombe et al. 2004; Jeronimo et al. 2004; Jeronimo et al. 2007). For this analysis, we systematically used the tandem affinity purification (TAP) tag placed at the C terminus of proteins, allowing the purification of protein complexes in native conditions (Rigaut et al. 1999). Indeed, the elution steps during the TAP were performed in the absence of detergent and high-salt concentrations, thus preserving the integrity of the purified protein complexes. In addition, near physiological levels of expression of the tagged protein in HEK 293 cells were achieved through the use of an ecdysone-inducible system that allows us to tune the expression of the tagged proteins by varying the dose of the inducer Ponasterone A (Jeronimo et al. 2004). This step was intended to prevent overexpression of tagged polypeptides, which sometimes generates spurious interactions. Following affinity purification, the TAP eluates were run on SDS gels and stained. Gel slices were excised and digested with trypsin. The resulting tryptic peptides were identified by liquid chromatography – tandem mass spectrometry (LC–MS/MS) with microcapillary reversed-phase high-pressure liquid chromatography coupled to an LCQ DecaXP (ThermoFinnigan), LTQ, or LTQ-Orbitrap (ThermoElectron) quadrupole ion trap mass spectrometer with a nano-spray interface.
As mentioned above, we used this technology to survey protein complexes containing basal transcription factors in the soluble compartment of human cells (Jeronimo et al. 2007). A number of newly identified partners, including RNA processing factors, were systematically tagged and submitted to the same procedure in reciprocal tagging experiments. Reciprocal tagging served to enrich the data set and to confirm many interactions by navigating through the network of protein complexes forming the transcription machinery (see Fig. 2 for a schematic representation). This semi-random procedure for selecting the proteins to be tagged is useful because it allows us to build relatively dense interaction networks, something that would not be possible at this early stage if we had proceeded randomly, because the coverage would have been much too low to produce reliable datasets.
Fig. 2.
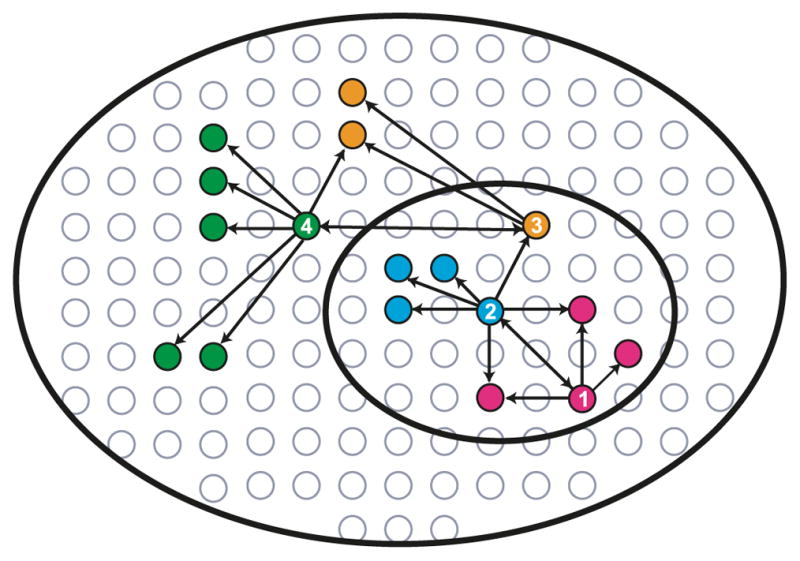
Strategy for the characterization of human protein–protein interaction networks. Affinity-tagged proteins (baits) are expressed at physiological levels in human cells, their complexes purified under native conditions, and their interaction partners (preys) identified using sensitive mass spectrometry (MS). Newly identified preys are iteratively tagged in reciprocal tagging experiments to navigate the network of protein complexes in human cells (1→2→3→4). This semi-random procedure for selecting the proteins to be tagged is useful because it allows us to build relatively dense interaction networks.
Our purification procedure was specifically designed to preserve the integrity of the purified complexes because they prospectively exist in live human cells. Affinity purification of tagged proteins theoretically allows the isolation of all protein complexes containing the tagged polypeptide. This method does not, however, allow for the direct determination of the abundance of the purified complexes. In addition, the high sensitivity of mass spectrometry requires the development of methods that discriminate between specific and spurious interactions (Patil and Nakamura 2005; Krogan et al. 2006; Gavin et al. 2006). In our case, this was accomplished through the development of an algorithm that selects high-confidence interactions by assigning interaction reliability (IR) scores to each protein–protein interaction (Jeronimo et al. 2007). After filtering out nonspecific interactions due to very abundant proteins, or proteins that bind nonspecifically to our affinity columns, the developed algorithm integrates data relating to the MS score of the interaction to data relating to the local topology of the network (e.g., bait 1 pulls down prey 2; bait 2 pulls down prey 1; bait 3 pulls down both prey 1 and 2, etc.) to calculate IR scores. The sensitivity and specificity of the algorithm was evaluated using literature-based classification of protein interactions. We selected as high-confidence interactions those for which the IR score exceeded a threshold (IR score above 0.6729) predicted to miss as false negatives only 17% of a set of literature-supported interactions while incorrectly retaining only 17% of a set of interactions without literature support as false positives. The selected protein–protein interactions were used to build protein interaction maps (see Fig. 3 for an example).
Fig. 3.

Map of a high-density network of high-confidence protein–protein interactions involving transcription and RNA processing factors. Affinity-tagged proteins (baits) and their copurified interaction partners (preys) are represented as coloured squares (nodes) and are connected using arrows. The color code is defined. M, MEPCE/BCDIN3; H, HEXIM1; C, P-TEFb subunits (CDK9 and CCNT1/Cyclin T1); A, hnRNPA1; P, RNA polymerase II subunits (Rpb2 and Rpb11); R, RPAPs; X, XAB1.
Mapping the topology of protein–protein interaction networks allows the determination of a putative function for previously uncharacterized proteins
Using the first-generation HuPI discovery engine with 32 tagged transcription and RNA processing factors, we defined a network of 805 interactions involving 436 different proteins (Jeronimo et al. 2007). This network reveals the connectivity of many protein complexes that form the core of the cell machinery involved in interpreting the genome (Fig. 3). By examining the topology of the network, one can easily conclude that the transcription and RNA processing machineries are tightly connected through many different interactions, forming a high-density network of connections in human cells. Because the protein–protein interaction data derived from this analysis mainly involves components of basal, “housekeeping” cellular machineries, namely the transcription and RNA processing machineries, we pose that the corresponding protein interaction maps represent a fundamental feature of mammalian cell function. We also believe that some interactions are specific to HEK 293 cells, thereby representing the signature of the physiological status of this cell line. Comparing this network with those derived from other cell lines is required to build a better understanding of the flexibility of human protein-interaction networks under various physiological and environmental conditions. Obtaining this information is a long-term goal of the HuPI.
Strikingly, our results identified a number of novel protein complexes containing transcription or RNA processing factors in association with proteins known to regulate the formation of protein complexes (see Fig. 3; green nodes). The term “formation of protein complexes” is specifically used here to describe the overall process leading from individual polypeptides at their site of synthesis to multicomponent complexes at their site of action; this general term is meant to include protein folding, assembly, and transport. These newly discovered partners define a novel class of regulatory factors that directly target the transcription and RNA processing machineries prior to their recruitment to active genomic loci or following their release from genomic DNA for recycling. Among these novel partners, some are previously uncharacterized proteins (Fig. 3; yellow nodes), for which we were able to determine a putative function according to their network connections (based on guilt by association) and, in some cases, perform functional analyses (see below).
Among the novel protein complexes unravelled by our proteomics analysis, two were characterized in further detail (Jeronimo et al. 2007). First, a group of four proteins, which we named RPAPs-XAB1 (the RNA polymerase II associated proteins-XAB1), and which are tightly connected to the enzyme RNA polymerase II itself, proved to form a molecular interface between the enzyme, the regulatory complex integrator, and a group of proteins with chaperone activity, including the prefoldins. These features, as well as their position at the interface of RNA polymerase II, regulatory complexes, and chaperones, suggest that they have a role in the formation of multicomponent transcription complexes. Experiments are in progress to determine the function of these novel regulatory proteins that promise to define novel regulatory mechanisms of gene expression and reveal some of the principles that govern protein complex assembly and (or) transport in eukaryotes.
A second part of the network captured our attention. Affinity purification of a tagged version of the splicing factor hnRNPA1 identified the previously uncharacterized protein BCDIN3. Bioinformatics analysis revealed the presence of a putative AdoMet-binding domain, AdoMet, being the methyl donor used by methyltransferases (Lu 2000). We then proceeded to the reciprocal tagging of BCDIN3 which, as expected, copurified with hnRNPA1 and other RNA processing factors. To our surprise, BCDIN3 also copurified with CDK9, CCNT1/Cyclin T1, and the HEXIMs, a set of cellular factors previously shown to regulate expression of the HIV-1 genome. CDK9 and CCNT1/Cyclin T1 form the P-TEFb elongation factor known to regulate HIV-1 transcription (Marshall and Price 1995; Zhou et al. 1998; Zhu et al. 1997; Mancebo et al. 1997), whereas the HEXIMs, in association with 7SK snRNA, regulate the activity of P-TEFb (Barboric et al. 2005; Blazek et al. 2005; Byers et al. 2005; Li et al. 2005; Yik et al. 2005).
To assess whether BCDIN3 is a bona fide methyltransferase and to start addressing its function, we searched for specific substrates within members of the 7SK-HEXIM-BCDIN3-P-TEFb-containing complex. Our results indicate that BCDIN3 can transfer methyl groups to the gamma phosphate of 7SK snRNA, thereby stabilizing 7SK in human cells. Together, these results indicate that our protein–protein interaction network led to the long-awaited discovery of the 7SK methylphosphate capping enzyme (MEPCE). BCDIN3 was renamed MEPCE.
In sum, the first generation of the HuPI discovery engine allowed us to characterize high-confidence, high-density protein–protein interaction networks for the human transcription machinery and to identify and determine a function for novel proteins that had not been previously characterized.
Moving the HuPI project forward: what are the next steps?
Keeping in mind that the HuPI is intended to develop systematic, unbiased experimental and computational tools for deciphering human protein interaction networks and to build comprehensive maps to be made publicly available, significant efforts have been devoted to pursue the development of the HuPI discovery engine. In some cases, these efforts have been directed towards improving some aspects of the technology pipeline that were found to be suboptimal in the previous version of the engine.
Increasing the number and variety of tagged proteins
As mentioned above, the first dataset of protein–protein interactions generated through affinity purification of tagged polypeptides used 32 baits, mainly within components of the transcription and RNA processing machineries. It is now important to increase the total number of baits for different reasons. First, it is essential to increase the coverage of the method and, consequently, to build networks that are as complete as possible. Second, reciprocal tagging of preys that have been identified as components of the network is required to improve the reliability score of the protein–protein interactions. Third, tagging of proteins involved in other machineries, such as chromatin remodelling factors, transcription elongation factors, DNA replication and repair factors, and others is needed to expand the current network to other biochemical processes. At the time of this writing, we have targeted 150 polypeptides to the HuPI discovery engine and will continue working towards expanding our human protein–protein interaction network.
As described above, our procedure, which uses an inducible expression system in stably transfected cell lines, has many advantages compared with those of transient expression assays. First, pilot experiments, which used tagged subunits of previously characterized proteins, including RNA polymerase II and some general transcription factors, revealed that transient expression assays can lead to the enrichment of subcomplexes during the purification, some of which lack essential subunits and are therefore inactive. Second, the use of an inducible system prevents the constitutive expression of the tagged protein, which may in some cases interfere in some ways with cell growth or have an adverse effect on the proteome of expressing cells. Finally, and although this cannot be done in a fully systematic manner, the use of the tuneable expression system helps to keep the expression level of the tagged proteins close to physiological.
Improving the sensitivity of MS
As MS is central to efficiently define protein–protein interactions (Aebersold and Mann 2003), we are developing novel MS methods that rely on the high sensitivity and high mass precision of new mass spectrometers, including the Thermo Fisher LTQ-Orbitrap equipments in place in the laboratory. Novel methods for sample fractionation that do not require gel analysis are also instrumental to the second generation of the HuPI discovery engine. Compared with the previous generation, we expect to increase our sensitivity by more than one order of magnitude. At the same time, and to increase the throughput of our MS procedure, we are currently strengthening the automation of both the MS procedure and the MS data analysis pipeline.
Adapting computational methods to increase the sensitivity and specificity of our high-confidence interaction dataset
As mentioned above, the first-generation HuPI discovery engine used a computational method capable of keeping the apparent rates of both false positives and false negatives between 15% and 20%. The implementation of more sensitive MS methods and the tagging of additional proteins require the development of more performing algorithms for filtering out false positives from noise, while keeping the sensitivity maximal (Patil and Nakamura 2005; Krogan et al. 2006; Gavin et al. 2006). The second generation of the technology pipeline has been developed in such a way that sensitivity and specificity are each expected to be above 90%.
Developing data integration tools
The development of computational and theoretical tools enabling the construction of relevant, useful maps is also a central effort. The complexity of the networks obtained is such that it can easily become overwhelming, thus requiring automated, goal-oriented network layout procedures facilitating the extraction of meaningful biological information.
Further developing the web-interfaced public database
A first version of the HuPI database was made publicly available by the end of 2007 (http://hupi.ircm.qc.ca). This database now contains our published data on human protein interactions (Jeronimo et al. 2007). In future versions, the database will contain comprehensive maps of protein interaction networks. Its interface will also allow the user to compare the data to those found in complementary databases, such as BIND, MIPS, and HPRD, as well as to access databases containing other types of biologically relevant information, such as expression profiles, SNPs, and others. These tools will help the user to evaluate the biological significance of our interaction data.
Integrating other types of interactions
As mentioned in the introductory paragraph, most proteins interact with other molecular components of the cell. To integrate protein–DNA, protein–RNA, and protein–metabolite interactions to our interaction maps, we are currently involved in various technology development projects. For example, our previous work has shown that TAP-tagged proteins can be used in chromatin immunoprecipitation (ChIP) experiments aimed at defining their location along the genome (Cojocaru et al. 2007; Jeronimo et al. 2004); when coupled with the identification of the immunoprecipi-tated DNA fragments using systematic ChIP-on-chip experiments (Lee et al. 2002; Ren et al. 2000), this method promises to reveal protein–DNA interactions that could then be integrated with protein–protein interactions. These efforts are also requiring the development of computational methods for the integration of the various datasets.
Assessing biological relevance
The direct identification by purification and mass spectrometry of protein interactions in HEK 293 cells is particularly relevant and powerful to study basal cellular machineries that are common to all (or most) cell types. The use of additional cell lines that can serve as models for various normal or diseased human tissues will be selected and studied to address the plasticity of human protein-interaction networks in various conditions. The use of cell lines is also advantageous because it provides access to protein interaction data rapidly at a moderate cost. However, the use of human cell lines also has disadvantages. The first relates to their availability. Cell lines are not available for many types of human cells and those that are available are usually transformed and far from normal. Because we expect many protein interactions to be different in different types of cells, many normally occurring interactions will be missed if the use of human cell lines is our sole method of identifying protein interactions. This problem can be solved in large part by turning to the mouse model for studying protein interactions. A partnership with groups studying protein interactions in the mouse is currently under discussion and will strengthen this aspect of the HuPI.
Making the Human Proteotheque Initiative international
Building a repertoire of comprehensive maps of protein interaction networks that will systematically enhance our understanding of both normal and disease conditions and, eventually, lead to the development of diagnosis tools and cures for important diseases, requires the concerted participation of large groups of scientists with relevant expertise. A significant number of groups in various countries have developed such expertise. Efforts are currently being made to leverage the HuPI into an international project that would unite many research groups into a collaborative venture that may well revolutionize biomedical research for years to come.
The first step of this international endeavour is the formation of a representative international consortium of scientists, which brings two types of expertise to the table. First, the consortium includes researchers with technical and conceptual expertise in the experimental characterization of protein interactions and in the creation of comprehensive maps of interaction networks using computational biology. Second, the consortium includes researchers with expertise in relevant biological and disease systems. The role of the consortium will be to select the cell lines and proteins to be targeted to the HuPI discovery engine, plan the deployment of the discovery platform at various sites, and design standard operation procedures for characterizing protein interactions using the HuPI discovery engine. This involvement of the community at all levels of the project will make the HuPI a highly innovative global project.
Conclusions
Building comprehensive, meaningful maps of human protein interaction networks and making them available to the scientific community through the internet is the main goal of the HuPI project. Recent progress that resulted in the publication of key papers reveals that this objective is at hand. In addition, to help infer putative functions to previously uncharacterized proteins and to increase our understanding of the proteome and its regulation, the HuPI maps provide a systems-based description of the relations between proteins and other molecules (proteins, DNA, RNA, metabo-lites) in human cells. We trust that such a complex molecular description of the physiological status of a cell or tissue will be invaluable to the development of a new generation of biomarkers that are both sensitive and specific because they rely on more accurate, multi-parameter indicators.
Acknowledgments
We thank all members of our laboratories for helpful discussions. This work was supported by grants from the Canadian Institutes for Health Research (CIHR), the Natural Sciences and Engineering Research Council of Canada (NSERC), Genome Canada and Génome Québec. C.J. holds studentships from the CIHR and the Fonds de la recherche en santé du Québec (FRSQ).
Footnotes
This paper is one of a selection of papers published in this Special Issue, entitled CSBMCB — Systems and Chemical Biology, and has undergone the Journal’s usual peer review process.
Contributor Information
Benoit Coulombe, Gene Transcription and Proteomics Laboratory and Proteomics Discovery Platform, Institut de recherches cliniques de Montréal, Montréal, Québec, Canada; Département de biochimie, Université de Montréal, Montréal, Québec, Canada.
Mathieu Blanchette, McGill Centre for Bioinformatics, McGill University, Montréal, Québec, Canada.
Célia Jeronimo, Gene Transcription and Proteomics Laboratory and Proteomics Discovery Platform, Institut de recherches cliniques de Montréal, Montréal, Québec, Canada.
References
- Aebersold R, Mann M. Mass spectrometry-based pro-teomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Alberts B. The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell. 1998;92:291–294. doi: 10.1016/S0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- Bader GD, Betel D, Hogue CW. BIND: the Biomo-lecular Interaction Network Database. Nucleic Acids Res. 2003;31:248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboric M, Kohoutek J, Price JP, Blazek D, Price DH, Peterlin BM. Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J. 2005;24:4291–4303. doi: 10.1038/sj.emboj.7600883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazek D, Barboric M, Kohoutek J, Oven I, Peterlin BM. Oligomerization of HEXIM1 via 7SK snRNA and coiled-coil region directs the inhibition of P-TEFb. Nucleic Acids Res. 2005;33:7000–7010. doi: 10.1093/nar/gki997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- Byers SA, Price JP, Cooper JJ, Li Q, Price DH. HEXIM2, a HEXIM1-related protein, regulates positive transcription elongation factor b through association with 7SK. J Biol Chem. 2005;280:16360–16367. doi: 10.1074/jbc.M500424200. [DOI] [PubMed] [Google Scholar]
- Cojocaru M, Jeronimo C, Forget D, Bouchard A, Bergeron D, Cote P, et al. Genomic location of the human RNA polymerase II general machinery: evidence for a role of TFIIF and Rpb7 at both early and late stages of transcription. Biochem J. 2008;409:139–147. doi: 10.1042/BJ20070751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe B, Jeronimo C, Langelier MF, Cojocaru M, Bergeron D. Interaction networks of the molecular machines that decode, replicate and maintain the integrity of the human genome. Mol Cell Proteomics. 2004;3:851–856. doi: 10.1074/mcp.R400009-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Ma Y, Jaramillo M, Bari H, Awan A, Yang S, et al. A map of human cancer signaling. Mol Syst Biol. 2007;3:152. doi: 10.1038/msb4100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusick ME, Klitgord N, Vidal M, Hill DE. Interac-tome: gateway into systems biology. Hum Mol Genet. 2005;14(Spec No 2):R171–R181. doi: 10.1093/hmg/ddi335. [DOI] [PubMed] [Google Scholar]
- Devos D, Russell RB. A more complete, complexed and structured interactome. Curr Opin Struct Biol. 2007;17:370–377. doi: 10.1016/j.sbi.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, et al. Functional organization of the yeast pro-teome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, et al. A protein interaction map of Drosophila melano-gaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- Han JD, Bertin N, Hao T, Goldberg DS, Berriz GF, Zhang LV, et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C, Langelier MF, Zeghouf M, Cojocaru M, Bergeron D, Baali D, et al. RPAP1, a novel human RNA polymerase II-associated protein affinity purified with recombinant wild-type and mutated polymerase subunits. Mol Cell Biol. 2004;24:7043–7058. doi: 10.1128/MCB.24.16.7043-7058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell. 2007;27:262–274. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Li Q, Price JP, Byers SA, Cheng D, Peng J, Price DH. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phos-phorylated at threonine 186. J Biol Chem. 2005;280:28819–28826. doi: 10.1074/jbc.M502712200. [DOI] [PubMed] [Google Scholar]
- Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, et al. Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- Lu SC. S-Adenosylmethionine. Int J Biochem Cell Biol. 2000;32:391–395. doi: 10.1016/S1357-2725(99)00139-9. [DOI] [PubMed] [Google Scholar]
- Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997;11:2633–2644. doi: 10.1101/gad.11.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;270:12335–12338. doi: 10.1074/jbc.270.44.26303. [DOI] [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Guldener U, Mannhaupt G, Mayer K, Mokrejs M, et al. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 2002;30:31–34. doi: 10.1093/nar/30.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S, Salwinski L, Kerrien S, Montecchi-Palazzi L, Oesterheld M, Stumpflen V, et al. The minimum information required for reporting a molecular interaction experiment (MIMIx) Nat Biotechnol. 2007;25:894–898. doi: 10.1038/nbt1324. [DOI] [PubMed] [Google Scholar]
- Patil A, Nakamura H. Filtering high-throughput protein-protein interaction data using a combination of genomic features. BMC Bioinformatics. 2005;6:100. doi: 10.1186/1471-2105-6-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri S, Navarro JD, Amanchy R, Kristiansen TZ, Jonnalagadda CK, Surendranath V, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–2371. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranish JA, Yi EC, Leslie DM, Purvine SO, Goodlett DR, Eng J, Aebersold R. The study of macromolecular complexes by quantitative proteomics. Nat Genet. 2003;33:349–355. doi: 10.1038/ng1101. [DOI] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Rives AW, Galitski T. Modular organization of cellular networks. Proc Natl Acad Sci USA. 2003;100:1128–1133. doi: 10.1073/pnas.0237338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dri-cot A, Li N, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Shoemaker BA, Panchenko AR. Deciphering protein-protein interactions. Part II Computational methods to predict protein and domain interaction partners. PLoS Comput Biol. 2007;3:e43. doi: 10.1371/journal.pcbi.0030043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spirin V, Mirny LA. Protein complexes and functional modules in molecular networks. Proc Natl Acad Sci USA. 2003;100:12123–12128. doi: 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Xia Y, Yu H, Jansen R, Seringhaus M, Baxter S, Greenbaum D, et al. Analyzing cellular biochemistry in terms of molecular networks. Annu Rev Biochem. 2004;73:1051–1087. doi: 10.1146/annurev.biochem.73.011303.073950. [DOI] [PubMed] [Google Scholar]
- Yik JH, Chen R, Pezda AC, Zhou Q. Compensatory contributions of HEXIM1 and HEXIM2 in maintaining the balance of active and inactive positive transcription elongation factor b complexes for control of transcription. J Biol Chem. 2005;280:16368–16376. doi: 10.1074/jbc.M500912200. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Chen D, Pierstorff E, Luo K. Transcription elongation factor P-TEFb mediates Tat activation of HIV-1 transcription at multiple stages. EMBO J. 1998;17:3681–3691. doi: 10.1093/emboj/17.13.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, et al. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11:2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]