

Abstract
Airway inflammation is the hallmark of many respiratory disorders, such as asthma and cystic fibrosis. Changes in airway gene expression triggered by inflammation play a key role in the pathogenesis of these diseases. Genetic linkage studies suggest that ESE-2 and ESE-3, which encode epithelium-specific Ets-domain-containing transcription factors, are candidate asthma susceptibility genes. We report here that the expression of another member of the Ets family transcription factors ESE-1, as well as ESE-3, is upregulated by the inflammatory cytokines interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) in bronchial epithelial cell lines. Treatment of these cells with IL-1β and TNF-α resulted in a dramatic increase in mRNA expression for both ESE-1 and ESE-3. We demonstrate that the induced expression is mediated by activation of the transcription factor NF-κB. We have characterized the ESE-1 and ESE-3 promoters and have identified the NF-κB binding sequences that are required for the cytokine-induced expression. In addition, we also demonstrate that ESE-1 upregulates ESE-3 expression and downregulates its own induction by cytokines. Finally, we have shown that in Elf3 (homologous to human ESE-1) knockout mice, the expression of the inflammatory cytokine interleukin-6 (IL-6) is downregulated. Our findings suggest that ESE-1 and ESE-3 play an important role in airway inflammation.
Keywords: epithelium, transcription factor, airway disease, asthma, gene regulation
Introduction
The airway epithelium is the first layer of lung tissue to encounter environmental stimuli such as microbes and allergens. In addition to acting as a physical barrier, airway epithelial cells are involved in the manifestation of airway inflammatory diseases [1–3]. In response to environmental perturbations, airway epithelia can produce and release a variety of inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), IL-6 and IL-8. In addition, airway epithelia are targets for inflammatory cytokines from a variety of sources. For example, TNF-α has been shown to alter epithelial cell permeability, as well as to stimulate IL-6, IL-8 and GM-CSF expression [3]. A number of studies have shown that the epithelium of patients who have airway inflammatory diseases is structurally and functionally altered [4–7], and that bronchial epithelial cells that are isolated from patients with asthma or cystic fibrosis (CF) express increased levels of cytokines [1, 3]. Therefore, it is important to fully understand gene regulation in airway epithelium in order to alleviate airway inflammatory diseases.
Over the past decade, a set of epithelium-specific Ets-like factors, ESE-1, ESE-2 and ESE-3, have been identified [8–10]. These genes encode proteins that belong to a large family of transcription factors, known as Ets transcription factors. Ets factors are defined by a highly conserved DNA-binding domain called the Ets domain which binds to a purine-rich GGAA/T core motif in enhancer and promoter regions of target genes [11]. ESE-1, ESE-2 and ESE-3 proteins, as well as some of the other Ets factors, also contain a pointed domain, which is involved in protein-protein interactions [12].
Many Ets family members are expressed in hematopoietic cells [13]. However, the three ESE transcription factors are expressed only in organs that have specialized epithelial cells, such as intestine, kidney, prostate, pancreas and lung [8, 9, 14–20]. ESE-1 is expressed in many different organs, whereas ESE-2 and ESE-3 expression is more restricted to the glandular organs, such as the salivary gland, mammary gland and prostate. ESE-1, ESE-2 and ESE-3 are expressed at high levels in the trachea. In the lung ESE-2 is expressed only at low levels, whereas both ESE-1 and ESE-3 are moderately expressed [8, 9, 14–20].
When Elf3, the murine homologue for the human ESE-1 gene, is disrupted, 30% of homozygous mutant mice die around embryonic day 11.5, indicating that there is a role for Elf3 in fetal development [21]. Surviving adult mice that lack Elf3 show a distinct phenotype in the small intestine that includes severe morphological alterations in the mucosa. In addition, Elf3 knockout mice develop a ‘wasting syndrome’ that is characterized by a malnourished physical appearance, watery diarrhea and lethargy. Defects in airway function have not been examined [21]. Knocking out Elf5, the murine homologue of the human ESE-2 gene, results in embryonic lethality, which indicates that Elf5 is essential for embryonic development. Heterozygous Elf5 mice show complete developmental arrest of the mammary gland in pregnant mice [22].
In addition to their role in development, these ESE transcription factors have been reported to be involved in inflammation. The expression ofESE-1 can be induced by β and TNF-α) in cultured non-epithelial cells, such as vascular smooth muscle cells, endothelial cells, monocytes[23]andfibroblasts[24, 25]. ESE-3 mRNA expression is induced in bronchial smooth muscle cells and fibroblasts and is constitutively expressed in human bronchial epithelial cells [26]. However, it is unknown whether these ESE genes are regulated by cytokines in airway epithelial cells. The induction of ESE-1 expression requires the activation of NF-κB [25]. The NF-κB pathway is one of the most important signaling cascades for the regulation of inflammation, which includes cyto-kine expression and immune responses [27, 28]. NF-κB is activated by stimuli that include cytokines such as IL-1β and TNF-α, bacterial lipopolysaccharide (LPS), and viral proteins. For example, binding of IL-1β and TNF-α to their cell surface receptors leads to activation of the cytokine-induced IκB kinase complex (IKK), and results in phos-phorylation and subsequent degradation of IκB (an NF-κB inhibitory protein), which allows NF-κB to translocate into the nucleus and activate target genes [28].
In this study, we have demonstrated that ESE-1 and ESE-3 expression is upregulated in human bronchial epithelial cells by inflammatory cytokines, and that this upregulation is mediated by the NF-κB pathway. We have also characterized the ESE-1 and ESE-3 promoters. We found that ESE-1 downregulates its own induction by cytokines. We also found that ESE-3 is regulated by ESE-1. Finally, we have demonstrated that IL-6 expression is downregulated in knockout mice lacking Elf3, and that the IL-6 promoter can be upregulated by ESE-1 overexpression.
Results
ESE-1 and ESE-3 are upregulated in bronchial epithelial cells in response to inflammatory cytokines
The airway epithelia are exposed to a variety of external substances that can induce inflammatory cytokine expression. To examine whether ESE-1 and ESE-3 expression in epithelial cells can be induced by IL-1β and/or TNF-α as in non-epithelial cells [23–26], we stimulated cultured human bronchial epithelial cells (BEAS-2B) with IL-1β and/or TNF-α. Using semi-quantitative RT-PCR (data not shown), we detected increased ESE-1 and ESE-3 but not ESE-2 mRNA expression after cytokine induction.
To quantitatively measure the level of induction, we carried out real-time RT-PCR analysis. As shown in Figure 1A, ESE-1 mRNA expression in BEAS-2B cells increased by more than 10-fold with the addition of TNF-α, more than 40-fold with the addition of IL-1β and more than 80-fold with a combination of IL-1β and TNF-α (2 h). The ESE-1 mRNA response was rapid, with maximal induction occurring 2 h after stimulation. By contrast, although IL-1β and TNF-α induced ESE-3 expression, the response time was slow (maximum at 24 h) compared with ESE-1. In addition, the magnitude of the response was smaller than the ESE-1 response (Figure 1B). For both ESE-1 and ESE-3, IL-1β induced a stronger response than TNF-α. This difference may partially reflect the availability of different membrane receptors or the potency of individual cytokines. mRNA expression of ESE-1 and ESE-3 was synergistically induced when both IL-1β and TNF-α were used. Therefore, we used both IL-1β and TNF-α for all subsequent experiments to examine cell stimulation. In agreement with its induction of mRNA expression, we observed a dramatic increase in ESE-1 protein (Figure 1C). As expected, the protein induction occurred after the RNA levels increased, peaked at 4 h and remained at a relatively high level for at least 6 h. These experiments were repeated in two other bronchial epithelial cell lines: IB3-1, cells derived from a patient with CF [29] and C38, CF-corrected IB3-1 [30], with similar results. Because ESE-3 protein was expressed at a much lower level, it was difficult to detect using western blot analysis (data not shown). In conclusion, endogenous ESE-1 and ESE-3 are upregulated by cytokines in bronchial epithelial cells.
Figure 1.

Induction of ESE-1 and ESE-3 mRNA expression, and ESE-1 protein expression by IL-1β and/or TNF-α in BEAS-2B human bronchial epithelial cells. (A, B) ESE-1 and ESE-3 mRNAs in BEAS-2B cells treated with IL-1β, TNF-α or a combination of IL-1β and TNF-α (Cyt), at 10 ng/ml each, for the indicated time periods (1–48 h). (C) ESE-1 protein in BEAS-2B cells treated with a combination of IL-1β and TNF-α, at 10 ng/ml each, for the indicated time periods. Nuclear extracts from treated and untreated cells were analyzed by western blotting with a polyclonal antibody specific for ESE-1. G3PDH was included as a protein loading control. (D, E). The effects of NF-κB inhibitors on cytokine-induced ESE-1 and ESE-3 mRNA expression. BEAS-2B cells were treated with increasing amounts of CAPE or Genistein (Gen.) as indicated for 2 h and then exposed to IL-1β and TNF-α (Cyt, at 10 ng/ml each) for 2 h (D) or 24 h (E). ESE-1 and ESE-3 mRNA expression was determined by real-time quantitative PCR. Values shown are the mean ± SD (n = 3), *P < 0.05 compared with the IL-1β+TNF-α group.
IL-1β and TNF-α are powerful activators in the NF-κB signaling pathway [28]. We investigated the involvement of NF-κB in the induction of ESE-1 and ESE-3 expression using caffeic acid phenethyl ester (CAPE), an NF-κB inhibitor [31], or genistein, a tyrosine kinase inhibitor [32–34]. As shown in Figure 1D and 1E, these inhibitors effectively blocked cytokine induction of both ESE-1 and ESE-3 expression as assessed by real-time RT-PCR. This suggests that NF-κB is involved in the induction of ESE-1 and ESE-3 expression.
NF-κB is directly involved in inducing ESE-1 and ESE-3 expression through binding to their promoters
To investigate whether NF-κB is directly involved in the induction of ESE-1 and ESE-3 expression, we examined their promoter regions for putative NF-κB binding sites using the computer program MatInspector 2.1 [35]. We identified4 and 3 putative NF-κB binding sites within the 1.2 kb promoter fragments of ESE-1 and ESE-3, respectively. These sites in the ESE-1 promoter region are GGA AAT CCC C (position −87 to −78), AGG AAT TTC C (−319 to −310), GGG TTT TTC C (−473 to −482) and GGG AAT AAC C (−1 014 to −1 005). The sites in the ESE-3 promoter region are GGG AAT TCC C (−119 to −110), GGG AAG GTC C (−674 to −683) and GGG AGT TAC C (−983 to −992). To test whether these putative NF-κB binding sites in the promoter regions are functional, we cloned both promoter fragments and used them to drive expression of the reporter gene SEAP2 (Figure 2A and 2B). We assessed the promoter activity in IB3-1 bronchial epithelial cells and A549 human lung carcinoma cells because both of these transfected than BEAS-2B celllines aremoreefficiently cells. As shown in Figure 2C–2F, both promoters can be upregulated by the cytokines, which indicates the presence of functional NF-κB binding sites in each of the promoter fragments.
Figure 2.

Involvement of NF-κB in cytokine-induced ESE-1 and ESE-3 expression. (A, B) Schematic diagrams of human ESE-1 and ESE-3 promoter SEAP2 reporter plasmids. The NF-κB binding sites that were analyzed are shown in the rectangle and the mutants that were introduced by PCR are shown as bold characters. (C–F) Transactivation of the ESE-1 and ESE-3 promoters by cytokines and requirement of the NF-κB binding site for transactivation. IB3-1 or A459 cells were transfected with 1.5 μg promoter-SEAP reporter plasmid containing either the vector alone or wild-type (wt) or mutated (gga) NF-κB sites for 24 h. The media were changed and the cells were then stimulated with IL-1β and TNF-α (at 10 ng/ml each) for 24 h. SEAP activity was measured as relative light units by a luminometer. Values shown are the mean ± SEM (n = 3) from one representative of three independent experiments. *P < 0.05.
To examine which NF-κB sites in the ESE-1 and ESE-3 promoters are essential for cytokine induction, we selected the NF-κB sites most proximal to the transcription start sites [25] and performed site-direct mutagenesis. We modified the ESE-1 NF-κB binding site GGA AAT CCCC (position –87 to –78) to GGA AAT CGGA and the ESE-3 NF-κB binding site GGG AAT TCCC (–119 to –110) to GGG AAT TGGA; transfection was performed as above. Both ESE-1 and ESE-3 promoters were fused to SEAP2 reporter vectors and transfected into IB3-1 and A549 cells. SEAP activity was measured in the presence and absence of cytokines in both cell lines. These mutations completely blocked upregulation of ESE-1 and ESE-3 by cytokines both in IB3-1 and in A549 cells (Figure 2C–2F). This indicates that the NF-κB binding site most proximal to the transcription start site in each promoter is required for cytokine induction of the promoter.
To investigate whether NF-κB binds to the ESE-1 and ESE-3 promoters during cytokine induction, we performed electrophoretic mobility shift assays (EMSA) using nuclear extracts from BEAS-2B cells with or without cytokine treatment. We used the NF-κB binding sites identified above, position –87 to –78 in the ESE-1 promoter and position –119 to –110 in the ESE-3 promoter, and their mutants as probes in the EMSA (Figure 3A). An NF-κB binding site from the IL-6 promoter was included as a positive control [36]. As indicated in Figure 3B, both of these ESE-1 and ESE-3 NF-κB binding sites can be shifted by nuclear extracts from cells stimulated with IL-1β and TNF-α (lanes 6 and 12). The three base pair mutations of the NF-κB sites completely abolished the binding activities (lanes 9 and 15). Interestingly, the same DNA/protein complexes are also presented in the nuclear extracts without cytokine stimulation, although at a much lower lever (lanes 2, 5 and 11). To verify the presence of NF-κB in these DNA/protein complexes, we performed supershift assays and found that antibodies against the NF-κB p65 and p50 subunits can supershift the complexes (Figure 3C, lanes 8, 9, 13 and 14). These experiments demonstrate that NF-kB binds to the ESE-1 and ESE-3 promoters.
Figure 3.

Binding of NF-κB to the ESE-1 and ESE-3 promoters. (A) Putative NF-κB binding sites (wt) and their mutants (gga) in human ESE-1 and ESE-3 promoters were analyzed in electrophoretic mobility shift assays (EMSA), using the NF-κB site on the IL-6 promoter as a positive control. (B) EMSA using oligonucleotide probes containing the indicated NF-κB sites (wt or gga) with 5 μg nuclear extract from BEAS-2B cells stimulated with IL-1β and TNF-α (at 10 ng/ml each) for 2 h, or nuclear extract from unstimulated cells. Brackets indicate the specific cytokine-inducible DNA-protein complexes.(C) Supershift and competition assays. Antibodies against the NF-κB subunits p65 and p50 were used to determine the specificity of their binding complexes. Arrows indicate supershift bands with the p65 or p50 subunits, ‘cold’ indicates competition with 500-fold of unlabelled probe.
To confirm whether NF-κB binds to the ESE-1 and ESE-3 promoters in vivo, we performed chromatin immunoprecipitation assays (ChIP) with antibodies against the NF-κB p65 or p50 subunits. We prepared extracts from BEAS-2B and A549 cells with cytokine stimulation for 4 or 24 h or without stimulation (0 h). PCR primers used to amplify genomic DNA fragments of the ESE-1 and ESE-3 promoters are illustrated in Figure 4A. In both cell lines there was an increase in p65 and p50 binding to both the ESE-1 and ESE-3 promoters from 0 to 24 h, with the association with p50 being particularly increased (Figure 4B and 4C). In the absence of cytokine stimulation, p65 and p50 bound weakly to both promoters in these two cell lines. This is consistent with the result that DNA/NF-κB complexes can be detected in non-stimulated BEAS-2B cells by a mobility shift assay (Figure 3B). These results suggest that NF-κB may play a role in the basal level of ESE-1 and ESE-3 gene expression in BEAS-2B and A549 cells. These results, together with the transfection analysis, indicate that NF-κB is directly involved in the induction of ESE-1 and ESE-3 expression by inflammatory cytokines through binding to their promoters.
Figure 4.

NF-κB binding to the ESE-1 and/or ESE-3 promoters in vivo. (A) Schematic diagrams of the PCR primer positions as indicated by arrows. Ovals represent the NF-κB binding sites. (B, C) Chromatin immunoprecipitation (ChIP) assays. Chromatin preparations that were made from BEAS-2B or A549 cells stimulated with IL-1β and TNF-α (at 5 ng/ml each) for the time periods indicated were precipitated using antibodies against NF-κB p65 or p50. ‘Pre’ represents pre-immune rabbit serum and ‘input’ represents a PCR control using chromatin without precipitation.
ESE-1 induction is downregulated by itself
Inflammatory processes can be controlled by feedback mechanisms [37]. Because ESE-1 mRNA peaked at 2 h in response to cytokines and then rapidly decreased (Figure 1A), we wanted to determine whether there is a feedback mechanism for this regulation. To this end, we performed co-transfection analysis of A549 cells with two plasmids one containing an ESE-1 promoter (–1 134 to +15) fused to the SEAP reporter gene and a second one overexpressing the ESE-1 protein. In our control experiments, the promoterless SEAP vector exhibited minimal background reporter activity in cells overexpressing ESE-1, which was in contrast to the promoterless SEAP2 vector (data not shown). Therefore, for co-transfection studies we used SEAP reporter constructs. We used A549 cells in the transfection studies because these cells are more efficiently transfected. By this co-transfection analysis, we found that overexpression of ESE-1 inhibited its own induction by cytokines (Figure 5A). Interestingly, overexpression of ESE-3 also inhibited ESE-1 induction; this implies that there is a feedback loop in control of cytokine-induced ESE-1 expression.
Figure 5.
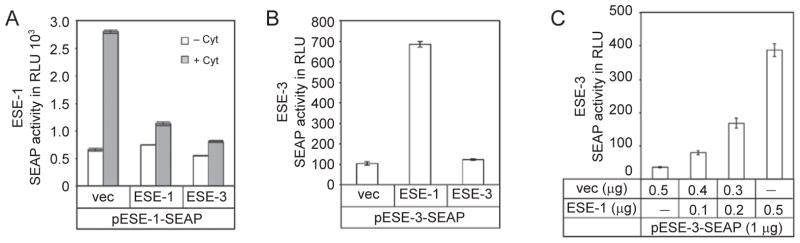
Overexpression of ESE-1 or ESE-3 affects promoter activity differently. (A) Overexpression of ESE-1 or ESE-3 down-regulates ESE-1 induction by cytokines. A549 cells were co-transfected with 1 μg of ESE-1 promoter reporter plasmid and 0.5 μg of pCDNA3 (vec), or pCDNA3 containing ESE-1 or ESE-3. At 24 h after transfection, the cells were cultured in new media with or without the presence of IL-1β and TNF-α (at 10 ng/ml each) for 24 h. (B) Overexpression of ESE-1 upregulates ESE-3 expression. A549 cells were co-transfected with 1 μg of ESE-3 promoter SEAP reporter plasmid and 0.5 μg of an expression vector, pCDNA3 (vec), or pCDNA3 containing ESE-1 or ESE-3. (C) Dose-dependent enhancement of ESE-3 expression by ESE-1. A549 cells were co-transfected with 1 μg of ESE-3 promoter reporter plasmid and different amount of ESE-1 expression plasmid as indicated. SEAP activity was measured as relative light units using a luminometer. Values shown are the mean ± SEM (n = 3) from one representative of three independent experiments.
ESE-3 is upregulated by ESE-1
Since ESE-1 expression is highly induced by inflammatory cytokines, and ESE-3 is implicated in asthma and other inflammatory diseases (Brooks-Wilson et al., US Patent #6087485), we tested whether ESE-1 regulates ESE-3 expression. We cloned the ESE-3 promoter fragment, as illustrated in Figure 2B, into the SEAP reporter plasmid. Co-transfection analysis in A549 cells showed that ESE-1 overexpression enhanced ESE-3 promoter-SEAP expression (Figure 5B). By contrast, overexpression of ESE-3 did not affect its own expression. We further demonstrated that the enhancement was dependent on the amount of ESE-1 plasmid used (Figure 5C). Note that the activity of SEAP is much lower than that of SEAP2 (compare Figure 2F with Figure 5B and 5C).
To further confirm the role of ESE-1 in its own regulation and the regulation of ESE-3, we designed an HD-Ad vector expressing two shRNAs to knock down ESE-1 expression (Figure 6A). These shRNAs were able to knock down both endogenous and exogenous ESE-1 expression as shown by western blotting and real-time RT-PCR in A549 cells (Figure 6B and 6C). The auto-inhibition of ESE-1 SEAP reporter expression that we observed (Figure 5A) was partially relieved by using shRNAs to target ESE-1 (Figure 6B). To examine whether ESE-1 is required for endogenous ESE-3 expression under constitutive conditions, we transduced ESE-1-expressing A549 cells with the ESE-1-RNAi virus for 3 days. Using these cells, we subsequently performed real-time quantitative RT-PCR in order to determine the levels of ESE-1 and ESE-3 transcripts. As expected, there was a dramatic decrease in the ESE-1 mRNA (~90%); this was consistent with the decrease in both endogenous and exogenous ESE-1 protein observed (Figure 6B). In the same experiment, the ESE-1-RNAi virus greatly reduced (58%) endogenous ESE-3 mRNA levels compared with the control virus (Figure 6C). The level of GAPDH mRNA expression was not significantly affected. We obtained similar results in BEAS-2B cells (data not shown). These results confirm a role for ESE-1 in its auto-inhibition and demonstrate that ESE-1 regulates endogenous ESE-3 expression.
Figure 6.

Effects of knocking down ESE-1 expression on ESE-1 promoter activity and endogenous ESE-3 expression. (A) Schematic diagrams of helper-dependent adenoviral (HDAd) vectors. HDAd-ESE-1-RNAi expresses two shRNAs from the murine U6 gene promoter. C4HSU is a control vector that does not express any transgene. ITR, inverted terminal repeat; ψ, packing signal. (B) Promoter activity analysis in ESE-1 knocked down cells. A549 cells were transduced with a control vector, C4SHU, or HDAd-ESE-1-RNAi overnight and then transfected and stimulated as before. Inhibition of cytokine-induced ESE-1 promoter activity by ESE-1 overexpression was partially relieved when ESE-1 was knocked down by HDAd-ESE-1-RNAi. ESE-1 expression (endogenous and/or exogenous) was undetectable when cells were transduced with HDAd-ESE-1-RNAi as shown using the western blotting technique. (C) Endogenous ESE-1 regulates constitutive ESE-3 gene expression. Cells were collected 3 days after virus transduction. Relative mRNA levels were calculated after normalizing to 18S using real-time RT-PCR. Data shown are the mean ± SD (n = 3) from one representative of three independent experiments.
Promoter analysis of the ESE-1 and ESE-3 genes
To further examine ESE-1 auto-regulation, we generated deletions in the ESE-1 promoter region as well as specific mutations in the Ets binding site and cloned them into the SEAP reporter plasmid (Figure 7A). Co-transfections with the ESE-1 expression vector and the ESE-1 promoter SEAP reporter were performed in A549 cells with or without cytokine stimulation. As shown in Figure 7B, removal of the region distal to −256 had a minimal effect on ESE-1 auto-inhibition, although the level of reporter expression induced by cytokines was reduced (compare pESE-1- with pN-, pS- and pB-SEAP). We then focused on the putative Ets binding site that was detected by the computer program MatIspector 2.1 (CCA GGA AAT C nucleotides −90 to −81, which overlap with the NF-κB site) in the proximal promoter region. Mutation at this site (nucleotides −86 to −85; pmEts-SEAP) completely abolished the induction of the ESE-1 reporter expression by cytokines (Figure 7B). Since these Ets sites overlap with the NF-κB site, it is possible that ESE-1 auto-inhibition is mediated through competitive binding of ESE-1 and NF-κB to the same site.
Figure 7.

Deletion and mutation analyses of the ESE-1 and ESE-3 promoters. (A) ESE-1 promoter deletion and mutant reporter plasmids. pSEAP is a vector plasmid containing a promoterless SEAP reporter gene. pESE-1-SEAP is a reporter plasmid containing the full-length promoter. pN, pS and pB are promoter deletion reporter plasmids that are derived from pESE-1-SEAP using restriction enzymes as indicated. pmEts is a full-length promoter reporter with the Ets/NF-κB site mutated. The oval and diamond represent the NF-κB and Ets binding sites, respectively. (B) ESE-1 reporter expression in A549 cells co-transfected with 1 μg of an ESE-1 promoter reporter plasmid and 0.5 μg of an ESE-1 expression plasmid or empty vector. At 24 h after the transfection, the cells were cultured in new media with or without the addition of IL-1β and TNF-α (at 10 ng/ml each) for 24 h. (C) Human ESE-3 promoter wild-type and mutant reporter plasmids. pESE-3-SEAP is a full-length ESE-3 promoter reporter plasmid. The oval and diamonds represent the NF-κB and Ets binding sites, respectively. pmEts1+2, pmEts3, pmEts3+4 and pmEts1+2+3+4 are mutant ESE-3 promoter plasmids with the mutated nucleotides shown in bold. △ indicates deletion of nucleotides. (D) ESE-3 reporter expression in A549 cells co-transfected with 1 μg of an ESE-3 promoter reporter plasmid and 0.5 μg of an ESE-1 expression plasmid or pCDNA3 vector. SEAP activity was measured as relative light units using a luminometer. Values shown are the mean ± SEM (n = 3) from one representative of three independent experiments with similar results. (E) EMSA of ESE-1 protein binding to ESE-1 or ESE-3 promoter fragments or double-stranded oligo nucleotides containing putative Ets binding sites. Full-length ESE-1 fusion protein with a His-tag was expressed and purified fromE. coli. E74 probe was used as a positive control for ESE-1 binding. Bracket indicates specific DNA-protein complexes. ‘Cold’ refers to the addition of a 500-fold unlabelled competitor probe. (F) Probes used in the EMSA. Gray diamonds represent the putative Ets sites that are mutated in (A) and (C), white diamonds represent the putative Ets sites (GGAA/T) that are not characterized, and ‘+’ and ‘−’ represent the top and bottom strands, respectively.
Next, we examined the ESE-3 promoter using deletions and mutations. Deletion of the ESE-3 promoter region from −1 152 to −210 did not affect upregulation by ESE-1 (data not shown); this suggests that critical binding sites are in the more proximal region. To further characterize putative Ets sites on the ESE-3 promoter for ESE-1 binding, we created a series of mutant ESE-3 promoter plasmids; all the putative Ets binding sites containing the core sequence (GGAA/T) from position −210 to +27 were mutated (Figure 7C). As shown in Figure 7D, individual mutations showed a slight effect on the activation of ESE-3-SEAP reporter expression by ESE-1 (compare pESE-3 with pmEts1+2, 3 and 3+4). In addition, the combination of Ets mutations 1+2+3+4 exhibited a much greater inhibitory effect on activation. This suggests that ESE-1 regulates ESE-3 expression at least partly through its binding to the ESE-3 promoter, and suggests that multiple Ets sites are used for ESE-1 binding.
To determine whether the ESE-1 protein could bind directly to the ESE-1 and ESE-3 promoters, we examined the same Ets sites (Figure 7F) that were mutated in the SEAP transfection analysis (Figure 7A and 7C). His-tagged ESE-1 recombinant protein was expressed and purified from Escherichia coli, and used in mobility shift assays. As shown in Figure 7E and 7F, both ESE-1 and ESE-3 proximal promoter fragments (position −256 to +15 and position −210 to +27, respectively) could be readily shifted by ESE-1 protein (lanes 2 and 5, respectively). The double-stranded DNA oligonucleotides containing the individual putative Ets sites could also be shifted (lanes 10, 12, 14 and 16), although to a lesser degree. Lane 8 showed complex formation of E74 (a well-characterized Ets binding site from Drosophila) and ESE-1 protein. This experiment demonstrates that ESE-1 can bind in vitro to both the ESE-1 and ESE-3 promoters.
Attenuated IL-6 induction by LPS in Elf3 knockout mice
To examine whether ESE-1 is involved in the regulation of inflammatory cytokine expression in the lung, we analyzed cytokine induction by LPS through intranasal instillation using an Elf3 knockout mouse model. We measured multiple cytokines in BALF, sera and lung tissue extracts 6 h after LPS induction using a FlowMetrix System [38]. We examined ten cytokines: IL-1β, IL-6, IL-10, TNF-α, IFNγ, GM-CSF, RANTES, MIP-1α, IP-10 and KC. Among those cytokines, GMCSF, IL-1β, IL-6, IP-10, KC, MIP-1, RANTES and TNF-α were dramatically induced by LPS compared to a saline control in the lung tissue of both wild-type and knockout mice. We found that IL-6 induction by LPS in Elf3 knockout mice was significantly reduced relative to their wild-type littermates; the induction of nine other cytokines was not significantly affected (data not shown). To confirm this result, we performed ELISA assays with an antibody against mouse IL-6. As shown in Figure 8A, the levels of IL-6 induction in BALF, lung tissue extracts and sera were significantly attenuated in Elf3 knockout mice. Without LPS stimulation, the IL-6 levels in wild-type and knockout mice did not vary significantly (P < 0.05). These results suggest that ESE-1 may play an important role in the process of airway inflammation.
Figure 8.
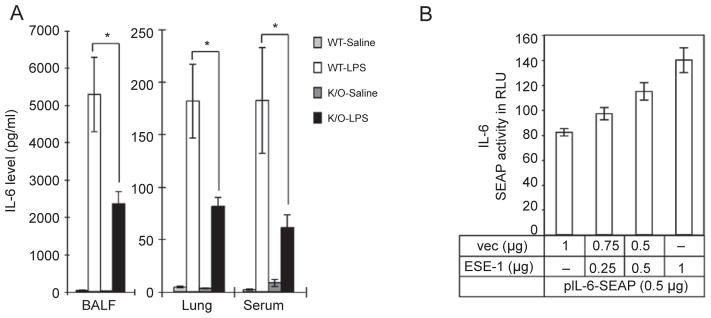
Regulation of IL-6 expression by ESE-1. (A) Levels of IL-6 in BAL fluid, lung tissue extracts and serum from wild-type and Elf3 knockout mice treated with LPS or saline (control) and analyzed with ELISA. Values are the mean ± SEM from 5–7 mice. *P < 0.05. (B) Overexpression of ESE-1 upregulates IL-6 promoter activity in a dose-dependent manner. A549 cells were co-transfected with 0.5 μg of the IL-6 promoter reporter plasmid and different amounts of the ESE-1 expression plasmid as indicated. SEAP activities were measured as relative light units using a luminometer. Values shown are the mean ± SEM (n = 3) from one representative of two independent experiments with similar qualitative results.
To further investigate whether ESE-1 directly regulates human IL-6 expression, we generated a human IL-6 promoter (~1.2 kb)-SEAP reporter plasmid and measured SEAP activity. Co-transfection analysis was performed in A549 cells with a plasmid expressing ESE-1 protein, and the IL-6 promoter SEAP reporter plasmid. As shown in Figure 8B, ESE-1 upregulated IL-6 SEAP reporter expression in a dose-dependent manner. This result, together with the downregulation of IL-6 observed in the Elf3 knockout mice, suggests that IL-6 is regulated directly by ESE-1.
Discussion
The control and regulation of the ESE subfamily of the Ets factors is largely unknown. Although the expression of ESEs is normally confined to epithelial cells, cytokine-regulated ESE expression exists in other cell types. For example, in response to inflammatory cytokines, ESE-1 is expressed in monocytes [23], chondrocytes and fibroblasts [24, 25]. We report here that the expression of ESE-1 and ESE-3 in airway epithelial cells can be dramatically enhanced by inflammatory cytokines. Such regulation is important because the airway epithelium is the first layer of lung cells to encounter external inflammatory stimuli, such as bacteria or viruses. A number of studies have suggested that airway epithelia play an active role in the development of chronic airway inflammatory diseases [1–3, 7, 39]. For example, the airway epithelia secrete a variety of cytokines to initiate immune responses [1, 3], and contribute to the irreversible structural changes of the airway that are referred to as ‘airway remodeling’, which occurs in patients with asthma [3, 7]. Our findings that bothESE-1 and ESE-3 are highly inducible in bronchial epithelial cells suggest that these transcription factors may be critical in the regulation of airway inflammation.
We have identifiedESE-3 as a downstream target gene of ESE-1. Like ESE-1, expression of ESE-3 is also induced by inflammatory cytokines in airway epithelial cells; the induction time course demonstrates that ESE-1 responds more rapidly to cytokine stimulation than ESE-3. This quick induction of ESE-1 is consistent with its role in regulating ESE-3. In addition, we demonstrate that ESE-1 is a positive regulator for constitutive ESE-3 expression.
Interestingly, a genetic linkage analysis of the inbred population of the remote island of Tristan da Cunha led to the discovery of ASTH1-I and ASTH1-J, two asthma candidate genes (Brooks-Wilson et al., US Patent #6087485). ASTH1-I and ASTH1-J are located close to each other on chromosome 11p12-15 and are now known as ESE-2 and ESE-3, respectively. Together with our finding thatESE-3 is upregulated by cytokines in bronchial epithelial cells, this supports the idea that ESE-3 may play a role in the pathogenesis of asthma. It has been shown that ESE-3 represses expression of the matrix metalloproteinase MMP-1 [18, 26]. MMPs mediate extracellular matrix remodeling during processes such as wound repair [40, 41]. Another member of the metalloproteinase family, ADAM-33, has been recently identified as an asthma gene [40–42]. Since MMP-1 is elevated in asthmatic airway tissue extracts [43], lack of ESE-3 function or a defective ESE-1 gene could increase susceptibility to the development of asthma.
Our finding that IL-6 expression is regulated by ESE-1 provides another piece of evidence for ESE-1 functioning as a regulator of the inflammatory process. IL-6 is a pleiotropic cytokine with a variety of biological activities not only in immune regulation, hematopoiesis and oncogenesis, but also in inflammation [44]. Moreover, IL-6 is considered a key modulator in chronic airway inflammatory disease such as asthma [1, 45, 46]. Airway epithelial cells can synthesize and release IL-6 in response to inflammatory stimuli. IL-6 induces T-cell activation and proliferation [1, 47, 48] and augments immunoglobulin production by B-lymphocytes [49]. However, a study using IL-6 knockout mice shows that IL-6 has an anti-inflammatory role during acute inflammatory responses by controlling the level of pro-inflammatoy cytokines such as TNF-α and MIP-2 [50]. IL-6 also stimulates hepatocytes to produce acute-phase proteins (such as α1-antitrypsin and α1-antichymotrypsin), which show anti-inflammatory properties in vivo [51]. Therefore, ESE-1, through its regulation of IL-6, could act as a dual function factor involved in inflammation. For example, ESE-1 is induced by inflammatory cytokines and it upregulates other inflammation mediators such as IL-6. By contrast, ESE-1 can downregulate its own expression and upregulate inflammation repressors such asESE-3. This type of regulation is critical for airway immunity. Upon infection by microorganisms, an inflammatory process may be required to fight the infection. Once the infectious organisms are eliminated, the inflammatory process must be shut down and any damage to tissues must be repaired. It is possible that both ESE-1 and ESE-3 play a role in the control of inflammatory process.
Materials and Methods
Cell culture and reagents
BEAS-2B, a cell line established from normal human bronchial epithelial cells [52], was obtained from the American Type Culture Collection. Cells were maintained in BEBM (referred to as basal medium) plus BEGM supplements (referred to as complete medium) (Clonetics, Walkersville, MD). A549, a human lung carcinoma cell line, was cultured in DMEM medium (Invitrogen Canada, Burlington, Ontario) with 10% fetal bovine serum.
Recombinant human IL-1β and TNF-α were obtained from R&D Systems (Minneapolis, MN) and reconstituted in PBS containing 0.1% bovine serum albumin (BSA). CAPE obtained from Sigma (St. Louis, MO) was prepared in 50% ethanol at 25 mM as stock solution and further dilutions were made in cell culture medium. Genistein obtained from Sigma was prepared in DMSO at a concentration of 100 mM as stock solution and further dilutions were made in cell culture medium.
Cell stimulation and inhibition of NF-κB activity
BEAS-2B cells were seeded at 1.25 × 105 cells per well in 6-well plates with the complete medium and then changed to the basal medium (containing no growth factor) the next day for 24 h. These media were serum free. CAPE or Genistein at various concentrations was added to the cells and incubated for 2 h before exposing the cells to IL-1β and TNF-α each at 10 ng/ml. The induced cells were then harvested for RNA isolation at various time points.
RNA analysis
Total RNA from cells was isolated using an RNeasy Micro Kit (Qiagen, Mississauga, ON) according to the manufacturer’s instructions. For TaqMan real-time RT-PCR, total RNA (1 μg) was reverse transcribed using random hexamers and SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA) following the manufacturer’s protocols. The resulting templates (20 ng cDNA) were used for each real-time PCR reaction (ABI Prism 7700, Applied Biosystems, Foster City, CA). Primers and TaqMan probes for human ESE-1 and ESE-3 were purchased from ABI. All measurements were performed as described [53]. For relative quantification, PCR signals were compared between groups after normalization using GAPDH (GAPDH Control Reagents, ABI) or 18S (Ribosomal RNA Control Reagents, ABI) as an internal reference. Fold change was calculated according to Livak and Schmittgen [53].
Plasmid construction
The human ESE-1 promoter fragment spanning nucleotide positions –1 134 to +15 (GenBank accession no. NT-004671) was amplified by PCR using genomic DNA following primers: 5′-CAA CGC GTA CAA CAC ACC TGC ATA CCT-3′ (forward) and 5′-CCA AGC TTC TAA ATA GAG CCT GGA G-3′ (reverse). PCR products were confirmed by sequencing before subcloning into a SEAP2 or SEAP reporter plasmid (Clontech, Palo Alto, CA). The mutant NF-κB binding site in the ESE-1 promoter (pESE-1-gga; position –80 to –78) were generated by PCR site-directed mutagenesis [54]. The three ESE-1 promoter deletion plasmids were generated by shortening the 5′ promoter region from pESE-1-SEAP using restriction enzymes. The mutation in the Ets site (pmEts; position –86 to –85, nucleotide change of GA to CT) was also generated by PCR-site directed mutagenesis.
The human ESE-3 promoter fragment spanning –1 152 to +27 (GenBank accession no. AF163685) was amplified genomic DNA from HeLa cells as a template with the primers 5′-CCA CGC GTC AAA GAA ATC TGC TTA GAC-3′ and 5′-CCA AGC TTG CAA TCT CAC CAG CTC CTA-3′, then cloned into a SEAP2 or SEAP reporter plasmid. The mutant NF-κB binding site (pESE-3-gga; position –112 to –110) and Ets mutation pmEts1+2 (position –117 to –110) was generated by PCR-site-directed mutagenesis. Three other Ets site mutations were generated by replacing the DNA sequences near the Ets sites with synthetic DNA fragments containing the mutations. For pmEts3, oligos 5′-AAT TCC CGG TTG GTG GCT TAT TGC TTA ACT AGT TAC AAA ATG ATT TAA AAT TAT TGT TAT ATG CA-3′ and 5′-TAT AAC AAT AAT TTT AAA TCA TTT TGT AAC TAG TTA AGC AAT AAG CCA CCA ACC GGG-3′ were annealed and cloned between the EcoRI and NsiI sites of pHESE-3-SEAP. For pmEts3+4, oligos 5′-TTT ATC TTC ACT CTG ATG AGG GCT CAG ACT TGA TAA CGC CCG TGG TGC CCG ATA TCT ATA GGA GCT GGT GAG ATT GCA-3′ and 5′-AGC TTG CAA TCT CAC CAG CTC CTA TAG ATA TCG GGC ACC ACG GGC GTT ATC AAG TCT GAG CCC TCA TCA GAG TGA AGA TAA ATG CA-3′ were annealed and cloned into pmEts3 plasmid between the NsiI to HindIII sites. For pmEts1+2+3+4, pmEts3+4 was further mutated by blunting the EcoRI site.
The human ESE-1 cDNA was amplified from a human intestinal cDNA library [55] by using primers 5′-GCG AAT TCA TGG CTG CAA CCT GTG AGA-3′ (forward) and 5′-CGT AAG CTT CGA GTG GTC CGT GAG TTT GGT-3′ (reverse). Mouse ESE-3 cDNA was amplified by RT-PCR using RNA isolated from 16.5d mouse embryo kidney and the primers 5′-CCG TCG ACC CTT GCA GAT CAT GAT TCT-3′ (forward) and 5′-GGG ATC CGG TTC TTC ATT GAT CAG A-3′ (reverse). The coding sequence of ESE-1 or ESE-3 was then cloned into the mammalian expression vector pcDNA3 (Invitrogen) and the bacterial expression vector pET28a (Novagen, Madison, WI). The human IL-6 promoter fragment spanning from –1 172 to +15 (GenBank accession no. AY170325) was amplified by PCR using human genomic DNA as a template with the following primer set: forward, 5′-CTG GTA CCT CCT GCA AGA GAC ACC ATC CT-3′, reverse, 5′-GGC TCG AGG GCA GAA TGA GCC TCA-3′, and then cloned into the SEAP reporter plasmid. All constructs containing PCR products were confirmed by sequencing.
Transient transfection and reporter assay
For transfection and co-transfection experiments, IB3-1 or A549 cells were seeded in 6-well plates and transfected at 50–60% confluency, using PolyFect transfection reagent (Qiagen) according to the manufacturer’s instructions. After 24 h, cells were cultured in new medium with or without IL-1β and TNF-α (10 ng/ml each). SEAP activity in the culture medium was measured using the Phospha-Light chemiluminescent assay system (Tropix, Bedford, MA) and a luminometer (EG&G Berthold, BadWildbad, Germany).
Electrophoretic mobility shift assays (EMSAs)
Nuclear extracts were prepared as described by Krämer and Keller [56]. EMSAs were performedusingdouble-strandedoligonucleotide four nucleotides (3 out of 4) such probes end-labeled with [γ-32P]-ATP by T4 polynucleotide kinase or [α-32P]-dCTP by Klenow for DNA fragments. For NF-κB EMSAs [57], a labeled probe (20 000 cpm/reaction) was incubated with 5 μg of nuclear extracts from BEAS-2B cells in 20 μl solution consisting of 10 mM Tris (pH 8.0), 15 mM KCl, 1 mM EDTA, 5% glycerol, 1 mM DTT, 1 mg BSA, 1 μg poly(dI-dC) at room temperature for 20 min. For Ets EMSAs, the labeled probes were incubated each with 2.5 μg of purified Ese-1 recombinant protein. In supershift assays, nuclear extracts were incubated with antibodies against NF-κB subunits p65 or p50 from Santa Cruz Biotechnology (Santa Cruz, CA) for 30 min at room temperature before incubation with the probes. The unbound oligonucleotides and protein-DNA complexes were separated on a 4% or 6% polyacrylamide gel (Acylamide/Bis, 29:1) in 0.5 × TBE.
Protein expression, antibody generation and western-blotting analysis
His-tagged full-length ESE-1 recombinant protein was expressed in E. coli BL21-CodonPlus (Stratagene, La Jolla, CA) with pET28a carrying ESE-1 cDNA. The protein was purified with Ni-NTA agarose beads under denaturing conditions, following the manufacturer’s instructions (Qiagen). The purified protein was used as antigen for the generation of antibodies in rabbits as described [55]. Nuclear extracts containing ~20 μg protein per lane were separated by electrophoresis on 10% SDS-PAGE gel and were transferred onto nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA). Blots were blocked in TBST with 5% non-fat milk and 1% BSA and then incubated with anti-ESE-1 polyclonal antibody at 1:2 000 dilution, washed in TBST, and then incubated with horseradish peroxidase-conjugated anti-rabbit IgG from donkey (Amersham Pharmacia Biotech, Piscataway, NJ). Detection of protein was performed with ECL chemiluminescence reagents (Amersham Pharmacia Biotech).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed as described by Boyd and Farnham [58] with minor modifications. In brief, cells were fixed with 1% formaldehyde for 10 min at room temperature and sonication was performed to break chromatin DNA into 100–500 bp fragments in nuclear lysis buffer. In each IP reaction, 3 × 107 cells were used. Antibodies against NF-κB subunits p65 or p50 used for immuno-precipitation were from Epitomics (Burlingam, CA). PCR primer sequences used were as follows: for the ESE-1 promoter, 5′-CCT GAT TTC CCA GCA CTG CCT-3′ (forward) and 5′-CCC TGC AGT AAC CTG AGC TG-3′ (reverse); and for the ESE-3 promoter, 5′-GCT GGG TAG CCA ATC AGA AGA G-3′ (forward) and 5′-CAC GGG CGT TAT CAA GTC TG-3′ (reverse).
Preparation of helper-dependent adenoviral (HD-Ad) vector for shRNA expression
To effectively knock down the endogenous ESE-1 expression, we designed an HD-Ad vector to express two shRNAs targeting the coding sequence of ESE-1. We selected the target sequences based on three criteria. First, we excluded sequences within 150 bp from the translation initiation site to avoid blocking the target sites by proteins involved in the translation. Second, we selected target sequences that were unique to the ESE-1 gene. We compared the potential target sequences with the human genome sequence in the Genbank using the BLASTN program. Finally, we selected the GC-rich target sequences in the first that the stem of the shRNAs would be more stable. The two target sequences were 5′-GGG CAA GAA GAG CAA GCA CGC-3′ and 5′-GGG AGA TCC TGG AAC GGG TGG-3′. A nine-nucleotide loop sequence (GTAAGCTTC) was used for the shRNAs. The design of the mouse U6-promoter-driven shRNA expression cassettes and viral vector was described by Gonzales-Santos et al. [59]. The viral vector was produced and purified as described [57].
Viral transduction
A549 cells were seeded at 2.5 × 105 cells per well in a 6-well dish. After incubation for 4 h, cells were transduced with a viral vector at 2 500 particles/cell (or 50 moi) in culture media without serum. We usually achieved nearly 100% transduction at this vector concentration [57, 59]. After 2 h, fetal bovine serum was added to a final RNA isolation in the following days.
Induction of mouse airway inflammation by LPS
A mouse knockout model of human ESE-1 was established by Ng et al. [21] and homozygous mutant mice and their wild-type littermates were used, with 5–7 mice in each group. In all, 5 μg LPS (E. coli 0111:B4, Sigma) per mouse in 25 μl of 0.9% NaCl was delivered to mouse lungs by nasal aspiration and the animals were sacrificed 6 h after treatment. Sera were collected and bronchoalveolar lavage fluid (BALF) was performed before the lungs were homogenized. Multiple cytokines (IL-1β, IL-6, IL-10, TNF-α, IFNγ, GM-CSF, RANTES, MIP-1α, IP-10 and KC) in BALF, sera, and lung tissue extracts were measured using mouse cytokine Lincoplex kit (Linco Research, Missouri) on a FlowMetrix System [38], Luminex100 (Luminex Corporation, TX). The levels of IL-6 in sera, BAL fluids and lung tissue extracts were measured by ELISA (Bender Medsystems, San Bruno, CA).
Statistical analysis
Data were analyzed using the two-tailed Student t-test comparing two means. P < 0.05 was considered statistically significant.
Acknowledgments
We thank Drs Yu-Hua Chow, Lin Ye and Anan Wang in Dr Hu’s lab for helpful discussions and technical help. We thank Jinxia Wang in Dr Post’s lab for help on real-time PCR. JH was a CCFF Scholar and holds a Premier’s Research Excellence Award of Ontario, Canada. This work was supported by Operating Grants from the Canadian Institutes of Health Research, from the Canadian Cystic Fibrosis Foundation and from the Foundation Fighting Blindness-Canada to JH.
References
- 1.Martin LD, Rochelle LG, Fischer BM, Krunkosky TM, Adler KB. Airway epithelium as an effector of inflammation: molecular regulation of secondary mediators. Eur Respir J. 1997;10:2139–2146. doi: 10.1183/09031936.97.10092139. [DOI] [PubMed] [Google Scholar]
- 2.Poynter ME, Irvin CG, Janssen-Heininger YM. A prominent role for airway epithelial NF-kappa B activation in lipopolysaccharide-induced airway inflammation. J Immunol. 2003;170:6257–6265. doi: 10.4049/jimmunol.170.12.6257. [DOI] [PubMed] [Google Scholar]
- 3.Takizawa H. Airway epithelial cells as regulators of airway inflammation (Review) Int J Mol Med. 1998;1:367–378. doi: 10.3892/ijmm.1.2.367. [DOI] [PubMed] [Google Scholar]
- 4.Bayram H, Devalia JL, Sapsford RJ, et al. The effect of diesel exhaust particles on cell function and release of inflammatory mediators from human bronchial epithelial cells in vitro. Am J Respir Cell Mol Biol. 1998;18:441–448. doi: 10.1165/ajrcmb.18.3.2882. [DOI] [PubMed] [Google Scholar]
- 5.Bayram H, Rusznak C, Khair OA, Sapsford RJ, Abdelaziz MM. Effect of ozone and nitrogen dioxide on the permeability of bronchial epithelial cell cultures of non-asthmatic and asthmatic subjects. Clin Exp Allergy. 2002;32:1285–1292. doi: 10.1046/j.1365-2745.2002.01435.x. [DOI] [PubMed] [Google Scholar]
- 6.Bucchieri F, Puddicombe SM, Lordan JL, et al. Asthmatic bronchial epithelium is more susceptible to oxidant-induced apoptosis. Am J Respir Cell Mol Biol. 2002;27:179–185. doi: 10.1165/ajrcmb.27.2.4699. [DOI] [PubMed] [Google Scholar]
- 7.Davies DE, Wicks J, Powell RM, Puddicombe SM, Holgate ST. Airway remodeling in asthma: new insights. J Allergy Clin Immunol. 2003;111:215–225. doi: 10.1067/mai.2003.128. [DOI] [PubMed] [Google Scholar]
- 8.Oettgen P, Alani RM, Barcinski MA, et al. Isolation and characterization of a novel epithelium-specific transcription factor, ESE-1, a member of the ets family. Mol Cell Biol. 1997;17:4419–4433. doi: 10.1128/mcb.17.8.4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oettgen P, Barcinski M, Boltax J, Stolt P, Akbarali Y, Libermann TA. Genomic organization of the human ELF3 (ESE-1/ESX) gene, a member of the Ets transcription factor family, and identification of a functionalpromoter. Genomics. 1999;55:358–362. doi: 10.1006/geno.1998.5681. [DOI] [PubMed] [Google Scholar]
- 10.Oettgen P, Kas K, Dube A, et al. Characterization of ESE-2, a novel ESE-1-related Ets transcription factor that is restricted to glandular epithelium and differentiated keratinocytes. J Biol Chem. 1999;274:29439–29452. doi: 10.1074/jbc.274.41.29439. [DOI] [PubMed] [Google Scholar]
- 11.Janknecht R, Nordheim A. Gene regulation by Ets proteins. Biochim Biophys Acta. 1993;1155:346–356. doi: 10.1016/0304-419x(93)90014-4. [DOI] [PubMed] [Google Scholar]
- 12.Seidel JJ, Graves BJ. An ERK2 docking site in the Pointed domain distinguishes a subset of ETS transcription factors. Genes Dev. 2002;16:127–137. doi: 10.1101/gad.950902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartel FO, Higuchi T, Spyropoulos DD. Mouse models in the study of the Ets family of transcription factors. Oncogene. 2000;19:6443–6454. doi: 10.1038/sj.onc.1204038. [DOI] [PubMed] [Google Scholar]
- 14.Arcasoy SM, Latoche J, Gondor M, et al. The effects of sialogly-coconjugates on adenovirus-mediated gene transfer to epithelial cells in vitro and in human airway xenografts. Chest. 1997;111(6 Suppl):142S–143S. doi: 10.1378/chest.111.6_supplement.142s. [DOI] [PubMed] [Google Scholar]
- 15.Bochert MA, Kleinbaum LA, Sun LY, Burton FH. Molecular cloning and expression of Ehf, a new member of the ets transcription factor/oncoprotein gene family. Biochem Biophys Res Commun. 1998;246:176–181. doi: 10.1006/bbrc.1998.8560. [DOI] [PubMed] [Google Scholar]
- 16.Kas K, Finger E, Grall F, et al. ESE-3, a novel member of an epithelium-specific ets transcription factor subfamily, demonstrates different target gene specificity from ESE-1. J Biol Chem. 2000;275:2986–2998. doi: 10.1074/jbc.275.4.2986. [DOI] [PubMed] [Google Scholar]
- 17.Kleinbaum LA, Duggan C, Ferreira E, Coffey GP, Buttice G, Burton FH. Human chromosomal localization, tissue/tumor expression, and regulatory function of the ets family gene EHF. Biochem Biophys Res Commun. 1999;264:119–126. doi: 10.1006/bbrc.1999.1493. [DOI] [PubMed] [Google Scholar]
- 18.Tugores A, Le J, Sorokina I, et al. The epithelium-specific ETS protein EHF/ESE-3 is a context-dependent transcriptional repressor downstream of MAPK signaling cascades. J Biol Chem. 2001;276:20397–20406. doi: 10.1074/jbc.M010930200. [DOI] [PubMed] [Google Scholar]
- 19.Tymms MJ, Ng AY, Thomas RS, et al. A novel epithelial-expressed ETS gene, ELF3: human and murine cDNA sequences, murine genomic organization, human mapping to 1q32. 2 and expression in tissues and cancer. Oncogene. 1997;15:2449–2462. doi: 10.1038/sj.onc.1201427. [DOI] [PubMed] [Google Scholar]
- 20.Zhou J, Ng AY, Tymms MJ, et al. A novel transcription factor, ELF5, belongs to the ELF subfamily of ETS genes and maps to human chromosome 11p13-15, a region subject to LOH and rearrangement in human carcinoma cell lines. Oncogene. 1998;17:2719–2732. doi: 10.1038/sj.onc.1202198. [DOI] [PubMed] [Google Scholar]
- 21.Ng AY, Waring P, Ristevski S, et al. Inactivation of the transcription factor Elf3 in mice results in dysmorphogenesis and altered differentiation of intestinal epithelium. Gastroenterology. 2002;122:1455–1466. doi: 10.1053/gast.2002.32990. [DOI] [PubMed] [Google Scholar]
- 22.Zhou J, Chehab R, Tkalcevic J, et al. Elf5 is essential for early embryogenesis and mammary gland development during pregnancy and lactation. EMBO J. 2005;24:635–644. doi: 10.1038/sj.emboj.7600538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudders S, Gaspar J, Madore R, et al. ESE-1 is a novel transcriptional mediator of inflammationthat interacts with NF-kappa B to regulate the inducible nitric-oxide synthase gene. J Biol Chem. 2001;276:3302–3309. doi: 10.1074/jbc.M006507200. [DOI] [PubMed] [Google Scholar]
- 24.Brown C, Gaspar J, Pettit A, et al. ESE-1 is a novel transcriptional mediator of angiopoietin-1 expression in the setting of inflammation. J Biol Chem. 2004;279:12794–12803. doi: 10.1074/jbc.M308593200. [DOI] [PubMed] [Google Scholar]
- 25.Grall F, Gu X, Tan L, et al. Responses to the proinflammatory cytokines interleukin-1 and tumor necrosis factor alpha in cells derived from rheumatoid synovium and other joint tissues involve nuclear factor kappaB-mediated induction of the Ets transcription factor ESE-1. Arthritis Rheum. 2003;48:1249–1260. doi: 10.1002/art.10942. [DOI] [PubMed] [Google Scholar]
- 26.Silverman ES, Baron RM, Palmer LJ, et al. Constitutive and cytokine-induced expression of the ETS transcription factor ESE-3 in the lung. Am J Respir Cell Mol Biol. 2002;27:697–704. doi: 10.1165/rcmb.2002-0011OC. [DOI] [PubMed] [Google Scholar]
- 27.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 28.Stancovski I, Baltimore D. NF-kappaB activation: the I kappaB kinase revealed? Cell. 1997;91:299–302. doi: 10.1016/s0092-8674(00)80413-4. [DOI] [PubMed] [Google Scholar]
- 29.Zeitlin PL, Lu L, Rhim J, et al. A cystic fibrosis bronchial epithelial cell line: immortalization by adeno-12-SV40 infection. Am J Respir Cell Mol Biol. 1991;4:313–319. doi: 10.1165/ajrcmb/4.4.313. [DOI] [PubMed] [Google Scholar]
- 30.Flotte TR, Afione SA, Solow R, et al. Expression of the cystic fibrosis transmembrane conductance regulator from a novel adeno-associated virus promoter. J Biol Chem. 1993;268:3781–3790. [PubMed] [Google Scholar]
- 31.Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci USA. 1996;93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang WC, Chen JJ, Chen CC. c-Src-dependent tyrosine phosphorylation of IKKbeta is involved in tumor necrosis factor-alpha-induced intercellular adhesion molecule-1 expression. J Biol Chem. 2003;278:9944–9952. doi: 10.1074/jbc.m208521200. [DOI] [PubMed] [Google Scholar]
- 33.Huang WC, Chen JJ, Inoue H, Chen CC. Tyrosine phosphorylation of I-kappa B kinase alpha/beta by protein kinase C-dependent c-Src activation is involved in TNF-alpha-induced cyclooxygenase-2 expression. J Immunol. 2003;170:4767–4775. doi: 10.4049/jimmunol.170.9.4767. [DOI] [PubMed] [Google Scholar]
- 34.Tabary O, Escotte S, Couetil JP, et al. Genistein inhibits constitutive and inducible NFkappaB activation and decreases IL-8 production by human cystic fibrosis bronchial gland cells. Am J Pathol. 1999;155:473–481. doi: 10.1016/s0002-9440(10)65143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ray A, Tatter SB, May LT, Sehgal PB. Activation of the human “beta 2-interferon/hepatocyte-stimulating factor/interleukin 6” promoter by cytokines, viruses, and second messenger agonists. Proc Natl Acad Sci USA. 1988;85:6701–6705. doi: 10.1073/pnas.85.18.6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winsauer G, de Martin R. Resolution of inflammation: intracellular feedback loops in the endothelium. Thromb Haemostasis. 2007;97:364–369. [PubMed] [Google Scholar]
- 38.Fulton RJ, McDade RL, Smith PL, Kienker LJ, Kettman JR., Jr Advanced multiplexed analysis with the FlowMetrix system. Clin Chem. 1997;43:1749–1756. [PubMed] [Google Scholar]
- 39.Choe MM, Sporn PH, Swartz MA. An in vitro airway wall model of remodeling. Am J Physiol Lung Cell Mol Physiol. 2003;285:L427–L433. doi: 10.1152/ajplung.00005.2003. [DOI] [PubMed] [Google Scholar]
- 40.Drazen JM, Weiss ST. Genetics: inherit the wheeze. Nature. 2002;418:383–384. doi: 10.1038/418383a. [DOI] [PubMed] [Google Scholar]
- 41.Shapiro SD, Owen CA. ADAM-33 surfaces as an asthma gene. New Engl J Med. 2002;347:936–938. doi: 10.1056/NEJMcibr022144. [DOI] [PubMed] [Google Scholar]
- 42.Van Eerdewegh P, Little RD, Dupuis J, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418:426–430. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- 43.Rajah R, Nachajon RV, Collins MH, Hakonarson H, Grunstein MM, Cohen P. Elevated levels of the IGF-binding protein protease MMP-1 in asthmatic airway smooth muscle. Am J Respir Cell Mol Biol. 1999;20:199–208. doi: 10.1165/ajrcmb.20.2.3148. [DOI] [PubMed] [Google Scholar]
- 44.Naka T, Nishimoto N, Kishimoto T. The paradigm of IL-6: from basic science to medicine. Arthritis Res. 2002;4(Suppl 3):S233–S242. doi: 10.1186/ar565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elias JA. Airway remodeling in asthma. Unanswered questions. Am J Respir Crit Care Med. 2000;161:S168–S171. doi: 10.1164/ajrccm.161.supplement_2.a1q4-4. [DOI] [PubMed] [Google Scholar]
- 46.O’Hara KA, Kedda MA, Thompson PJ, Knight DA. Oncostatin M. an interleukin-6-like cytokine relevant to airway remodelling and the pathogenesis of asthma. Clin Exp Allergy. 2003;33:1026–1032. doi: 10.1046/j.1365-2222.2003.01714.x. [DOI] [PubMed] [Google Scholar]
- 47.Holsti MA, Raulet DH. IL-6 and IL-1 synergize to stimulate IL-2 production and proliferation of peripheral T cells. J Immunol. 1989;143:2514–2519. [PubMed] [Google Scholar]
- 48.Mattoli S, Miante S, Calabro F, Mezzetti M, Fasoli A, Allegra L. Bronchial epithelial cells exposed to isocyanates potentiate activation and proliferation of T-cells. Am J Physiol. 1990;259:L320–L327. doi: 10.1152/ajplung.1990.259.4.L320. [DOI] [PubMed] [Google Scholar]
- 49.Kishimoto T. The biology of interleukin-6. Blood. 1989;74:1–10. [PubMed] [Google Scholar]
- 50.Xing Z, Gauldie J, Cox G, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–320. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tilg H, Dinarello CA, Mier JW. IL-6 and APPs: anti-inflammatory and immunosuppressive mediators. Immunol Today. 1997;18:428–432. doi: 10.1016/s0167-5699(97)01103-1. [DOI] [PubMed] [Google Scholar]
- 52.Reddel RR, Ke Y, Gerwin BI, et al. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 1988;48:1904–1909. [PubMed] [Google Scholar]
- 53.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods (Duluth) 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 54.Chow YH, O’Brodovich H, Plumb J, et al. Development of an epithelium-specific expression cassette with human DNA regulatory elements for transgene expression in lung airways. Proc Natl Acad Sci USA. 1997;94:14695–14700. doi: 10.1073/pnas.94.26.14695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang A, Forman-Kay J, Luo Y, et al. Identification and characterization of human genes encoding Hprp3p and Hprp4p, interacting components of the spliceosome. Hum Mol Genet. 1997;6:2117–2126. doi: 10.1093/hmg/6.12.2117. [DOI] [PubMed] [Google Scholar]
- 56.Kramer A, Keller W. Preparation and fractionation of mammalian extracts active in pre-mRNA splicing. Methods Enzymol. 1990;181:3–19. doi: 10.1016/0076-6879(90)81107-6. [DOI] [PubMed] [Google Scholar]
- 57.Cao HB, Wang A, Martin B, et al. Down-regulation of IL-8 expression in human airway epithelial cells through helper-dependent adenoviral-mediated RNA interference. Cell Res. 2005;15:111–119. doi: 10.1038/sj.cr.7290275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boyd KE, Farnham PJ. Coexamination of site-specific transcription factor binding and promoter activity in living cells. Mol Cell Biol. 1999;19:8393–8399. doi: 10.1128/mcb.19.12.8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gonzalez-Santos JM, Cao H, Wang A, et al. A complementation method for functional analysis of mammalian genes. Nucleic Acids Res. 2005;33:e94. doi: 10.1093/nar/gni093. [DOI] [PMC free article] [PubMed] [Google Scholar]