

Abstract
Identification of protein–protein interactions (PPI) by affinity purification (AP) coupled with tandem mass spectrometry (AP-MS/MS) produces large data sets with high rates of false positives. This is in part because of contamination at the AP level (due to gel contamination, nonspecific binding to the TAP columns in the context of tandem affinity purification, insufficient purification, etc.). In this paper, we introduce a Bayesian approach to identify false-positive PPIs involving contaminants in AP-MS/MS experiments. Specifically, we propose a confidence assessment algorithm (called Decontaminator) that builds a model of contaminants using a small number of representative control experiments. It then uses this model to determine whether the Mascot score of a putative prey is significantly larger than what was observed in control experiments and assigns it a p-value and a false discovery rate. We show that our method identifies contaminants better than previously used approaches and results in a set of PPIs with a larger overlap with databases of known PPIs. Our approach will thus allow improved accuracy in PPI identification while reducing the number of control experiments required.
Keywords: bioinformatics, protein–protein interactions, artificial intelligence, contaminants, affinity purification, mass spectrometry, computational biology, proteomics, Bayesian statistics
Introduction
The study of protein–protein interactions (PPI) is crucial to the understanding of biological processes taking place in cells.1 Affinity purification (AP) combined with mass spectrometry (MS) is a powerful method for the large scale identification of PPIs.2–7 The experimental pipeline of AP consists in first tagging a protein of interest (bait) by genetically inserting a small peptide sequence (tag) onto the recombinant bait protein. The bait protein is affinity purified, together with its interacting partners (preys), which are identified using MS. However, this type of experiment is prone to false positive identifications for various reasons,8 which can seriously complicate the downstream analyses. In the context of affinity purification, contamination of manually handled gel bands, inadequate purification, purification of specific complexes from abundant proteins, and nonspecificity of the tag antibody used are some of the many ways contaminants can be introduced in the experimental pipeline before the mass spectrometry (MS) phase. These contaminants, added to the already large set of valid preys of a given bait, create even longer lists of proteins to analyze. While common contaminants can be identified easily by a trained eye, sporadic contaminants can be considered erroneously as true positive interactions. In addition to contaminants, false positive PPIs can be introduced at the tandem mass spectrometry phase (MS/MS) step.9 For example, peptides of proteins with low abundance or involved in transient interactions can be difficult to identify because of the lack of spectra. Such peptides can be misidentified by database searching algorithms such as Mascot10 or SEQUEST.11 Although many approaches have been proposed to limit the number of mismatched MS/MS spectra (e.g., Peptide Prophet12 and Percolator13), the modeling and detection of contaminants, which is the problem we consider in this paper, has received much less attention.
Related Work
A number of experimental and computational approaches have been proposed to reduce the rate of false-positive PPIs. Several steps in the experimental pipeline can be optimized to minimize contamination. In-cell near physiological expression of the tagged proteins is preferred to overexpression to prevent spurious PPIs. Also, additional purifications could be performed in order to remove contaminating proteins from affinity purified eluate. The drawback of an increased number of purifications is a loss of sensitivity, as transient or weak PPIs will be more likely to be disrupted.2 When performing gel-based sample separation methods before MS/MS, manual gel band cutting can introduce contaminants such as human keratins in the sample. This can be addressed by robot gel cutting, although this increases equipment cost. As an alternative, gel-free protocols simply use liquid chromatography to separate the peptide mixture before MS/MS. However, depending on the complexity of the mixture, less separation might result in an important decrease in sensitivity. Finally, liquid chromatography column contamination from previous chromatographic runs is also important to consider. Although it is possible to wash the column to eluate peptides from previous chromatographic runs, very limited washing is typically done because its time consumption.
Several computational methods have been used to identify the correct PPIs from AP-MS/MS data.14,15 Some involve the use of the topology of the network formed by the PPIs (e.g., number of times two proteins are observed together in a purification to assign a Socio-affinity index4 or a Purification Enrichment score).16 Others used various combinations of data features such as mass spectrometry confidence scores, network topology features, and reproducibility data with machine learning approaches to assign probabilities that a given PPI is a true positive.2,5,17,18 However, with each of these methods, contaminants would often be classified as true interactions because of their high database matching scores and reproducibility. Such sophisticated machine learning procedures can be prone to overfitting and the use of small, manually curated, but often biased training set, such as MIPS complexes19 as used by Krogan et al.5 or a manually selected training set as used by our previous approach,2,18 can be problematic depending on the nature of the data analyzed. Finally, Chua et al. combined PPI data obtained from several different experimental techniques in an effort to reduce false-positives.20 Although such methods will be very efficient at filtering contaminants, they will typically suffer of poor sensitivity.
All of these methods attempt to model simultaneously several sources of false positive identifications including contamination but also, for example, misidentification of peptides at the mass spectrometry level. For instance, scoring methods relying on the topology of PPI networks will tend to assign low scores to proteins being observed as preys in several experiments because they are likely contaminants, while also scoring poorly proteins largely disconnected from the network, which are potential database misidentifications. However, none of these attempt to directly model and filter out contaminants resulting from AP experiments. Deconvoluting the modeling of false identification into AP contaminants modeling and database matching of MS/MS spectra will potentially lead to methods identifying PPIs with higher accuracy. To date, most computational methods aiming specifically at filtering out likely contaminants have been quite simplistic. Several groups maintain a manually assembled list of contaminants and then systematically reject any interactions involving these proteins.8 However, it is possible that a contaminant for one bait is a true interaction for another, suggesting that a finer model of contaminant level would be beneficial. Recently, the Significance Analysis of Interactome (SAINT), a sophisticated statistical approach attempting to filter out contaminant interactions resulting from AP-MS/MS experiments, was introduced.7 SAINT assesses the significance of an interaction according to the semiquantitative peptide count measure of the prey. It discriminates true from false interactions using mixture modeling with Bayesian statistical inference. However, the currently available version of SAINT (1.0) lacks the flexibility to learn contaminant peptide count distributions from available control data and requires a considerable number of baits (15 to 20) in order to yield optimal performances. Moreover, although not necessary, manual labeling of proteins as hubs or known contaminants is required to achieve the best possible accuracy. (Another version of SAINT is currently under development and promises to address many of these issues.)
Once the tagging performed, some affinity purification methods require the vector of the bait to be induced so that the tagged protein is expressed. An alternate method to identify likely contaminant PPIs for a given bait is to perform a control experiment where the expression of the tagged protein is not induced prior to immunoprecipitation. It is then possible to compare mass spectrometry confidence scores (e.g., from Mascot10) for the preys from both the control and induced experiments. For example, in Jeronimo et al.,18 only preys with a Mascot score at least 5 times larger in the induced experiment than the control experiment were retained, the others being considered as likely contaminants. There are limitations to such false positive filtering procedure. First, this method is expensive in terms of time and resources, since the cost is doubled for each bait studied. Second, because of the noisy nature of MS scores for low-abundance preys, comparing a single induced experiment to a single noninduced experiment is problematic. Pooling results from several noninduced experiments could be beneficial. Third, some baits will show leaky expression of the noninduced vector. Depending on the level of leakiness, several true interactions may be mistakenly categorized as false positives.
Here, we propose a confidence assessment algorithm (Decontaminator) using only a limited number of high quality controls sufficient to the proper identification of contaminants obtained from AP-MS/MS experiments without prior knowledge about neither hubs nor contaminants. By pooling control experiments, one-to-one comparisons of induced and noninduced experiment Mascot scores are avoided. Our fast computational method thus provides accurate modeling of contaminants while limiting resource usage.
Methods
We propose a Bayesian approach called Decontaminator that makes use of a limited number of noninduced control experiments in order to build a model of contaminant levels as well as to analyze the noise in the measurements of Mascot scores. Decontaminator then uses this model to assign a p-value and an associated false discovery rate (FDR) to the Mascot score obtained for a given prey. We start by describing the AP-MS/MS approach used to generate the data, and then describe formally our contaminant detection algorithm. Alternate approaches are also considered and their accuracy is compared in the Results section.
Biological Data Set
The Proteus database contains the results of tandem affinity purification (TAP) combined with MS/MS experiments performed for a set E of 89 baits, both in noninduced and induced conditions.2,18,21,22 The baits selected revolve mostly around the transcriptional and splicing machineries. The set of proteins identified as preys by at least one bait (in either the induced or noninduced experiments) consists of 3619 proteins. Detailed TAP-MS/MS methodology has been described elsewhere.2 Briefly, a vector expressing the TAP-tagged protein of interest was stably transfected in HEK 293 cells. Following induction, the cells were harvested and lysed mechanically in detergent-free buffers. The lysate was cleared of insoluble material by centrifugation and the tagged protein complexed with associated factors were purified twice using two sets of beads each targeting a different component of the TAP tag. The purified protein complexes were separated by SDS-PAGE and stained by silver nitrate. The acrylamide gel was then cut in its entirety in about 20 slices that were subsequently trypsin-digested. Identification of the tryptic peptides obtained was performed using microcapillary reversed-phase high pressure liquid chromatography coupled online to a LTQ-Orbitrap (Thermo Fisher Scientific) quadrupole ion trap mass spectrometer with a nanospray device. Proteins were identified using the Mascot software10(Matrix Science) (see Supporting Information for software information). For some of the baits tested, the promoter was leaky, which resulted in the expression, at various levels, of the tagged protein, even when it was not induced. These baits were identified by detection of the tagged protein in the noninduced samples and these samples were excluded from our analysis. A set B ⊂ E of 14 nonleaky baits was selected for this study: B = {b1, …, b14} ={SFRS1, NOP56, TWISTNB, PIH1D1, UXT, MEPCE, SART1, RP11 – 529I10.4, TCEA2, PDRG1, PAF1, KIAA0406, POLR1E, KIN}. The set P of preys they detected in at least one of these 28 induced and noninduced experiments contains 2415 proteins. Out of these, 1067 proteins were unique to a single noninduced bait and 808 were detected more than once in the set of 14 controls, while 540 were only observed in induced experiments. We denote by the Mascot score obtained for prey p in the experiment where bait b is not induced, and by the analogous score in the induced experiment. Note that for most pairs (b,p), where b ∈ B and p ∈ P, p was not detected as a prey for b, in which case we set the relevant Mascot score to zero.
In the noninduced experiments, the number of preys detected for each bait varies from 206 to 626, with a mean of 316. These preys are likely to be contaminants, as the tagged bait is not expressed. In induced experiments, the number of preys per bait goes from 5 to 516, with a mean of 135. It may appear surprising that there are on average more preys detected in the noninduced experiments than in the induced ones. This is likely due to the fact that the presence of high-abundance preys in the induced experiments masks the presence of lower-abundance ones, including contaminants. Still, a significant fraction of the proteins detected in the induced condition are likely contaminants.
Computational Analysis
An ideal model of contaminants would specify, for each prey p, the distribution of the MS scores (in our case, Mascot score10) in noninduced experiments, which we call the null distribution for p. However, accurately estimating this distribution would require a large number of noninduced experiments and the cost would be prohibitive. Instead, we use a small number of noninduced experiments and make the assumption that preys with similar average Mascot scores have a similar null distribution (this assumption is substantiated in the Discussion section). This allows us to pool noninduced scores from different preys (if they have similar Mascot score averages) to build a more accurate noise model. Thus, results from a few control experiments are sufficient to build a contaminant model that can then be used to analyze the results of any number of induced experiments performed under the same conditions. Figure 1 summarizes our approach.
Figure 1.

Decontaminator workflow. First, control and induced experiments are pooled to build a noise model for each. These noise models are then used to assign a p-value to each prey obtained upon the induction of a bait. Finally, a false discovery rate is calculated for each p-value.
Our goal is now to use the data gathered from induced and noninduced AP-MS/MS experiments to build a model of noise in Mascot score measurements and eventually be able to assess the significance of a given Mascot score . Let be the unobserved true mean of Mascot scores for prey p in noninduced experiments, which is defined as the mean of the Mascot scores of an infinite number of noninduced biological replicates. can never be observed, but its posterior distribution can be obtained if a few samples are available. Similarly, define as the average of the Mascot scores of p of an infinite number of biological replicates of experiments where b is induced. The essence of our approach is to calculate the posterior distribution of , the true mean Mascot score of prey p given our data from noninduced experiments, and to compare it to the posterior distribution of , the true mean Mascot score of prey p in an induced experiment, given our observed data . If the second distribution is significantly to the right of the first, prey p is a likely bona fide interaction of bait b. If not, p is probably a contaminant and should be discarded.
Before describing our method in details, we give a few examples that illustrate how it works. Figure 2a shows an example of an interaction accepted by Decontaminator. POLR2E obtained a Mascot score of 320 in the induced experiment of RPAP3 and was only detected twice in control experiments (Mascot scores 38 and 48). From this figure, it can clearly be seen that the resulting posterior distribution of is significantly to the right of the posterior distribution of . Therefore, the RPAP3-POLR2E interaction obtains a small p-value (0.00018) and FDR (0.013) and is considered a valid interaction. This prediction is consistent with the literature about this interaction,18 which is present in databases such as BioGrid.23 Conversely, Figure 2b shows the posterior distribution of of SAMD1 for the induced experiment of RPAP3 (Mascot score: 102). The distribution is largely overlapping with the posterior distribution of , which obtained Mascot scores of 95, 53, 147, and 164 in control experiments but was unobserved in 10 other controls. Clearly the RPAP3-SAMD1 interaction is not very reliable because its Mascot score does not exceed by enough some of the observed noninduced Mascot scores. Decontaminator assigns a large p-value (0.18) and FDR (>0.9) and labels the interaction as a contaminant. Of note, this interaction would often have been incorrectly classified by methods based on a direct comparison of Mascot scores between matched induced and noninduced experiments (as used, for example, in Jeronimo et al.18), as SAMD1 shows up in control experiments only four out of 14 times. Finally, Figure 2c shows the results for the KPNA2-ACTB interaction. ACTB, like other actin proteins, is a known contaminant for our experimental pipeline, obtaining Mascot scores varying from 35 to 210 for eight of the controls. However, the Mascot score observed for this interaction (793) is sufficiently higher to allow the interaction to be predicted as likely positive (p-value 0.005, FDR 0.17). Beta-actin (ACTB) is known to shuttle to and from the nucleus,24 a process which may directly require karyopherin alpha import protein KPNA2. Beta-actin and actin-like proteins have been shown to be part of a number of chromatin remodeling complexes25 and analysis of the KPNA2 purification revealed a strong presence for such complexes (TRRAP/TIP60 and SWI/SNF-like BAF complexes). Therefore, it is possible that these remodelers are assembled in the cytoplasm and imported together to the nucleus via KPNA2. This example shows how the method can differentiate specific interactions from classic contamination.
Figure 2.
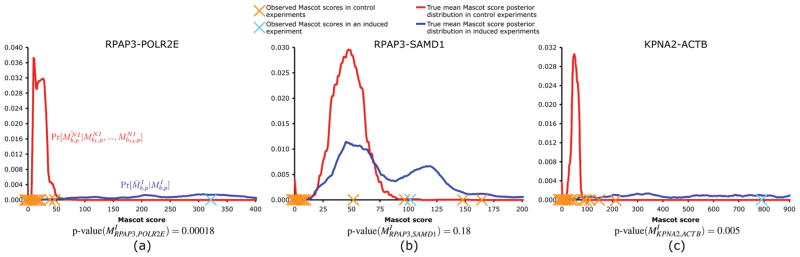
Plots of both the posterior distributions of and posterior distributions of for three different interactions. The blue curve is the posterior distribution of and the red curve is the posterior distribution of . Orange X’s positions on the x-axis are observed Mascot scores of the prey in control experiments. When the prey was not detected for a given control, an X is drawn on the x-axis close to the origin. The blue X’s position on the x-axis is the Mascot score of the prey in the induced experiment for the corresponding bait. (a) RPAP3-POLR2E interaction is an example of an interaction considered valid by the algorithm. (b) RPAP3-SAMD1 interaction is an example of an interaction where the prey is considered as a contaminant. (c) KPNA2-ACTB is an example of an interaction that is predicted as positive, even though ACTB is often observed as a contaminant (but with lower Mascot scores) in control experiments.
The Decontaminator approach involves four steps (see Figure 1):
Use noninduced experiments for different baits as biological replicates and obtain a noise model defined by and .
For each protein p ∈ P, calculate the posterior distribution of given . Similarly, calculate the posterior distribution of , given .
- For each pair (b,p) ∈ B × P, assign a p-value to :
Using the noninduced and full induced data sets, assign a false discovery rate (FDR) to each p-value.
Each step is detailed further below.
Step 1: Building a Noise Model from Noninduced Experiments
The set of 14 TAP-MS/MS experiments where the bait’s expression was not induced can be seen as a set of biological replicates of the null condition. We use these measurements to assess the amount of noise in each replicate, i.e. to estimate , the probability of a given observation , given its true mean Mascot score . This distribution is estimated using a leave-one-out cross-validation approach on the set of 14 noninduced experiments. Specifically, for each bait b ∈ B, we compare to μ≠b,p, the corrected average (see Supporting Information) of the 13 Mascot scores of p in all noninduced experiments except where bait b was used. μ≠b,p provides a good estimate of . Let C(i,j) be the number bait-prey pairs for which and ⌊μ≠b,p⌋ = j. Then, a straightforward estimator is
Note that C is a fairly large matrix (the number of rows and columns is set to 1000; larger Mascot scores are culled to 1000). In addition, aside from the zero-th column C(*,0), it is quite sparsely populated, as the sum of all entries is 40 306. Thus, the above formula yields a very poor estimator. Matrix C therefore needs to be smoothed to matrix Cs using a k -nearest neighbors smoothing algorithm.26 Specifically, let Nδ(i,j) = {(i′,j′): |i − i′|≤δ,|j − j′|≤δ} be the set of neighboring matrix cells to entry i,j, for some distance threshold δ. For each entry (i,j) in the matrix, we choose δ in such a way that Sδ(i,j) = Σ(i′,j′) ∈ Nδ(i,j)C(i′,j′)≤ k and Sδ+1 = Σ(i′,j′) ∈ Nδ+1(i,j)C(i′,j′) > k. Then, Cs is obtained as:
where
In our experiments k = 10 has produced the best results.
Finally, we obtain our estimate of the noise in each measurement as
| (1) |
The approach proposed so far is appropriate to model noise in noninduced experiments. However, it does not correctly model noise in induced experiments for the following reason. The distribution of Mascot scores observed in noninduced and induced experiments differ significantly, with many more high scores observed in the induced experiments. High Mascot scores in noninduced data are rare and, when they do occur, relatively often correspond to cases where a prey p obtains very low scores with most noninduced baits, but a fairly high score with one particular bait, resulting in a large difference between and . Large Mascot scores are thus seen as particularly unreliable, which is the correct conclusion for noninduced experiments, but not for induced experiments. In a situation where high are frequent, for example, when b is induced, this does not properly reflect the uncertainty in the true Mascot score. To address this problem, a correction factor is computed. Let I(i) be the fraction of (b,p) pairs such that and let NI(i) be the fraction of preys with . The I and NI distributions are first smoothed using a k-nearest neighbor approach, then the corrected matrix Cc for the noise model of Mascot scores for induced experiments is obtained as:
The correction is performed by multiplying each entry (i,j) of Cs by the ratio of the smoothed probabilities of the induced mascot score i and of the true mean Mascot score j. This correction allows a better estimation of the noise in induced experiments by putting more weight in the matrix Cc for larger Mascot scores. (See Supporting Information section for correction factor derivation.)
Step 2: Posterior Distribution of and
We are now interested in obtaining the posterior distribution of the mean Mascot score , given the set of observations . This is readily obtained using Bayes rule and the conditional independence of the observations, given their means:
| (2) |
where ζ is a normalizing constant and .
Since observed Mascot scores of induced experiments are also noisy, we estimate the noise of each Mascot score from an induced experiment as:
Step 3: P-Value Computation
Given an observed Mascot score for prey p, Decontaminator can now assign a p-value to this score, which represents the probability that , the true Mascot score of p in noninduced experiments, is larger than or equal to , the true Mascot score for the induced experiment:
| (3) |
where the required terms are obtained from Steps 1 and 2.
Step 4: False Discovery Rate Estimation
Although our p-values are in principle sufficient to assess the confidence in the presence of a given PPI, they may be biased if some of the assumptions we make were violated. A hypothesis-free method to assess the accuracy of our predictions is to measure a false discovery rate (FDR) for each PPI. For a given p-value threshold t, FDR(t) is defined as the expected fraction of predictions (interactions with p-values below t) that are false positives (i.e., due to contaminants). We use a leave-one-out strategy to estimate the FDR: For every bait b in B, we compute for all preys (the set of preys detected when b was the control bait) and for all preys (the set of preys detected when b was induced), based on the set of noninduced experiments excluding bait b. Then, we obtain
where 1c = 1 if c is true and 0 otherwise.
Five Alternate Approaches
We considered five alternate approaches to compare to Decontaminator:
Significance Analysis of Interactome (SAINT),7 without any prior knowledge about hubs or contaminants. SAINT was executed with abundance, sequence length and bait coverage normalization and the following parameters: burn-in period 2000, iterations 20 000, empirical frequency threshold 0.1.
SAINT with the same set of parameters except that hubs were manually labeled. All 24 tagged proteins in the data set that were identified as preys in 10 or more other induced bait experiments were labeled as hubs.
-
The MScore method simply computes
and reports the interaction as true positive if r(b,p) > t, for some threshold t. This is the approach that was used as primary filtering by Jeronimo et al.18 and Cloutier et al.2
The MRatio method computes . It reports the interaction as true positive if r′(b,p) > t′, for some threshold t′. We note the MScore and MRatio approaches are only applicable if both induced and noninduced experiments are performed for all baits of interest.
The Z-score method assigns a Z-score to each Mascot score, as compared to the prey-specific mean and standard deviation for prey p across all baits: . Compared to our Bayesian approach, the Z-score method may be advantageous if the Mascot score variance of contaminants with similar averages varies significantly from prey to prey. Our approach, by pooling all noninduced Mascot score observations from different preys, makes the assumption that such variation is low. If this is not the case the modeling accuracy of contaminants will be negatively affected. However, the Z-score approach works under the assumption that noise in Mascot scores is normally distributed which is not always the case. In addition, the mean and variance estimates are obtained from only as many data points as there are control experiments available, as no pooling is performed.
Implementation and Availability
The proposed methods are implemented in a fast, platform independent Java program. Given a representative set of AP-MS/MS control experiments our software computes FDRs for all interactions identified in induced experiments as we describe here. Note that any protein identification mass spectrometry confidence scores (SEQUEST Xcorr, spectral counts, peptide counts, etc.) could replace the Mascot scores used in our software package by applying simple modifications. The Java program is available at: http://www.mcb.mcgill.ca/~blanchem/Decontaminator.
Results
Contaminant Detection Accuracy
We start by studying the ability of Decontaminator to tease out contaminants from true interactions. Because neither contaminants nor true interactions are known before hand, one way to assess our method and compare it to others is to consider the number of bait-prey pairs from induced experiments that achieve a p-value at most t to the number of such pairs in noninduced experiments. The ratio of these two numbers, the false discovery rate FDR(t), indicates the fraction of predictions from the induced experiments that are expected to be due to contaminants. One can thus contrast two prediction methods by studying the number of bait-prey pairs that can be detected at a given level of FDR. Figure 3 shows the cumulative distributions of FDRs for data from induced experiments, for Decontaminator as well as the Z-score approach described in the Methods section. Since the MScore and MRatio approaches require matched induced and noninduced experiments for every bait, FDRs cannot be computed for them. FDRs comparison is also not possible with SAINT because it scores the entirety of the input data set without possibility of leaving out a subset of the data. It can be observed that at low FDRs, Decontaminator always yield a larger set of predicted interactions than the Z-score approach. For example, at 1, 3, and 15% FDRs, our approach yields 140, 74, and 43% more interactions than the Z-score approach. Alternatively, for the same number of interactions predicted, say 2000, the FDR of Decontaminator (1%) is more than three times lower than that of the Z-score approach (3.4%).
Figure 3.

Cumulative distributions of the FDRs obtained from Decontaminator and the Z-score approaches. Each curve shows the number of interactions that can be predicted positive, as a function of the false discovery rate tolerated.
Number of Control Experiments Required
The required number of control noninduced experiments needed to build an accurate model of contaminants is an important issue to consider. So far, we used all 14 available controls to filter out contaminants. Figure 4 shows the cumulative distributions of the FDRs obtained from Decontaminator with different number of controls. It can be observed that there is an increase in the number of high confidence (<20% FDR) predictions when using a larger number of control baits. For example at 2% FDR we obtain 2855 interactions using the set of 14 control experiments but only 2440 with 12 controls, 2169 with 10 controls, 1858 with 8 controls, and 1620 with 6 controls. These results show that the more high quality control baits are available, the better our algorithm will perform. It can also be observed that the prediction accuracy decreases significantly when fewer than 8 controls are used. However, this deterioration is not striking and therefore shows that even with very few control experiments it is possible to use Decontaminator to detect contaminants in AP-MS/MS experiments reasonably accurately. This aspect is particularly important for small scale studies where only a few baits are analyzed and for laboratories where the number of control experiments that can be performed is limited.
Figure 4.

Cumulative distributions of the FDRs obtained from Decontaminator with 14, 12, 10, 8, and 6 controls. For sets of controls of size smaller than 14, we show the average cumulative distributions over 100 randomly selected subsets of controls.
External Validation
To evaluate the quality of our contaminant detection method, we compared it to the five other methods described above on the basis of their ability to detect known PPIs or PPIs involving pairs of proteins of similar function.
We first used the union of two high-quality PPI databases, BioGrid23 and HPRD27 (both downloaded on Feb 1st, 2010), to assess the quality of the predictions made by each method. Note that BioGrid PPIs that originated from the curation of our own previously published data set18 were excluded from the analysis. Since a small fraction of all PPIs are known, we do not expect a large fraction of our predictions to be found in these databases. However, clearly, the size of the overlap is a good indication of the accuracy of the methods. Figure 5 shows the fraction of positively predicted PPIs present in the merged database for the six methods described above, as a function of the number of PPIs predicted. For all six methods, high-confidence predictions overlap the two databases significantly more than low-confidence predictions. However, at any high confidence FDR level, the set of predicted interactions produced by Decontaminator always has a larger overlap than any of the alternate methods. The Z-score method also outperforms the other four alternate methods. For example, with a predicted set of 2000 PPIs (FDR of 1% for Decontaminator), 7.1% of the interactions overlap with the merged database for Decontaminator, 6.6% for the Z-score approach, 5.8% for SAINT, 5.7% for SAINT without manual labeling of hubs, 5.8% for the MScore, and only 4.9% for the MRatio methods. On average, Decontaminator results in a predicted set of PPIs with ~40% more overlap with known PPIs than that obtained with the MRatio methods from Jeronimo et al.,18 ~20% more than both types of runs of SAINT, ~15% more than the MScore method, and ~5% more than the Z-score method. We assessed the statistical significance of the differences between the overlaps obtained by each method using a two sample z-test. Decontaminator’s performance significantly exceed those of the two SAINT variants, MScore, and MRatio (p-value ≤ 0.05), but is not significantly better than that of the Z-Score approach (p-value = 0.26).
Figure 5.

Fraction of positive predictions present in the HPRD and BioGrid merged databases (y-axis), for a varying number of predicted interactions (x-axis) by six filtering methods (MRatio, MScore, Z-score, SAINT without hub labeling, SAINT with hub labeling and Decontaminator).
The Gene Ontology (GO) annotation database28 associates a set of terms to characterized proteins, describing their functions, localization, and the biological processes in which they are involved. We say that a GO term is x%-specific if less than x% of the proteins in our data set are annotated with this term.2 We assume that if two interacting proteins are sharing a x%-specific term (for x relatively small), they have greater chances to be truly interacting. Therefore, we applied the six filtering methods on our data set and compared the fraction of the positively predicted interactions for which the proteins were sharing at least one 10%-specific GO term (Figure 6). The PPIs predicted by our Bayesian approach are consistently more supported by shared GO annotations than those made by the five other approaches. For a set of 2000 positively predicted PPIs, 28.6% of the PPIs predicted by Decontaminator had both proteins sharing at least one 10%-specific GO term, compared to 26.7% with the Z-score, 26.3% with the MScore, 23.8% with SAINT, 23.7% with SAINT without hub labeling and 23.5% with the MRatio. The advantage of Decontaminator is statistically significant for all comparisons (p-value <0.05; two-sample z-test), except against the Z-Score approach, for which the advantage is marginal (p-value = 0.095). Similar patterns are observed for most other values of GO term specificity (x). Even though sharing a specific molecular function, biological process or cellular component does not guarantee that two proteins are interacting, we believe that this improved enrichment for shared GO terms reflects the higher quality of our contaminant detection method.
Figure 6.

Fraction of positively predicted PPIs for which the interacting partners share at least a 10%-specific GO term (y-axis), for a given number of predicted interactions (x-axis) by six filtering methods (MRatio, MScore, Z-score, SAINT without hub labeling, SAINT with hub labeling and Decontaminator).
Overall, one might argue that the differences between Decontaminator and the Z-score approach are not striking. However, when referring back to Figure 3, it is clear that Decontaminator yields a much larger set of predictions for a given FDR when compared to the Z-score approach. It is possible that the external validation sets chosen are too small to demonstrate the importance of the improvement of Decontaminator over the Z-score approach as easily as it can be seen in Figure 3.
Discussion
Decontaminator shows an improvement in accuracy for the detection of contaminant PPIs in our data set when compared to currently used alternate approaches. We expect that this decrease in false positive interactions will facilitate the analysis of PPI networks and ease the characterization of novel biological pathways. At the same time, our approach will greatly reduce experimental costs by cutting the number of most experimental manipulations almost in half. This expense reduction is due to the much smaller number of control experiments needed by our algorithm compared to methods such as that described in Jeronimo et al.,18 where each induced experiment requires a matched noninduced experiment for its interactions to be classified. It is also worth noting that in theory, the control experiments provided as input to the algorithm could all be performed with the same bait protein. However, we used noninduced experiments produced from different baits, by different experimentalists at different time periods. These biological and technical replicates allow us to factor in the noise resulting from the change of baits in TAP-MS/MS experiments and technical variation.
On the Impact of the Protein–Protein Interaction Discovery Experimental Design
PPIs are often viewed and studied as a network. Different strategies have been applied in order to decide which proteins should be tagged first in order to build this network. Some groups use a molecular function centric approach and choose as baits a set of proteins performing a specific type of molecular functions, such as kinases, methyltransferases, or phosphatases.7 Others use a complex-centric approach and orient their study around specific biological processes and complexes, such as the RNA polymerase II,2,18,29 tagging several interacting partners to obtain a dense, focused network. Other impressive projects focused on whole interactome mapping of particular organisms by tagging most of their known proteins to analyze the protein complexes present in their interaction networks.4,5,17 The PPIs obtained with those strategies form networks that have drastically different topologies. Networks obtained by complex-centric or whole interactome mapping approaches will tend to be more connected than those obtained by a molecular function centric strategy. This will largely influence the type of computational analysis that should be used in order to filter contaminants and assign confidence scores to interactions. Several algorithms use the topology of PPI networks to determine whether an interaction is a likely true or false positive,4,16,18 reasoning that truly interacting proteins are likely to interact with similar sets of other proteins. However, this method only applies to networks obtained from complex-centric and whole interactome mapping studies, as molecular function centric approaches yield loosely connected networks, making shared neighborhood a rarity. Thus, for small or sparse networks, topology is of little use, making approaches such as SAINT or Decontaminator the only alternative.
Advantages
Decontaminator is an alternative that does not rely on topological data and is not affected by the type or size of the network being analyzed. Obviously, in cases where the network topology is informative, our Bayesian FDR scores can be integrated into more complex predictors such as our Interaction Reliability Scores (IRS),2 the Socio-affinity index4 or the Purification Enrichment score.16 Even though we applied our algorithm on a large scale data set in this paper, we showed that it can be applied to much smaller scale studies since a small number of controls are required to accurately model contaminants and score interactions.
Another important factor to consider is the need for prior knowledge in training the prediction program. Methods such as the Interaction Reliability Scores (IRS)2 or the machine learning pipeline used by Krogan et al.5 require a fairly large training set of examples of true positive and possibly true negative interactions. These training sets are difficult to assemble, prone to errors, and often not representative of the set of interactions one is really seeking (e.g., true positive interactions come from very strong complexes such as RNA polymerase II, whereas interactions of interest are weaker or more transient). On the other hand, SAINT requires no labeled training set, but it is reported to perform better when known contaminants and hubs are manually labeled. Such labeling might be hard to perform on smaller networks where hubs and contaminants are hard to differentiate. Thus, methods requiring no prior knowledge, such as Decontaminator, offers significant advantages.
Decontaminator can easily be incorporated in a mass spectrometry analysis computational pipeline. Because it provides, for each predicted PPI, a FDR, which can be interpreted as the probability that the PPI is involving a contaminant, our method can be added as a contaminant filter at the end of the typical computational pipelines involving identification of the preys via database searches (Mascot10 and SEQUEST11) and validation of the identification (Percolator13 and Protein Prophet30).
Limitations
Our approach works under the assumption that all experiments are performed using the same protocol, in such a way that the distribution of contaminants does not change over time (note that this does not mean that the observed levels of contaminants are constant, but rather that the true levels are). Clearly, whenever the experimental pipeline of the approach is modified (e.g., changes to the experimental protocol, the mass spectrometer, the tag, or the chromatography column used), new control experiments need to confirm the validity of the model or to build a new one. Therefore, in a context where the experimental pipeline would be rapidly evolving or when bait-specific antibodies are used for the pull down, Decontaminator may yield little or no cost benefit compared to the approach where each induced experiment is paired with its control counterpart. However, our results indicate that the number of control experiments required to obtain a good contaminant model is relatively modest, so unless changes are extremely common, significant benefits should be achievable. Finally, even when no obvious changes to the experimental pipeline have taken place, periodic control experiments should be performed to ensure that the set of control experiments used by the Bayesian approach remains representative.
Another limitation is that Decontaminator cannot differentiate true positive interactions from contaminants obtained from inefficient purifications. Instead, the Mascot scores obtained for contaminants that are usually excluded at the purification step are likely to be significantly larger than what was observed in control experiments. However, other approaches can be used to detect such problematic purifications. For example, typically the distribution of Mascot scores observed in a faulty purification is significantly shifted to the right (greater number of high Mascot scores). This can be explained by the high abundance of various proteins, causing more peptides to be detected and therefore increasing the overall Mascot score of each protein. Also, a GO analysis can be performed to accomplish the same goal: preys interacting with a bait for which the purification was faulty will often not be enriched for specific GO terms. On the contrary, one would expect that when an efficient purification is performed, the preys interacting with a given bait will be more likely to share functions, biological processes, or cellular components with it.
Mascot scores, which are used in the current version of Decontaminator, are not always accurate in the identification of peptides and quantification of their abundance. Peptide misidentifications will affect the accuracy of our method in several ways. For example, a contaminant that would have been misidentified in many of the control experiments may be predicted as a true interaction when correctly identified in an induced experiment. In addition, Mascot scores, like peptide and spectral counts, are influenced by the length of the protein and the detectability of its peptides, resulting in some true interactions obtaining relatively low scores. These problems can be overcome with the utilization of software like Peptide/Protein Prophet,12,30 which give a probability of the protein presence based on database search scores like Mascot. Other measures like peptide retention time and precursor ion intensity have also shown to be valuable information and could be used in conjunction with database search scores to yield more accurate protein identifications.31–33 These more accurate measures of protein identication/abundance could easily replace the Mascot score in Decontaminator.
Finally, our approach will work best if different preys with the same true mean Mascot score have the same Mascot score distribution in the noninduced condition, that is, that the variability in the observed Mascot scores is not dependent on the identity of the prey. Manual inspection confirms that this is the case for the vast majority of the preys in our data set. Should this assumption fail, that is, if the Mascot score of a certain protein had a much larger variance than other proteins with the same mean Mascot score, the consequences would be that its p-value would tend to be assessed incorrectly (above average Mascot scores would obtain unduly low p-values, and below average scores would obtain unduly high p-values). Indeed, some of the errors made by our approach are due to this type of proteins, such as CKAP5, INF2, and TUBB2A. This large variability could be in part explained by the large size (>1200 a.a.) of these proteins. These preys could also be suffering of an undersampling by the mass spectrometer and be masked due to the presence of more abundant proteins. However, those are rare cases that can be treated separately by flagging, from the set of noninduced experiments, proteins with unexpectedly high variance, and analyzing them separately.
Conclusion
Several methods have been proposed to identify false positive PPIs in AP-MS/MS experiments. However, very few have considered the modeling of contaminants resulting only from the AP experimental pipeline. We hypothesized that a more accurate model of contaminants would yield higher accuracy of PPI identification. We have shown here that using Decontaminator, only a few representative control experiments are necessary to accurately discard the vast majority of contaminants while allowing the detection of true PPIs involving preys that, in other experiments, may be contaminants. These findings will allow significant reductions in expenses and a greater number of experiments to be conducted with higher accuracy.
Supplementary Material
Acknowledgments
This work was funded by a CIHR operating grant to BC and MB and NSERC Vanier and CGS scholarships to MLA. The authors thank Christian Poitras for his help in the software development process, Denis Faubert for the mass spectrometry analysis, and Ethan Kim for useful suggestions.
Abbreviations
- AP
affinity purification
- FDR
false discovery rate
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- PPI
protein–protein interaction
- TAP
tandem affinity purification
Footnotes
Supporting Information Available: Protein and peptide identification software information, computation of corrected averages for Mascot scores, and Cc correction factor derivation. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.von Mering C, Krause R, Snel B, Cornell M, Oliver S, Fields S, Bork P. Nature. 2002:399–404. doi: 10.1038/nature750. [DOI] [PubMed] [Google Scholar]
- 2.Cloutier P, Al-Khoury R, Lavallée-Adam M, Faubert D, Jiang H, Poitras C, Bouchard A, Forget D, Blanchette M, Coulombe B. Methods. 2009;48:381–386. doi: 10.1016/j.ymeth.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gavin A, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick J, Michon A, Cruciat C, et al. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 4.Gavin A, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen L, Bastuck S, Dumpelfeld B, et al. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 5.Krogan N, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis A, et al. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 6.Ho Y, Gruhler A, Heilbut A, Bader G, Moore L, Adams S, Millar A, Taylor P, Bennett K, Boutilier K, et al. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 7.Breitkreutz A, Choi H, Sharom J, Boucher L, Neduva V, Larsen B, Lin Z, Breitkreutz B, Stark C, Liu G, et al. Sci STKE. 2010;328:1043. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gingras A, Aebersold R, Raught B. J Physiol. 2005;563:11. doi: 10.1113/jphysiol.2004.080440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boutilier K, Ross M, Podtelejnikov A, Orsi C, Taylor R, Taylor P, Figeys D. Anal Chim Acta. 2005;534:11–20. [Google Scholar]
- 10.Perkins D, Pappin D, Creasy D, Cottrell J, et al. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Eng J, McCormack A, Yates J, et al. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 12.Keller A, Nesvizhskii A, Kolker E, Aebersold R. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 13.Kall L, Canterbury J, Weston J, Noble W, MacCoss M. Nat Methods. 2007;4:923–926. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 14.Sardiu M, Cai Y, Jin J, Swanson S, Conaway R, Conaway J, Florens L, Washburn M. Proc Natl Acad Sci USA. 2008;105:1454. doi: 10.1073/pnas.0706983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sowa M, Bennett E, Gygi S, Harper J. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins S, Kemmeren P, Zhao X, Greenblatt J, Spencer F, Holstege F, Weissman J, Krogan N. Mol Cell Proteomics. 2007;6:439. doi: 10.1074/mcp.M600381-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Ewing R, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson M, O’Connor L, Li M, et al. Mol Syst Biol. 2007:3. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Thérien C, Bergeron D, Bourassa S, Greenblatt J, et al. Mol Cell. 2007;27:262–274. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mewes H, Amid C, Arnold R, Frishman D, Guldener U, Mannhaupt G, Munsterkotter M, Pagel P, Strack N, Stumpflen V, et al. Nucleic Acids Res. 2004;32:D41. doi: 10.1093/nar/gkh092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chua H, Sung W, Wong L. Bioinformatics. 2006;22:1623. doi: 10.1093/bioinformatics/btl145. [DOI] [PubMed] [Google Scholar]
- 21.Forget D, Lacombe A, Cloutier P, Al-Khoury R, Bouchard A, Lavallée-Adam M, Faubert D, Jeronimo C, Blanchette M, Coulombe B. Mol Cell Proteomics. 2010;9:2827. doi: 10.1074/mcp.M110.003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krueger B, Jeronimo C, Roy B, Bouchard A, Barrandon C, Byers S, Searcey C, Cooper J, Bensaude O, Cohen E, et al. Nucleic Acids Res. 2008;36:2219. doi: 10.1093/nar/gkn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stark C, Breitkreutz B, Reguly T, Boucher L, Breitkreutz A, Tyers M. Nucleic Acids Res. 2006;34:D535. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wada A, Fukuda M, Mishima M, Nishida E. EMBO J. 1998;17:1635–1641. doi: 10.1093/emboj/17.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olave IA, Reck-Peterson SL, Crabtree GR. Annu Rev Biochem. 2002;71:755–81. doi: 10.1146/annurev.biochem.71.110601.135507. [DOI] [PubMed] [Google Scholar]
- 26.Fix E, Hodges J., Jr . Project 21-49-004, Report No. 11, under Contract No. AF41(148)-31E. USAF School of Aviation Medicine; 1952. [Google Scholar]
- 27.Prasad T, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A. Nucleic Acids Res. 2008;37:D767. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashburner M, Ball C, Blake J, Botstein D, Butler H, Cherry J, Davis A, Dolinski K, Dwight S, Eppig J, et al. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosley A, Pattenden S, Carey M, Venkatesh S, Gilmore J, Florens L, Workman J, Washburn M. Mol Cell. 2009;34:168–178. doi: 10.1016/j.molcel.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nesvizhskii A, Keller A, Kolker E, Aebersold R, et al. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 31.Strittmatter E, Kangas L, Petritis K, Mottaz H, Anderson G, Shen Y, Jacobs J, Camp D, II, Smith R. J Proteome Res. 2004;3:760–769. doi: 10.1021/pr049965y. [DOI] [PubMed] [Google Scholar]
- 32.Kawakami T, Tateishi K, Yamano Y, Ishikawa T, Kuroki K, Nishimura T. Proteomics. 2005;5:856–864. doi: 10.1002/pmic.200401047. [DOI] [PubMed] [Google Scholar]
- 33.Havilio M, Haddad Y, Smilansky Z. Anal Chem. 2003;75:435–444. doi: 10.1021/ac0258913. [DOI] [PubMed] [Google Scholar]
- 34.Bernholtz B. Stat Papers. 1977;18:2–12. [Google Scholar]
- 35.Persson T, Rootzen H. Biometrika. 1977;64:123. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.