

Abstract
Mutations in PRPF3, a gene encoding the essential pre-mRNA splicing factor Hprp3p, have been identified in patients with autosomal dominant retinitis pigmentosa type 18 (RP18). Patients with RP18 have one of two single amino acid substitutions, Pro493Ser or Thr494Met, at the highly conserved Hprp3p C-terminal region. Pro493Ser occurs sporadically, whereas Thr494Met is observed in several unlinked RP families worldwide. The latter mutation also alters a potential recognition motif for phosphorylation by casein kinase II (CKII). To understand the molecular basis of RP18, we examined the consequences of Thr494Met mutation on Hprp3p molecular interactions with components of the U4/U6.U5 small nuclear ribonucleoprotein particles (snRNPs) complex. Since numerous mutations causing human diseases change pre-mRNA splice sites, we investigated whether Thr494Met substitution affects the processing of PRPF3 mRNA. We found that Thr494Met does not affect PRPF3 mRNA processing, indicating that the mutation may exert its effect primarily at the protein level. We used small hairpin RNAs to specifically silence the endogenous PRPF3 while simultaneously expressing HA-tagged Thr494Met. We demonstrated that the C-but not N-terminal region of Hprp3p is indeed phosphorylated by CKII in vitro and in cells. CKII-mediated Hprp3p phosphorylation was significantly reduced by Thr494Met mutation. Consequently, the Hprp3p C-terminal region is rendered partially defective in its association with itself, Hprp4p, and U4/U6 snRNA. Our findings provide new insights into the biology of Hprp3p and suggest that the loss of Hprp3p phosphorylation at Thr494 is a key step for initiating Thr494Met aberrant interactions within U4/U6 snRNP complex and that these are likely linked to the RP18 phenotype.
INTRODUCTION
Retinitis pigmentosa (RP) refers to a group of inherited disorders characterized by degeneration of retina cells (1,2). The disease can be inherited as autosomal dominant (adRP), autosomal recessive (arRP) and X-linked (xlRP) (3,4). Four (PRPC8, PRPF31, PRPF3 and PAP-1) of the at least 17 genes associated with adRP encode ubiquitously expressed pre-mRNA splicing proteins (2,5–9). Pre-mRNA splicing is one of the essential RNA processing events by which pre-mRNA introns are excised and exons ligated forming a mature mRNA, which is translated into functional proteins (10). It is catalyzed by the spliceosome, a large RNA protein complex that contains a pre-mRNA, four essential small nuclear ribonucleoprotein particles (snRNP) U1, U2, U5 and U4/U6, and numerous non-snRNP splicing factors (11–14). The spliceosome assembles de novo on each pre-mRNA to be spliced through the formation of intermediate complexes that recognize both conserved and degenerate sequences on each intron and surrounding exons (15,16).
We previously identified, along with others, PRPF3, the gene that encodes Hprp3p (17–19). Within the U4/U6 snRNP, Hprp3p and Hprp4p form a heterotrimer protein complex with PPIH, the 20 kDa cyclophilin H (20). Hprp3p interacts directly with Hprp4p while PPIH associates with Hprp4p, but not with Hprp3p (21,22). Hprp3p is present in the early pre-spliceosome or complex B, that is not catalytically active, but absent from the activated spliceosome (6,14,23). Hprp3p is highly conserved throughout evolution, in particular at its C-terminal region (19). In accord, this region is both necessary and sufficient for binding to Hprp4p and U4/U6 snRNA (21). Additionally, it contacts the U4/U6 snRNA recycling factor p110 (24) as well as U5 snRNP associated proteins: Hprp6 and Hsnu66 (25). The C-terminal region is also needed for binding to PAP-1, a protein that associates with the U4/ U6.U5 tri-snRNP complex, and is also implicated in adRP (26).
Given the importance of Hprp3p C-terminal region, it is not surprising to find that two missense mutations within this region lead to adRP type 18 (Pro493Ser or Thr494Met; RP18). The amino acid changes result from C-to-T mutations at positions 1478 and 1482 within the exon 11 of PRPF3 gene. Pro493Ser mutation has been observed only in sporadic cases; whereas, Thr494Met segregates perfectly with the disease in several unlinked families worldwide (7,27,28) supporting the presence of a hot spot at position 1482 of PRPF3 gene. Thr494Met causes a mild late-onset and less severe RP phenotype than the mutations found in other splicing factors, i.e. PRPF31 and PRPC8 (27). Moreover, amino acids 494–497 (TKVE) of Hprp3p form a potential recognition motif for phosphorylation by casein kinase II (CKII), that is altered due to the T494M mutation (15,16).
The molecular consequences of RP18 mutations on Hprp3p function are unknown. Here we show that Hprp3p is indeed phosphorylated both in vitro and in cell culture. Most importantly, substitution of threonine for methionine at codon 494 of Hprp3p (henceforth T494M) results in reduction of Hprp3p phosphorylation by CKII. Consequently, T494M is partially defective in its interaction with Hprp3p, Hprp4p and U4/U6 snRNA. In short, our findings provide new insights into the biology of Hprp3p and suggest that the disruption of Hprp3p C-terminal region alters multiple interactions within U4/U6 snRNP particle resulting in the development of RP18 phenotype.
RESULTS
PRPF3 mRNA processing is not impaired by C1482T substitution on exon 11
Up to 50% of mutations causing human diseases have their effect, not solely through the amino acid substitution, but also by altering pre-mRNA splice sites (29–31). Impaired mRNA processing events may lead to exon skipping, intron retention or usage of cryptic splice sites (32). Bioinformatics analysis of exon 11 sequence of PRPF3 [using a software that detects exon splicing enhancer (ESE) sites] (33) indicated that the C-to-T substitution in codon 494 destroys predicted ESE recognized by the arginine and serine rich (SR) proteins ASF/SF2 and SRp40 (34–36) (Fig. 1A). To test whether the bioinformatics outcome agrees with in vivo settings, a minigene approach was used due to the lack of clinical samples. Two minigenes were constructed spanning exons 10, 11 and 12 of PRPF3 gene and their corresponding flanking intronic sequences. They contained either the wild-type (PRPF3) or the C-to-T substitution on exon 11 (T494M) of PRPF3. To distinguish between endogenous PRPF3 gene from the one produced by the minigenes, the forward primer sequence used during PCR amplification was present only in the pcDNA vector, while the reverse one, complemented a region of exon 12 (Fig. 1B).
Figure 1.
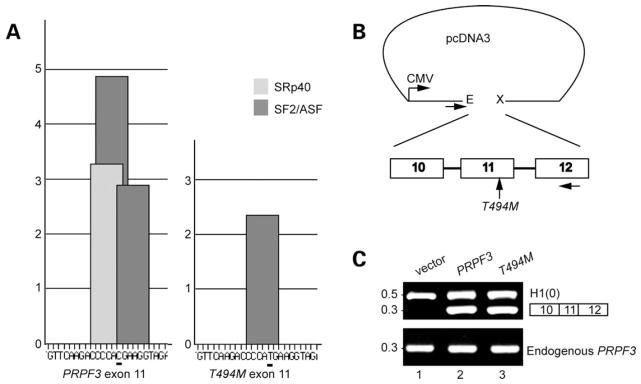
Putative high-score SR protein motifs in exon 11 of PRPF3 are abrogated by C1482T mutation. (A) ESE motif scores for exon 11 of PRPF3 wild-type (left) and T494M (right) were calculated by ESEfinder (5,6,9,53). High-score motifs are in dark gray for SF2/ASF, and light gray for SRp40. The height of each bar indicates the score value, the position along the x axis indicates its location along the exon 11 and the width of the bar represents the length of the motif. The C at position 1482 in PRPF3 is underlined. The T at the same position in T494M causes both SF2/ASF and SRp40 scores to fall below threshold (2.87 to −0.075 and 3.26–1.54, respectively). Thresholds and maximal values are different for different SR proteins (1.95 for SF2/ASF and 2.67 for SRp40). (B) Schematic of the minigenes used in this study. PRPF3 and T494M minigenes were cloned into pcDNA3, using EcoRI (E) and XhoI (X) restriction endonucleases. Horizontal arrows under pcDNA3 and exon 12 indicate forward and reverse primers for PCR, respectively. (C) Analysis of PRPF3 and T494M minigenes. RT–PCR was carried out to assess pre-mRNA splicing from PRPF3 (lane 2), and T494M (lane 3) minigene constructs or empty vector (lane 1). The RT–PCR products were resolved by 1.5% agarose. Histone 1(0) gene served as an internal control. DNA size loading marker is shown on the left.
Pre-mRNA processing was assessed by RT–PCR amplification of RNA products from differentially transfected cells. Both PRPF3 and T494M minigenes generated only the cDNAs of the expected three exon size (300 bp) (Fig. 1C, lanes 2 and 3, respectively), suggesting that the splicing machinery recognized the genomic structure of these constructs. The intronless Histone 1(0) gene served as an internal control (Fig. 1C, lane 1). The RT–PCR products were generated in similar ratios independently of which minigene was used in the experiments (t-test, P < 0.05). Experiments with HeLa and A549 cells produced similar results (data not shown).
T494m substitution does not affect expression or nuclear/ cytoplasmic distribution of Hprp3p in cells
Since substitution of T494M does not impair PRPF3 mRNA processing, we suspected that such mutation may affect the function of Hprp3 protein. To investigate this idea, we utilized a complementation method, previously developed by our group (37). This method employs an helper-dependent adenoviral (HD-Ad) vector to express small hairpin RNAs (shRNAs) that knock down the expression of the endogenous PRPF3 gene by targeting both 5′-and 3′-UTRs of the gene. Simultaneously, this vector expresses the corresponding gene HA-tagged (wild-type: HD-Ad-F3iplus or T494M mutant: HD-Ad-F3iT494M), without the sequences targeted by the shRNAs, under the control of the human Ubiquitin C promoter (Fig. 2A). Since there are no photoreceptor cell lines available, we used ARPE19 cells which exhibit behavior, that is, similar to that of adult human retinal pigment epithelium (RPE) (38–40). Cells transduced with these viruses were harvested 4 days post-transduction, a time point when 90% of endogenous Hprp3p is silenced (37). Cell nuclear extracts were then prepared and used for the detection of the expression levels of Hprp3p and T494M by immunoblotting analysis. Both HD-Ad-F3iplus and HD-Ad-F3iT494M demonstrated similar levels of expression of total Hprp3p and T494M suggesting that the presence of the T494M mutation does not substantially alter the production of Hprp3p molecules (Fig. 2B, lanes 2 and 3, respectively).
Figure 2.

Expression of wild-type and mutant Hprp3p in cells when the endogenously expressed Hprp3p is knocked down. (A) Schematic of the complementation system designed to silence the endogenous PRPF3 gene while expressing its wild-type or mutant versions in cells. An HD-Ad vector was designed to produce two shRNAs, which silence expression of the endogenous PRPF3 by targeting both the 5′- and 3′-UTRs of its mRNA. The vector also expresses either the exogenous wild-type PRPF3: HD-Ad-F3iplus (upper) or T494M: HD-Ad-F3iT494M (lower), both lacking the targeted sequences. (B) Expression of T494M 4 days post-transduction with HD-Ad viral vector. Western blots were performed with antibodies against Hprp3p (upper) or HA (middle). β-actin was used as loading control (lower). exo, exogenous HA-Hprp3p; en, endogenous Hprp3p. Numbers on the left refer to size in kDa. (C) Indirect immunostaining micrographs show expression and subcellullar localization of the HA-Hprp3p protein in cells 4 day post-transduction. Vectors used for transduction are indicated on the right: empty vector (HD-Ad; a and b), HD-Ad-F3i which expresses shRNAs only (c and d), HD-Ad-F3iplus (e and f), and HD-Ad-F3iT494M (g and h). Identical fields are shown for DAPI (left) and FITC (right) channels. The nuclear DNA is visualized as blue fluorescence (a, c, e and g). The localization of the HA-tagged protein is visualized as green immunofluorescence (f and h). Mock-transfected cells did not display any immunostaining signal under the FITC channel (b and d). Fluorescence micrographs were recorded using a 100× objective.
Hprp3p is a U4/U6 snRNP associated protein normally localized in the cell nucleus. It is unknown whether T494M mutation causes subcellular mislocalization of the mutant protein. To investigate the intracellular distribution of T494M protein, we performed indirect immunostaining of cells expressing different HD-Ad viral vectors 4 days post-transduction. A primary antibody that recognizes the HA-tagged exogenous Hprp3p proteins (Hprp3p and T494M) was used along with FITC-conjugated secondary antibody to visualize the location of protein expression while DAPI staining was used to mark the cell nuclei by fluorescence microscopy (Fig. 2C). Both Hprp3p (positive control) (Fig. 2C, panels e and f) and T494M (Fig. 2C, panels g and h) were localized to the cell nucleus suggesting that the mutant amino acid on Hprp3p does not disrupt Hprp3p nuclear translocation. An empty HD-Ad vector (Fig. 2C, panels a and b) and HD-Ad-F3i, which silences endogenous Hprp3p (Fig. 2C, panels c and d), were employed as negative controls. This result does not exclude possible T494M subnuclear mislocalization.
Hprp3p phosphorylation by CKII is weakened by T494M mutation
Protein phosphorylation plays a central role in controlling the assembly and disassembly of spliceosome intermediate complexes. For example, addition of the serine/threonine phosphatases to nuclear extract inhibits spliceosome assembly at a very early step (41–43). Likewise, phosphorylation of SR proteins is required for progression from the A to the B complex (42). It is not known whether Hprp3p is phosphorylated and whether T494M mutation leads to any phosphorylation deficiency. Nevertheless, Hprp3p does contain several potential consensus sequences for phosphorylation by serine/threonine kinases. At least five of these potential phosphorylation sites match CKII phosphorylation consensus motifs, (S/T)XX(E/D), as predicted by NetPhos (44) and Scansite programs (45) (Fig. 3A). Moreover, substitution of threonine 494 for methionine abrogates one of the potential CKII phosphorylation sites present between amino acids 494–497 (TKVE). Since T494M mutation occurs in a region of Hprp3p, which mediates binding to proteins and snRNAs, dephosphorylation on Thr494 may render Hprp3p defective in such function. To examine whether Hprp3p is phosphorylated in mammalian cells, nuclear extracts from cells expressing HA-Hprp3p were immunoprecipitated with anti-HA antibody. The precipitated proteins were then separated by SDS–PAGE, immunoblotted and visualized by using antibody against phosphothreonine antibody. Fig. 3B (top) shows that Hprp3p is indeed phosphorylated on threonine residues in cells (lane 2). Hprp3p phosphorylation was partly blocked in the presence of heparin (lane 3), a strong CKII inhibitor, and was not seen when the kinase was replaced with reaction buffer (lane 1). More importantly, Hprp3p phosphorylation on threonines was significantly reduced by T494M to levels similar to those observed in Hprp3p upon heparin treatment (lane 4). Heparin did not completely block phosphorylation of either wild-type or T494M Hprp3p suggesting that other threonine kinases contribute to Hprp3p phosphorylation in cells (lanes 3 and 5).
Figure 3.

CKII-mediated Hprp3p phosphorylation is weakened by substitution of Thr494 for Met. (A) Diagram of Hprp3p, T494M and truncated constructs (mutants I, and IV) shows potential threonine phosphorylation sites as predicted by NetPhos and Scansite programs. (B) CKII mediates Hprp3p phosphorylation in cells. Nuclear extracts, transduced with HA-tagged Hprp3p or T494M, were subjected to immunoprecipitation using antibody against HA in the absence (lanes 1, 2 and 4) or presence (lanes 3 and 5) of heparin. As negative control, nuclear extract from non-transduced cells was used (lane 1). Threonine phosphorylation was detected by western blotting with antibodies against phosphothreonine (upper). For loading control, membranes were stripped and reprobed with anti-HA antibody (lower). Results are representative of at least three experiments. *IgG heavy chains. (C and D) CKII phosphorylates Hprp3p in vitro. His-tagged Hprp3p, T494M (C) or Hprp3p truncated versions—ΔI, ΔIII and ΔIII-T494M (D) were incubated with CKII in the presence of GTP. Representative bar graph (C, lower) shows the decrease levels on threonine phosphorylation of T494M mutant protein relative to Hprp3p (*P < 0.01). The proteins were separated on 8–10% SDS–PAGE and visualized by western blotting with antibodies specific to phosphothreonine antibody (upper). Membranes were stripped and reprobed with anti-HA antibody (lower). *background bands. anti-HA, anti-HA antibody; anti-P-thr, anti-phosphothreonine antibody.
In order to map the phosphorylation sites on Hprp3p, His-tagged Hprp3p, T494M and deletions (Fig. 3A) were subjected to in vitro kinase assays. None of these proteins produced in bacteria is phosphorylated (data not shown). However, wild-type Hprp3p (Fig. 3C, lane 1), and mutant ΔIII (Fig. 3D, lane 2) served as CKII substrates when incubated with this enzyme in vitro. Strong phosphorylation was observed in the C-terminal (ΔIII: 194–683 residues) but not in the N-terminal (ΔI: 1–194 residues) region of Hprp3p (Fig. 3D, lane 2 and 1, respectively). More importantly, Hprp3p phosphorylation on threonines was significantly reduced by substituting Thr494 for Met (Fig. 3C, lane 2 and 3D, lane 3). Since Hprp3p also contains potential PKA and CaMKII phosphorylation sites, similar experiments using these enzymes were performed. We did not detect any threonine phosphorylation in these experiments (data not shown).
Hprp3p mutation leading to RP18 weakens the association of Hprp3p with Hprp4p but not with Hprp6p
Next, we examined the potential molecular consequences on protein/protein interactions of the reduced phosphorylation of T494M mutant protein. Both in vitro and in cell culture U4/U6-specific proteins Hprp3p and Hprp4p are stably associated with the U4/U6 snRNP in 1 M KCl (17,21). In vitro, Hprp3p and Hprp4p co-immunoprecipitate using highly purified proteins, suggesting they associate directly independent of RNA or other protein(s). Hprp3p middle and C-terminal regions (amino acids 195–683) contain the domain sufficient for its interaction with Hprp4p since deletion of this region abolishes its interaction with Hprp4p (21,25).
Given that protein phosphorylation and dephosphorylation may alter protein conformation and create docking sites for protein/protein interaction, we examined whether T494M mutation strengthens, weakens or has no effect on Hprp3p/ Hprp4p interaction. Thus, co-immunoprecipitation (co-IP) studies were performed using nuclear extracts from cells expressing either HA-Hprp3p or HA-T494M. The Hprp3p proteins were immunoprecipitated using HA antibodies, the immunocomplexes separated by SDS–PAGE and Hprp4p was detected using anti-Hprp4p antibodies. T494M shows reduced ability to associate with Hprp4p (up to 40% reduction) relative to Hprp3p and Hprp4p (Fig. 4A). Similar outcomes were obtained from alternative co-IP analysis using anti-Hprp4p or anti-Hprp3p, as the precipitating antibody (data not shown). Fig. 4B corresponds to one-fourth of the nuclear extract used for IP and shows the level of expression of the protein used for the immunoprecipitation.
Figure 4.

Effect of T494M mutation on Hprp3p binding to Hprp4p and Hprp6p. (A) Hprp3p/Hprp4p interactions is weakened by substitution of Thr494 for Met. Nuclear extracts were prepared from ARPE-19 cells 4 days after transduction with HD-Ad viral vectors that silence endogenous Hprp3p and express intact HA-Hprp3p (upper, lane 2), HA-T494M (lane 3). The extracts were immunoprecipitated using anti-HA monoclonal antibody bound to protein G-agarose beads. 20% of immunoprecipitates were resolved by 8% SDS–PAGE and immunoblotted using anti-Hprp4p polyclonal antibody. Control (lane 1) represents cells transduced with empty HD-Ad vector. The blot shown in A (upper) was stripped and reprobed with anti-HA antibody (lower). *IgG heavy chains. (B) The level of total nuclear proteins was analyzed by direct immunoblotting. 5% of nuclear extract input, used for immunoprecipitation in A, was resolved as above and probed with anti-Hprp4p (upper), Hprp3p (second, arrows indicate size difference between endogenous Hprp3p and HA-Hprp3p), anti-HA antibodies (third) or anti-β-actin as a loading control (lower). (C) Percentage of Hprp4 bound to T494M relative to Hprp3p was calculated by NIH Image software (v 1.62). (D) Phosphorylation of Hprp3p increases its binding to Hprp4p. E. coli expressed Hprp3p and T494M were phosphorylated in vitro using recombinant CKII and GTP (see Fig. 3C). After phosphorylation, the proteins were mixed with Hprp4p and co-IP using anti-Hprp4p antibody. In parallel, unphosphorylated His-tagged proteins were co-immunoprecipitated in a similar way. The immunoprecipitates were resolved by 8% SDS–PAGE and immunoblotted using anti-HA antibodies (top). The blots were stripped and reprobed with anti-Hprp4p antibody (bottom). (E) Hprp3p/Hprp6p association is not affected by T494M mutation. co-IP of HA-tagged Hprp3p and T494M by cMyc-Hprp6p. Cells co-expressing cMyc-Hprp6p and HA-Hprp3p or HA-T494M were co-IP with anti-cMyc antibody and the immunocomplexes fractionated by 8% SDS–PAGE (top). Nuclear extracts from cells transfected with empty vectors or expressing Hprp6p alone were used as controls (lanes 1 and 2, respectively).
Notably, we did not detect measurable protein/protein interaction differences when Escherichia coli expressed proteins (Hprp3p, T494M and Hprp4p) instead of nuclear extracts were used in the IP assays (Fig. 4D, left). This suggests that post-translational modification(s) on Hprp3p may play a significant role in the decrease of the interaction observed between T494M and Hprp4p in nuclear extracts. Furthermore, given that T494M mutation significantly reduces Hprp3p phosphorylation on threonines, we speculated that phosphorylation may also mediate the interaction between wild-type Hprp3p and Hprp4p. To test this idea, E. coli expressed proteins were in vitro phosphorylated by CKII and subjected to co-IP analyses. In parallel, similar co-IPs were performed with non-phosphorylated proteins (control). The ability of Hprp4p to interact with either phosphorylated or non-phosphorylated Hprp3p and T494M was assayed by immunobloting analysis using antibodies against HA tag. Fig. 4D shows that phosphorylation of Hprp3p (lane 3, top) by CKII enhances its binding to Hprp4p when compared with the non-phosphorylated version (lane 1, top); substitution of Thr494 with Met weakened this association (lane 2, top). More importantly, in vitro phosphorylation of Hprp3p-T494M did not improve its binding ability to Hprp4p (lane 4, top), indicating that the lack of a threonine at position 494 may be responsible for the weakening of T494M/Hprp4p association observed in the cell nuclear extracts.
U4/U6 snRNP interacts with U5 snRNP, presumably via protein/protein interactions, to form a tri-snRNP complex, that is, recruited into the spliceosome. Previous studies using both yeast two-hybrid screening system and pull-down experiments have shown that Hprp3p also interacts with Hprp6p, the 102 kDa protein associated specifically with U5 snRNP (25). In particular, C-terminal Hprp3p (amino acids 417–683) but not the N-terminal fragment of Hprp3p suffices for binding to Hprp6p (25). Moreover, Hprp6p has been proposed to serve as a bridge between U4/U6 and U5 snRNP that allows proper formation and stability of the U4/U6.U5 tri-snRNP complex (25). Hence, we examined the possibility that the association between Hprp3p and Hprp6p could be affected upon T494M mutation. To do that, c-Myc-tagged Hprp6p and HA-tagged Hprp3p were co-expressed in cells and nuclear extracts were prepared for co-IP assays using antibodies against c-Myc tag. This procedure confirmed the association between Hprp3p and Hprp6p; however, we did not observe any difference between T494M/Hprp6p and Hprp3p/Hprp6p under our experimental conditions (Fig. 4E). Similar outcomes were obtained from co-IP analysis using anti-HA antibody (data not shown).
Hprp3p self-association is mediated by its C-terminal region and weakened by T494M mutation
In addition to heteromeric associations, many proteins require the formation of homodimeric-like structures for proper function (46,47). Previously, we noticed that upon purification, Hprp3p self-associates in a concentration dependent manner (at >1 mg/ml). Similarly, truncated versions of Hprp3p, containing only the central and C-terminal regions, show a behavior similar to that of the wild-type protein (21). Moreover, Hprp3p yeast homologue has the capacity to form homodimers in vivo (25,48). Given that, we examined whether Hprp3p self-association indeed occurs in human cells. We performed co-IP assays using nuclear extracts from cells expressing similar levels of endogenous Hprp3p and either HA-Hprp3p or HA-T494M (Fig. 5A, top). Proteins were co-immunoprecipitated with anti-HA antibodies and the presence of the endogenous protein detected by antibodies against Hprp3p. HA-tagged Hprp3p (lane 2) was able to immunoprecipitate around 30% of endogenous Hprp3p (5A, top) from the nuclear extracts (5A, lower) suggesting Hprp3p ability to self-associate, although weakly. T494M ability to immunoprecipitate the endogenous Hprp3p was markedly reduced (5A, lane 3) in comparison with that of HA-Hprp3p.
Figure 5.

Hprp3p self-association is affected by T494M substitution. (A) Hprp3p self-association is mediated by RNA in cells. NE from ARPE-19 cells transduced with HD-Ad-F3iplus, HD-Ad-F3iT494M or empty vector were immunoprecipitated with anti-HA antibody and the precipitants analyzed by western blotting with anti-Hprp3p antibody (top). Similar immunoprecipitation and immunoblotting analysis were performed using NE pretreated with RNase A (middle). The levels of each protein in the cell extracts are shown (lower). (B) Cross-linker induces Hprp3p self-association in cells and in animal tissue. DFDNB cross-linking of nuclear extracts from cells expressing Hprp3p (top left), T494M (top center) or animal tissue (top right). Middle and lower panels represent the same blots as above after probing with anti-Hprp4p or anti-U5-116 antibody as controls. (C) Hprp3p self-association is not mediated by Hprp4p. His-tagged, E. coli translated Hprp3p pulled-down either HA-Hprp3p or T494M from ARPE-19 NE in which Hprp4p was immunoblocked (top) or from NE pretreated with RNase A (lower). (D) T494M mutant protein does not associate with Hprp3p C-terminal region. Co-IP using His-tagged, E. coli translated truncated Hprp3p versions (mutants I, II, III and IV) and NE from ARPE-19 cells expressing either HA-Hprp3p or HA-T494M (arrows on the right). Input and precipitated proteins were resolved by 8 or 10% SDS–PAGE and probed with antibody against Hprp3p. NE, nuclear extracts; co-IP, co-immunoprecipitation; IN, input control showing 10% of the amount of the nuclear extract used in the co-IP. (E) Schematic shows Hprp3p, T494M and truncated Hprp3p versions (mutants I, II, III and IV).
To further validate Hprp3p self-association, we performed chemical cross-linking experiments using nuclear extracts from cells expressing HA-Hprp3p or HA-T494M as well as animal tissue expressing endogenous Hprp3p (Fig. 5B). We chose the cross-linking reagent 1,5-difluoro-2,4-dinitroben-zene (DFDNB), that spans a distance of only 3Å, and thus can only cross-link residues that are in close proximity to each other. DFDNB was added to intact cells to prevent the disruption of native molecular interactions upon rupture of cellular and nuclear membranes. Nuclear extracts were prepared after quenching the cross-linking reactions. By immunoblotting, we detected that in addition to the 90 kDa protein (the monomer form of Hprp3p) an extra band of a relative molecular mass of 180 kDa appeared after cross-linking (Fig. 5B, top). The same blots were then reprobed with antibodies against Hprp4p (middle) or U5-116 kDa protein (bottom), no additional bands were detected. These experiments indicate that DFDNB cross-links Hprp3p to itself more readily than to Hprp4p or to the U5-116 kDa protein. Hprp4p and U5-116 were used as internal controls. Other cross-linking reagents [dithiobis[succinimidylpropionate] (DSP), formaldehyde and glutaraldehyde) were also tested; however, these reagents induce the formation of large complexes between Hprp3p and other spliceosome components that are difficult to resolve by electrophoresis (data not shown).
Since Hprp3p functions as part of a protein/RNA complex, we next examined the involvement of U4/U6-snRNP factors in Hprp3p self-association. To do that, we first investigated whether Hprp4p, the closest physical and functional partner of Hprp3p, was required for Hprp3p self-association. This was assayed by using His-tagged Hprp3p to pull down HA-Hprp3p or HA-T494M from nuclear extracts in which Hprp4p was immunoblocked. Fig. 5C (top) shows that the self-association ability of Hprp3p remains intact in the absence of Hprp4p.
Hprp3p self-association cannot be detected by SDS–PAGE under non-reducing conditions, suggesting it is neither very stable nor mediated by disulfide bonds (data not shown). Since self-association via their RNA-binding regions has been observed in RNA-binding proteins (49–51), we next tested the possibility of RNA involvement in Hprp3p self-interaction in cell nuclear extracts. Hprp3p self-interaction was partly abrogated after RNase A pretreatment of nuclear extracts suggesting that RNA has a cooperative effect on Hprp3p self-association (Fig. 5C, lower).
Finally, we asked whether Hprp3p self-association is mediated by any specific region of the protein. For this purpose, we used truncated versions of His-tagged Hprp3p (Fig. 5E) to pull down HA-Hprp3p or HA-T494M from nuclear extracts 4 days post-transduction. Interestingly, the C-terminal tail alone (residues 443–683) was not sufficient to interact with T494M even under the least stringent conditions tested (Fig. 5D). This result suggests that the interaction between Hprp3p and T494M is disrupted by the RP18-causing mutation. The molecular basis for the weaker protein interaction may result from the disruption of T494M association with factors such as RNAs that mediate Hprp3p self-association or due to the loss of phosphorylation on T494.
Hprp3p mutation leading to RP18 affects the interaction of Hprp3p with U4/U6 snRNA
Previous studies have shown that Hprp3p is a dsRNA-binding protein that specifically contacts U4/U6 snRNA through both its central and C-terminal regions (13,21); in addition, this region is also critical for its interaction with the non-RNA-binding protein Hprp4p (21). It is therefore likely that the T494M mutation might weaken both the Hprp3p-RNA association as well as the Hprp3p/Hprp4p interaction (see above). To test the effect of T494M on its ability to bind to U4/U6 snRNAs, total RNA was extracted from aliquots of the immunocomplexes precipitated from nuclear extracts with anti-HA antibodies (as in Fig. 4A) and the presence of U4/ U6 snRNAs was detected by northern blotting (Fig. 6A) and primer extension analysis (Fig. 6B). Consistent with our previous analysis (21), U4/U6 snRNAs co-immunoprecipitated with HA-Hprp3p (Fig. 6A, lane 2; Fig. 6B, lanes 3 and 4); however, significantly lower levels of U4/U6 were detected associated with T494M (Fig. 6A, lane 3; Fig. 6B, lanes 5 and 6; Fig. 6C). Notably, the relative levels of U4 and U6 snRNAs in cells are unaffected by the mutation on Hprp3p (Fig. 6D). As a negative control, immunoprecipitation was performed with nuclear extracts from cells transduced with the HD-Ad-LacZ. In this case, there was no specific precipitation of U4 or U6 snRNA by the anti-HA antibodies. Since RNA-bound forms of Hprp3p have increased self-interaction, it is possible that the reduced RNA-binding activity of T494M weakens its interaction with several other proteins.
Figure 6.

U4 and U6 snRNAs association with Hprp3p is weakened by T494M mutation. (A and B) Binding of T494M mutant protein to U4/U6 snRNA. From the remaining 80% of immunoprecipitates mentioned in Fig. 4A, total RNA was extracted and the presence of U4 and U6 snRNAs detected by northern blot (A) and primer extension analysis (B). The migration position of U4 (138 or 82 nucleotides) and U6 (105 or 91nucleotides) are indicated on the right. *partial extension product from U4 snRNA. (C) Percentage of U4/U6 snRNAs bound to T494M relative to Hprp3p as detected by northern blot was calculated by NIH Image software (v 1.62). (D) Levels of U1, U2, U4, U5 and U6 snRNAs in 5 μg of total RNA isolated from ARPE-19 nuclear extracts used for immunoprecipitation are shown. SnRNAs were separated on 8% polyacrylamide-urea (8 M) gel, hybridized to oligonucleotide probes complementary to U1, U2, U4, U5 and U6 snRNAs and hybridization signals detected using a phosphoImager. RNAs were visualized by phosphoImager, analyzed using NIH Image software (v 1.62) and normalized to HA-Hprp3 proteins (for immunocomplexes) or the internal U1 control (for RNA inputs).
T494M missense mutation causes reduction in cell proliferation but does not affect global pre-mRNA splicing
During the course of our experiments, we observed that some of the cells expressing T494M mutant protein detached and floated in the culture medium. Control cells and those expressing exogenous Hprp3p were completely attached to the flask. The loss of cell adherence was strongly dependent on the length of time of expression of T494M. The 3-4,5-(dimethylthyazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay (metabolically active cells), which is based upon the conversion by viable cells of a tetrazolium salt into a blue formazan product (52), was utilized to test the effect of T494M on cell proliferation (Fig. 7A). The cytotoxic effect of T494M on cells was also determined by the trypan blue exclusion assay (Fig. 7B) and by measuring the release of lactate dehydrogenase (LDH) from the cytosol of damaged cells into the extracellular medium (53) (Fig. 7C). Over 30% reduction in the proliferation of cells expressing T494M but not of those expressing Hprp3p occurred within 6 days post-transduction with corresponding HD-Ad viral vector (Fig. 7A). Furthermore, expression of T494M in the absence of Hprp3p ultimately causes significant cell death (35%) (Fig. 7B and C). In contrast, most cells survived when exogenous PRPF3 was expressed in those cells. Interestingly, the mutant effect on cell phenotype was not observed when wild-type and mutant Hprp3 proteins were co-expressed within 4 days post-transduction (Fig. 7A–C).
Figure 7.

Effect of T494M missense mutation on cell proliferation, viability and global pre-mRNA splicing. Cell proliferation and viability were assessed by the MTT (A), trypan blue exclusion (B) and LDH cytotoxicity assay (C), after 2, 4 or 6 days post-transduction with HD-Ad-LacZ, HD-Ad-Fi, HD-Ad-F3iplus or HD-Ad-F3iT494M. Each experiment (mean ± SEM) was carried out in triplicate (n = 4). Statistical significance was assessed by one-way ANOVA and Holm’s multiple comparison test (*P < 0.05). The result is expressed in MTT activity (A), cell viability (B) and LDH release (C) relative to the corresponding treatment at day 0. (D) Expression of T494M 6 day post-transduction with HD-Ad viral vector. Western blots were performed with antibodies against Hprp3p (upper), HA (middle) or β-actin (bottom). exo, exogenous HA-Hprp3p; en, endogenous Hprp3p. (E) Indirect immunostaining micrographs show expression and subcellular localization of the HA-Hprp3p protein in ARPE-19 cells 6 day post-transduction with HD-Ad-F3iplus (see Fig. 2C, for labels). (F) Expression of RPE-specific and house-keeping genes in cells expressing wild-type or T494M mutant protein. RT–PCR was performed with total RNA isolated from ARPE-19 cells 6 day post-transduction. RPL18, EEF1A1, RPE65 and H1(0) mRNAs transcripts are shown. The DNA size marker is shown on the right.
These results indicate that expression of T494M damages the cell membrane integrity and causes cell death by necrosis rather than apoptosis. Furthermore, T494M induces DNA smear on analysis by agarose gel electrophoresis (supplementary Figure). This observation is typically associated with necrosis. The nuclei of cells expressing T494M were indistinguishable from those expressing endogenous Hprp3p or its HA-tagged exogenous wild-type version (Fig. 7E, panels e and f and g and h, respectively) as showed by indirect immunostaining. Since the cytoplasm and plasma membrane are the first target of the necrotic process, the relatively well preserved nuclear appearance, at least concerning its inner constituents further points toward a necrotic rather than apoptotic process.
We next investigated the effect of T494M expression on the pre-mRNA splicing of both RPE-specific (RPE65) and house-keeping (RPL18 and EEF1A1) genes. RT–PCR of total RNA isolated from cells transduced with HD-Ad, HD-Ad-F3iplus or HD-Ad-F3iT494M (6 days post-transduction) was performed to measure mRNA levels (Fig. 7F). Primers were designed in regions of the DNA located in different exons to avoid amplification of genomic DNA and to distinguish between unspliced and spliced forms. The expected mature mRNAs (RPL18: 250 bp; RPE65: 900 bp; EEF1A1: 300 bp) were detected in T494M (lane 3) at levels similar to those from control (lane 1) and PRPF3 expressing cells (lane 2). The level of Histone 1(0), an intronless gene, was used as internal control. Experiments with HeLa and A549 cells produced similar results (data not shown).
DISCUSSION
The genetic complexity of RP has been recently highlighted by the discovery of RP mutations in pre-mRNA splicing genes (PRPC8, PRPF31, PRPF3 and PAP-1) that function not only in the retina, but also throughout the human body (5,6,9,54). Coincidently, all of these gene products form part of the U4/U6.U5 snRNP whose proper assembly is essential for the transition from an inactive to a catalytically active spliceosome. The mechanism by which these genes cause RP remains unknown. However, given that all these genes encode essential splicing proteins, it is likely that the dynamic of the various protein/protein and protein/RNA interactions required for the formation and stability of U4/U6.U5 snRNP is perturbed by the resulting mutant proteins.
In this study, we attempted to understand the molecular basis for the occurrence of RP18 as a result of the substitution of T494M at the highly conserved Hprp3p C-terminal region (amino acids 442–683). We first confirmed that the processing of PRPF3 mRNA is not affected upon T494M mutation in cells. Hence, we studied the consequences of T494M mutation on Hprp3p potential phosphorylation, and on its known interactions with components of the U4/U6.U5 snRNP complex, i.e. Hprp4p, Hprp6p and U4/U6 snRNA. We showed for the first time that the human Hprp3 protein is able to self-associate in cells. We demonstrated that Hprp3p is indeed phosphorylated by CKII in vitro and in cell culture. Moreover, CKII-mediated Hprp3p phosphorylation was significantly reduced by substituting Thr494 for Met, indicating that amino acids 494–497 (TKVE) form a recognition motif for phosphorylation by CKII.
Hprp3p phosphorylation on Thr494 appears to have distinct effects on its interactions with various U4/U6 snRNP factors. For example, as a result of the T494M mutation, binding of the mutant protein to wild-type-Hprp3p, Hprp4p and U4/U6 snRNA is impaired, leading to a reduction in cell proliferation and viability. Previous results have shown that 90% depletion of PRPF3 interferes not only with cell viability, but also leads to deficiencies in pre-mRNA splicing (35). However, substitution of Thr494 for Met on Hprp3p did not block pre-mRNA splicing at a measurable level, indicating that T494M mutant protein might be largely functional.
The reduction on Hprp3p phosphorylation observed in T494M may also have implications for the mechanism by which the protein’s subnuclear localization is determined. Recently, it was shown that T494M mutation causes Hprp3p to mislocalize in the nucleolus of photoreceptor cells (55). Several studies have provided evidence that phosphorylation–dephosphorylation controls the localization and subnuclear trafficking of nuclear proteins (41,56–58). Our observation that partial dephosphorylation of Hprp3p decreases the protein’s interaction with itself, Hprp4p and U4/U6 snRNA provides a possible mechanistic explanation of how phosphorylation and dephosphorylation cycle can modulate Hprp3p subnuclear trafficking. The lack of these functions may impair the normal accumulation of the protein into speckles (sites of storage of mRNA splicing factors) (57,59) and account for the nucleolar accumulation observed in photoreceptor cells. On the other hand, the nucleolar targeting of the T494M protein may also result from its association with photoreceptor-specific nucleolar components.
Based on our results and those of other groups (17,21,25,48), we propose the following model to explain the possible role of Hprp3p in RP18 development (Fig. 8). In our model, Hprp3p C-terminal region contains the protein functional core that mediates essential protein/protein and protein/RNA interactions, ultimately required for the formation of a stable U4/U6 snRNP. This C-terminal region is highly conserved during evolution, and particularly residues 494–497 are 100% identical from yeast to human. Hprp3p interacts with both proteins and U4/U6 snRNAs through its C-terminal region. Moreover, Hprp3p interaction with Hprp4p is essential for cell viability (21,48). Hprp3p binding to RNA brings Hprp3p molecules into close proximity thereby facilitating Hprp3p contacts with other molecules through its C-terminal region. Hprp3p association with the stem II of U4/U6 snRNA may also stabilize the U4/U6 base-paired structure (13,24) and Hprp3p tertiary conformation. Hprp3p C-terminal region binds specific proteins present in U5 snRNP, i.e. Hprp6p and Hsnu66 (25). By doing so, Hprp3p may serve as one of the factors that bring together and/or stabilize U4/U6.U5 snRNP. Expression of T494M mutant protein may partially destabilize the assembly of the U4/U6 snRNP complex by weakening its association with Hprp3p, Hprp4p and U4/U6 snRNA. U4/U6 snRNP recruitment to the pre-mRNA could be a rate-limiting step in spliceosome assembly. Hence, any minor mutation in one of its core proteins could manifest primarily in highly metabolically active tissue such as the retina.
Figure 8.

Thr494 modulates Hprp3p association with the U4/U6-snRNP complex during the assembly of the B complex. The schematic shows a representation of the spliceosome assembly cycle, indicating the proposed role for Hprp3p in promoting the addition of the U4/U6 snRNP to the A complex through its association with other components of the spliceosome (thick arrow on the left). Substitution of Thr494 for Met may destabilize U4/U6 snRNP assembly by weakening Hprp3p association with U4/U6 snRNP components (thin arrow on the right). RNA, U4/U6 snRNA; U1, U2 and U5 indicate the corresponding snRNP; E, exon; A, intron branch site.
Our experimental system speeds up the mild, late onset phenotype that takes place in clinical settings. Cellular and molecular effects on cells primarily manifest once the wild-type protein is absence or expressed at a lower level than the mutant one. Since RP18 is an autosomal dominant disease, it is possible that the presence of a wild-type allele may compensate for the partially functional mutant one. This idea is supported by the fact that T494M causes a late onset and less severe phenotype than RP-causing mutations found in other splicing factors, i.e. PRPF31 and PFPC8 (27). Considering that retina cell metabolism is highly active, the splicing turnover rates might also be more active here than in other cells; therefore, retina cells might be primarily affected by mild mutations in the splicing factor genes.
MATERIALS AND METHODS
Minigene construction and expression
Wild-type sequences of PRPF3 exon 10, 11 and 12, and their corresponding flanking intronic sequences were amplified from human genomic DNA, using primer sequences complementary to exons 10 (nucleotides 1256–1283; 5′-AAACTTCGGAG-ACAAACAAGGAGGGAAG-3′) and 12 (nucleotides 1709–1738: 5′-ACCCCCTGTGAAATGTCTTCTTTAAGCTTT-3′). For T494M minigene, C-to-T substitution at position 1482 (5′-CTGAGCTCTAGAGTGGGCTTCTACCTTCaTGGGGTC-TTGAACAGC-3′) was introduced in exon 11 by overlap extension PCR. The constructs were assembled into pcDNA3 and transiently transfected in ARPE-19 cells with polyfect transfection reagent (QIAGEN, Inc., Mississauga, ON, Canada). After 48 h, total RNA was extracted with TRIzol (Life Technologies, Rockville, MD, USA) and cDNA was synthesized using Superscript II H−(Life Technologies) according to the instructions provided. Expression of minigenes was detected by PCR using primer sequences present in pcDNA3 (5′-AAAGGTACCA-GATCTACCATGGGTGGGCGGATCTTTTAC-3′) and exon 12 of PRPF3.
HD-Ad preparation
HD-Ad-F3i and HD-Ad-F3iplus were created as previously described (37). HD-Ad-F3iT494M was constructed by introducing the HA-tagged PRPF3 coding sequence carrying T493M mutation, under control of the Ubiquitin C promoter and followed by the bovine growth hormone poly(A) signal, into HD-Ad-F3i. HD-Ad-F3i vector knocks down PRPF3 expression by targeting its 5′- and 3′-UTR region with shRNAs under the control of the murine U6 promoter (37,60). The control virus contains the lacZ gene under the control of the K18 promoter (37,60).
Cell culture
ARPE19 cells were cultured in DMEM-F12, supplemented with 10% fetal bovine serum (FBS) (Invitrogen Canada Inc., Burlington, ON, Canada). Cells were transduced at 40–60% confluency with virus (5000 particles/cell) under serum-free conditions for 2 h, followed by the addition of media to a final concentration of 10% FBS.
Co-IP, immunoblotting and immunostaining
Cells expressing different versions of Hprp3p were harvested 2, 4 or 6 days post-transduction, and used for nuclear extract preparation. For immunoprecipitation, nuclear extracts (21) were added to protein G-agarose beads pre-bound with anti-HA antibody or protein A-agarose beads pre-bound with anti-Hprp4p antibody in 500 μl of buffer containing 100 mM potassium phosphate, 500 mM KCl, 1× protease inhibitor mixture and 0.1% (v/v) Tween 20, pH 7.5 for 1 h at 4°C with constant mixing. Hprp4p or Hprp3p were detected in the pull-down complexes by immunoblotting. Rabbit antisera against Hprp3p, Hprp4p and U5-116 kDa protein were generated in our animal facility. The mouse monoclonal anti-HA, anti-cMyc and anti-β-actin antibodies were purchased from COVANCE (Richmond, Canada), Clontech Canada Inc. (Mississauga,. ON, Canada) and Abcam (Cambridge, MA, USA), respectively. Cells for immunostaining were prepared as described previously (21) and examined with a fluorescence microscope (Leica DM IRB; Leica Microsystems, Richmond Hill, ON, Canada) using a 100×oil objective lens. Fluorescence micrographs were recorded using a CCD camera (DFC 300F) (Leica Microsystems). Photoshop 7.0 software (Adobe) was used for minor adjustments of contrast and overlaying.
Phosphorylation of Hprp3p by CKII
Phosphorylation of His-tagged Hprp3p, T494M and of truncated versions by CKII was performed in 30 μl of reaction mixture containing 20 mM Tris–HCl, pH 7.5, 50 mM KCl, 10 mM MgCl2, 1 mM DTT, 100 μM GTP and 1 μg of His-tagged Hprp3p or Hprp3p versions. The reactions were started by the addition of purified CKII (New England BioLabs Inc., Mississauga, ON, Canada) and incubated for 30 min at 30°C. Phosphorylation inhibitor, heparin (Sigma Chemical Co., St. Louis, MO, USA), was used at a final concentration of 5 μg/ml. The samples were then separated on 8 or 10% SDS–PAGE and detected by immunoblotting using antibodies against phosphothreonine (Cell signaling Technology, Danvers, MA, USA). To detect Hprp3p phosphorylation in cells, HA-tagged Hprp3p or T494M were immunoprecipitated from nuclear extracts with G-agarose beads pre-bound with anti-HA antibody in 500 μl of buffer containing 100 mM potassium phosphate, 500 mM KCl, 1×protease inhibitor mixture, 1 mM Na3VO4, 50 mM NaF and 0.1% (v/v) Tween 20, pH 7.5 for 1 h at 4°C. The immunoprecipitated proteins obtained were then immunoblotted and detected with anti-phosphothreonine antibody. Changes in Hprp3p phosphorylation level were calculated by NIH ImageQuant analysis.
Cell viability and proliferation assays
Cell proliferation upon transduction with HD-Ad-LacZ, HD-Ad-F3i, HD-Ad-F3iplus or HD-Ad-F3iT494M was evaluated by a colorimetric MTT assay as previously described (37,52). The toxicity of T494M was also determined by the trypan blue exclusion assay (haemocytometer counting) and by measuring the percentage of total cellular release of LDH release (Roche Diagnostics, Laval, Québec, Canada).
PRPF6 cloning
A fragment of 3070 bp containing the full-length PRPF6 cDNA (Accession AF221842) was amplified from a human ORF clone (Open Biosystems, Hunstville, AL, USA) using the primers: 5′-AAAAGAATTCTTATGAACAAGAAGAA GAAACCGTTCCTAG-3′ and 5′-TTTGCGGCCGCTCAGA AGGTGTTCTTGATGCGG-3′ which include EcoRI and NotI restriction sites, respectively, and cloned into pCMV-myc (Clontech Canada Inc.).
Northern blotting and primer extension
The presence of U4 and U6 snRNAs in immunocomplexes precipitated with anti-HA antibodies was determined by northern blot and primer extension (21). For northern blotting detection of snRNAs associated with Hprp3p versions, total cellular RNA extracted from immunocomplexes precipitated with anti-HA antibody (see co-IP) was separated on a 8% polyacrylamide-urea (8 M) gel, transferred to nylon membrane (Roche Diagnostics), and UV cross-linked. The membrane was hybridized using ExpressHyb™ (BD Biosciences, Mississauga, ON, Canada) to oligonucleotide probes complementary to U1, U2, U4, U5 and U6 snRNAs at 37°C. The oligonucleotides listed below were used as probes after being labeled at the 5′ end with [γ-30P]-ATP using T4 polynucleotide kinase (MBI Fermentas Inc. Burlington, ON, Canada):
U1-(56–80): 5′-ACATCCGGAGTGCAATGGATAAG CC-3′
U2-(96–123): 5′-CTTCTATTCCATCTCTCTGCTCCA AAAATC-3′
U4-(82–109): 5′-GGTATTGGGAAAAGTTTTCAATT AG-3′
U5-(73–99): 5′-GCTCAAAAAATTGGGTTAAGACT CAG-3′
U6-(66–91): 5′-TCACGAATTTGCGTGTCATCCTT GC-3′
For primer extension, primers that yield extension products specific to human U4 (82 nucleotides) and U6 (91 nucleotides) snRNAs, respectively, were used as described previously (21).
Chemical cross-linking
Cells or animal tissue lysates were incubated with 1 mM solution of a cross-linking reagent in DMSO or DMSO alone for 2 h at 4°C. DMSO was used as the solvent for DFDNB at concentrations not exceeding 2% of total reaction mixtures (v/v). The cross-linking reagents tested included: DSP and DFDNB (Pierce, Rockford, IL, USA). The cross-linking was quenched by adding 100 mM glycine and incubating at 4°C for 5 min. The nuclear fraction was extracted and subjected to 8% SDS–PAGE under reducing conditions.
Real-time polymerase chain reaction
Studies of mRNA splicing were done by semiquantitative reverse transcription RT–PCR during 25 cycles. Primers were designed in regions of the cDNA located in different exons to avoid amplification of genomic DNA and to distinguish between unspliced and spliced forms. Primers were as follows:
-
RPL18—Homo sapiens RPL8 gene for ribosomal protein L8 (Accession AB061821):
5′-ACATCATCCACGACCCGGGCCGCGG-3′/5′-TGGCA TAGTTCCCTGATGCCC-3′;
H1 (0)—H. sapiens histone family, member 0 (Accession BC000145) 5′-CAGACATGATCGTGGCTGCCATCCA-3′/ 5′-GGCACTGGACTTTGCTTTGGGTTTC-3′;
EEF1A1—H. sapiens eukaryotic translation elongation factor 1 alpha 1 (Accession NM_001402)5′-TGGGAAAGG AAAAGACTCATATCAA-3′/5′-TGAGATGTCCCTGTAAT CATGTTTT-3′;
RPE65—H. sapiens RPE-specific protein 65 kDa (Accession NM_000329.2) 5′-GTGGTTACAAGAAACTGTTTGA AACT-3′/5′-AAAGGAAGTACATATCTCCTAACTTC-3′.
Statistical analysis
Student’s t-test or one-way ANOVA followed by Holm’s multiple comparison was used to compare data pairs or sets, respectively. Data are presented as mean ± SEM. P < 0.05 was considered significant.
Acknowledgments
We would like to thank Dr Gail Otulakowski, Dr Deborah J. Field and Attila Fust for helpful insights and review of this manuscript.
FUNDING
This work was supported by operating grants from The Foundation Fighting Blindness of Canada (FFB-C), Canadian Institutes of Health Research (CIHR) and Canadian Cystic Fibrosis Foundation (CCFF) to J.H. J.M.G. is a recipient of a FFB-C studentship award.
Footnotes
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
Conflict of Interest statement. None declared.
References
- 1.Wang Q, Chen Q, Zhao K, Wang L, Traboulsi EI. Update on the molecular genetics of retinitis pigmentosa. Ophthalmic Genet. 2001;22:133–154. doi: 10.1076/opge.22.3.133.2224. [DOI] [PubMed] [Google Scholar]
- 2.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125:151–158. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 4.Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. doi: 10.1186/1750-1172-1-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKie AB, McHale JC, Keen TJ, Tarttelin EE, Goliath R, van Lith-Verhoeven JJ, Greenberg J, Ramesar RS, Hoyng CB, Cremers FP, et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum Mol Genet. 2001;10:1555–1562. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- 6.Makarova OV, Makarov EM, Liu S, Vornlocher HP, Luhrmann R. Protein 61K, encoded by a gene (PRPF31) linked to autosomal dominant retinitis pigmentosa, is required for U4/U6*U5 tri-snRNP formation and pre-mRNA splicing. Embo J. 2002;21:1148–1157. doi: 10.1093/emboj/21.5.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakarova CF, Hims MM, Bolz H, Abu-Safieh L, Patel RJ, Papaioannou MG, Inglehearn CF, Keen TJ, Willis C, Moore AT, et al. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- 8.Maita H, Kitaura H, Keen TJ, Inglehearn CF, Ariga H, Iguchi-Ariga SM. PAP-1, the mutated gene underlying the RP9 form of dominant retinitis pigmentosa, is a splicing factor. Exp Cell Res. 2004;300:283–296. doi: 10.1016/j.yexcr.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 9.Mordes D, Luo X, Kar A, Kuo D, Xu L, Fushimi K, Yu G, Sternberg P, Jr, Wu JY. Pre-mRNA splicing and retinitis pigmentosa. Mol Vis. 2006;12:1259–1271. [PMC free article] [PubMed] [Google Scholar]
- 10.Soller M. Pre-messenger RNA processing and its regulation: a genomic perspective. Cell Mol Life Sci. 2006;63:796–819. doi: 10.1007/s00018-005-5391-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Will CL, Luhrmann R. Spliceosomal UsnRNP biogenesis, structure and function. Curr Opin Cell Biol. 2001;13:290–301. doi: 10.1016/s0955-0674(00)00211-8. [DOI] [PubMed] [Google Scholar]
- 12.Will CL, Luhrmann R. Protein functions in pre-mRNA splicing. Curr Opin Cell Biol. 1997;9:320–328. doi: 10.1016/s0955-0674(97)80003-8. [DOI] [PubMed] [Google Scholar]
- 13.Nottrott S, Urlaub H, Luhrmann R. A hierarchical assembly pathway common to U4/U6 and U4atac/U6atac snRNPs. Sixth Annual meeting of the RNA Society; Alberta, Canada. 2001. p. 5. [Google Scholar]
- 14.Brow DA. Allosteric cascade of spliceosome activation. Annu Rev Genet. 2002;36:333–360. doi: 10.1146/annurev.genet.36.043002.091635. [DOI] [PubMed] [Google Scholar]
- 15.Konarska MM, Sharp PA. Association of U2, U4, U5 and U6 small nuclear ribonucleoproteins in a spliceosome-type complex in absence of precursor RNA. Proc Natl Acad Sci USA. 1988;85:5459–5462. doi: 10.1073/pnas.85.15.5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Behzadnia N, Hartmuth K, Will CL, Luhrmann R. Functional spliceosomal A complexes can be assembled in vitro in the absence of a penta-snRNP. RNA. 2006;12:1738–1746. doi: 10.1261/rna.120606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horowitz DS, Kobayashi R, Krainer AR. A new cyclophilin and the human homologues of yeast Prp3 and Prp4 form a complex associated with U4/U6 snRNPs. RNA. 1997;3:1374–1387. [PMC free article] [PubMed] [Google Scholar]
- 18.Lauber J, Plessel G, Prehn S, Will CL, Fabrizio P, Groning K, Lane WS, Luhrmann R. The human U4/U6 snRNP contains 60 and 90kD proteins that are structurally homologous to the yeast splicing factors Prp4p and Prp3p. RNA. 1997;3:926–941. [PMC free article] [PubMed] [Google Scholar]
- 19.Wang A, Forman-Kay J, Luo Y, Luo M, Chow YH, Plumb J, Friesen JD, Tsui LC, Heng HH, Woolford JL, Jr, et al. Identification and characterization of human genes encoding Hprp3p and Hprp4p, interacting components of the spliceosome. Hum Mol Genet. 1997;6:2117–2126. doi: 10.1093/hmg/6.12.2117. [DOI] [PubMed] [Google Scholar]
- 20.Teigelkamp S, Achsel T, Mundt C, Gothel SF, Cronshagen U, Lane WS, Marahiel M, Luhrmann R. The 20 kD protein of human [U4/U6. U5] tri-snRNPs is a novel cyclophilin that forms a complex with the U4/U6-specific 60 kD and 90 kD proteins. RNA. 1998;4:127–141. [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez-Santos JM, Wang A, Jones J, Ushida C, Liu J, Hu J. Central region of the human splicing factor hprp3p interacts with hprp4p. J Biol Chem. 2002;277:23764–23772. doi: 10.1074/jbc.M111461200. [DOI] [PubMed] [Google Scholar]
- 22.Reidt U, Reuter K, Achsel T, Ingelfinger D, Luhrmann R, Ficner R. Crystal structure of the human U4/U6 small nuclear ribonucleoprotein particle-specific SnuCyp-20, a nuclear cyclophilin. J Biol Chem. 2000;275:7439–7442. doi: 10.1074/jbc.275.11.7439. [DOI] [PubMed] [Google Scholar]
- 23.Makarova OV, Makarov EM, Urlaub H, Will CL, Gentzel M, Wilm M, Luhrmann R. A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. Embo J. 2004;23:2381–2391. doi: 10.1038/sj.emboj.7600241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medenbach J, Schreiner S, Liu S, Luhrmann R, Bindereif A. Human U4/U6 snRNP recycling factor p110: mutational analysis reveals the function of the tetratricopeptide repeat domain in recycling. Mol Cell Biol. 2004;24:7392–7401. doi: 10.1128/MCB.24.17.7392-7401.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu S, Rauhut R, Vornlocher HP, Luhrmann R. The network of protein-protein interactions within the human U4/U6. U5 tri-snRNP. RNA. 2006;12:1418–1430. doi: 10.1261/rna.55406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maita H, Kitaura H, Ariga H, Iguchi-Ariga SM. CIR, a corepressor of CBF1, binds to PAP-1 and effects alternative splicing. Exp Cell Res. 2005;303:375–387. doi: 10.1016/j.yexcr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Martinez-Gimeno M, Gamundi MJ, Hernan I, Maseras M, Milla E, Ayuso C, Garcia-Sandoval B, Beneyto M, Vilela C, Baiget M, et al. Mutations in the pre-mRNA splicing-factor genes PRPF3, PRPF8 and PRPF31 in Spanish families with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2003;44:2171–2177. doi: 10.1167/iovs.02-0871. [DOI] [PubMed] [Google Scholar]
- 28.Wada Y, Itabashi T, Sato H, Tamai M. Clinical features of a Japanese family with autosomal dominant retinitis pigmentosa associated with a Thr494Met mutation in the HPRP3 gene. Graefes Arch Clin Exp Ophthalmol. 2004;242:956–961. doi: 10.1007/s00417-004-0923-x. [DOI] [PubMed] [Google Scholar]
- 29.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 31.Cooper TA. Use of minigene systems to dissect alternative splicing elements. Methods. 2005;37:331–340. doi: 10.1016/j.ymeth.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 32.Liu HX, Cartegni L, Zhang MQ, Krainer AR. A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet. 2001;27:55–58. doi: 10.1038/83762. [DOI] [PubMed] [Google Scholar]
- 33.Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet. 2006;78:63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005;122:365–378. doi: 10.1016/j.cell.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Smith PJ, Krainer AR, Zhang MQ. Distribution of SR protein exonic splicing enhancer motifs in human protein-coding genes. Nucleic Acids Res. 2005;33:5053–5062. doi: 10.1093/nar/gki810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Y, Zhang Y, Zhang J. Distribution of exonic splicing enhancer elements in human genes. Genomics. 2005;86:329–336. doi: 10.1016/j.ygeno.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez-Santos JM, Cao H, Wang A, Koehler DR, Martin B, Navab R, Hu J. A complementation method for functional analysis of mammalian genes. Nucleic Acids Res. 2005;33:e94. doi: 10.1093/nar/gni093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- 39.Philp NJ, Wang D, Yoon H, Hjelmeland LM. Polarized expression of monocarboxylate transporters in human retinal pigment epithelium and ARPE-19 cells. Invest Ophthalmol Vis Sci. 2003;44:1716–1721. doi: 10.1167/iovs.02-0287. [DOI] [PubMed] [Google Scholar]
- 40.Bazan NG. Survival signaling in retinal pigment epithelial cells in response to oxidative stress: significance in retinal degenerations. Adv Exp Med Biol. 2006;572:531–540. doi: 10.1007/0-387-32442-9_74. [DOI] [PubMed] [Google Scholar]
- 41.Shi Y, Reddy B, Manley JL. PP1/PP2A phosphatases are required for the second step of Pre-mRNA splicing and target specific snRNP proteins. Mol Cell. 2006;23:819–829. doi: 10.1016/j.molcel.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 42.Cao W, Jamison SF, Garcia-Blanco MA. Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA. 1997;3:1456–1467. [PMC free article] [PubMed] [Google Scholar]
- 43.Mermoud JE, Cohen PT, Lamond AI. Regulation of mammalian spliceosome assembly by a protein phosphorylation mechanism. Embo J. 1994;13:5679–5688. doi: 10.1002/j.1460-2075.1994.tb06906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 45.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neet KE, Timm DE. Conformational stability of dimeric proteins: quantitative studies by equilibrium denaturation. Protein Sci. 1994;3:2167–2174. doi: 10.1002/pro.5560031202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends Biochem Sci. 2004;29:618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Ayadi L, Callebaut I, Saguez C, Villa T, Mornon JP, Banroques J. Functional and structural characterization of the prp3 binding domain of the yeast prp4 splicing factor. J Mol Biol. 1998;284:673–687. doi: 10.1006/jmbi.1998.2183. [DOI] [PubMed] [Google Scholar]
- 49.Pellizzoni L, Charroux B, Dreyfuss G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc Natl Acad Sci USA. 1999;96:11167–11172. doi: 10.1073/pnas.96.20.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen T, Damaj BB, Herrera C, Lasko P, Richard S. Self-association of the single-KH-domain family members Sam68, GRP33, GLD-1 and Qk1: role of the KH domain. Mol Cell Biol. 1997;17:5707–5718. doi: 10.1128/mcb.17.10.5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hashiyama K, Takeuchi A, Makino O. Effects of single amino acid substitutions at the predicted coiled-coil or hydrophobic region on the self-assembly of phi29 replication protein, gp1. Biochem Biophys Res Commun. 2005;331:1310–1316. doi: 10.1016/j.bbrc.2005.04.056. [DOI] [PubMed] [Google Scholar]
- 52.Gerlier D, Thomasset N. Use of MTT colorimetric assay to measure cell activation. J Immunol Methods. 1986;94:57–63. doi: 10.1016/0022-1759(86)90215-2. [DOI] [PubMed] [Google Scholar]
- 53.Legrand C, Bour JM, Jacob C, Capiaumont J, Martial A, Marc A, Wudtke M, Kretzmer G, Demangel C, Duval D, et al. Lactate dehydrogenase (LDH) activity of the cultured eukaryotic cells as marker of the number of dead cells in the medium [corrected] J Biotechnol. 1992;25:231–243. doi: 10.1016/0168-1656(92)90158-6. [DOI] [PubMed] [Google Scholar]
- 54.Maita H, Kitaura H, Ariga H, Iguchi-Ariga SM. Association of PAP-1 and Prp3p, the products of causative genes of dominant retinitis pigmentosa, in the tri-snRNP complex. Exp Cell Res. 2005;302:61–68. doi: 10.1016/j.yexcr.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 55.Comitato A, Spampanato C, Chakarova C, Sanges D, Bhattacharya SS, Marigo V. Mutations in splicing factor PRPF3, causing retinal degeneration, form detrimental aggregates in photoreceptor cells. Hum Mol Genet. 2007;16:1699–1707. doi: 10.1093/hmg/ddm118. [DOI] [PubMed] [Google Scholar]
- 56.Misteli T, Caceres JF, Spector DL. The dynamics of a pre-mRNA splicing factor in living cells. Nature. 1997;387:523–527. doi: 10.1038/387523a0. [DOI] [PubMed] [Google Scholar]
- 57.Carrero G, Hendzel MJ, de Vries G. Modelling the compartmentalization of splicing factors. J Theor Biol. 2006;239:298–312. doi: 10.1016/j.jtbi.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 58.Lai MC, Tarn WY. Hypophosphorylated ASF/SF2 binds TAP and is present in messenger ribonucleoproteins. J Biol Chem. 2004;279:31745–31749. doi: 10.1074/jbc.C400173200. [DOI] [PubMed] [Google Scholar]
- 59.Lamond AI, Spector DL. Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol. 2003;4:605–612. doi: 10.1038/nrm1172. [DOI] [PubMed] [Google Scholar]
- 60.Chow YH, O’Brodovich H, Plumb J, Wen Y, Sohn KJ, Lu Z, Zhang F, Lukacs GL, Tanswell AK, Hui CC, et al. Development of an epithelium-specific expression cassette with human DNA regulatory elements for transgene expression in lung airways. Proc Natl Acad Sci USA. 1997;94:14695–14700. doi: 10.1073/pnas.94.26.14695. [DOI] [PMC free article] [PubMed] [Google Scholar]