

Abstract
The success of all-trans retinoic acid (ATRA) therapy in acute promeylocytic leukemia (APL) has spawned numerous attempts to translate the paradigm of differentiation therapy to non-APL acute myelocytic leukemia (AML). However, the results of clinical trials have been overall disappointing. In this review we discuss the mechanism of retinoic acid signaling and the results of major clinical trials that have attempted to incorporate ATRA into AML regimens. We discuss recent evidence that indicate that the retinoic acid signaling pathway may be dysfunctional in AML. Preliminary studies suggest that targeting the pathways that modify retinoic acid receptor activity may reactivate the dormant retinoic acid-signaling pathway. Such strategies may revive the ability of ATRA to induce myeloid differentiation and apoptosis in non-APL AML.
Keywords: ATRA, differentiation therapy, AML
1. Introduction
Through binding to the retinoic acid receptor (RAR), All-trans retinoic acid (ATRA, Figure 1) activates transcription of genes necessary for myeloid cell differentiation (reviewed in Melnick and Licht1). Conversely, ATRA treatment fails to induce differentiation in cells with dysfunctional RAR signaling. The most notable example of this occurs in acute promyelocytic leukemia (APL), in which an RAR fusion protein acts as a dominant-negative inhibitor of retinoic acid-mediated transcription2, 3. Pharmacologic doses of ATRA induce activation and degradation of the APL fusion protein4, leading to re-expression of the myeloid differentiation program, terminal differentiation and apoptosis of the leukemic cells, and ultimate remission of the leukemia. For almost forty years investigators have hoped that ATRA therapy would similarly impact non-APL acute myeloid leukemia (AML). Results from clinical trials have been mixed, and overall, disappointing. In this review we explore potential reasons for the disappointing results, as well as recent molecular studies that bring new excitement to the field.
Figure 1.
Structure of all-trans retinoic acid.
2. Retinoic acid Receptor
ATRA is synthesized from retinol (Vitamin A) by oxidation to retinaldhyde, and then to retinoic acid (for review, see Melnick and Licht1). Other forms of retinoic acid – 9-cis, 11-cis, or 13 cis – can be converted through isomerization into ATRA. Cells protect themselves from transient changes in levels of retinoic acid via CRABP (cytoplasmic retinoic acid binding protein) and CRBP II (cytosolic retinoid binding protein) which function as high capacity/low affinity buffers. ATRA not only induces expression of these proteins, but also the p450 enzymes responsible for its own metabolism. Within the nucleus, ATRA serves as the physiologic ligand for RAR. Three proteins comprise the RAR family of nuclear receptors – alpha, beta, and gamma. All are structurally similar, and differ primarily in tissue and developmental expression (gamma being expressed at higher levels in skin; alpha more so in hematopoietic cells). The RARs are members of the steroid nuclear receptor family, and most closely share homology with the nuclear receptors for vitamin D and thyroid hormone5. RARs bind DNA primarily as heterodimers with the cognate receptor RXR to target promoters that contain direct repeats of the consensus sequence C/AGGAA/T separated by 5 bases6.
Like other members of the nuclear hormone receptor family, RAR has a modular structure: an N-terminal AF-1 activating domain, a DNA-binding zinc-finger motif, a coiled-coil domain that primarily mediates dimerization with RXR, and a large C-terminal domain that mediates ligand binding as well as interaction with co-repressor and co-activator partners5. In contradistinction from the estrogen or glucocorticoid receptors, RAR is primarily DNA-bound in both the presence and absence of ligand. In the absence of ligand, RAR tethers to target promoters in complex with co-repressor proteins that contain histone decetylase activity, to modulate target chromatin structure and actively repress transcription7. Upon binding ATRA, RAR undergoes a major change in conformation8. As a result the co-repressor complex is released, and a domain is exposed that allows RAR to interact with a co-activator complex (Figure 2). This reverses the chromatin-silencing effects of the co-repressor, and results in recruitment of RNA polymerase to initiate gene transcription.
Figure 2.

Schematic demonstrating interaction of RAR with Co-repressor and Co-activator complexes in absence (panel A) and presence (panel B) of ATRA.
3. ATRA and myeloid differentiation
Though regulation of myeloid development is complex, it is clear that many RAR target genes are involved in regulation of myelopoiesis9, 10, including C/EBPepsilon, PU.1 and HOX proteins; many others are involved in regulation of the cell cycle, and the intrinsic and extrinsic apoptotic pathways, such as p21, c-myc, several of the cyclin proteins, and FAS and FASL.
Early empiric work of Breitman et al. established the ability of ATRA to induce differentiation in myeloid leukemia cells, most notably APL cells11. With the cloning of the receptor for retinoic acid in 198712, genetic approaches established the critical role of the retinoic acid-signaling pathway in regulation of myeloid differentiation. Tsai et al., for instance, showed that transfection of a dominant-negative RARα blocked myeloid differentiation of the FDCP1 cell line13. Surprisingly, murine knock-out models of RARα did not show a dramatic reduction in myelopoiesis10. This may be attributed to redundancy between the three isoforms of RAR–α, β, and γ. Double knockout of both RARα and RARγ is necessary to demonstrate impaired myelopoiesis14. The strongest evidence for the involvement of RAR in myelopoiesis has come from detailed studies of PML-RAR, the fusion protein expressed as a result of t(15;17)(q22;q21) in APL15-17. PML-RAR serves as a dominant-negative for RAR signaling, both through sequestration of RXR and through competition for binding to RAR target promoters, where PML-RAR blocks transcription due to its increased avidity for co-repressor molecules.
3.1 ATRA effects on primary AML cultures
Preclinical observations in tissue culture cells identified numerous AML and myeloid cell lines that undergo differentiation upon exposure to ATRA, including HL-60, THP-1, MOLM-14, HF-6, and U937 18. However, the effects of ATRA on primary leukemic cells have been mixed. Most reports indicate induction of apoptosis rather than differentiation, with effects demonstrable in approximately 50% of samples. For instance, Bradbury et al. found that in vitro culture of primary AML cells with 1 micromolar ATRA resulted in decreased expression of Bcl-2 in 8 of 25 samples19. Similarly, Sakashita et al. 20 reported that ATRA decreased in vitro clonal growth in 14 of 25 primary AML samples: none underwent differentiation. Lehman et al. found that ATRA accelerated in vitro daunorubicin-induced apoptosis and decreased growth of primary blasts in more than half of 23 samples 21.
Looking at the cellular and molecular effects of a short in vivo course of treatment with ATRA (22.5 mg/m2 twice daily for two days), Ryningen et al. found no morphologic changes in circulating leukemic cells, nor alteration in expression of CD34, CD11b, CD15, or CD71 differentiation markers22. Several patients showed increased expression of p21 and decreased levels of GATA-2 in their blasts. Minor changes were observed in Bcl-2 expression. Decreased levels of NFκB p65 were also observed. Consistent with these molecular changes, an increased population of cells in G0/G1 was observed, suggesting cell cycle arrest22.
3.2 ATRA in clinical trials
3.2.1 Single agent
Few clinical trials in AML have utilized single agent ATRA; most monotherapy trials have focused on MDS, with few responders. Griggs et al. described one patient with normal cytogenetic-AML who was refractory to standard induction chemotherapy, but who entered a remission upon treatment with ATRA 45 mg/m2/day for 34 days23. Qian et al. reported the effects of single-agent ATRA in four t(8;21) AML patients who were originally misdiagnosed as APL24. In one case, a patient was treated with arsenic trioxide for 50 days with no response; he was thereafter transitioned to 40 mg/d ATRA, and cleared his marrow of blasts within one week, but relapsed three weeks later. Case 2 was an 8 year old boy with AML treated with 30 mg/d ATRA: promyelocytes decreased from 70% to 21% at 20 days, but increased again to 62% after 40 days of ATRA. Case 3 was a 16 year old treated with 60 mg/d ATRA; she achieved a remission at day 32, after which therapy was discontinued, but one month later she relapsed. The fourth case was a 16 year old boy treated with 60 mg/d ATRA (for the first seven days he was also given hydrea); remission was obtained by day 35, after which he was treated with consolidation chemotherapy. Though tantalizing, these cases are few, and the responses mixed and short-lived.
3.2.2 ATRA as part of induction chemotherapy
Studies combining ATRA with induction chemotherapy have yielded disparate and controversial results. Several large randomized trials failed to observe an advantage to adding ATRA to induction chemotherapy. In a Phase II randomized study of patients with relapsed or refractory AML Belhabri et al. combined idarubicin 10 mg/d x3d with cytarabine 1000 mg/m2 q 12 h for 6 days, with or without ATRA 45 mg/m2/d from day 1 until remission25. With 47-48 subjects in each arm, they found no significant difference in outcomes, with an overall remission rate of 57%. In a larger study, the MRC AML-HR group26 randomized 405 patients with high-risk AML to 2 courses of ADE (cytarabine 100 mg/m2 q12 d 1-10; daunrobuicin 50 mg/m2 d 1,3,5; etoposide 100 mg/m2 qd d1-5) vs 2 courses of FLA (fludarabine 30 mg/m2 d 1-5; cytarabine 1 or 2 gm/m2 qd d1-5), +/− ATRA 45 mg/m2 to a maximum of 90 days, +/− G-CSF. No advantage to ATRA (nor G-CSF) was observed in remission rate, relapse rate, disease-free or overall survival. Studying a population deemed not fit for induction chemotherapy, Burnett et al.27 randomized patients to receive either low dose cytarabine (20 mg sq bid x 10d every 4-6 weeks) or hydrea, +/− ATRA 45 mg/m2 qd for 60 days. They found no significant benefit in survival or remission rate upon addition of ATRA. In the MRC AML12 trial, Burnett et al. randomized 1075 patients to induction therapy with daunorubicin 50 gm/m2 d 1,3,5; cytarabine 100 or 200 mg/m2 d1-10 q12 h; and thioguanine 100 mg/m2 d1-10; followed by a second induction cycle of 8 day duration, with or without ATRA at a dose of 45 mg/m2 day 1-628. They found no effect from the addition of ATRA on remission rate, overall survival, toxicity, nor kinetics of hematologic recovery. Estey et al.29 randomized high risk patients to receive fludarabine 30 mg/m2 qd x 4 plus cytarabine 2 gm/m2/d d 1-4, and idarubicin 12 mg/m2 days 2-4 +/− G-CSF +/− ATRA 45 mg/m2/d day −2 though d7, with 53-55 patients in each arm. The addition of G-CSF did not affect outcomes. Initial univariate analysis suggested an advantage to the ATRA arm in remission rate and survival, but after accounting for other covariances (age, platelets, treatment in protected environment, performance status, treated as emergency, and unfavorable cytogentics) multivariate analysis failed to indicate superior outcomes29.
In contradistinction from these studies, the ULM Study Group HD98B trial demonstrated an advantage to receiving ATRA with induction chemotherapy30. In this Phase III trial, 242 elderly AML patients were randomized to receive either conventional chemotherapy for induction and consolidation, or the same regimen with ATRA. The induction regimen consisted of idarubicin 12 mg/m2 d 1 and 3, cytarabine 100 mg/m2 d 1-5, and etoposide 100 mg d 1 and 3. ATRA was given at a dose of 45 mg/m2 day3-5 followed by a lower dose of 15 mg/m2 day 6-28. Patients achieving a complete remission received a second round of the same regimen. Consolidation consisted of cytarabine 0.5 gm/m2/q12 d 1-3; and mitoxantrone 10 mg/m2 d 2 and 3, with or without ATRA 15 mg/m2 d 3-28. Those who did not undergo allogeneic transplant were then randomized to a second intensive consolidation regimen, or oral maintenance therapy (without further ATRA). Patients in the ATRA arm had a statistically significant improvement in remission rate (38.0% versus 27.5% in the ATRA versus standard arms, respectively) and overall survival (estimated median survival 11.3 versus 7.0 months). Sub-analysis indicated that the benefit of ATRA was limited to the population with NPM1 mutation without FLT3-ITD. The reason for the association with NPM mutation is unclear: reanalysis of the Estey trial failed to show similar association31, nor did subgroup analysis of the AML12 trial find a difference in the population with NPM1 mutation28. Based on the data from the HD98B study, the German-Austrian group developed AML 07-04, targeting a younger population of AML patients with NPM1 mutation; preliminary analysis32 suggests a statistically significant increase in response rate, event-free survival, and overall survival in the ATRA-treated cohort.
The discrepant results between the two ULM studies and the trials led by Burnett, Belharbi, and Estey are not easily explained, but it should be noted that the age, entry criteria, chemotherapy regimens, dose, and duration of administration of ATRA differed amongst all the trials, making it difficult to make comparisons. None of the clinical trials explored mechanism.
4. Is RAR functional in AML?
The results of clinical trials utilizing ATRA have been inconsistent and, overall, disappointing. This raises the question of whether the retinoic acid receptor transcriptional mechanism is functional in AML blasts, and susceptible to activation by ATRA.
4.1 Inhibition of RAR activity by leukemic oncoproteins
A panoply of genes are mutated or aberrantly expressed in AML, many of which impact RAR either directly or indirectly. For instance, SKI, a nuclear oncogene frequently overexpressed in AML, binds RAR directly to mute RAR transcriptional activity33. RAR serves as a component of the macromolecular complex formed by the t(8;21) fusion protein RUNX1-CBFA2T1,which suppresses RAR activity by both sequestering RAR and potentially recruiting corepressor activities to RAR target genes34. MN1, another commonly overexpressed oncogene in AML, directly binds to RAR to inhibit retinoic acid signaling35. Bullinger et al. have recently reported overexpression of PRAME1, a member of the RAR-associated co-repressor complex, in AML36: interestingly, high levels of PRAME were associated with ATRA responsiveness in the ULM AML 07-04 trial.
4.2 Epigenomic changes that affect RAR signaling
Many proteins that are aberrantly expressed in AML affect the epigenome37. Among these, DNMT1, 3A, and 3B alter DNA methylation; MLL as well as TET1 and 2 and IDH 1 and 2 modulate histone methylation; AML1-ETO aberrantly recruits histone deacetylases, to name but a few. Indeed, AML blasts show global changes in the epigenome38. Several groups have hypothesized that such epigenetic changes could silence the promoters of RAR target genes to the transactivating effects of liganded-RAR, thus protecting the blasts from the differentiating and apoptosis inducing effects of ATRA.
Investigating the potential of reversing the chromatin modifications induced by histone deacetylation of RAR targets, Ferrara et al.39 utilized the pan-histone decetylase inhibitor trichostatin A. Co-culture of primary blasts from 23 AML patients with ATRA plus trichostatin A for 5 days resulted in increased expression of a series of RA-responsive genes and enhanced differentiation of all the samples. Trus et al.40 studied the effects of the clinically available histone deacetylase inhibitor valproic acid in combination with ATRA, and found that valproic acid restored the ability of ATRA to induce expression of retinoic response genes in the AML cell line OCI/AML-2. The combination also induced expression of p21, which correlated with induction of cell cycle arrest. Similar effects were observed in culture of 6 AML primary samples.
Fortified by in vitro data suggesting that valproic acid could “prime” targets for ATRA-induced transcription, several clinical studies were begun with a focus on the combination of ATRA plus valproic acid. Ciminao et al.,41 in a study of eight patients, found that sequential valproic acid-ATRA therapy resulted in a decrease in bone marrow blasts and an increase in expression of myeloid and monocytoid differentiation markers, but only two patients showed hematological improvement. Several larger studies failed to show a dramatic effect of the combination on clinical outcomes. Kuendgen et al.,42 for instance, found a 5% response in a study of 58 patients treated with valproic acid and ATRA. Fredly et al.,43 in a study of 36 AML patients treated with valproic acid, ATRA, and ara-C, observed that only two obtained a remission, though nine showed stable disease. Ryningen et al.44 reported hematologic improvement in 4 of 22 patients treated with the combination of ATRA, valproic acid, and theophylline. Bug et al. treated 26 patients with valproic acid and ATRA45; only two demonstrated an objective response. Recently, Tassara et al. reported the results of the AMLSG 06-04 study, which added valproic acid to induction therapy of idarubicin, cytarbine, and ATRA46. They found a statistically significant increase in relapse-free survival in patients who received valproic acid, though no change in overall survival was observed; remission rates were actually lower in the group receiving valproic acid because of increased pulmonary toxicity and early death. In sum, the results of such trials focusing on histone modification have not yielded dramatic improvements in patient responses.
An alternative strategy has been to reverse DNA methylation through the use of the DNA methyltransferase inhibitor 5-azacytidine as a ‘priming’ agent for ATRA. Fazi et al. showed that in vitro alteration of methylation by 5-azacytidine restored retinoic acid-mediated differentiation in RUNX1-CBFA2T1 blasts34. In an attempt to target both DNA methylation and histone deacetylation, Soriano et al. developed a Phase II trial of the combination of 5-azacytidine with valproic acid and ATRA in 53 patients47, and obtained an overall response rate of 42%. Oddly, clinical outcomes did not correlate with degree of hypomethylation, histone acetylation, or p21 expression. The trial was not powered to determine an advantage to the addition of ATRA and valproic acid to treatment with single agent 5-azacytidine, which has been reported to generate similar response rates48.
Many commonly mutated genes in AML are enzymes that modify target gene expression through alteration of histone methylation. Schenk et al. found that several RAR target genes are epigenetically silenced in AML49. The promoters of these genes were characterized by decreased methylation of H3K4 (H3K4me2). H3K4 methylation is regulated by the LSI1 demethylase, which is commonly overexpressed in AML. Indeed, this group found that inhibition of LSI1 enhanced ATRA-driven gene expression, supporting the hypothesis that nonspecific changes in histone methylation could lead to silencing of RAR target genes49 (Figure 3). Reactivation of ATRA signaling by the LSI inhibitor tranylcypromine enhanced differentiation of ATRA-sensitive cell lines. Moreover, the combination of ATRA and LSI1 inhibition induced primary AML cells to differentiate in culture, and decreased AML engraftment in xenotransplantation models.
Figure 3.

Model of epigenetic modulation on activity of RAR: Alteration of histone methylation or acetylation (depicted as red balls) of histones may allow the RAR transcriptional complex to form, but prevents activation of transcription of target genes.
4.3 Targeting post-translational modification of RAR
A further mechanism that could affect RAR functionality is through direct post-translational modification of RAR to suppress its activity. For instance, P38alphaMAPK, a downstream mediator of the MEK/ERK pathway, associates with RARα and directly phosphorylates it at Ser369, leading to degradation of the RAR protein50. The serine/threonine kinase JNK phosphorylates RARα at T181, S445, and S461, which results in targeting RAR for proteosomal degradation51. AKT suppresses RARα transactivation through phosphorylation of Ser96 within the RAR DNA binding domain; AKT does not alter RAR DNA binding, but most probably results in modification of a region necessary for cofactor binding52. PKC phosphorylates RAR at Ser157 to inhibit dimerization with RXR, and thereby decreases transcriptional activity 53. Many of these enzymatic cascades are downstream from pathways commonly activated in AML, such as FLT3-ITD, Src family kinases, or Ras, and thus are activated in the majority of AML.
Several groups have begun to investigate the hypothesis that inhibition of these post-translational mechanisms might enhance RAR activity. Gianni et al. utilized pharmacologic inhibitors of p38MAPK and found that inhibition of RARα S369 phosphorylation in myeloid cells inhibited proteasomal degradation of RARα, and increased ATRA-mediated differentiation50. Hoshikawa et al., studying hepatic cell lines, found that inhibition of JNK activation blocked RARα degradation, leading to enhanced RAR activity54. Srinivas et al., in a lung cancer model, showed that AKT inhibition reactivated suppressed RARα52. Though not yet in clinical trials, the concept that activation of RAR can be achieved through inhibition of these pathways merits investigation.
Glycogen synthase kinase 3 (GSK3) is a serine/threonine kinase that is often overexpressed in AML55. It has been shown to directly associate with and phosphorylate the N-terminus of RAR at Ser443, 445, and 44955. Phosphorylation of these sites decreases the activity of RAR; conversely, inhibition of GSK3 activity blocks phosphorylation of these sites, and increases RAR activity56. In addition, GSK3 inhibition leads to increased levels of RAR expression, potentially by enhancing stability of the RAR protein56. Gupta et al. have shown that GSK3 inhibitors synergize with ATRA in induction of differentiation of a panel of non-APL AML cell lines, including HL-60 (M2 phenotype) and OCl-AML3 (M4 phenotype)56. Interestingly, lithium is a potent inhibitor of GSK3, and shows significant synergy with ATRA in inducing differentiation of AML cell lines in vitro as well as in a xenotransplantation model of leukemia56. A clinical trial assessing the synergy of lithium carbonate and ATRA as differentiation therapy for AML has begun at Case Western Reserve (clinicaltrials.gov NCT01820624).
Our group has focused on the role of activation of Src family kinases (SFKs) in AML. We have found that Lyn kinase, which is aberrantly activated in AML, suppresses the activity of RARα57. The degree of inhibition of RAR transcriptional activity correlates well with Lyn expression levels. SFK inhibitors reactivate the silenced RAR, and synergize with ATRA to enhance RAR-mediated transcription58 (Figure 4). Treating AML model cells lines or primary AML samples with a combination of SFK inhibitor and ATRA leads to differentiation in vitro57. Studies to assess the detailed pathways through which SFKs modulate RAR activity are underway. Dasatinib is an FDA-approved SFK-inhibitor, and we have shown that dasatinib similarly enhances ATRA-mediated differentiation in AML cell lines and primary samples in vitro. At the University of Pittsburgh Medical Center we have initiated a clinical trail of the combination of dasatinib plus ATRA for non-APL AML (clinicaltrials.gov identifier NCT00892190).
Figure 4.
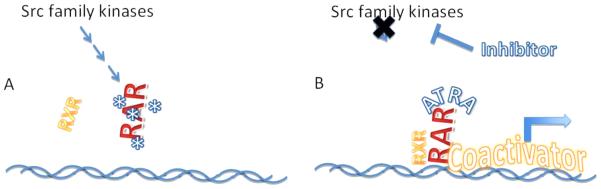
Proposed model of the mechanism underlying synergy between Src Family Kinase inhibition and ATRA. Panel A: activation of SFK tyrosine kinases in AML promotes, either directly or indirectly leads to phosphorylation of RAR, inhibiting the ability of RAR to bind to RXR and/or DNA. Panel B: SKF inhibition blocks the ability of SFKs to promote phosphorylate RAR, allowing RAR to bind normally RXR and DNA targets, and in the presence of ATRA, to regain its ability to function as a transcriptional activator.
4.4 Enhancing RAR expression
As mentioned above, GSK3 leads to decreased expression of RAR, and GSK3 inhibitors increase the level of RAR protein, thus contributing to the observed synergistic activity with RAR56. RAR is degraded in cells through ubiquination and targeting to the proteasome59-61. With this in mind, Luo et al.62 tested the concept that proteasome inhibition could enhance the activity of ATRA. In a neuroblastoma cell model, they found that bortezomib, a clinically available inhibitor of the 26S proteasome, increased RAR activity and enhanced the ability of ATRA to induce neurite differentiation 500-fold. Recently, the same group reported enhancement of ATRA-mediated differentiation of human AML cell lines by bortezomib63.
4.5 Other novel approaches
Aldoketoreductase AKR1C3 is a multifunctional NADPH-dependent oxidoreductase that is involved in the metabolism of androgens, estrogens, retinoids and xenobiotics64. Elevated expression of AKR1C3 has been shown in primary AML. Inhibition of AKR1C3 would be expected to increase endogenous levels of ATRA, and increase RAR activity. Though the exact mechanism has not been reported, medroxypogesterone, a potential inhibitor of AKR1C3, has been used in combination with a PPAR agonist, and found to induce differentiation of AML cell lines64, 65.
Inosine 5’-monophosphate dehydrogenase (IMPDH) is the rate-controlling enzyme in synthesis of guanine nucleosides. IMPDH is expressed at high levels in AML blasts66. Though the mechanism by which guanine nucleoside levels might affect RAR activity has not been determined, treatment of ATRA-sensitive cell lines with ATRA and an IMPDH inhibitor results in a greater degree of differentiation than with either agent alone 67, 68. Mycophenolate mofetil, commonly used as an immunomdulating agent, is an FDA-approved IMPDH inhibitor – it is tempting to speculate that it might show synergistic activity with ATRA in AML.
5. Conclusions
Despite its importance in controlling myeloid differentiation and apoptotic pathways, ATRA has yet to prove itself as a useful agent in the armamentarium of AML therapeutics. The explanation probably lies in the fact that RAR is expressed, but of limited functionality in AML blasts. This likely stems from a combination of epigenetic changes that prevent RAR from activating transcription of target genes, as well as post-translation modifications of RAR itself, which limit its functionality and stability. Genetic alterations affecting the epigenome, as well as RAR post-translational modification, are common in AML. The work of our group, along with the work of many others, indicates that silenced RAR functionality can be re-established in AML cells under the appropriate circumstances. Because of silencing of RAR activity in AML, single agent ATRA would not be expected to have a major impact, and indeed it has not. It may well prove necessary to address more than one of the pathways that regulate RAR activity in order to allow ATRA to induce the (silenced) expression of RAR target genes in AML, and allow ATRA to emerge as a therapeutic modality for non-APL AML.
Practice Points.
The retinoic acid receptor controls expression of multiple gene pathways necessary for myeloid differentiation
Multiple abnormal pathways in AML may impact retinoic acid receptor-mediated transcription
ATRA, either as single agent or combined with chemotherapy, has had limited success in treatment of AML
Research Agenda.
Identification of pathways that modify activity of the retinoic acid receptor
Development of clinical trials of ATRA in conjunction with agents that may reactivate the silenced retinoic acid receptor in AML
Acknowledgements
This work was supported by LLS Translational Research Award 6028-10 and NIH grant P30 CA047904.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors report no conflicts of interest.
References
- 1.Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999;93:3167–215. [PubMed] [Google Scholar]
- 2.de The H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 1991;66:675–84. doi: 10.1016/0092-8674(91)90113-d. [DOI] [PubMed] [Google Scholar]
- 3.Kakizuka A, Miller WH, Jr., Umesono K, Warrell RP, Jr., Frankel SR, Murty VV, et al. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell. 1991;66:663–74. doi: 10.1016/0092-8674(91)90112-c. [DOI] [PubMed] [Google Scholar]
- 4.Nasr R, Guillemin MC, Ferhi O, Soilihi H, Peres L, Berthier C, et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat Med. 2008;14:1333–42. doi: 10.1038/nm.1891. [DOI] [PubMed] [Google Scholar]
- 5.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–95. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell. 1991;65:1255–66. doi: 10.1016/0092-8674(91)90020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, et al. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–80. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 8.Nagy L, Kao HY, Love JD, Li C, Banayo E, Gooch JT, et al. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev. 1999;13:3209–16. doi: 10.1101/gad.13.24.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 10.Chambon P. A decade of molecular biology of retinoic acid receptors. Faseb J. 1996;10:940–54. [PubMed] [Google Scholar]
- 11.Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci U S A. 1980;77:2936–40. doi: 10.1073/pnas.77.5.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giguere V, Ong ES, Segui P, Evans RM. Identification of a receptor for the morphogen retinoic acid. Nature. 1987;330:624–9. doi: 10.1038/330624a0. [DOI] [PubMed] [Google Scholar]
- 13.Tsai S, Bartelmez S, Heyman R, Damm K, Evans R, Collins SJ. A mutated retinoic acid receptor-alpha exhibiting dominant-negative activity alters the lineage development of a multipotent hematopoietic cell line. Genes & development. 1992;6:2258–69. doi: 10.1101/gad.6.12a.2258. [DOI] [PubMed] [Google Scholar]
- 14.Mendelsohn C, Lohnes D, Decimo D, Lufkin T, LeMeur M, Chambon P, et al. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994;120:2749–71. doi: 10.1242/dev.120.10.2749. [DOI] [PubMed] [Google Scholar]
- 15.Ablain J, de The H. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood. 2011;117:5795–802. doi: 10.1182/blood-2011-02-329367. [DOI] [PubMed] [Google Scholar]
- 16.Nasr R, de The H. Eradication of acute promyelocytic leukemia-initiating cells by PML/RARA-targeting. Int J Hematol. 2010;91:742–7. doi: 10.1007/s12185-010-0582-0. [DOI] [PubMed] [Google Scholar]
- 17.de The H, Chen Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer. 2010;10:775–83. doi: 10.1038/nrc2943. [DOI] [PubMed] [Google Scholar]
- 18.Collins SJ. The role of retinoids and retinoic acid receptors in normal hematopoiesis. Leukemia. 2002;16:1896–905. doi: 10.1038/sj.leu.2402718. [DOI] [PubMed] [Google Scholar]
- 19.Bradbury DA, Aldington S, Zhu YM, Russell NH. Down-regulation of bcl-2 in AML blasts by all-trans retinoic acid and its relationship to CD34 antigen expression. Br J Haematol. 1996;94:671–5. doi: 10.1046/j.1365-2141.1996.d01-1838.x. [DOI] [PubMed] [Google Scholar]
- 20.Sakashita A, Kizaki M, Pakkala S, Schiller G, Tsuruoka N, Tomosaki R, et al. 9-cis-retinoic acid: effects on normal and leukemic hematopoiesis in vitro. Blood. 1993;81:1009–16. [PubMed] [Google Scholar]
- 21.Lehmann S, Bengtzen S, Broberg U, Paul C. Effects of retinoids on cell toxicity and apoptosis in leukemic blast cells from patients with non-M3 AML. Leuk Res. 2000;24:19–25. doi: 10.1016/s0145-2126(99)00153-8. [DOI] [PubMed] [Google Scholar]
- 22.Ryningen A, Stapnes C, Paulsen K, Lassalle P, Gjertsen BT, Bruserud O. In vivo biological effects of ATRA in the treatment of AML. Expert Opin Investig Drugs. 2008;17:1623–33. doi: 10.1517/13543784.17.11.1623. [DOI] [PubMed] [Google Scholar]
- 23.Griggs JJ, Henley SE, Rowe JM. Treatment of refractory undifferentiated acute myelogenous leukemia with all-trans-retinoic acid. Am J Hematol. 1994;45:177–80. doi: 10.1002/ajh.2830450215. [DOI] [PubMed] [Google Scholar]
- 24.Qian SX, Li JY, Hong M, Qiu HR, Fan L, Xu W. Acute myeloid leukemia in four patients with t(8;21) treated with all-trans retinoic acid as a single agent. Leuk Lymphoma. 2008;49:998–1001. doi: 10.1080/10428190801959018. [DOI] [PubMed] [Google Scholar]
- 25.Belhabri A, Thomas X, Wattel E, Chelghoum Y, Anglaret B, Vekhoff A, et al. All trans retinoic acid in combination with intermediate-dose cytarabine and idarubicin in patients with relapsed or refractory non promyelocytic acute myeloid leukemia: a phase II randomized trial. Hematol J. 2002;3:49–55. doi: 10.1038/sj.thj.6200141. [DOI] [PubMed] [Google Scholar]
- 26.Milligan DW, Wheatley K, Littlewood T, Craig JI, Burnett AK. Fludarabine and cytosine are less effective than standard ADE chemotherapy in high-risk acute myeloid leukemia, and addition of G-CSF and ATRA are not beneficial: results of the MRC AML-HR randomized trial. Blood. 2006;107:4614–22. doi: 10.1182/blood-2005-10-4202. [DOI] [PubMed] [Google Scholar]
- 27.Burnett AK, Milligan D, Prentice AG, Goldstone AH, McMullin MF, Hills RK, et al. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer. 2007;109:1114–24. doi: 10.1002/cncr.22496. [DOI] [PubMed] [Google Scholar]
- 28.Burnett AK, Hills RK, Milligan DW, Goldstone AH, Prentice AG, McMullin MF, et al. Attempts to optimize induction and consolidation treatment in acute myeloid leukemia: results of the MRC AML12 trial. J Clin Oncol. 2010;28:586–95. doi: 10.1200/JCO.2009.22.9088. [DOI] [PubMed] [Google Scholar]
- 29.Estey EH, Thall PF, Pierce S, Cortes J, Beran M, Kantarjian H, et al. Randomized phase II study of fludarabine + cytosine arabinoside + idarubicin +/− all-trans retinoic acid +/− granulocyte colony-stimulating factor in poor prognosis newly diagnosed acute myeloid leukemia and myelodysplastic syndrome. Blood. 1999;93:2478–84. [PubMed] [Google Scholar]
- 30.Schlenk RF, Frohling S, Hartmann F, Fischer JT, Glasmacher A, del Valle F, et al. Phase III study of all-trans retinoic acid in previously untreated patients 61 years or older with acute myeloid leukemia. Leukemia. 2004;18:1798–803. doi: 10.1038/sj.leu.2403528. [DOI] [PubMed] [Google Scholar]
- 31.Nazha A, Bueso-Ramos C, Estey E, Faderl S, O'Brien S, Fernandez MH, et al. The Addition of All-Trans Retinoic Acid to Chemotherapy May Not Improve the Outcome of Patient with NPM1 Mutated Acute Myeloid Leukemia. Front Oncol. 2013;3:218. doi: 10.3389/fonc.2013.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlenk RF, Dohner K, Krauter J, Giadzik VI, Paschka P, Heuser M, et al. All-trans Retinoic Acid Improves Outcome in Younger Adult Patients with Nucleophosmin-1 Mediated Acute Myeloid Leukemia - Resutls of the AMLSG 07-04 Randomized Treatment Trial. American Society of Hematology Annual Meeting. 2011:80A. [Google Scholar]
- 33.Ritter M, Kattmann D, Teichler S, Hartmann O, Samuelsson MK, Burchert A, et al. Inhibition of retinoic acid receptor signaling by Ski in acute myeloid leukemia. Leukemia. 2006;20:437–43. doi: 10.1038/sj.leu.2404093. [DOI] [PubMed] [Google Scholar]
- 34.Fazi F, Zardo G, Gelmetti V, Travaglini L, Ciolfi A, Di Croce L, et al. Heterochromatic gene repression of the retinoic acid pathway in acute myeloid leukemia. Blood. 2007;109:4432–40. doi: 10.1182/blood-2006-09-045781. [DOI] [PubMed] [Google Scholar]
- 35.Heuser M, Argiropoulos B, Kuchenbauer F, Yung E, Piper J, Fung S, et al. MN1 overexpression induces acute myeloid leukemia in mice and predicts ATRA resistance in patients with AML. Blood. 2007;110:1639–47. doi: 10.1182/blood-2007-03-080523. [DOI] [PubMed] [Google Scholar]
- 36.Bullinger L, Schlenk RF, Gotz M, Botzenhardt U, Hofmann S, Russ AC, et al. PRAME-induced inhibition of retinoic acid receptor signaling-mediated differentiation--a possible target for ATRA response in AML without t(15;17) Clinical cancer research. 2013;19:2562–71. doi: 10.1158/1078-0432.CCR-11-2524. [DOI] [PubMed] [Google Scholar]
- 37.Melnick AM. Epigenetics in AML. Best Pract Res Clin Haematol. 2010;23:463–8. doi: 10.1016/j.beha.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 38.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrara FF, Fazi F, Bianchini A, Padula F, Gelmetti V, Minucci S, et al. Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer research. 2001;61:2–7. [PubMed] [Google Scholar]
- 40.Trus MR, Yang L, Suarez Saiz F, Bordeleau L, Jurisica I, Minden MD. The histone deacetylase inhibitor valproic acid alters sensitivity towards all trans retinoic acid in acute myeloblastic leukemia cells. Leukemia. 2005;19:1161–8. doi: 10.1038/sj.leu.2403773. [DOI] [PubMed] [Google Scholar]
- 41.Cimino G, Lo-Coco F, Fenu S, Travaglini L, Finolezzi E, Mancini M, et al. Sequential valproic acid/all-trans retinoic acid treatment reprograms differentiation in refractory and high-risk acute myeloid leukemia. Cancer research. 2006;66(8903):11. doi: 10.1158/0008-5472.CAN-05-2726. [DOI] [PubMed] [Google Scholar]
- 42.Kuendgen A, Knipp S, Fox F, Strupp C, Hildebrandt B, Steidl C, et al. Results of a phase 2 study of valproic acid alone or in combination with all-trans retinoic acid in 75 patients with myelodysplastic syndrome and relapsed or refractory acute myeloid leukemia. Ann Hematol. 2005;84(Suppl 1):61–6. doi: 10.1007/s00277-005-0026-8. [DOI] [PubMed] [Google Scholar]
- 43.Fredly H, Ersvaer E, Kittang AO, Tsykunova G, Gjertsen BT, Bruserud O. The combination of valproic acid, all-trans retinoic acid and low-dose cytarabine as disease-stabilizing treatment in acute myeloid leukemia. Clin Epigenetics. 2013;5:13. doi: 10.1186/1868-7083-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryningen A, Stapnes C, Lassalle P, Corbascio M, Gjertsen BT, Bruserud O. A subset of patients with high-risk acute myelogenous leukemia shows improved peripheral blood cell counts when treated with the combination of valproic acid, theophylline and all-trans retinoic acid. Leukemia research. 2009;33:779–87. doi: 10.1016/j.leukres.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, et al. Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer. 2005;104:2717–25. doi: 10.1002/cncr.21589. [DOI] [PubMed] [Google Scholar]
- 46.Tassara M, Dohner K, Brossart P, Held G, Gotze K, Horst HA, et al. Valproic acid in combination with all-trans retinoic acid and intensive therapy for acute myeloid leukemia in older patients. Blood. 2014;123:4027–36. doi: 10.1182/blood-2013-12-546283. [DOI] [PubMed] [Google Scholar]
- 47.Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110:2302–8. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 48.van der Helm LH, Scheepers ER, Veeger NJ, Daenen SM, Mulder AB, van den Berg E, et al. Azacitidine might be beneficial in a subgroup of older AML patients compared to intensive chemotherapy: a single centre retrospective study of 227 consecutive patients. J Hematol Oncol. 2013;6:29. doi: 10.1186/1756-8722-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nature medicine. 2012;18:605–11. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gianni M, Peviani M, Bruck N, Rambaldi A, Borleri G, Terao M, et al. p38alphaMAPK interacts with and inhibits RARalpha: suppression of the kinase enhances the therapeutic activity of retinoids in acute myeloid leukemia cells. Leukemia. 2012;26:1850–61. doi: 10.1038/leu.2012.50. [DOI] [PubMed] [Google Scholar]
- 51.Srinivas H, Juroske DM, Kalyankrishna S, Cody DD, Price RE, Xu XC, et al. c-Jun N-terminal kinase contributes to aberrant retinoid signaling in lung cancer cells by phosphorylating and inducing proteasomal degradation of retinoic acid receptor alpha. Mol Cell Biol. 2005;25:1054–69. doi: 10.1128/MCB.25.3.1054-1069.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Srinivas H, Xia D, Moore NL, Uray IP, Kim H, Ma L, et al. Akt phosphorylates and suppresses the transactivation of retinoic acid receptor alpha. Biochem J. 2006;395:653–62. doi: 10.1042/BJ20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delmotte MH, Tahayato A, Formstecher P, Lefebvre P. Serine 157, a retinoic acid receptor alpha residue phosphorylated by protein kinase C in vitro, is involved in RXR.RARalpha heterodimerization and transcriptional activity. J Biol Chem. 1999;274:38225–31. doi: 10.1074/jbc.274.53.38225. [DOI] [PubMed] [Google Scholar]
- 54.Hoshikawa Y, Kanki K, Ashla AA, Arakaki Y, Azumi J, Yasui T, et al. c-Jun N-terminal kinase activation by oxidative stress suppresses retinoid signaling through proteasomal degradation of retinoic acid receptor alpha protein in hepatic cells. Cancer Sci. 2011;102:934–41. doi: 10.1111/j.1349-7006.2011.01889.x. [DOI] [PubMed] [Google Scholar]
- 55.Si J, Mueller L, Collins SJ. GSK3 inhibitors enhance retinoic acid receptor activity and induce the differentiation of retinoic acid-sensitive myeloid leukemia cells. Leukemia. 2011;25:1914–8. doi: 10.1038/leu.2011.171. [DOI] [PubMed] [Google Scholar]
- 56.Gupta K, Gulen F, Sun L, Aguilera R, Chakrabarti A, Kiselar J, et al. GSK3 is a regulator of RAR-mediated differentiation. Leukemia. 2012;26:1277–85. doi: 10.1038/leu.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kropf PL, Wang L, Zang Y, Redner RL, Johnson DE. Dasatinib promotes ATRA-induced differentiation of AML cells. Leukemia. 2010;24:663–5. doi: 10.1038/leu.2009.267. [DOI] [PubMed] [Google Scholar]
- 58.Miranda MB, Redner RL, Johnson DE. Inhibition of Src family kinases enhances retinoic acid induced gene expression and myeloid differentiation. Mol Cancer Ther. 2007;6:3081–90. doi: 10.1158/1535-7163.MCT-07-0514. [DOI] [PubMed] [Google Scholar]
- 59.Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 2004;328:1–16. doi: 10.1016/j.gene.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 60.Boudjelal M, Voorhees JJ, Fisher GJ. Retinoid signaling is attenuated by proteasome-mediated degradation of retinoid receptors in human keratinocyte HaCaT cells. Exp Cell Res. 2002;274:130–7. doi: 10.1006/excr.2001.5450. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka T, Rodriguez de la Concepcion ML, De Luca LM. Involvement of all-trans-retinoic acid in the breakdown of retinoic acid receptors alpha and gamma through proteasomes in MCF-7 human breast cancer cells. Biochem Pharmacol. 2001;61:1347–55. doi: 10.1016/s0006-2952(01)00600-1. [DOI] [PubMed] [Google Scholar]
- 62.Luo P, Lin M, Li L, Yang B, He Q. The proteasome inhibitor bortezomib enhances ATRA-induced differentiation of neuroblastoma cells via the JNK mitogen-activated protein kinase pathway. PLoS One. 2011;6:e27298. doi: 10.1371/journal.pone.0027298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ying M, Zhou X, Zhong L, Lin N, Jing H, Luo P, et al. Bortezomib sensitizes human acute myeloid leukemia cells to all-trans-retinoic acid-induced differentiation by modifying the RARalpha/STAT1 axis. Mol Cancer Ther. 2013;12:195–206. doi: 10.1158/1535-7163.MCT-12-0433. [DOI] [PubMed] [Google Scholar]
- 64.Desmond JC, Mountford JC, Drayson MT, Walker EA, Hewison M, Ride JP, et al. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003;63:505–12. [PubMed] [Google Scholar]
- 65.Khanim FL, Hayden RE, Birtwistle J, Lodi A, Tiziani S, Davies NJ, et al. Combined bezafibrate and medroxyprogesterone acetate: potential novel therapy for acute myeloid leukaemia. PLoS One. 2009;4:e8147. doi: 10.1371/journal.pone.0008147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brown G, Hughes P. Retinoid differentiation therapy for common types of acute myeloid leukemia. Leuk Res Treatment. 2012;2012:939021. doi: 10.1155/2012/939021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meli M, Tolomeo M, Grifantini M, Franchetti P, Cappellacci L, Simoni D, et al. The synergistic apoptotic effects of thiophenfurin, an inosine monophosphate dehydrogenase inhibitor, in combination with retinoids in HL60 cells. Oncol Rep. 2007;17:185–92. [PubMed] [Google Scholar]
- 68.Moosavi MA, Yazdanparast R, Lotfi A. 3-Hydrogenkwadaphnin induces monocytic differentiation and enhances retinoic acid-mediated granulocytic differentiation in NB4 cell line. J Biochem Mol Biol. 2006;39:722–9. doi: 10.5483/bmbrep.2006.39.6.722. [DOI] [PubMed] [Google Scholar]