

Abstract
Background
The endonuclease ARTEMIS, encoded by the DCLRE1C gene, is a component of the non-homologous end-joining (NHEJ) pathway, and participates in hairpin opening during the V(D)J recombination process and repair of a subset of DNA double strand breaks. Patients with ARTEMIS deficiency usually present with severe combined immunodeficiency (SCID) and cellular radiosensitivity, but hypomorphic mutations may cause milder phenotypes (leaky SCID).
Objective
We sought to correlate the functional impact of human DCLRE1C mutations on phenotypic presentation in patients with ARTEMIS deficiency.
Methods
We studied recombination and DNA repair activity of 41 human DCLRE1C mutations in Dclre1c−/− v-abl kinase transformed pro-B cells retrovirally engineered with a construct that allows quantification of recombination activity by flow-cytometry. For assessment of DNA repair efficacy, resolution of γH2AX accumulation was studied after ionizing radiation.
Results
Low or absent activity was detected for mutations causing a typical SCID phenotype. Most of leaky SCID patients were compound heterozygous for one loss of function (LOF) and one hypomorphic allele with significant residual levels of recombination and DNA repair activity. Deletions disrupting the C-terminus result in truncated, but partially functional proteins, and are often associated with leaky SCID. Overexpression of hypomorphic mutants may improve the functional defect.
Conclusions
Correlation between the nature and location of DCLRE1C mutations, functional activity, and the clinical phenotype, has been observed. Hypomorphic variants that have been reported in the general population may be disease-causing if combined in trans with a LOF allele. Therapeutic strategies aimed at inducing overexpression of hypomorphic alleles may be beneficial.
Keywords: V(D)J recombination, non-homologous end-joining, DNA repair, ARTEMIS deficiency, DCLRE1C mutations, severe combined immunodeficiency
Introduction
The endonuclease ARTEMIS, encoded by the gene DCLRE1C (NG_007276.1; ENSG00000152457), is an essential component of the V(D)J recombination machinery during T and B cell development, and plays an important role in the non-homologous end joining (NHEJ)-mediated DNA double-strand break (DSB) repair pathway 1.
The V(D)J recombination process is initiated by the recombination-activating gene products 2 RAG1 and RAG2, which bind to recombination signal sequences (RSS) adjacent to the coding V-, D-, and J-segments and induce a DNA double strand break, leaving hairpin structure at coding ends. Upon phosphorylation by the DNA protein kinase catalytic subunit (DNA-PKcs) complex, Artemis is recruited, and mediates hairpin opening via its endonuclease activity 3. The open ends at the overhangs can be modified by ARTEMIS’ endo- and exonuclease activities before the XRCC4/XLF/Ligase 4 complex seals the DNA strands. Defects in ARTEMIS result in aberrant hairpin opening and give rise to increased numbers of P-nucleotides in the coding joints 4. In contrast to what observed in defects of other members of the NHEJ pathways, the blunt ends formed at the RSSs cleavage sites (yielding excision circles or inversions) rejoin normally in the absence of ARTEMIS.
The ARTEMIS protein is a member of the metallo-β-lactamase superfamily and is organized into an N-terminal region containing the β-lactamase homology domain, a central β-CASP domain, and a C-terminal region. The first two domains have critical catalytic function, whereas the C-terminus plays a role in protein stabilization and regulation of function 5, 6.
ARTEMIS is also involved in an ATM-dependent slow-kinetic NHEJ DNA repair pathway, where it is required to modify non-ligatable ends of DNA breaks before they can be rejoined 7, 8. This type of breaks occur in a small fraction (around 10%) of DNA DSB after exposure to ionizing irradiation and require more time for repair, resulting in delayed repair kinetics.
By affecting V(D)J recombination, mutations of the DCLRE1C gene impair T and B cell development, leading to T−B− severe combined immunodeficiency (SCID), associated with a mild form of cellular radiosensitivity 9.
The most common causes for DCLRE1C loss-of-function (LOF) alleles are large deletions affecting the first 4 exons, and a nonsense founder mutation which has been described in Athabascan-speaking Native Americans 10. However, missense mutations affecting highly conserved residues can also abrogate protein function 11, 12. On the other hand, hypomorphic mutations in DCLRE1C retaining residual function have been reported in patients with B−/low Tlow “leaky” SCID or Omenn syndrome (OS)13.
We have recently reported correlation between recombination activity of naturally occurring RAG1 mutant proteins and clinical and immunological phenotype associated with RAG1 mutations 14. In that study, RAG1 recombination activity was studied in murine Rag1−/− v-abl kinase transformed pro-B cell lines retrovirally engineered with a recombination construct and transduced with RAG1-expressing vectors, thus allowing GFP expression as a read-out of recombination proficiency. Using a similar approach, we have attempted to measure V(D)J recombination and DNA repair activity levels of 41 naturally occurring mutated DCLRE1C products caused by point mutations or small deletions.
Methods
Patient selection based on genotype
Clinical and molecular data from patients with ARTEMIS deficiency caused by missense mutations or small deletions in DCLRE1C were collected from previous reports, or were provided by physicians from Europe and the United States, according to protocols approved by local institutional review boards. Based on the clinical and immunological presentation, each patient was assigned to subgroups of T−B−SCID or a leaky/atypical presentation referred to as “leaky SCID”, the latter including patients with OS, according to criteria defined by the Primary Immune Deficiency Treatment Consortium 15.
Determination of relative recombination activity levels associated with mutations in DCLRE1C
Analysis of recombination activity was performed in a cellular system of murine Dclre1c−/− Abelson murine leukemia virus (A-MuLV)-transformed pro-B cells (Fig. E1, A) 16, as previously described 17,14. Cells were retrovirally transduced to insert an inverted GFP cassette flanked by two RSS, and subjected to limiting dilution to isolate pro-B cells containing only one copy of such cassette (Fig. E1, B). Finally, in vitro mutagenized hDCLRE1C cDNAs were individually introduced into engineered Dclre1c−/− A-MuLV-pro-B cells using retroviral vectors. Efficient mutagenesis was confirmed by Sanger sequencing in constructs and transduced cells.
Targeting of the RSS contained in the recombination cassette allows GFP expression as a functional read-out of recombination activity that can be quantified by flow-cytometry upon gating on cells expressing both hCD4 and hCD2. For assessment of recombination and DNA repair activities, Dclre1c−/− abl pro-B cells were blocked in G0/G1 cell cycle phases for 72h by culture in 3μM of the v-abl kinase inhibitor imatinib (STI-571) (Novartis, Basel, Switzerland), which allows the Rag1/Rag2 complex to initiate the recombination process with higher efficiency (Fig. E2). Experiments were carried out in wild-type (WT), mock transduced cells and sets of up to 8 mutants at a time and were run in triplicates. Recombination activity levels were assessed as percentages of GFP expression detected in cells transduced with WT-DCLRE1C, after subtraction of background activity observed in mock-transduced cells.
Determination of DNA repair activity levels
NHEJ-mediated DNA repair was assessed in WT, mock-transduced and mutant-transduced cells at 1h, 8h, 24h, and 36h after ionizing radiation (IR) with 10Gy. Phosphorylation of the histone protein H2AX (γH2AX) was quantified by flow-cytometry (Phospho Histo H2A.X, (Ser139) (20E3), Cell Signaling, 5763S; Rabbit (DA1E) IgG XP isotype control, Cell Signaling, 5742). Since chromosomal replication is known to cause formation of γH2AX foci 18, also for this study cell cycles were blocked in G0/G1 for 72h. DNA repair efficiencies were calculated based on mean fluorescent intensities (MFI) at 36h after IR. For cells transduced with either WT or mutant constructs of DCLRE1C, the γH2AX MFI was subtracted from mock MFI, and activities were assessed as percentages of WT construct performance at 36h after exposure to 10Gy IR.
Southern Blotting
To assess the number of DCLRE1C vector integrations in transduced cells, 15μg genomic DNA extracted from Dclre1c−/− abl pro B cells were digested with EcoR1, HindIII, and NcoI (NEB, Ipswich, MA), run on TBE gels and blotted on a 0.45μm nylon membrane (Whatman Turbo Blotter Transfer System, Fisher Scientific, Waltham, MA). Membranes were hybridized with digoxigenin-labeled probes detecting hCD4 and hCD2 respectively, and signal detection was performed using an anti-dioxigenin antibody based system (Roche, Basel, Switzerland) following manufacturers instructions.
Quantitative real-time PCR
Quantitative PCR (qPCR) was performed using the ΔΔCT method, and normalized to WT gene expression. Additional information and primer sequences are listed in the online repository material.
Statistical analysis
Recombination and DNA repair activity levels of Artemis proteins were analyzed with the Mann-Whitney U test. Gene expression levels were compared using the unpaired two-tailed students t test, and p≤ 0.05 was considered significant. Correlation between recombination and DNA repair activity levels was assessed with the Pearson product-moment correlation coefficient (r), two-tailed, 95% confidence interval (CI).
Results
Recombination and DNA repair activity in ARTEMIS mutated proteins
We analyzed recombination and DNA repair activity levels for 22 missense and 6 nonsense mutations, 2 insertions (max. 8bp) and 11 small deletions (max. 17bp) detected in the DCLRE1C gene in a cohort of 40 ARTEMIS deficient patients (Table 1) 1, 3, 4, 9–11, 13, 19–28.
Table 1.
Genotype and clinical presentation of ARTEMIS-deficient patients
| Allele 1 | Allele 2 | Protein | Age at diagnosis |
Infections | Autoimmunity/ Lymphoproliferation |
Malignancy | Immunology | Radiosensitivity | Reference |
|---|---|---|---|---|---|---|---|---|---|
| SCID | |||||||||
| c.47T>C | c.356C>G | I16T; S119* | 1mo | LRI, oral thrush | T−B−NK+ | sensitive | 20 | ||
| c.82G>C | c.82G>C | A28P | T−B−NK+ | sensitive | 11 | ||||
| c.95C>T | del Ex 1-3 | S32F | 19 | ||||||
| c.95C>G | 2N/A | S32C | 1 | ||||||
| c.110A>G | c.110A>G | D37G | 2mo | moniliasis, gastroenteritis | T−B−NK+ | sensitive | 4 | ||
| c.241C>T | c.241C>T | R81* | 9 | ||||||
| c.281delC | c.281delC | S94fs*2 | TmatB−NK+ | 11 | |||||
| c.353G>T | c.353G>T | G118V | 5mo | LRI, PJP, CMV | T−B−NK+ | sensitive | 22 | ||
| c.353G>T | c.353G>T | G118V | T−B−NK+ | 11 | |||||
| c.362T>C | del Ex 1-3 | M121T | 8mo | LRI | T−B−NK+ agamma | unpublished | |||
| c.404G>A | c.404G>A | G135E | 4mo | VZV | sensitive | 22 | |||
| c.493G>A | c.493G>A | D165N | 3 | ||||||
| c.494A>T | c.494A>T | D165V | T−B−NK+ | 11 | |||||
| c.571C>T | N/A | R191* | unpublished | ||||||
| c.597C>A | c.597C>A | Y199* | oral and genital ulcers | 10 | |||||
| c.682C>A | c.682C>A | H228N | T−B−NK+ | sensitive | 11 | ||||
| c.716delC | c.716delC | P239Lfs*46 | 8mo | recurrent URI, oral thrush, chronic diarrhea | T−B−NK+ | unpublished | |||
| c.761A>T | c.309delG (splice site) | H254L; K103fs | 25 | ||||||
| c.781delG | c.781delG | A261Qfs*24 | 9 | ||||||
| c.1050delA | c.1050delA | V381Lfs*5 | T−B−NK+ | 11 | |||||
| c.1140_1146delAGTTCAC | c.1140_1146delAGTTCAC | V383Efs*19 | TmatB−NK+ | sensitive | 11 | ||||
| c.1147C>T | c.1147C>T | R383* | TmatB−NK+ | 11 | |||||
| c.1167_1168insAG | c.delEx3 | D390Rfs*13 | 2mo | URI | T−B−NK+ hypogam | sensitive | 26 | ||
| c.1179delT | c.1179delT | F393Lfs*9 | 3mo | LRI, PJP, oral thrush, CMV | T−B−NK+ hypogam | sensitive | 20 | ||
| c.1391_1395delGAATC | c.1391_1395delGAATC | G464Afs*18 | 1mo | LRI, GI | T−B−NK+ | sensitive | 4 | ||
| Omenn Syndrome | |||||||||
| c.2T>C | c.103C>G | M1T; H35D | 5mo | sepsis, BCG | alopecia, erythrodermia, lymphadenopathy, hepato/splenomegaly, granuloma | lymphoma | ThiB−NK+ hypogam TCR repertoire 25% of WT |
sensitive | 13 |
| Leaky SCID | |||||||||
| c.1A>C | c.401C>G | M1V; T134R | 6mo | pharyngitis, stomatitis, gastroenteritis | T−B−NK+ | unpublished | |||
| c.19C>T | c.457G>A | Q7*; G153R | 73% CD3 3% CD19 of MNC in BM |
19 | |||||
| c.194C>T | N/A | T65I | 18mo | pulmonary TB | T−B−NK+ | unpublished | |||
| c.211A>C | del Ex 1-3 | T71P | 10mo (onset) | URI, Pseudomonas soft tissue infection | AIHA, neutropenia, thrombocytopenia, lymphadenopathy hepato/splenomegaly | TloBloNK+ low IgA and IgG2; absent response to vaccine (Hib and tetanus); proliferation to PHA initially normal, later absent | 21 | ||
| c.211A>C | del Ex 1-3 | T71P | birth | URI, oral candidiasis | AIHA | TloB−NK+ low IgG, absent IgA; decreased TRECs; poliferation to PHA initially normal, later absent | 21 | ||
| c.207_209delGTT | c.377G>A | L70del; G126D | 4.5y | URI, Candida | AIHA | TloB+NK+ hypogam no specific Ab response | sensitive | 23 | |
| c.400A>G | del Ex 1+ (not specified) | T134A | lymphadenopathy | Tlo, polyclonal T cells, proliferation to PHA >30% of normal | unpublished | ||||
| c.512C>G | c.insInt2b | P171R | 27y | Candida, HPV, fungal brain abscess | neutropenia, thrombocytopenia | bilateral carcinoma in situ of nipples | TloBloNK+ abnormal T cell proliferation | mildly sensitive | 24 |
| c.512C>G | c.1299_1306dupAGGATGCT | P171R; C436* | 7y | URI, LRI | lymphadenopathy, granulomatous inflammation | Hodgkin lymphoma | TloB−NK+ absent IgA | 24 | |
| c.1290_1306delACAAACCCCAGGATGCT | c.1290_1306delACAAACCCCAGGATGCT | T432fs*16 | 4y | LRI, GI | cholangitis and liver cirrhosis (died at 16y) | TloBlo hypogam | 27 | ||
| c.1353_1359delTTGTGAA | c.delEx1-3 | D451Kfs*11 | birth | LRI, Candida | AIHA, ITP, cervical lymphadenopathy | lymphoma (9mo) | T−B−NK+ monoclonal IgG, absent IgA | 27 | |
| c.1353_1359delTTGTGAA | c.delEx1-3 | D451Kfs*11 | birth | LRI, Candida | lymphoma | TloBlo hypogam | 27 | ||
| c.1353_1359delTTGTGAA | c.delEx1-3 | D451Kfs*11 | 1y | URI, LRI, CNS Toxo | TloB−NK+ hypogam | sensitive | 27 | ||
| c.1464delG | c.1464delG | Q488fs | 1mo | URI, LRI, CMV | oral ulcers, sclerosing cholangitis, thrombocytopenia, hepatosplenomegaly | TloB−NK+ LGL cells, hypogam | 28 | ||
Ab, antibody; agamma, agammaglobulinemia; AIHA, autoimmune hemolytic anemia; BCG, Bacillus Calmette-Guérin; BM, bone marrow; CMV, cytomegalovirus; CNS, central nervous system; GI, gastrointestinal infection; HPV, human papillomavirus; hypogam, hypogammaglobulinemia; ITP, idiopathic thrombocytopenic purpura; LGL, large granular lymphocytes; LRI, lower respiratory infections; MNC, mononuclear cells; N/A, not available; PJP, Pneumocystis jirovecii pneumonia; PHA, phytohemagglutinin; TB, Tuberculosis; TCR, T cell receptor; Tmat, maternal T cells; Toxo, Toxoplasmosis; TRECs, T cell receptor excision circles; URI, upper respiratory infection; VZV, varicella zoster virus
Chromosomal V(D)J recombination activity was tested in a cellular model previously described 14, 29. V-abl kinase transformed murine Dclre1c−/− A-MuLV pro-B cells 16, from now on referred to as Dclre1c−/− abl pro-B cells, were engineered with a stable single integration of the pMX-INV GFP cassette flanked by two coding RSSs (Fig.1, A; Fig. E1, B), followed by transduction with a retroviral vector containing WT, mock or mutagenized hDCLRE1C cDNAs. Subsequently, GFP expression was analyzed as a read-out of recombination activity (Fig. 1, B) and γH2AX levels were assessed at 1h, 8h, 24h and 36h after IR with 10Gy to measure DNA repair activity (Fig. 1, C).
FIG. 1. Schematic representation of the experimental outline and readout.

A, Dclre1c−/− abl pro-B cells were retrovirally engineered with a single construct containing an inverted GFP cassette flanked by two RSS and the hCD4 sequence for positive selection. Subsequently, cells were transduced with a second construct containing either WT or mutated hDCLRE1C (isoform a) and the hCD2 sequence. Sufficient targeting of RSS by the Rag1/Rag2 complex and completion of the recombination process, which involves Artemis, allows the GFP cassette to flip into sense orientation. B, GFP expression can be used for quantification of V(D)J recombination activity in cells expressing both vectors (CD4+CD2+). C, To assess DNA repair activity, cells were irradiated with 10Gy and γH2AX de-phosphorylation was quantified by flow-cytometry at 1h, 8h, 24h, and 36h in cells expressing the DCLRE1C-hCD2 vector. The difference in GFP expression and down-regulation of γH2AX is shown for Dclre1c−/− abl pro B-cells transduced with WTa DCLRE1C or a mock plasmid, respectively.
Due to alternative splicing, 16 hDCLRE1C transcript isoforms are known, where isoform a, b, and c encode for proteins with 692, 577 and 572 amino acids, respectively. In a preliminary set of experiments, we tested the functional activity of each of these products, by transducing Dclre1c−/− abl pro-B cells with DCLRE1C transcript isoforms a, b, and c (NM_001033855; NM_022487; NM_001033858). Only variant a (further referred to as WTa), which encodes for the 692aa canonical sequence, induced high levels of GFP expression, as did also the murine Dclre1c transcript isoform a (Fig. E3). Consequently, isoform WTa was used for mutagenesis experiments. Expression of DCLRE1C transcript was confirmed by qPCR (Fig. E4).
In contrast to what observed with Rag1−/− abl pro B-cells 14, minimal levels of GFP expression were observed in mock-transduced Dclre1c−/− abl pro-B cells (Fig. 1, B), indicating occurrence of construct rejoining in a small fraction of cells even in the absence of Artemis.
While DNA breaks are repaired over time, progressive de-phosphorylation of the H2AX histone occurs. Since ARTEMIS is only required for processing of a small fraction of DNA breaks repaired in a slow-kinetic pathway 7, differences in phosphorylated H2AX (γH2AX) expression in Dclre1c−/− abl pro-B cells expressing WT or mutant ARTEMIS constructs can be best appreciated at later time points, such as 24h–36h (Fig. 2).
FIG. 2.
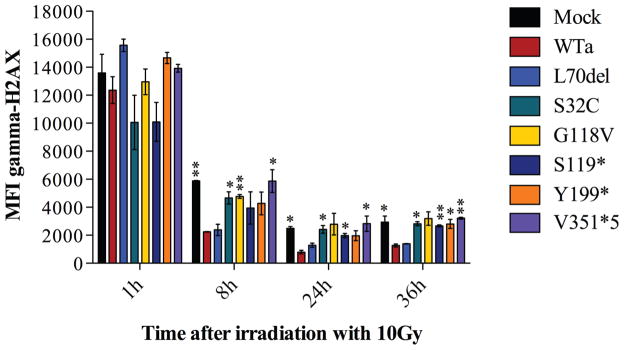
DNA repair activities were analyzed in Dclre1c−/− abl pro-B cells blocked in G0/G1 cell cycle phases. Mean fluorescence intensities (MFIs) for γH2AX at 1h, 8h, 24h and 36h after irradiation with 10Gy are shown for one representative experiment testing 6 ARTEMIS mutants compared to WTa and mock (A–G). Error bars indicate standard deviations, * p≤0.05; **p≤0.01; ***p≤0.001.
Correlation between functional activity and clinical phenotype
ARTEMIS mutants were divided into two major subgroups: a) missense mutations and in frame deletion; and, b) frameshift mutations. When recombination and DNA repair activity were plotted against location of the individual mutations along the protein, significant variability of protein function was observed for missense/inframe mutations affecting the β-lactamase or the β-CASP domain, including missense mutations within the same codon (eg, T134A and T134R, or D165N and D165V) (Fig. 3, A and B; Table 2). Frameshift mutations disrupting these domains almost completely abrogated ARTEMIS function. By contrast, frameshift mutations disrupting the C-terminus consistently retained high activity levels when expressed in Dclre1c−/− abl pro-B cells (Table 2), consistent with the less severe phenotype in patients harboring these mutations.
FIG. 3. Correlation of V(D)J recombination with DNA repair activities.

Recombination and DNA repair activity levels of 41 genetic DCLRE1C variants were studied in 23 missense mutations and in frame deletions (A) and 18 frameshift mutations (B) affecting various domains: Metallo-β-lactamase domain (amino acids 1–155), β-CASP domain (amino acids 156–385) and the C-terminal region (amino acids 386–692) 5.
V(D)J recombination activity levels of mutations affecting the β-lactamase (C), β-CASP (D) and C-terminal domain (E) of the protein are plotted against DNA repair activities.
Activity levels are expressed as percentages of WTa performance. Error bars indicate standard deviations, ***p≤0.001. r, Pearson correlation coefficient.
Table 2.
Recombination and DNA repair activity levels of DCLRE1C mutated products
| Mutations in DCLRE1C | Activity levels in % of WT activity | ||||
|---|---|---|---|---|---|
| Genomic | Protein | Recombination | DNA repair (36h after IR) | ||
| Mean | SD | Mean | SD | ||
| Metallo-β-Lactamase Domain | |||||
| c.1A>G | M1V | 86.32 | 1.67 | 95.35 | 11.60 |
| c.2T>C | M1T | 80.41 | 4.77 | 76.04 | 6.43 |
| c.19C>T | Q7* | 0.00 | 0.35 | 13.52 | 6.66 |
| c.47T>C | I16T | 79.67 | 0.27 | 80.11 | 18.11 |
| c.82G>C | A28P | 3.00 | 0.15 | 5.13 | 11.95 |
| c.95C>T | S32F | 5.14 | 0.34 | 20.95 | 6.17 |
| c.95C>G | S32C | 0.00 | 0.40 | 6.56 | 8.73 |
| c.103C>G | H35D | 0.00 | 0.30 | 27.29 | 16.57 |
| c.110A>G | D37G | 5.24 | 0.65 | 19.15 | 13.04 |
| c.194C>T | T65I | 83.75 | 1.32 | 89.45 | 14.05 |
| c.207_209delGTT | L70del | 90.01 | 0.77 | 92.63 | 0.96 |
| c.211A>C | T71P | 89.52 | 0.80 | 81.03 | 4.18 |
| c.241C>T | R81* | 19.48 | 1.56 | 16.05 | 4.77 |
| c.281delC | S94*2 | 0.00 | 1.01 | 31.86 | 0.04 |
| c.353G>T | G118V | 0.00 | 0.62 | 0.00 | 28.89 |
| c.356C>G | S119* | 0.00 | 0.27 | 15.59 | 4.51 |
| c.362T>C | M121T | 51.87 | 1.45 | 59.88 | 21.87 |
| c.377G>A | G126D | 27.40 | 0.21 | 50.80 | 3.31 |
| c.400A>G | T134A | 89.04 | 2.43 | 111.18 | 2.53 |
| c.401C>G | T134R | 0.00 | 0.44 | 13.21 | 4.25 |
| c.404G>A | G135E | 11.57 | 0.54 | 42.84 | 29.68 |
| c.457G>A | G153R | 101.82 | 0.45 | 110.04 | 7.03 |
| β-CASP Domain | |||||
| c.493G>A | D165N | 53.26 | 1.28 | 78.20 | 8.48 |
| c.494A>T | D165V | 0.00 | 0.62 | 17.71 | 4.91 |
| c.512C>G | P171R | 95.79 | 0.62 | 104.12 | 4.91 |
| c.571C>T | R191* | 0.00 | 0.87 | 0.00 | 12.11 |
| c.597C>A | Y199* | 0.00 | 1.21 | 7.46 | 19.56 |
| c.682C>A | H228N | 74.00 | 1.31 | 63.14 | 5.13 |
| c.716delC | P239*46 | 0.00 | 0.73 | 18.30 | 8.29 |
| c.761A>T | H254L | 39.89 | 1.73 | 20.85 | 4.11 |
| c.781delG | A261*24 | 12.69 | 0.61 | 60.92 | 11.34 |
| c.1050delA | V351*5 | 0.00 | 0.29 | 0.00 | 4.21 |
| c.1140_1146delAGTTCAC | V381*19 | 0.00 | 0.71 | 42.18 | 14.54 |
| c.1147C>T | R383* | 35.84 | 1.78 | 67.60 | 5.46 |
| C-Terminal Domain | |||||
| c.1167_1168insAG | D390*13 | 0.00 | 0.30 | 36.84 | 9.00 |
| c.1179delT | F393*9 | 0.00 | 0.42 | 18.07 | 19.99 |
| c.1290_1306del17 | T432*16 | 82.49 | 0.12 | 88.29 | 1.09 |
| c.1299_1306dupAGGATGCT | C436* | 77.26 | 0.42 | 98.89 | 1.26 |
| c.1353_1359delTTGTGAA | D451*11 | 88.57 | 1.64 | 77.43 | 6.53 |
| c.1391_1395delGAATC | G464*18 | 39.21 | 0.71 | 55.22 | 7.76 |
| c.1464delG | Q488*56 | 89.38 | 0.46 | 86.14 | 2.59 |
Although there was a trend for slightly higher DNA repair activities, particularly in the β-CASP domain, recombination activities correlated well with DNA repair activities (rb-lact= 0.964, p<0.001; rb-CASP= 0.818, p= 0.001; rC-term= 0.968, p<0.001) (Fig. 3, C–E). This may reflect the notion that ARTEMIS plays a more critical role in V(D)J recombination than in NHEJ-mediated DNA repair.
To correlate recombination and DNA repair activity levels of mutated ARTEMIS proteins with the clinical presentation, patients were divided into two cohorts: a) typical SCID: and, b) milder, atypical presentation (leaky SCID). Definition of SCID was done in accordance to PIDTC criteria 15, which included T cell count below 300/μl and/or presence of maternal T cell engraftment. In cases where this information was not available, we assumed the interpretation of the authors of the original report.
As expected, most of the patients in the SCID group presented with severe and opportunistic infections early in life; by contrast, patients with leaky SCID often developed autoimmune cytopenias (6/15), other autoimmune manifestations (2/15), lymphoproliferation (5/15), or malignancies in the form of lymphoma (4/15) or squamous cell carcinoma (1/15) (Table 1). Two patients in this group had mutations affecting the DCLRE1C translation initiation site, one presenting with OS, the other one with leaky SCID, but without skin manifestations.
To attempt correlation with the clinical phenotype, activity levels of the mutated proteins were analyzed in patients with SCID vs. leaky SCID. For patients with compound heterozygous mutations, we considered the mutant with the higher activity level. As shown in Fig. 4, A and B, both recombination and DNA repair activity levels were significantly higher for mutations observed in patients with leaky SCID (p≤ 0.001). However, while robust levels of protein function were uniformly recorded for mutations associated with leaky SCID, significant variability was observed for mutations associated with SCID. Importantly, patients with leaky SCID were either homozygous for frameshift mutations affecting the C-terminus and that retained residual activity, or were compound heterozygous for a hypomorphic mutation (or a putative polymorphism) and a LOF allele (Fig. 4C).
FIG. 4.
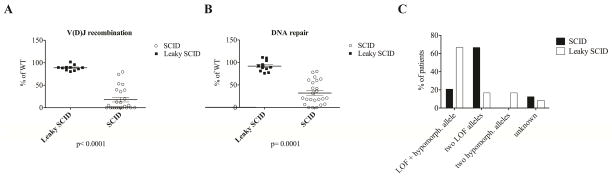
The activity levels for recombination (A) and DNA repair (B) of the mutations with the highest activity (in case of compound heterozygosity) found in patients presenting with SCID and leaky SCID are shown (p< 0.0001, and p=0.0001). The percentages of patients being heterozygous or homozygous for a LOF or a hypomorphic DCLRE1C allele are shown for SCID and leaky SCID patients (C). LOF, loss of function.
Correlation of predicted pathogenicity with functional activity
To predict the pathogenicity of the DCLRE1C mutations analyzed, we used the online bioinformatics tools PolyPhen-230, and SIFT31 (Table E1).
All, but three genetic variants (G153R, T71P and P171R) were predicted as disease causing by all tools. The variant P171R is a known polymorphism and has been reported before 24. All three variants showed high activity levels in our study.
To investigate the frequency of putatively damaging DCLRE1C genetic variants in the general population, we analyzed data of the Exome Aggregation Consortium (ExAC) of the Broad Institute Cambridge, MA (URL: http://exac.broadinstitute.org) [12/2014], that includes whole exome sequencing data from over 60,000 individuals. Most of the mutations reported in patients with SCID or leaky SCID and analyzed in this manuscript were not reported in the ExAC database. However, two of them (G153R and P171R) were reported at rather high frequency (0.01 and 0.09). Furthermore, homozygosity for these variants was demonstrated in a considerable number of individuals (12 and 612, respectively) in the ExAC database (Table E1). The fully normal VDJ activity of these variants is consistent with their polymorphism status.
The mutations M1T and M1V, which had been identified in compound heterozygous patients presenting with OS and leaky SCID (13 and unpublished), were predicted by Mutation T@ster 32 to use an alternative translation initiation site 7 amino acids downstream. We observed high activity levels for both mutants in our system suggesting residual function.
Protein overexpression restores activity of hypomorphic Artemis variants
A feature of the cellular platform described here is that retrovirus-mediated transduction typically results in generation of a population of cells harboring a different number of copies of DCLRE1C cDNA. The number of vector copies in the pool of unselected cells harboring these mutations consistently ranged between 4–8 per cell (data not shown). To investigate whether high levels of activity could reflect protein overexpression, we selected the hypomorphic mutants M1T and T432*16, and the polymorphism P171R, all of which showed high recombination and DNA repair activities when tested in bulk culture of Dclre1c−/− abl pro-B cells (80% – 100% of wild-type DCLRE1C, Table 2). Upon limiting dilution subcloning of transduced Dclre1c−/− abl pro-B cells harboring these mutations, we selected clones with more than six copies of vector, and compared their activity levels to that of clones with two vector copies (Fig. E5). Both the recombination and the DNA repair activities of the M1T and T432*16 mutants were significantly lower in clones harboring 2 vector copies than in those with 6 copies. By contrast, high activity was observed for the P171R, even in clones with two copies of this variant (Fig. 5). These data are consistent with P171R being a polymorphism. Furthermore, they indicate that overexpression can partially restore functional activity of hypomorphic ARTEMIS mutants.
FIG. 5. Recombination and DNA repair activity levels of hypomorphic DCLRE1C variants with various vector copy numbers.
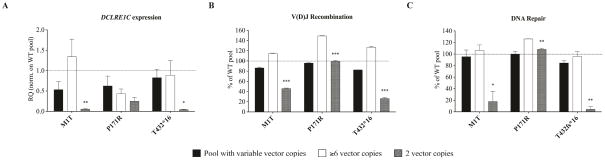
Expression levels of two hypomorphic DCLRE1C variants and one polymorphism were titrated by varying vector copy numbers to ≥6 or 2. Expression of human DCLRE1C was assessed by quantitative PCR in cDNA samples and was normalized to WTa (indicated by dashed line) (A). Activity levels for V(D)J recombination (B) and DNA repair (C) were compared between ≥6 or 2 vector copies, or the pool containing variable copy numbers per cell. Error bars indicate standard deviations. * p≤0.05; **p≤0.01; ***p≤0.001.
Discussion
The endo- and exonuclease ARTEMIS has a crucial function in opening the hairpin-sealed coding ends of V, D and J elements during V(D)J recombination in developing T and B lymphocytes 3, and in processing overhangs that occur in a small fraction of DNA double strand breaks 8. The severity of ARTEMIS deficiency depends on the amount of residual nuclease activity. While lack of protein expression or function completely abrogates T and B cell development, hypomorphic mutant proteins allow V(D)J recombination, albeit with reduced efficacy. This results in a milder immunodeficiency, but also in susceptibility to develop autoimmunity, lymphoproliferation, and malignancy. In this regard, the clinical phenotype of leaky ARTEMIS deficiency resembles that observed in patients with hypomorphic RAG1 or RAG2 defects 33. To gain more insights into genotype-phenotype correlation in ARTEMIS deficiency, we analyzed recombination and DNA repair activity in a large series of naturally occurring ARTEMIS mutations, and we correlated these results with the clinical phenotype. To this purpose, we used a similar cellular platform to the one we have recently reported to investigate genotype-proteotype-phenotype correlation in RAG1 deficiency 14. Importantly, an advantage of the Dclre1c−/− abl pro B-cell system is that it allows at the same time to analyze recombination activity and DNA repair.
While ARTEMIS plays a critical role in V(D)J recombination, its major function as a component of the NHEJ pathway of DNA repair is to process specific DNA double strand breaks, which are repaired in a slow-kinetic process. By analyzing the kinetics of H2AX histone dephosphorylation in pro-B cell lines expressing mutant (or WT) ARTEMIS at various time points after exposure to IR, we observed different levels of DNA repair activity for the various mutants analyzed, with good correlation with the data observed in the recombination assay. However, slightly higher levels of DNA repair than of recombination were observed in mutants with absent or low recombination activity, which was particularly the case for mutations affecting the β-CASP domain. It has been proposed that the β-CASP domain participates with the β-lactamase region in forming the catalytic core of ARTEMIS 5. Our findings suggest that ARTEMIS may play a less critical role in DNA repair than in V(D)J recombination.
Consistent with previous observations 12, 25, we found that mutations affecting highly conserved residues in the catalytically active β-lactamase or β-CASP domains completely abrogate function, and are associated with typical features of SCID. Mutations affecting other residues in the same domains may cause conformational changes that may variably affect protein function, as indicated by heterogeneous levels of activity even for missense mutations involving the same codon.
Mutations disrupting the C-terminus take a special position. In contrast to the β-lactamase and β-CASP domain, this region is dispensable for the catalytic function 5, but plays a critical role in regulation of V(D)J recombination and DNA repair through the interaction with DNA-PKcs and DNA Ligase 4 6, 34. It has recently been reported that ARTEMIS also interacts with PTIP, a BRCT-domain containing effector protein in the 53BP1 pathway, which antagonizes the homologous recombination DNA repair pathway 35. The importance of this region for genomic stability has been studied in detail in a mouse model harboring a C-terminal 7-nt deletion resulting in a frameshift mutation at D449 (corresponding to D451*11 in humans) 36. This mutation was shown to cause aberrant intra- and interchromosomal V(D)J joining events predisposing to lymphoma 37. Five out of 8 patients reported with deletions in this region presented with leaky SCID, four had features of autoimmunity and two developed EBV-associated lymphomas 4, 20, 26–28. Except for two mutations affecting the most proximal part of the C-terminal domain and reported in SCID patients, we observed very high activity levels for all other truncating C-terminal mutations. Interestingly, 50% of the leaky SCID patients with C-terminal mutations were homozygous for such defects. This contrasts with the observation that the remainder of the patients with leaky SCID carried a hypomorphic missense mutation on one allele, and a deletion on the second allele.
Our system allows to assess functional activity of ARTEMIS mutated proteins on an intrachromosomal substrate in a relevant cell type (pro-B cells), which represents a stage of B cell development where V(D)J recombination occurs physiologically. Other approaches to test ARTEMIS activity are based on co-transfection of a reporter construct and RAG1 and RAG2 expression plasmids into the cytoplasm of ARTEMIS mutated fibroblast cell lines 5, 12. However, it is important to mention that none of the cellular platforms available allow to fully model functional activities as they occur in patients. In particular, in the cellular platform described here, ARTEMIS expression is driven under a viral promoter and influenced by random genomic integration and by the number of vector copies. All of these factors may contribute to variable expression, including overexpression. To adjust for this limitation, we normalized mutant activities to WT activity and subtracted background activities obtained in mock-transduced cells. Furthermore, to assess the impact of protein overexpression on recombination and DNA repair activity we performed limiting dilution analysis of vector copy numbers. By titrating the expression levels of the hypomorphic mutations M1T and T432*16, we observed that overexpression was associated with improved functional activity. This was not the case for the P171R variant, for which high levels of activity were consistently observed, even in clones harboring only two vector copies. Our data differ from previous observations that had reported that the P171R variant had significantly lower than normal activity, which could be partially restored by increased product expression 24. Importantly, the observation that overexpression of the M1T and T432*16 mutants was associated with improved functional activity in Dclre1c−/− abl pro-B cells raises the interesting hypothesis that novel therapeutic approaches, aimed at enhancing gene transcription, might be efficacious in patients with hypomorphic DCLRE1C mutations. However, overexpression of DCLRE1C has also been associated with cytotoxicity 38, suggesting that regulated control of increased expression may be necessary for therapeutic purposes.
In summary, we report on a cellular platform that permits to analyze both recombination activity and DNA double strand break repair function of naturally occurring ARTEMIS mutations. Good correlation was observed between the functional activity of the mutated protein and the clinical phenotype of affected patients. Use of this cellular system may allow to assess the possible disease causing role of novel DCLRE1C genetic variants identified in patients. Finally, the observation that overexpression of hypomorphic mutants may significantly rescue function, opens the way to possible future therapeutic interventions based on modulation of gene expression for patients with ARTEMIS deficiency.
Supplementary Material
Key messages.
We describe a cellular model that allows rapid analysis of functional activity of recombination and DNA repair associated with human DCLRE1C mutations.
Phenotypic diversities in ARTEMIS deficiency depend on residual activity levels on both alleles, which, for hypomorphic variants, can be increased with higher levels of expression.
These findings can potentially help guide gene therapy strategies
Acknowledgments
Sources of Funding: This work was partially supported by grants P01 AI076210-05 and R01AI00887 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (to LDN). KF received funding from the German Research Foundation (FE 1253/1-1). JpdeV lab is partly funded by Ligue Nationale contre le Cancer (Equipe Labellisée La LIGUE). EM is supported by MZCR 00064203 and CZ.2.16/3.1.00/21540.
Abbreviations
- A-MulV
Abelson murine leukemia virus
- ATM
Ataxia telangiectasia mutated protein
- CID
Combined immunodeficiency
- DSB
Double strand break
- DNA-PKcs
DNA protein kinase catalytic subunit
- GFP
Green fluorescent protein
- IR
Ionizing radiation
- LOF
Loss of function
- MFI
Mean fluorescent intensity
- NHEJ
Non-homologous end-joining
- OS
Omenn syndrome
- RAG
Recombination activation gene
- RSS
Recombination signal sequence
- SCID
Severe combined immunodeficiency
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Le Deist F, Poinsignon C, Moshous D, Fischer A, de Villartay JP. Artemis sheds new light on V(D)J recombination. Immunol Rev. 2004;200:142–55. doi: 10.1111/j.0105-2896.2004.00169.x. [DOI] [PubMed] [Google Scholar]
- 2.Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 3.Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–94. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 4.van der Burg M, Verkaik NS, den Dekker AT, Barendregt BH, Pico-Knijnenburg I, Tezcan I, et al. Defective Artemis nuclease is characterized by coding joints with microhomology in long palindromic-nucleotide stretches. Eur J Immunol. 2007;37:3522–8. doi: 10.1002/eji.200737624. [DOI] [PubMed] [Google Scholar]
- 5.Poinsignon C, Moshous D, Callebaut I, de Chasseval R, Villey I, de Villartay JP. The metallo-beta-lactamase/beta-CASP domain of Artemis constitutes the catalytic core for V(D)J recombination. J Exp Med. 2004;199:315–21. doi: 10.1084/jem.20031142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malu S, De Ioannes P, Kozlov M, Greene M, Francis D, Hanna M, et al. Artemis C-terminal region facilitates V(D)J recombination through its interactions with DNA Ligase IV and DNA-PKcs. J Exp Med. 2012;209:955–63. doi: 10.1084/jem.20111437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–24. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 8.Darroudi F, Wiegant W, Meijers M, Friedl AA, van der Burg M, Fomina J, et al. Role of Artemis in DSB repair and guarding chromosomal stability following exposure to ionizing radiation at different stages of cell cycle. Mutat Res. 2007;615:111–24. doi: 10.1016/j.mrfmmm.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 9.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–86. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Moshous D, Zhou Y, Wang J, Xie G, Salido E, et al. A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking Native Americans. J Immunol. 2002;168:6323–9. doi: 10.4049/jimmunol.168.12.6323. [DOI] [PubMed] [Google Scholar]
- 11.Pannicke U, Honig M, Schulze I, Rohr J, Heinz GA, Braun S, et al. The most frequent DCLRE1C (ARTEMIS) mutations are based on homologous recombination events. Hum Mutat. 2010;31:197–207. doi: 10.1002/humu.21168. [DOI] [PubMed] [Google Scholar]
- 12.Pannicke U, Ma Y, Hopfner KP, Niewolik D, Lieber MR, Schwarz K. Functional and biochemical dissection of the structure-specific nuclease ARTEMIS. EMBO J. 2004;23:1987–97. doi: 10.1038/sj.emboj.7600206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ege M, Ma Y, Manfras B, Kalwak K, Lu H, Lieber MR, et al. Omenn syndrome due to ARTEMIS mutations. Blood. 2005;105:4179–86. doi: 10.1182/blood-2004-12-4861. [DOI] [PubMed] [Google Scholar]
- 14.Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. 2014;133:1099–108. doi: 10.1016/j.jaci.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol. 2014;133:1092–8. doi: 10.1016/j.jaci.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bredemeyer AL, Helmink BA, Innes CL, Calderon B, McGinnis LM, Mahowald GK, et al. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature. 2008;456:819–23. doi: 10.1038/nature07392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, et al. Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood. 2010;116:1263–71. doi: 10.1182/blood-2010-02-267583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin OA, Ivashkevich A, Choo S, Woodbine L, Jeggo PA, Martin RF, et al. Statistical analysis of kinetics, distribution and co-localisation of DNA repair foci in irradiated cells: cell cycle effect and implications for prediction of radiosensitivity. DNA Repair (Amst) 2013;12:844–55. doi: 10.1016/j.dnarep.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 19.Lagresle-Peyrou C, Benjelloun F, Hue C, Andre-Schmutz I, Bonhomme D, Forveille M, et al. Restoration of human B-cell differentiation into NOD-SCID mice engrafted with gene-corrected CD34+ cells isolated from Artemis or RAG1-deficient patients. Mol Ther. 2008;16:396–403. doi: 10.1038/sj.mt.6300353. [DOI] [PubMed] [Google Scholar]
- 20.Musio A, Marrella V, Sobacchi C, Rucci F, Fariselli L, Giliani S, et al. Damaging-agent sensitivity of Artemis-deficient cell lines. Eur J Immunol. 2005;35:1250–6. doi: 10.1002/eji.200425555. [DOI] [PubMed] [Google Scholar]
- 21.Lee PP, Woodbine L, Gilmour KC, Bibi S, Cale CM, Amrolia PJ, et al. The many faces of Artemis-deficient combined immunodeficiency - Two patients with DCLRE1C mutations and a systematic literature review of genotype-phenotype correlation. Clin Immunol. 2013;149:464–74. doi: 10.1016/j.clim.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 22.Noordzij JG, Verkaik NS, van der Burg M, van Veelen LR, de Bruin-Versteeg S, Wiegant W, et al. Radiosensitive SCID patients with Artemis gene mutations show a complete B-cell differentiation arrest at the pre-B-cell receptor checkpoint in bone marrow. Blood. 2003;101:1446–52. doi: 10.1182/blood-2002-01-0187. [DOI] [PubMed] [Google Scholar]
- 23.Evans PM, Woodbine L, Riballo E, Gennery AR, Hubank M, Jeggo PA. Radiation-induced delayed cell death in a hypomorphic Artemis cell line. Hum Mol Genet. 2006;15:1303–11. doi: 10.1093/hmg/ddl050. [DOI] [PubMed] [Google Scholar]
- 24.Woodbine L, Grigoriadou S, Goodarzi AA, Riballo E, Tape C, Oliver AW, et al. An Artemis polymorphic variant reduces Artemis activity and confers cellular radiosensitivity. DNA Repair (Amst) 2010;9:1003–10. doi: 10.1016/j.dnarep.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 25.de Villartay JP, Shimazaki N, Charbonnier JB, Fischer A, Mornon JP, Lieber MR, et al. A histidine in the beta-CASP domain of Artemis is critical for its full in vitro and in vivo functions. DNA Repair (Amst) 2009;8:202–8. doi: 10.1016/j.dnarep.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi N, Agematsu K, Sugita K, Sako M, Nonoyama S, Yachie A, et al. Novel Artemis gene mutations of radiosensitive severe combined immunodeficiency in Japanese families. Hum Genet. 2003;112:348–52. doi: 10.1007/s00439-002-0897-x. [DOI] [PubMed] [Google Scholar]
- 27.Moshous D, Pannetier C, Chasseval Rd R, Deist Fl F, Cavazzana-Calvo M, Romana S, et al. Partial T and B lymphocyte immunodeficiency and predisposition to lymphoma in patients with hypomorphic mutations in Artemis. J Clin Invest. 2003;111:381–7. doi: 10.1172/JCI16774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bajin IY, Ayvaz DC, Unal S, Ozgur TT, Cetin M, Gumruk F, et al. Atypical combined immunodeficiency due to Artemis defect: a case presenting as hyperimmunoglobulin M syndrome and with LGLL. Mol Immunol. 2013;56:354–7. doi: 10.1016/j.molimm.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–70. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- 30.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 32.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 33.Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. 2008;358:2030–8. doi: 10.1056/NEJMoa073966. [DOI] [PubMed] [Google Scholar]
- 34.Soubeyrand S, Pope L, De Chasseval R, Gosselin D, Dong F, de Villartay JP, et al. Artemis phosphorylated by DNA-dependent protein kinase associates preferentially with discrete regions of chromatin. J Mol Biol. 2006;358:1200–11. doi: 10.1016/j.jmb.2006.02.061. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Aroumougame A, Lobrich M, Li Y, Chen D, Chen J, et al. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014;28:2693–8. doi: 10.1101/gad.252478.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Y, Giblin W, Kubec M, Westfield G, St Charles J, Chadde L, et al. Impact of a hypomorphic Artemis disease allele on lymphocyte development, DNA end processing, and genome stability. J Exp Med. 2009;206:893–908. doi: 10.1084/jem.20082396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobs C, Huang Y, Masud T, Lu W, Westfield G, Giblin W, et al. A hypomorphic Artemis human disease allele causes aberrant chromosomal rearrangements and tumorigenesis. Hum Mol Genet. 2011;20:806–19. doi: 10.1093/hmg/ddq524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Multhaup M, Karlen AD, Swanson DL, Wilber A, Somia NV, Cowan MJ, et al. Cytotoxicity associated with artemis overexpression after lentiviral vector-mediated gene transfer. Hum Gene Ther. 2010;21:865–75. doi: 10.1089/hum.2009.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.