

Summary
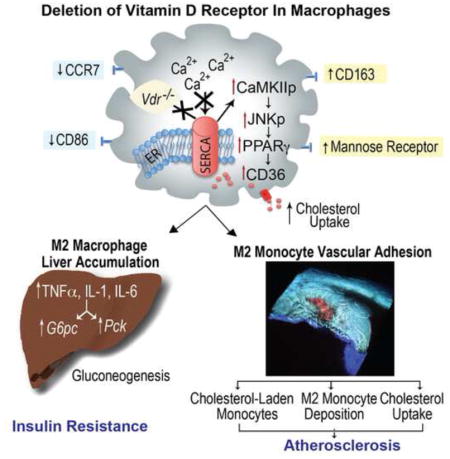
Intense effort has been devoted to understanding predisposition to chronic systemic inflammation as this contributes to cardiometabolic disease. We demonstrate that deletion of the macrophage vitamin D receptor (VDR) in mice (KODMAC) is sufficient to induce insulin resistance by promoting M2 macrophage accumulation in the liver, as well as increase cytokine secretion and hepatic glucose production. Moreover, VDR deletion increases atherosclerosis by enabling lipid-laden M2 monocytes to adhere, migrate, and carry cholesterol into the atherosclerotic plaque, and by increasing macrophage cholesterol uptake and esterification. Increased foam cell formation results from lack of VDR-SERCA2b interaction, causing SERCA dysfunction, activation of ER stress-CaMKII-JNKp-PPARγ signaling, and induction of the scavenger receptors CD36 and SR-A1. BM transplant of VDR-expressing cells into KODMAC mice improved insulin sensitivity, suppressed atherosclerosis, and decreased foam cell formation. The immunomodulatory effects of vitamin D in macrophages are thus critical in diet-induced insulin resistance and atherosclerosis in mice.
Graphical Abstract
Introduction
The combination of type 2 diabetes (T2DM) and cardiovascular disease (CVD) is the most common cause of morbidity and mortality in Western populations. Despite the known link between chronic inflammation, insulin resistance, and accelerated vascular disease, little is known about the mechanisms causing this immune activation. Environmental conditions that regulate macrophage differentiation and infiltration within metabolically active tissues play a role in disease progression (Olefsky and Glass, 2010). In high-fat-feeding-induced obese mice, classical M1 macrophage infiltration into adipose and liver generates pro-inflammatory cytokines and reactive oxygen intermediates that accelerate additional immune cell recruitment and promote insulin resistance (Lumeng et al., 2008). In early atherosclerotic plaque formation, retention of cholesterol in the subendothelial space triggers monocyte recruitment (Hansson and Hermansson, 2011). Both M1 and M2 macrophage phenotypes interchange dynamically depending on the plaque environment (Bouhlel et al., 2007). M1 macrophages, stimulated by the T-helper 1 cytokine, IFNγ, or by cholesterol crystals, are present early in the atherosclerotic plaque, promoting more inflammation, but they have lower scavenger receptor expression and cholesterol deposition and migrate more easily out of the plaque (Mantovani et al., 2009; Murray et al., 2014). In contrast, T-helper 2 cytokines or activation of lipid sensing nuclear receptors by polyunsaturated fatty acids promotes M2 macrophage differentiation. This heterogeneous M2 macrophage subtype displays a spectrum of inflammatory responses (both pro- and anti-inflammatory), but is consistently characterized by expression of IL-10, arginase 1, and mannose receptor (MR) that contribute to tissue repair. Interestingly, these cells have increased scavenger receptor expression facilitating foam cell formation, suggesting that modified lipid deposition and M2 macrophage differentiation perpetuate one another within the atherosclerosis plaque (Chinetti-Gbaguidi et al., 2011; Oh et al., 2012). Therefore, understanding the environmental conditions that govern monocyte recruitment and subsequent macrophage immune programming and cholesterol deposition is key to the development of novel therapeutic strategies.
Endoplasmic reticulum (ER) stress is an important cellular adaptation linking immune responses to obesity, insulin resistance, and atherosclerosis (Hotamisligil, 2010). Stimulation of ER stress induces responses to improve protein folding, but persistent stress triggers further inflammation through NF-kB and c-Jun N-terminal kinase activation (JNK) activation, increasing foam cell formation and inducing plaque necrosis in advanced atherosclerotic lesions (Hotamisligil, 2006; Ozcan and Tabas, 2010). Vitamin D is a natural macrophage ER stress reliever (Riek et al., 2012). Vitamin D receptor (VDR) is present in almost all cells of the immune system, and vitamin D deficiency is widely prevalent and has emerged as a potential contributor to the pathophysiology of T2DM and CVD (Holick, 2007; Norman and Powell, 2014; Veldman et al., 2000). Active vitamin D [1,25(OH)2D3] reduces adipocyte monocyte recruitment and inflammatory responses, improving adipocyte glucose uptake (Gao et al., 2013). In contrast, diet-induced vitamin D deficiency in mice results in hypertension and accelerated atherosclerosis due to increased plaque ER stress (Weng et al., 2013). Similarly, total body VDR knockout increases blood pressure and accelerates atherogenesis, possibly by local activation of the renin angiotensin system in macrophages (Szeto et al., 2012). In vitamin D-deficient diabetic patients, suppression of monocyte/macrophage ER stress by activating VDR decreases adhesion to endothelial cells and migration (Riek et al., 2013a; Riek et al., 2013b; Riek et al., 2012). Furthermore, 1,25(OH)2D3 suppresses macrophage cholesterol uptake and foam cell formation through downregulation of ER stress and subsequent reduction of JNK phosphorylation, peroxisome proliferator activated receptor gamma (PPARγ) expression, and scavenger receptor CD36 and SR-A1 expression. Together, these data suggest that regulation of ER stress by vitamin D exerts a pivotal role in macrophage cholesterol deposition and monocyte infiltration to critical metabolic tissues. This study was designed to determine the in vivo effects of VDR deletion on macrophage ER stress, chronic inflammation, insulin resistance, and atherosclerosis.
Results
Efficiency of macrophage VDR deletion
Animals with specific inactivation of VDR in myeloid cells (KnockOut of vitamin D receptor in MACrophages; KODMAC) were obtained by crossing Vdrfl/fl mice (Masuyama et al., 2006) with transgenic mice expressing Cre recombinase under the control of the Lyzosomal M promoter (Lyz2Cre+/−) in Ldlr−/− or Apoe−/− backgrounds, models of diet-induced insulin resistance and atherosclerosis, to produce KODMAC-L and KODMAC-E mice, respectively (Figure 1A). All animals were fed chow diet for 6 wks after weaning, then high fat diet (HFD) for an additional 8 wks in Apoe−/− and 10 wks in Ldlr−/− mice unless otherwise specified.
Figure 1. Deletion of Vdr in macrophages in Ldlr−/− mice.

(A) Schematic representation of generation of KODMAC mice. (B) qRT-PCR analysis of relative Vdr mRNA expression normalized to Mrpl32 expression in CD11b+ BMDMs, monocytes, and peritoneal macrophages (n=3 per group). (C) Western blot analysis of VDR from KODMAC-L and control-L tissues. (D) qRT-PCR analysis of Cyp24a1 mRNA expression normalized to Mrpl32 in cultured peritoneal macrophages with or without stimulation with 1,25(OH)2D3 10−8 M (n=6 per group). (E) Weight before and after high fat diet (HFD) (n=16–20 per group). (F) Body fat and (G) lean body mass percentage after HFD (n=18–20 per group). Fasting serum (H) cholesterol, (I) triglycerides, and (J) glucose before and after HFD (n=12 per group). *P<0.05 vs. control-L. Data presented as mean ± SEM. See also Figure S1.
The efficiency of macrophage-specific Vdr inactivation in KODMAC-L was demonstrated with Vdr mRNA levels in bone-marrow-derived macrophages (BMDM), peripheral blood monocytes, and peritoneal macrophages and VDR protein levels in multiple tissues (Figure 1B–1C). Similar results were found for KODMAC-E (data not shown). Incubation of KODMAC-L peritoneal macrophages with 1,25(OH)2D3 confirmed suppressed expression of the VDR target gene Cyp24a1 (Figure 1D). KODMAC-L mice had similar body weight and composition compared to control littermates (control-L) (Figure 1E–1G). Cholesterol and triglyceride levels were also similar before and after HFD (Figure 1H–1I). In contrast, fasting glucose was higher in KODMAC-L before and after HFD (Figure 1J). Similar results were found in KODMAC-E mice compared to littermates (control-E) (Figures S1A–S1D).
Deletion of macrophage Vdr induces insulin resistance
To characterize the altered glucose metabolism in KODMAC-L, we performed glucose tolerance tests (GTT) and insulin tolerance tests (ITT) after 6 weeks on chow and after an additional 10 weeks of HFD. KODMAC-L developed glucose intolerance before and after HFD with no compensatory increase in insulin secretion (Figure 2A and S2A). In addition, KODMAC-L mice were more resistant to glucose lowering by exogenous insulin, suggesting insulin resistance (Figure 2B and S2B). KODMAC–E also showed insulin resistance before and after 8 wks on HFD (Figures S2C–F). To clarify the primary site of insulin resistance in KODMAC-L, we performed hyperinsulinemic-euglycemic clamps in mice before HFD. KODMAC-L had significantly reduced glucose infusion rate and increased hepatic glucose production, with no difference in disposal rate, consistent with increased hepatic gluconeogenesis (Figure 2C–2E). mRNA expression of two key gluconeogenic enzymes, phosphoenolpyruvate carboxykinase (Pck1) and glucose 6-phosphatase (G6pc), was higher in KODMAC-L hepatocytes (Figure 2F), and hepatic AKT phosphorylation was diminished in KODMAC-L mice after inferior vena cava insulin injection, confirming hepatic insulin resistance (Figure 2G). To determine whether KODMAC-L macrophages directly modulate hepatic glucose production, we co-cultured primary hepatocytes from controlL mice with peritoneal macrophages from KODMAC-L or control-L and found increased hepatocyte expression of Pck1 and G6pc after incubation with KODMAC-L macrophages. Moreover, culture of primary control-L hepatocytes with KODMAC-L macrophage media recapitulated the induction of gluconeogenic enzymes (Figure 2H), implying a macrophage secretory factor affecting hepatic gluconeogenesis. Media from the KODMAC-L co-culture had a higher concentration of TNFα IL-1β, and IL-6, all cytokines reported to induce hepatic gluconeogenesis (Figure 2I). Liver histologic analysis showed increased macrophage accumulation and cholesterol deposition in KODMAC-L mice (Figure 2J–2L), suggesting increased cytokine secretion from hepatic macrophage infiltration induces gluconeogenic enzymes. We next explored the macrophage subtype in KODMAC-L livers. Interestingly, KODMAC-L liver macrophages showed an M2 pattern of membrane markers by flow cytometry with a consistent M2 gene signature (Figure 2M and S2G-S2H). To clarify whether the M2 macrophage subtype differentiation was liver-specific, we analyzed BMDM and peritoneal macrophages from KODMAC-L mice, with all cells demonstrating an M2 membrane and mRNA expression pattern (Figure 2N–2O and S2I–S2L), suggesting that macrophage Vdr deletion increased M2 macrophages and cytokine activation of hepatic insulin resistance.
Figure 2. Deletion of macrophage Vdr induced insulin resistance.

KODMAC-L and control-L mice after 6 wks of chow were assessed for glucose metabolism and insulin resistance after overnight fast. (A) Glucose tolerance test with 30 min plasma insulin levels (inset) (n=35 per group) and (B) insulin tolerance test (n=16 per group). From hyperinsulinemic euglycemic clamps, (C) glucose infusion rate, (D) hepatic glucose production after insulin stimulation, and (E) glucose disposal rate (n=7–8 per group). (F) qRT-PCR analysis of relative Pck1 and G6pc mRNA expression from hepatocytes, normalized to Mrpl32 (n=6 per group). (G) Western blot analysis of phosphorylated AKT from hepatocytes (n=3 per group). (H) qRT-PCR analysis of relative Pck1 and G6pc mRNA expression from control hepatocytes co-cultured with KODMAC-L or control-L macrophages or their media, normalized to Mrpl32 (n=6 per group). (I) Cytokine concentrations from co-culture media (n=4 per group). (J) Hematoxylin and eosin stain (left panels) and F4/80 immunohistochemistry (right panels) of liver (representative of n=3 per group). Arrows show macrophage infiltrates. Scale bar represents 250 μm (100 μm for inset). (K) Percentage of F4/80-positive cells by manual counting of light microscopy fields (n=3 per group). (L) Liver macrophage cholesterol content (n=6 per group). Flow cytometry expression of membrane M1 markers CCR7 and CD86 and M2 markers MR and CD163 in (M) CD11b+ liver macrophages, (N) BMDMs, and (O) peritoneal macrophages (n=4 per group). *P<0.05 vs. control-L. Data presented as mean ± SEM. See also Figure S2.
Deletion of macrophage Vdr increases foam cell formation and accelerates atherosclerosis
Macrophage infiltration and lipid accumulation in the vessel wall are critical in atherosclerosis initiation and progression. In type 2 diabetics, active vitamin D promotes macrophage differentiation in vitro and suppresses cytokine secretion (Giulietti et al., 2007). To determine whether the immunomodulatory effects of macrophage VDR signaling play a role in atherosclerosis, we measured atherosclerotic lesions in KODMAC mice after HFD. KODMAC-L and -E had ~3-fold and ~2-fold greater median plaque area, respectively, in all aortic regions compared to control-L and control-E (Figure 3A and S3A) and to Cre+/−Ldlr−/− littermates (data not shown). Moreover, KODMAC-L and KODMAC-E mice on prolonged chow diet only without exposure to HFD had 4- to 10-fold increased plaque area across all aortic regions (Figure S3B and S3C), suggesting that defective macrophage VDR signaling is critical to atherosclerosis progression.
Figure 3. Deletion of macrophage Vdr accelerated atherosclerosis.

KODMAC-L and control-L mice were assessed for atherosclerosis and macrophage cholesterol metabolism after HFD. (A) Atherosclerotic plaque area of pinned aortas (n=16 per group). Lines indicate median values. In peritoneal macrophages, (B) representative image of Oil Red O staining (scale bar represents 10 μm); (C) free and (D) total cholesterol content (n=6 each group); (E) cholesterol binding and (F) cholesterol uptake after stimulation with DiI-oxLDL for 6 h; (G) cholesteryl ester formation after stimulation with oxLDL and 3H oleic acid for 6 h; and (H) cholesterol efflux after incubation with 3H cholesterol for 24 h and stimulation with HDL or apolipoprotein AI for 6 h (n=6 per group). *P<0.05 vs. control-L for A–H. (I) Immunofluorescent staining for ADRP to identify lipid droplets (red, left column), M1 marker CCR7 or M2 marker MR (green, middle column), and co-localization (yellow, right column) in the proximal aorta after HFD. Blue shows nuclei stained with DAPI. Scale bar represents 50 μm. (J) Quantification of CCR7 and MR as a percentage of total plaque area and (K) lipid co-localization with M1 or M2 macrophages assessed as the percentage of total ADRP co-localizing with CCR7 or MR (n=6 per group). *P<0.05 vs. same receptor in KODMAC-L, **P<0.05 vs. KODMAC-L CCR7. Data presented as mean ± SEM. See also Figure S3.
We next explored whether alterations in KODMAC-L macrophage cholesterol metabolism contribute to increased atherosclerosis. Peritoneal macrophages from KODMAC-L mice after 10 wks of HFD had increased foam cell formation by Oil-Red-O staining and higher free and total cholesterol content (Figure 3B–3D). Foam cell formation was mediated by increasing oxidized (ox)LDL cholesterol binding and uptake and cholesterol ester formation with no differences in cholesterol efflux (Figure 3E–3H). To determine whether the M1/M2 macrophage profile induced in KODMAC-L correlates with the deposition of atherosclerotic plaque foam cells, we examined frozen sections of the aortic root from KODMAC-L mice after 10 wks on HFD. Immunofluorescent staining revealed increased vessel wall infiltration with predominantly M2 macrophages that co-localized with fat staining by adipose differentiation-related protein (ADRP) compared to control-L, consistent with our previous observation that M2 macrophages induced by vitamin D deficiency accumulate more cholesterol (Figure 3I–K) (Weng et al., 2013).
CaMKII links ER stress to increased cholesterol uptake in KODMAC-L macrophages
In diabetic patients, ER stress activation induced by vitamin D deficiency triggers M2 macrophage differentiation and foam cell formation by increasing scavenger-receptor-mediated cholesterol uptake (Oh et al., 2009; Riek et al., 2013a). Therefore, we explored whether the effects of vitamin D on foam cell formation are ER stress-VDR-dependent in vivo. Consistent with our previous data, peritoneal macrophages from KODMAC-L mice after 6 wks of chow showed upregulation of ER stress, JNK phosphorylation, PPARγ, and scavenger receptors CD36 and SRA-1, regardless of vitamin D conditions (Figure 4A). KODMAC-L macrophages also demonstrated increased acetylated (Ac)LDL cholesterol binding and uptake (Figure S4A–S4B). We then confirmed that the effects of Vdr deletion on macrophage subtype are ER-stress-dependent. Incubation of KODMAC-L macrophages with ER stress inhibitor, 4-phenylbutyrate (PBA), shifted them toward an M1 expression pattern similar to that of control-L cells (Figure S4C–SD). In previous studies in cholesterol-loaded macrophages, increased cytosolic calcium resulting from ER stress activates calmodulin-dependent protein kinase II (CaMKII), linking ER stress to JNK-mediated macrophage apoptosis (Timmins et al., 2009). We studied type 2 diabetic human macrophages cultured in vitamin D-deficient or –1,25(OH)2D3-supplemented conditions and chow-fed KODMAC-L or control-L peritoneal macrophages to explore whether CaMKII activation links impaired vitamin D signaling to macrophage cholesterol uptake. Both KODMAC-L and vitamin D-deficient human macrophages had increased CaMKII phosphorylation compared to control-L or 1,25(OH)2D3-supplemented human macrophages, respectively, but no differences in total CaMKII expression (Figure 4B). To determine the effects of CaMKII inhibition on macrophage cholesterol uptake, we used KN-93, a CaMKII inhibitory agent, in KODMAC-L and control-L peritoneal macrophages under vitamin D-deficient or 1,25(OH)2D3-supplemented conditions. Vitamin D-deficient control-L macrophages and KODMAC-L macrophages in both vitamin D conditions all had significantly higher oxLDL cholesterol uptake compared to 1,25(OH)2D3-supplemented control-L macrophages, but CaMKII inhibition abolished this increased cholesterol uptake in all circumstances (Figure 4C), suggesting that CaMKII activation is key in macrophage cholesterol uptake induced by defective vitamin D signaling. Moreover, inhibiting CaMKIIp in KODMAC-L or control-L macrophages under vitamin D-deficient conditions suppressed JNKp, PPARγ, CD36, and ER stress (Figure 4D), suggesting that activation of CaMKII links vitamin-D-deficiency-induced ER stress to JNK-mediated macrophage cholesterol uptake.
Figure 4. Deletion of macrophage Vdr increased ER stress and accelerated cholesterol uptake by activation of the CaMKII signaling pathway.

Peritoneal macrophages from KODMAC-L or control-L before HFD and/or human monocyte-derived macrophages from type 2 diabetics were cultured for 5 d in vitamin D-deficient or 1,25(OH)2D3-supplemented conditions. (A) Western blot of ER stress and cholesterol signaling proteins). (B) Western blot of SERCA2b and CaMKII activation (representative of n=4 per group), (C) Cholesterol uptake after DiI-oxLDL stimulation in murine macrophages with or without CaMKII inhibitor KN-93 for 6 h (n=7 per group) (*P<0.001 by ANOVA vs. all others without KN-93, **P<0.05 vs. same cells without KN-93). (D) Western blot of activated CaMKII and JNK, PPARγ, CD36, and ER stress protein CHOP in murine macrophages with or without KN-93 (representative of n=4 per group). SERCA2b activity from (E) murine macrophages cultured in 1,25(OH)2D3-supplemented media (n=4 per group), (F) human macrophages cultured in vitamin D-deficient or 1,25(OH)2D3-supplemented conditions (n=5 per group), and (G) human macrophages cultured in 1,25(OH)2D3-supplemented conditions and infected with Vdr-siRNA or control siRNA (n=5 per group) (*P<0.05 vs. control-L or vitamin D-supplemented for all). (H) SERCA2b expression in murine control-L macrophage lysates immunoprecipitated with VDR antibody and (I) Immunofluorescent staining for VDR (green, left), SERCA2b (red, middle), and co-localization (yellow, right) in murine control-L macrophages. Scale bar represents 10 μm. (J) Schematic depicting VDR-driven modulation of SERCA2b activity and induction of foam cell formation. Data expressed as mean ± SEM. See also Figure S4.
VDR signaling facilitates SERCA2b function and suppresses ER stress
Sarcoendoplasmic reticulum calcium ATPase 2b (SERCA2b) is a critical enzyme maintaining high ER calcium levels to optimize protein production and folding. Induction of macrophage ER stress by SERCA inhibitor Thapsigargin in control-L macrophages shifts macrophages toward an M2 expression pattern similar to that of KODMAC-L (Figure S4E–S4F), implying similar effects of Vdr deletion and SERCA dysfunction. To clarify whether Vdr deletion induces ER stress by changing SERCA2b expression or function, we again studied peritoneal macrophages from KODMAC-L and control-L mice after 6 wks of chow and human type 2 diabetic macrophages cultured in vitamin D-deficient or 1,25(OH)2D3-supplemented conditions. We found no differences in SERCA2b protein expression between all groups (Figure 4B). However, SERCA2b activity in KODMAC-L and vitamin D-deficient human macrophages was decreased by at least 20% compared to control-L and 1,25(OH)2D3-supplemented macrophages, respectively, suggesting that VDR activates SERCA2b to maintain normal ER functionality (Figure 4E and 4F). To confirm the effects of deletion of macrophage VDR on SERCA2b function in diabetic patients, we infected human macrophages cultured in 1,25(OH)2D3- supplemented media with lentivirus containing either siRNA VDR hairpins or control siRNA. VDR-siRNA–infected macrophages showed significantly lower SERCA2b activity compared to control siRNA-infected macrophages (Figure 4G). Finally, we immunoprecipitated KODMAC-L and control-L peritoneal macrophage lysates with VDR antibody, and blotted for SERCA2b, identifying expression in control-L but not KODMAC-L cells, suggestive of VDR-SERCA2b interaction (Figure 4H). Co-localization of SERCA2b with VDR by immunofluorescence in control-L peritoneal macrophages confirmed a mechanism of VDR interaction with SERCA2b to optimize ER function (Figure 4I and 4J).
Deletion of VDR signaling enables monocyte cholesterol transport into the atherosclerotic plaque
Monocyte adhesion to the endothelium and migration into the subendothelial space are critical to atherosclerotic plaque macrophage deposition and foam cell formation (Gautier et al., 2009). However, recent studies suggest that non-classical monocytes can become cholesterol-laden in the circulation, and may be a potential source of vascular cholesterol deposition in addition to differentiated plaque macrophages (Mosig et al., 2009; Wu et al., 2009). We evaluated cholesterol content in KODMAC-L circulating monocytes after 3 wks of HFD, demonstrating higher cholesterol content and oxLDL uptake (Figure 5A–5B). Moreover, KODMAC-L monocytes demonstrated higher adhesion to fibronectin and increased migration in response to monocyte chemoattractant protein 1 (MCP-1), and accordingly, increased mRNA expression of adhesion membrane receptor β1 integrin and MCP-1 receptor (CCR2) (Figure 5C and 5D; Figure S5A and S5B), indicating that Vdr deletion induces a pro-atherogenic phenotype that might enable monocytes to carry cholesterol into the atherosclerotic plaque. To study this potential mechanism directly, we incubated KODMAC-L or controlL monocytes from mice after 6 wks of chow with stably-labeled, non-naturally-occurring cholesterol-d7 and infused them into chow-fed 6-mo-old LDLR−/− recipient mice to determine if cholesterol–d7 was detectable in the recipient mouse aorta. We first confirmed cholesterol-d7 uptake into cells. As expected, KODMAC-L monocytes had nearly doubled cholesterol-d7 esterification compared to control-L monocytes (Figure 5E). After transfusion, we found minimal enrichment of cholesterol-d7 in the plasma at multiple time points up to 24 h, suggesting that cholesterol efflux out of monocytes was not a significant contributor to vessel wall cholesterol-d7 deposition (data not shown). Strikingly, 24 h after infusion, aortas from mice receiving KODMAC-L monocytes had ~150-fold higher enrichment of cholesterol-d7 compared to plasma and more than 3-fold higher enrichment than control-L monocyte recipient aortas (Figure 5F), suggesting that monocyte cholesterol transport into the vessel wall may explain the increased atherosclerosis in KODMAC-L. We confirmed this monocyte capability with an additional experiment where we isolated CD11b+ monocytes from KODMAC-L mice after 6 wks on chow, incubated with labeled DiI-oxLDL cholesterol, and infused them daily for 3 d into high-fat-fed LDLR−/− mice. Two-photon microscopy from explanted aortas showed localization of DiI-oxLDL inside the vascular wall in the subendothelial space, implying accumulation of lipid-laden monocytes (Figure 5G), confirming an LDL-independent pathway for vascular cholesterol deposition.
Figure 5. Deletion of macrophage Vdr increased lipid-laden M2 cells in the atherosclerotic plaque.

Peripheral blood monocytes from KODMAC-L and control-L mice after 3 wks of HFD were assessed for (A) total cellular cholesterol (n=6 per group), (B) cholesterol uptake after DiI-oxLDL stimulation for 6 h (n=4 per group), (C) adhesion to fibronectin (n=6 per group) and (D) transwell migration in response to MCP-1 stimulation (n=12 per group). For E and F, KODMAC-L and control-L monocytes were incubated with stably-labeled cholesterol-d7 for 24 h and transfused into 6-mo-old Ldlr−/− mice on chow. Cholesterol-d7 ester enrichment in (E) monocytes (n=5 per group) and (F) aortic arch relative to plasma (n=3 per group). (G) Representative image of cross-section of aortic plaque by two-photon microscopy from high-fat-diet-fed Ldlr−/− mice after 3 d of daily transfusions with DiI-oxLDL-cholesterol-incubated KODMAC-L monocytes. Red shows DiI-oxLDL, green shows CD31+ endothelium, and blue shows matrix autofluorescence. Scale bar represents 100 μm. From peripheral monocytes, flow cytometry expression of (H) monocyte markers Ly6C and CCR2 and (I) macrophage M1 markers (CCR7 and CD86) and M2 markers (CD163 and MR) (n=4 per group). *P<0.05 vs. control-L for A–I. (J) Representative image of immunofluorescent staining for ADRP to identify lipid droplets (red, left column), M1 marker CCR7 or M2 marker MR (green, middle column), and co-localization (yellow, right column) in the proximal aorta after 3 wks of high fat diet. Blue shows nuclei stained with DAPI. Scale bar represents 50 μm. Quantification of immunostaining of the proximal aorta for (K) M1 and M2 macrophage markers as a percentage of total plaque area and for (L) lipid co-localization with M1 or M2 macrophages assessed as the percentage of total ADRP co-localizing with CCR7 or MR (n=6 per group). *P<0.05 vs. same receptor in KODMAC-L, **P<0.05 vs. KODMAC CCR7. Data presented as mean ± SEM. See also Figure S5.
To characterize the circulating monocytes associated with cholesterol deposition in KODMAC-L, we used flow cytometry to evaluate traditional murine monocyte markers. Monocytes from KODMAC-L mice after 3 wks of HFD had significantly more Ly6clo cells and fewer Ly6c− cells compared to control-L cells. However, there was no difference in Ly6chi cells between the groups. In addition, there were significantly more cells positive for CCR2 in KODMAC-L, and this was driven by the shift toward Ly6clo cells (Figure 5H), suggesting that deletion of VDR induces a non-classical subtype of monocytes that express membrane markers facilitating vascular invasion. In our previous work, M1/M2 macrophage markers assessed in diabetic monocytes correlate better with adhesion and migration than the typical markers used to classify human monocytes (CD14/CD16). Therefore, we tested whether the M1/M2 expression phenotype of circulating monocytes would reflect the early plaque macrophage subtype. KODMAC-L monocytes expressed predominantly M2 membrane markers with mRNA expression typical of M2 polarization (Figure 5I and S5C–S5D). Frozen sections of early atherosclerotic plaques in the aortic root from KODMAC-L mice after 3 wks on HFD also showed a predominance of M2 macrophages with more ADRP co-localization compared to M1 macrophages (Figure 5J–L), similar to KODMAC-L after HFD. Therefore, deletion of monocyte VDR promotes a pro-atherogenic M2 monocyte phenotype, resulting in M2-predominant plaque macrophages with increased cholesterol deposition that persist throughout plaque progression.
Cardiometabolic abnormalities from Vdr deletion are transplantable
To determine whether introduction of normal macrophage VDR signaling by bone marrow (BM) transplant could prevent metabolic dysregulation and atherosclerosis in KODMAC-L, we transplanted previously-irradiated KODMAC-L mice with BM from KODMAC-L (KOD/KOD) or control-L (Con/KOD) animals. After 6 wks of chow diet, Con/KOD mice had decreased fasting glucose, improved insulin sensitivity with decreased expression of hepatic gluconeogenic enzymes, and liver macrophages shifted toward an M1-predominant phenotype (Figure 6A–6D). After HFD, Con/KOD mice also had increased insulin sensitivity (Figure S6A–S6C) and decreased atherosclerotic area in the proximal and thoracic aorta compared to KOD/KOD (Figure 6E). Moreover, peritoneal macrophages had decreased cholesterol uptake and cholesteryl ester formation with no differences in cholesterol efflux (Figure 6F–6H). Conversely, we transplanted control-L mice with BM from KODMAC-L (KOD/Con) or control-L (Con/Con) animals to induce the metabolic phenotype. As expected, KOD/Con mice demonstrated cardiometabolic dysregulation compared to Con/Con, with increased glucose levels, insulin resistance, atherosclerosis, M2 macrophages, and foam cell formation with greater cholesterol uptake (Figure 6I–6P; Figure S6D–S6F). There was no differential effect of BM transplantation on fasting cholesterol or triglycerides (Figure S7G–S7J). These data confirm the fundamental role of macrophage VDR signaling in chronic inflammation, affecting metabolic function and atherosclerosis progression.
Figure 6. Deletion of macrophage Vdr was sufficient to induce insulin resistance and atherosclerosis.

Irradiated KODMAC-L mice were transplanted with BM from KODMAC-L (KOD/KOD) or control-L (Con/KOD) animals to rescue the phenotype (A–H), and irradiated control mice were transplanted with BM from KODMAC-L (KOD/Con) or control-L (Con/Con) animals to induce the phenotype (I–P). All experiments except E and M were performed before HFD. (A and I) Glucose tolerance test with 30 min plasma insulin levels (inset) and (B and J) insulin tolerance test (n=7–11 per group). (C and K) qRT-PCR analysis of relative Pck1 and G6pc mRNA expression from hepatocytes, normalized to Mrpl32 (n=3 per group). (D and L) Flow cytometry expression of membrane M1 and M2 markers in liver macrophages (n=4 per group). (E and M) Atherosclerotic plaque area of pinned aortas after HFD (n=8–11 per group). (F and N) Cholesterol uptake after DiI-oxLDL stimulation (n=6 per group) and (G and O) cholesteryl ester formation after stimulation with oxLDL and 3H oleic acid (n=4–6 per group) and (H and P) cholesterol efflux after incubation with 3H cholesterol and stimulation with HDL or apolipoprotein AI in peritoneal macrophages (n=6 per group). *P<0.05 vs. KOD/KOD or Con/Con, respectively. Data presented as mean ± SEM. See also Figure S6.
Discussion
Little is known about environmental immunomodulators affecting the chronic inflammation in metabolic tissues that contribute to vascular disease. In this work, we demonstrate that Vdr deletion in a single cell lineage in KODMAC mice is sufficient to induce insulin resistance and accelerate atherosclerosis. KODMAC-L mice had accelerated M2 macrophage infiltration and cholesterol deposition in the liver, increasing cytokine secretion and hepatic glucose production. Moreover, KODMAC-L animals were predisposed to plaque cholesterol deposition via both M2 monocyte transport of cholesterol into the vessel wall and cholesterol uptake by M2 plaque macrophages. Increased cholesterol deposition precipitated by impaired VDR-SERCA interaction induced ER stress activation of CaMKII, triggering the JNK-PPARγ pathway and upregulation of scavenger receptors CD36 and SR-A1 to facilitate foam cell formation. BM transplant of VDR-expressing cells into KODMAC-L mice improved insulin sensitivity, decreased foam cell formation, and suppressed atherosclerosis, confirming the critical role of macrophage VDR signaling in diet-induced insulin resistance and atherosclerosis.
Previous evidence reveals the role of liver macrophage infiltration in hepatic insulin resistance. In rats, selective depletion of liver-resident macrophages enhances hepatic insulin sensitivity after high-fat diet (Lanthier et al., 2010). In mice, macrophage ablation of an upstream activator of NF-κB enhances hepatic and peripheral insulin sensitivity (Arkan et al., 2005). Accordingly, we found that deletion of macrophage Vdr increased monocyte adhesion and migration, promoting hepatic cholesterol deposition, macrophage accumulation, and glucose production through cytokine release. Interestingly, hepatic macrophage subtypes also play a key role in hepatic gluconeogenesis. BM transplant from mice deficient for Pparδ, a transcription factor that facilitates IL-4-induced M2 macrophage differentiation, causes hepatic infiltration and increased gluconeogenesis (Odegaard et al., 2008). In contrast, we found that deletion of macrophage Vdr favors hepatic accumulation of M2 cells that stimulate gluconeogenesis. The transplantability of this phenotype confirms that VDR signaling links macrophage subtype to hepatic gluconeogenesis. These contrasting findings imply that the M1-M2 paradigm to describe the inflammatory profile of macrophage subtypes as they contribute to hepatic insulin resistance may be oversimplified.
The recruitment of circulating monocytes into the vessel wall is critical in the pathophysiology of vascular complications (Hansson and Hermansson, 2011). Recent studies in patients with familial hypercholesterolemia and in mouse models of atherosclerosis show that non-classical monocytes can become cholesterol-laden in the circulation, suggesting they may carry cholesterol into the vessel wall (Mosig et al., 2009; Wu et al., 2009). We found that VDR signaling regulates atherosclerosis initiation by modifying circulating monocyte cholesterol transport and plaque macrophage phenotype. Monocyte Vdr deletion induced a cholesterol-laden M2 phenotype with increased adhesion to and migration into the vessel wall, facilitating an LDL-independent pathway for cholesterol transport into the early atherosclerotic plaque.
Macrophages are key cells in atherosclerosis progression. The cellular microenvironment directs macrophages into different subtypes with distinct cholesterol metabolism phenotypes. In IL-4-induced M2 macrophages, CD36 signaling activation by long-chain free fatty acids generates small lipid droplets and increased β-oxidation (Huang et al., 2014). In contrast, CD36 signaling activation by oxLDL results in increased cholesterol deposition and nuclear sequestration of AMPK, diminishing β–oxidation (Oh et al., 2009; Samovski et al., 2014). In vivo, the location of M2 macrophages in atherosclerotic plaque affects cholesterol metabolism. Increased IL-4 overlying the lipid core induces M2 macrophages (CD68+ and MR+) with increased cholesterol ester formation, reduced cholesterol efflux, and small lipid droplets (Chinetti-Gbaguidi et al., 2011). In contrast, in areas of previous intraplaque hemorrhage, M2 macrophages (MR+ and CD163+) produce IL-10 and have increased cholesterol efflux and lower cholesterol content (Finn et al., 2012), highlighting the effect of the microenvironment on the interplay between macrophage subtype and functionality. In our study, KODMAC-L mice had increased atherosclerosis with a persistent predominance of cholesterol-laden M2 cells as plaques progressed. Similar results of increased atherosclerosis have been demonstrated in Vdr total knockout mice; these animals also have increased ex vivo macrophage foam cell formation in response to modified LDL cholesterol (Szeto et al., 2012). Our model shows that deletion of VDR is sufficient to program a sustained, pro-atherogenic monocyte/macrophage phenotype, confirmed by the transplantability of the cardiometabolic outcomes.
Chronic excess of cytokines, glucose, and lipids triggers ER stress, provoking inflammatory signaling, macrophage cholesterol uptake or macrophage death, key mechanisms in insulin resistance and atherosclerosis (Hummasti and Hotamisligil, 2010; Oh et al., 2012; Tabas and Ron, 2011). Vitamin D acts as an ER stress reliever that suppresses pro-atherogenic macrophage properties and decreases atherosclerosis. This work demonstrates that the macrophage VDR interacts with SERCA2b to improve its function, identifying a mechanism by which vitamin D decreases ER stress. Macrophage accumulation of modified lipids impairs SERCA2b expression, increasing cytosolic calcium activation of the CaMKII-JNK pathway to cause macrophage apoptosis and plaque instability (Liang et al., 2012; Timmins et al., 2009). Similarly, we found that macrophage VDR signaling activation diminished CaMKII-JNK activation and suppressed cholesterol accumulation to break the cycle of ER stress activation.
We recognize that the KODMAC model of Vdr deletion in monocyte/macrophages is an extreme model of vitamin D deficiency, and may not directly represent animal and human physiology. However, in animal models of insulin resistance and in patients with T2DM, adequate dietary vitamin D also promotes a monocyte/macrophage phenotype with lower adhesion, migration, and foam cell formation, as well as decreasing atherosclerosis (Oh et al., 2009; Riek et al., 2012; Weng et al., 2013). It is also important to acknowledge that these metabolic changes induced by macrophage-specific Vdr deletion might represent just a component of the myriad of cardiometabolic effects linked to vitamin D deficiency in humans, such as hypertension, thrombosis and systemic inflammation. However, the KODMAC model highlights that VDR-dependent signaling in a single cell linage is a critical pathway integrating inflammation, insulin resistance, and atherosclerosis.
Experimental Procedures
Further details and references are provided in Supplemental Methodology.
Experimental models
Vdrfl/fl mice were backcrossed into C57BL/6 for 10 generations (Masuyama et al., 2006). Inactivation of VDR in myeloid cells (KnockOut of vitamin D receptor in MACrophages; KODMAC) was generated by crossing Vdrfl/fl mice with transgenic mice expressing Cre recombinase under the control of the Lyzosomal M promoter (Lyz2Cre+/) in Ldlr−/− (KODMAC-L) or Apoe−/− (KODMAC-E) backgrounds. Littermate mice Lyz2Cre−/−Vdrfl/flLdlr−/− mice (Control-L) and Lyz2Cre−/−Vdrfl/flApoe−/− (Control-E) were used as controls. Mice received normal chow diet (Harlan TD87095) for 6 wks after weaning, then vitamin D-sufficient high fat diet (HFD) for 8 wks in the Apoe−/− and for 10 wks in the Ldlr−/− background (Harlan TD88137). Additional mice were maintained on chow for 1 yr for atherosclerosis assessment. For BM transplants, lethally irradiated 8-wk-old chow-fed KODMAC-L or control-L recipient mice were reconstituted with ~5 × 106 BM cells from 8-wk-old chow-fed KODMAC-L or control-L mice via injection. Recipient mice were given 6 wks of chow, then 10 wks of HFD. All experiments included male and female animals and were approved by the Washington University Animal Studies Committee. Adult subjects with T2DM were voluntarily recruited and provided with written informed consent, approved by the Human Research Protection Office of Washington University School of Medicine.
Isolation of murine peritoneal, hepatic, and BMDMs and peripheral monocytes
Peritoneal macrophages were collected immediately following injection of PBS into the peritoneum and cultured for 3 h in vitamin D-deficient media [deficient in both 25(OH)D and 1,25(OH)2D; obtained by DMEM plus 10% charcoal/dextran-treated FBS] with or without 1,25(OH)2D3 (Sigma, 10−8 M) prior to assessment. Cells were subjected to CD11b microbeads (Miltenyi Biotec) prior to flow cytometry. For hepatic macrophage isolation, livers were resected, minced, treated with collagenase, and filtered (70 μm cell strainer, BD Bioscience) to remove hepatocytes. Hepatic macrophages were selected for CD11b (present in macrophages but not Kupffer cells) with microbeads. For BMDMs, BM was flushed from femurs and tibias and subjected to erythrocyte lysis, then CD11b microbeads prior to 5 d of culture with 100 ng/ml of M-CSF in vitamin D-deficient media plus 1,25(OH)2D3 (10−8 M). Peripheral blood monocytes were selected by CD11b microbeads and stabilized for 3 h in DMEM plus 10% serum from the original mouse to mimic in vivo conditions prior to any procedures.
Isolation of human monocyte-derived macrophages
Peripheral monocytes were isolated by standard Ficoll isolation techniques and selected by CD14 microbeads (Miltenyi Biotec) with CD14+/CD11b+ cell purity of 97% by flow cytometry (FACStar Plus). For monocyte-derived macrophages, monocytes were cultured with 100 ng/ml of M-CSF for 5 days in vitamin D–deficient media with or without supplementation of 1,25(OH)2D3 (10−8 M). For ER stress suppression or induction, differentiated macrophages were cultured with PBA (10 mM) or Thapsigarin (0.25 μM), respectively, for an additional 18 h. For lentiviral infection, after 5 days of differentiation in 1,25(OH)2D3-supplemented conditions, macrophages were infected with lentivirus containing either Vdr-siRNA or control-siRNA, with assays performed 72 h later.
Metabolic assessment
Mice underwent metabolic characterization after 6 wks of chow and after an additional 8–10 wks of HFD (Apoe−/− or Ldlr−/− background, respectively). Body composition was performed by dual energy X-ray absorptiometry (PIXImus, GE Corporation). Serum glucose, cholesterol, and triglycerides, and insulin were measured after 6 h of fasting using commercially available kits. For glucose and insulin tolerance tests, mice were fasted overnight prior to peritoneal injection with 10% D-glucose (1 g/kg) or insulin (0.75 U/kg, Humulin R). Hyperinsulinemic euglycemic clamps were performed in conscious mice after 6 h of fasting with a clamped glucose level of 120 mg/dl. For hepatic insulin signaling, livers were isolated and snap frozen 2 min after inferior vena cava insulin injection in fasted mice, then homogenized and immunoblotted for phospho-AKT (Ser473).
Hepatocyte isolation and macrophage co-culture
Livers of anesthetized 8-wk-old control-L mice were perfused with collagenase-containing media (Invitrogen) and resected. The dissociated hepatocytes were allowed to recover for 24 h. Peritoneal KODMAC-L and control-L macrophages were cultured for 24 h in 1,25(OH)2D -supplemented media prior to co-culture onto hepatocyte cultures at a 1:2 macrophage:hepatocyte ratio for 18 h. TNFα (BD bioscience) and IL-1β and IL-6 (R&D Biosystems) concentrations were measured by ELISA in co-culture media. For liver histology, sections of frozen livers were stained with hematoxylin and eosin or with macrophage-specific antibody F4/80 (1:100, Santa Cruz Biotechnology). Nuclei were manually counted and cells were identified as positive or negative for F4/80 in 10 fields at 10X magnification per animal.
Flow cytometry
Monocytes and hepatic, BM-derived, and peritoneal macrophages were selected for CD11b with microbeads, then stained with antibodies directed against CCR7, CD86, Ly6C, and CD115 (E-Bioscience), CD163 (Bioss USA), and MR and CCR2 (R&D Systems). Monocytes and macrophages were gated for live-cells, and monocytes were additionally gated for CD115+ for analysis.
Murine atherosclerosis
For atherosclerosis, aortae were prepared using the en face technique, with results reported as percentage involvement of the arch, thoracic, and abdominal regions as analyzed using Image J. For co-localization of plaque macrophages with cholesterol deposition, cryosections of the aortic root were stained with antibodies specific for CCR7 (M1 marker) or MR (M2 marker) and adipocyte differentiation-related protein (ADRP). Plaque M1 or M2 macrophages were assessed as the percentage of total plaque area with staining for CCR7 or MR. Lipid co-localization with M1 or M2 macrophages was measured by the percentage of total ADRP-staining area co-localizing with staining for CCR7 or MR.
Cellular cholesterol metabolism
Foam cell formation was assessed with fixation and staining with Oil-red-O. For cellular total or free cholesterol, lipids were extracted with chloroform:methanol (1:2 v/v), dried, and reconstituted for enzymatic assays using commercial reagents. For cholesterol binding and uptake, cells were incubated for 6 h with 10 μg/mL DiI-labeled oxidized LDL (DiI-oxLDL) at 4°C (binding) or at 37°C (binding and uptake), with uptake calculated as the difference. For cholesterol uptake assessments with CaMKII inhibitor KN93, macrophages were incubated with KN93 (10 uM) for 24h with DiI-oxLDL added for the final 6 h. For cholesterol ester, macrophages were incubated with oxLDL (200 μg/mL) with 3H oleic acid (0.1 mM) for 6 h prior to lipid extraction and radioactivity assessment. For cholesterol efflux, macrophages were incubated with oxLDL (100 μg/mL) with 5 mCi of 3H cholesterol for 24 h, then with apolipoprotein AI (25μg/mL) or HDL (50 μg/mL) for 6 h, after which supernatant and cells were assessed for radioactivity.
SERCA2b activity
SERCA2 activity was assayed by isolating macrophage ER fractions with differential centrifugation, incubating with radiolabeled CaCl2 for 15 min at 37°C, and measuring radioactivity.
Immunofluorescence
Peritoneal macrophages were fixed on slides with 4% formalin, blocked with 5% goat serum, and incubated with SERCA2 (1:250, Abcam) and VDR (1:200, Santa Cruz) overnight, then incubated with secondary antibody 488-conjugated goat anti-rat (green) and 539-conjugated goat anti-rabbit (red). Images were captured on a confocal microscope (Olympus Fv1000).
Monocyte adhesion and migration
Monocytes were added to fibronectin-coated plates and incubated for 4 h at 37°C before adhered cells were stained and quantified. Transwell migration assays were performed by addition of monocytes to the upper chamber with 8 h of MCP-1 (100 ng/well) stimulation in the lower chamber. Cells migrated into the lower chamber were manually counted.
Monocyte cholesterol transport
The deuterium-labeled cholesterol-d7 tracer was added to 20% Intralipid, generating a cholesterol-d7-containing emulsion. Monocytes were incubated with the emulsion for 6 h prior to infusion into recipient mice. Neutral lipids from monocytes, plasma at 6, 12, and 24 h, and recipient aortas at 24 h after infusion were extracted with chloroform:methanol (1:2 v/v). Underivatized sterols were injected into a Thermo TSQ 8000 GC/MS/MS instrument for quantification. Monocyte cholesterol enrichment as the ratio of cholesterol-d7/natural cholesterol ester and aortic cholesterol ester enrichment relative to the plasma cholesterol-d7/natural cholesterol ester were reported. Two-photon microscopy was performed by the In Vivo Imaging Core at Washington University. Aortic explants from mice after infusions with DiI-oxLDL-incubated monocytes were placed in CO2-independent medium and stained with anti-CD31. Optical sections were acquired by taking 60 sequential z steps at 2.5 μm spacing, with each plane representing an image of 220 μm (x) by 240 μm (y) (at 2 pixels/μm). Imaging was performed using a custom-built video-rate 2P microscope equipped with a Chameleon Vision II Ti:Sapphire laser.
Gene expression
qRT-PCR was performed using the TaqMan or SYBR® Green reagent kit, with relative expression levels of mRNAs calculated by the comparative threshold cycle method using L32 (Mrpl32) as an internal control.
Immunoblotting and immunoprecipitation
Cellular proteins were immunoblotted for: VDR (Santa Cruz); ER stress proteins [pPERK and PERK (Cell Signaling), CHOP (Santa Cruz)]; proteins involved in cholesterol signaling [JNKp (Cell Signaling), PPARγ, CD36, and SR-A1 (Santa Cruz)]; SERCA2 (ABcam); and pCAMKII and CAMKII (Cell Signaling), and β-actin (Cell Signaling) was used as a loading control. For immunoprecipitation, macrophage lysates from control-L mice after 6 wks of chow were incubated with monoclonal anti-VDR (ABcam), then blotted with anti-SERCA2 (ABcam).
Statistical analysis
Experiments were carried out in duplicate or triplicate. Parametric data are expressed as the mean ± standard error of the mean and analyzed by t-tests, paired or unpaired as appropriate, or by one-way analysis of variance (ANOVA) and Tukey’s post-test for more than 2 groups. Non-parametric data (atherosclerosis) are presented as the median and analyzed using the Mann-Whitney test. Differences were considered statistically significant if p ≤ 0.05. Statistical analysis was carried out using GraphPad Prism.
Supplementary Material
Acknowledgments
This work was supported by NIH R01HL094818-0, Children’s Discovery Institute CH-II-2012-209 and the American Diabetes Association 1-12-CT-08. AER was supported by K12HD001459 and the Washington University ICTS, UL1 TR000448 and KL2 TR000450 from NCATS. KF was supported by T32 DK007120 and P60 DK20579. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of NIH. We would like to thank Dr. Clay F. Semenkovich for his thoughtful guidance, Dr. Mark Sands for his insightful comments and for sharing his expertise in BM transplant, and Dr. Nicholas Davidson for his assistance in hepatocyte isolation.
Footnotes
Author Contributions: All authors assisted in drafting and revising the work, approved the final version for publication, and agree to be accountable for all aspects of the work. Individual contributions are as follows: JO–conceived and designed overall work; acquired data. AER–conceived and designed overall work; analyzed and interpreted data. ID–designed glucose metabolism experiments; acquired data. KF–designed glucose metabolism and SERCA function experiments; acquired, analyzed, and interpreted data. JSS and KC–designed atherosclerosis experiments; acquired data. OS–designed hepatic inflammation experiments; acquired, analyzed, and interpreted data. GC–conceived and designed overall work. REO–conceived and designed monocyte cholesterol transport experiments; acquired, analyzed, and interpreted data. CBM–conceived and designed overall work; analyzed and interpreted data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, Derudas B, Mayi T, Bories G, Tailleux A, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res. 2011;108:985–995. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, Yazdani S, Otsuka F, Davis T, Habib A, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. 2012;59:166–177. doi: 10.1016/j.jacc.2011.10.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Trayhurn P, Bing C. 1,25-Dihydroxyvitamin D3 inhibits the cytokine-induced secretion of MCP-1 and reduces monocyte recruitment by human preadipocytes. Int J Obes (Lond) 2013;37:357–365. doi: 10.1038/ijo.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Jakubzick C, Randolph GJ. Regulation of the migration and survival of monocyte subsets by chemokine receptors and its relevance to atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1412–1418. doi: 10.1161/ATVBAHA.108.180505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulietti A, van Etten E, Overbergh L, Stoffels K, Bouillon R, Mathieu C. Monocytes from type 2 diabetic patients have a pro-inflammatory profile. 1,25-Dihydroxyvitamin D(3) works as anti-inflammatory. Diabetes Res Clin Pract. 2007;77:47–57. doi: 10.1016/j.diabres.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nature immunology. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16:396–399. doi: 10.1038/nm0410-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nature immunology. 2014;15:846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res. 2010;107:579–591. doi: 10.1161/CIRCRESAHA.110.225698. [DOI] [PubMed] [Google Scholar]
- Lanthier N, Molendi-Coste O, Horsmans Y, van Rooijen N, Cani PD, Leclercq IA. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2010;298:G107–116. doi: 10.1152/ajpgi.00391.2009. [DOI] [PubMed] [Google Scholar]
- Liang CP, Han S, Li G, Tabas I, Tall AR. Impaired MEK signaling and SERCA expression promote ER stress and apoptosis in insulin-resistant macrophages and are reversed by exenatide treatment. Diabetes. 2012;61:2609–2620. doi: 10.2337/db11-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008;57:3239–3246. doi: 10.2337/db08-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29:1419–1423. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- Masuyama R, Stockmans I, Torrekens S, Van Looveren R, Maes C, Carmeliet P, Bouillon R, Carmeliet G. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. The Journal of clinical investigation. 2006;116:3150–3159. doi: 10.1172/JCI29463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosig S, Rennert K, Krause S, Kzhyshkowska J, Neunubel K, Heller R, Funke H. Different functions of monocyte subsets in familial hypercholesterolemia: potential function of CD14+ CD16+ monocytes in detoxification of oxidized LDL. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2009;23:866–874. doi: 10.1096/fj.08-118240. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman PE, Powell JT. Vitamin D and cardiovascular disease. Circ Res. 2014;114:379–393. doi: 10.1161/CIRCRESAHA.113.301241. [DOI] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, Chawla A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, Cella M, Bernal-Mizrachi C. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem. 2012;287:11629–11641. doi: 10.1074/jbc.M111.338673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Weng S, Felton SK, Bhandare S, Riek A, Butler B, Proctor BM, Petty M, Chen Z, Schechtman KB, et al. 1,25(OH)2 vitamin d inhibits foam cell formation and suppresses macrophage cholesterol uptake in patients with type 2 diabetes mellitus. Circulation. 2009;120:687–698. doi: 10.1161/CIRCULATIONAHA.109.856070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annual review of physiology. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Tabas I. Pivotal role of calcium/calmodulin-dependent protein kinase II in ER stress-induced apoptosis. Cell cycle. 2010;9:223–224. doi: 10.4161/cc.9.2.10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek AE, Oh J, Bernal-Mizrachi C. 1,25(OH)2 vitamin D suppresses macrophage migration and reverses atherogenic cholesterol metabolism in type 2 diabetic patients. J Steroid Biochem Mol Biol. 2013a;136:309–312. doi: 10.1016/j.jsbmb.2012.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek AE, Oh J, Darwech I, Moynihan CE, Bruchas RR, Bernal-Mizrachi C. 25(OH) vitamin D suppresses macrophage adhesion and migration by downregulation of ER stress and scavenger receptor A1 in type 2 diabetes. J Steroid Biochem Mol Biol. 2013b doi: 10.1016/j.jsbmb.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek AE, Oh J, Sprague JE, Timpson A, de las Fuentes L, Bernal-Mizrachi L, Schechtman KB, Bernal-Mizrachi C. Vitamin D suppression of endoplasmic reticulum stress promotes an antiatherogenic monocyte/macrophage phenotype in type 2 diabetic patients. J Biol Chem. 2012;287:38482–38494. doi: 10.1074/jbc.M112.386912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samovski D, Sun J, Pietka T, Gross RW, Eckel RH, Su X, Stahl PD, Abumrad NA. Regulation of AMPK activation by CD36 links fatty acid uptake to beta-oxidation. Diabetes. 2014 doi: 10.2337/db14-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto FL, Reardon CA, Yoon D, Wang Y, Wong KE, Chen Y, Kong J, Liu SQ, Thadhani R, Getz GS, et al. Vitamin d receptor signaling inhibits atherosclerosis in mice. Mol Endocrinol. 2012;26:1091–1101. doi: 10.1210/me.2011-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. The Journal of clinical investigation. 2009;119:2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman CM, Cantorna MT, DeLuca HF. Expression of 1,25-dihydroxyvitamin D(3) receptor in the immune system. Arch Biochem Biophys. 2000;374:334–338. doi: 10.1006/abbi.1999.1605. [DOI] [PubMed] [Google Scholar]
- Weng S, Sprague JE, Oh J, Riek AE, Chin K, Garcia M, Bernal-Mizrachi C. Vitamin d deficiency induces high blood pressure and accelerates atherosclerosis in mice. PLoS ONE. 2013;8:e54625. doi: 10.1371/journal.pone.0054625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Gower RM, Wang H, Perrard XY, Ma R, Bullard DC, Burns AR, Paul A, Smith CW, Simon SI, et al. Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119:2708–2717. doi: 10.1161/CIRCULATIONAHA.108.823740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.