

Abstract
To date, most genetic engineering approaches coupling the type II Streptococcus pyogenes clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system to lambda Red recombineering have involved minor single nucleotide mutations. Here we show that procedures for carrying out more complex chromosomal gene replacements in Escherichia coli can be substantially enhanced through implementation of CRISPR/Cas9 genome editing. We developed a three-plasmid approach that allows not only highly efficient recombination of short single-stranded oligonucleotides but also replacement of multigene chromosomal stretches of DNA with large PCR products. By systematically challenging the proposed system with respect to the magnitude of chromosomal deletion and size of DNA insertion, we demonstrated DNA deletions of up to 19.4 kb, encompassing 19 nonessential chromosomal genes, and insertion of up to 3 kb of heterologous DNA with recombination efficiencies permitting mutant detection by colony PCR screening. Since CRISPR/Cas9-coupled recombineering does not rely on the use of chromosome-encoded antibiotic resistance, or flippase recombination for antibiotic marker recycling, our approach is simpler, less labor-intensive, and allows efficient production of gene replacement mutants that are both markerless and “scar”-less.
INTRODUCTION
Since its inception in 1998, phage recombinase-mediated homologous recombination, also known as recombineering, has revolutionized bacterial genetics, synthetic biology, and metabolic engineering (1, 2). Relying on either RecET from the Rac prophage (2) or the bacteriophage lambda Red proteins, Exo, Beta, and Gam (1), recombineering allows simple and efficient construction of gene knockout mutants via homologous recombination of a double-stranded DNA (dsDNA) PCR product with bacterial chromosomes (3). This work has led to the construction of an Escherichia coli K-12 single-gene knockout library (the Keio collection [4]), enabled widespread use of chromosome-encoded expression platforms for metabolic and strain engineering (5–11), and simplified conventional recombinant DNA technology through the advent of ligation-independent DNA cloning (2, 12). Automated, high-throughput, and multiplexed genome editing applications have also been envisioned, including multiplexed automated genome engineering (MAGE), generating up to 4.3 billion genomic mutants per day (13). Finally, phage-mediated recombineering systems have been identified and exploited for use in an array of bacterial genera (14–19).
Recombineering can be carried out using either single-stranded DNA (ssDNA) or dsDNA substrates. Since recombineering occurs through an entirely ssDNA intermediate (20), ssDNA recombineering requires only an ssDNA recombinase (RecT or Beta) for recombination (21), whereas recombination of dsDNA substrates requires both an exonuclease (RecE or Exo) and its associated recombinase (RecT or Beta). For this reason, recombination of ssDNA substrates, such as oligonucleotides, is simpler, more efficient, and better understood than dsDNA recombineering (21, 22). Until recently, a fundamental limitation of oligonucleotide recombineering lay in the inability to identify and select cells carrying the target mutation. Recombination of dsDNA PCR products allows the introduction of antibiotic resistance markers directly into the host chromosome (3), while ssDNA recombineering utilizes short synthetic oligonucleotides (≤90 nucleotides [nt]), excluding the prospect of recombination-coupled antibiotic selection. Although successful ssDNA recombinants can be identified in the absence of selection using laborious pooled colony PCR screening (21), recombination efficiency is often too low (e.g., ≤1% [23]), rendering oligonucleotide recombineering largely impractical. To overcome this limitation, Jiang et al. (24) recently coupled the powerful clustered regularly interspaced short palindromic repeat (CRISPR) selection system from Streptococcus pyogenes to lambda Red oligonucleotide recombineering, leading to efficient selection of ssDNA recombinants. This CRISPR system, consisting of the CRISPR-associated (Cas) protein, Cas9, a trans-activating CRISPR RNA (tracrRNA), and a programmable CRISPR targeting RNA (crRNA), generates a Cas9-mediated double-strand break (DSB) at almost any target DNA locus (25). Since the DSB is lethal, cells possessing specific modifications generated via homologous recombination of a plasmid, PCR product, or synthetic oligonucleotide can easily be selected (24). Using the CRISPR/Cas9 system for selection of E. coli genome editing, up to 65% of cells were found to possess the desired chromosomal mutation specified by the synthetic oligonucleotide (24), rendering CRISPR/Cas9 recombineering the most powerful methodology for engineering bacterial genomes to date (26). In addition to E. coli and Streptococcus (24, 27), this technology has recently been adapted for use with lactic acid bacteria (28), Streptomyces (29), and Clostridium (30).
To date, most studies of E. coli CRISPR/Cas9 selection have involved precise mutation of only a few nucleotides using ssDNA oligonucleotide recombineering (24). On the other hand, replacement of native chromosomal genes with foreign metabolic pathways or new cellular capabilities represents a central facet of metabolic engineering and synthetic biology (31). Owing to the immense selective utility harnessed by the CRISPR/Cas9 system, we hypothesized that it could simplify current protocols for carrying out gene replacement in E. coli, the workhorse of molecular biology and a biotechnological host of interest for the production of biological fuels and chemicals (32–34). In the present study, we outlined a simplified strategy for chromosomal gene replacement by coupling the powerful CRISPR/Cas9 system with conventional lambda Red recombineering. We performed preliminary gene replacements to demonstrate the utility of this approach, extensively probed its capacity for generating large chromosomal deletions and insertions, and examined its advantages over traditional recombineering approaches in the absence of CRISPR/Cas9 selection (3, 10). This method overcomes the dependence on flippase (Flp) recombination, avoids the creation of chromosomal “scar” sites (i.e., Flp recognition target [FRT] scar sites), is adaptable to multiplexing (27), and can be performed in a shorter period than previous protocols. Accordingly, clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 recombineering simplifies the construction of markerless gene replacement mutants of E. coli and serves as an effective tool for strain optimization, metabolic engineering, and synthetic biology.
MATERIALS AND METHODS
Bacterial strains, plasmids, oligonucleotides, and growth conditions.
Bacterial strains, plasmids, and oligonucleotides utilized in this work are listed in Tables 1 and 2. E. coli DH5α was employed for both vector construction and genome editing purposes. Lambda Red recombineering donor plasmid pKD46 (3) was obtained from The E coli Genetic Stock Center (CGSC) at Yale University (New Haven, CT). Addgene (Cambridge, MA) supplied pCas9 (identifier [ID] 42876) and pCRISPR (ID 42875) donor plasmids (24). Plasmid pTH18cr (35) was kindly provided by Tamotsu Hashimoto-Gotoh. Oligonucleotides were synthesized by Integrated DNA Technologies (IDT; Coralville, IA) at the 25-nm scale using standard desalting.
TABLE 1.
Strains and plasmids employed in this study
| Strain or plasmid | Relevant characteristic(s) | Source and/or reference |
|---|---|---|
| Strain | ||
| Escherichia coli DH5α | F− endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG ϕ80dlacZΔM15 Δ(lacZYA-argF)U169, hsdR17(rK− mK+) λ− | Laboratory stock |
| Plasmids | ||
| pTH18cr | E. coli cloning vector (pSC101 ori; Cmr; lacZ′) | 35 |
| pUC19 | E. coli cloning vector (pMB1 ori; Apr; lacZ′) | Laboratory stock and reference 43 |
| pKD46 | E. coli lambda Red recombineering expression vector (pSC101ts; Apr; araC-ParaB-gam-bet-exo) | E coli Genetic Stock Center and reference 3 |
| pCas9 | E. coli vector containing the S. pyogenes cas9 gene and tracrRNA (p15A ori; Cmr; cas9; tracrRNA) | Addgene and reference 24 |
| pCRISPR | E. coli vector containing a crRNA expression cassette for Cas9 targeting (pBR322 ori; Knr; crRNA) | Addgene and reference 24 |
| pCRISPR::dbpA | Derived by targeting the pCRISPR crRNA region to the E. coli dbpA gene | This study |
| pCRISPR::ctrl | Derived by inserting a nonhomologous control crRNA sequence into pCRISPR | This study |
TABLE 2.
Oligonucleotides employed in this study
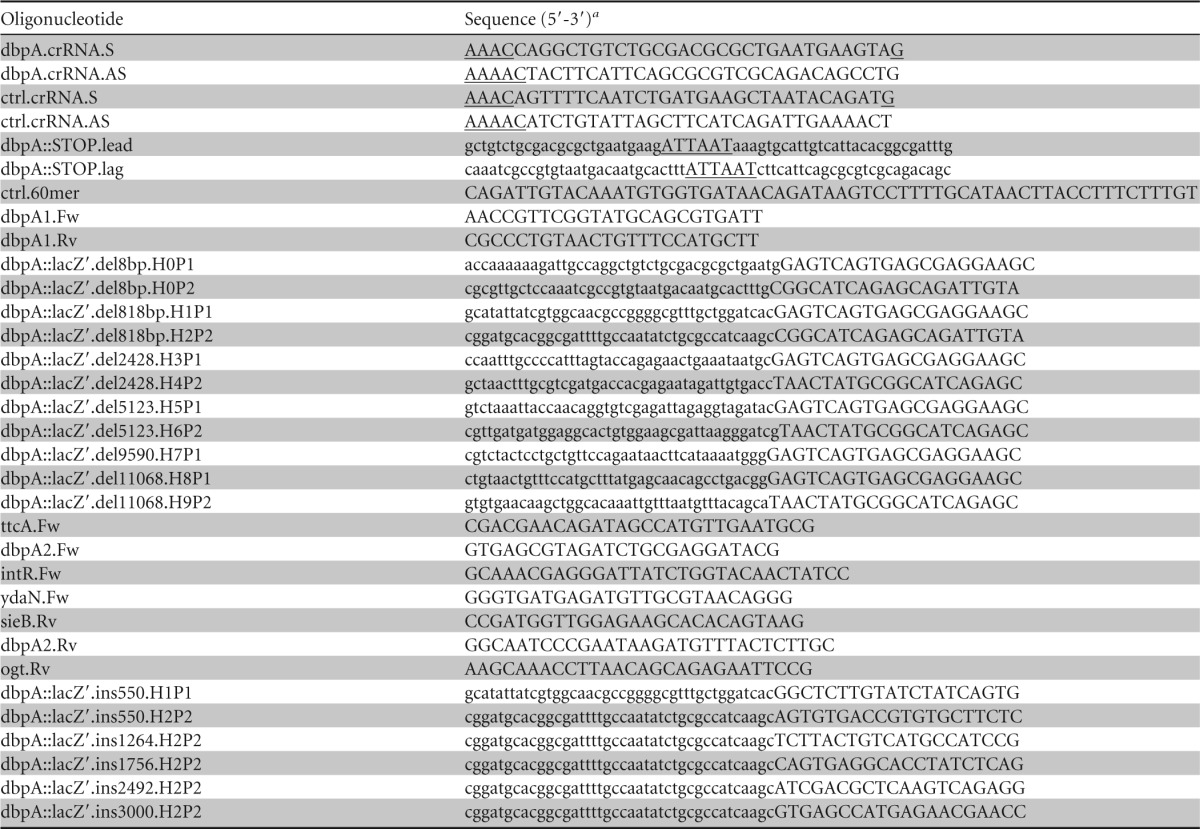
Underline indicates restriction recognition sequence or overhang; lowercase indicates homology regions.
E. coli strains were grown aerobically in shake flask cultures at 37°C and 250 rpm in lysogeny broth (LB) unless stated otherwise. Recombinant strains were selected with ampicillin, chloramphenicol, or kanamycin at a concentration of 100, 34, or 30 μg ml−1, respectively. Antibiotic concentrations were reduced by half for selection of strains harboring two plasmids. Recombinant strains were stored in 15% glycerol at −80°C.
DNA isolation, manipulation, and plasmid construction.
Purification, gel extraction, and cleanup of plasmid DNA and PCR products were performed using commercial spin column kits from Bio Basic Inc. (Markham, ON, Canada) according to the manufacturer's instructions. Restriction endonucleases, a Quick ligation kit, and Phusion and Taq DNA polymerases were obtained from New England BioLabs (Beverly, MA). Chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and Bio Basic Inc. Plasmid pCRISPR::dbpA was constructed by annealing oligonucleotides dbpA.crRNA.S and dbpA.crRNA.AS and ligating the resulting product with a 2,398-bp BsaI restriction fragment of pCRISPR. Compatible BsaI cohesive DNA ends were generated upon oligonucleotide annealing. Similarly, pCRISPR::ctrl was constructed by annealing oligonucleotides ctrl.crRNA.S and ctrl.crRNA.AS and ligating the resulting product with BsaI-digested pCRISPR.
Preparation of recombinogenic DNA.
For ssDNA oligonucleotide recombineering experiments, 60-nt recombinogenic oligonucleotides (dbpA::STOP.lead or dbpA::STOP.lag) were utilized. Six consecutive point mutations corresponding to all 3 nt of the dbpA protospacer adjacent motif (PAM) and 3 nt of the protospacer were placed roughly in the middle of the oligonucleotide sequences. Oligonucleotides were also designed to introduce two consecutive stop codons into the dbpA open reading frame (ORF), in addition to a unique AseI restriction recognition sequence to facilitate screening of transformants for successful recombination.
PCR primers used to generate recombinogenic dsDNA PCR products were designed to possess 20-nt priming regions (P1 or P2) for amplification of the lacZ′ cassette of pUC19 or pTH18cr and 40-nt homology regions (Hn) for homologous recombination with the E. coli DH5α chromosome. To generate chromosomal deletions of various sizes encompassing the dbpA DSB, a 560-bp lacZ′ PCR product was amplified from pUC19 using primer sets dbpA::lacZ′.del8bp.H0P1 plus dbpA::lacZ′.del8bp.H0P2 (8-bp deletion), dbpA::lacZ′.del818bp.H1P1 plus dbpA::lacZ′.del818bp.H2P2 (818-bp deletion), dbpA::lacZ′.del2428bp.H3P1 plus dbpA::lacZ′.del2428bp.H4P2 (2,428-bp deletion), dbpA::lacZ′.del5123bp.H5P1plus dbpA::lacZ′.del5123bp.H6P2 (5,123-bp deletion), dbpA::lacZ′.del9590bp.H7P1 plus dbpA::lacZ′.del2428bp.H4P2 (9,590-bp deletion), dbpA::lacZ′.del11068bp.H8P1 plus dbpA::lacZ′.del11068bp.H9P2 (11,068-bp deletion), and dbpA::lacZ′.del9590bp.H7P1 plus dbpA::lacZ′.del11068bp.H9P2 (19,378-bp deletion). To generate PCR products of various sizes for deletion of an 818-bp region of the dbpA gene, pUC19 was used as the template along with primer sets dbpA::lacZ′.del818bp.H1P1 plus dbpA::lacZ′.del818bp.H2P2 (560-bp insertion), dbpA::lacZ′.del818bp.H1P1 plus dbpA::lacZ′.ins1264.H2P2 (1,264-bp insertion), dbpA::lacZ′.del818bp.H1P1 plus dbpA::lacZ′.ins1756.H2P2 (1,756-bp insertion), and dbpA::lacZ′.del818bp.H1P1 plus dbpA::lacZ′.ins2492.H2P2 (2,492-bp insertion), or pTH18cr was used as the template along with primer sets dbpA::lacZ′.ins550.H1P1 plus dbpA::lacZ′.ins550.H2P2 (550-bp insertion) and dbpA::lacZ′.ins550.H1P1 plus dbpA::lacZ′.ins3000.H2P2 (3,000-bp insertion). All recombinogenic PCR products were treated with 5 U of DpnI for 2 h prior to gel electrophoresis, extraction, and purification.
CRISPR/Cas9-coupled lambda Red recombineering.
E. coli DH5α harboring pKD46 and pCas9 was used as the host strain for all CRISPR/Cas9 recombineering experiments. Cultures for electroporation were grown at 30°C in LB medium. A 10 mM concentration of l-arabinose was added at an optical density at 600 nm (OD600) of 0.3 to 0.4 for induction of the lambda Red recombineering operon of pKD46 (3). Cells were harvested at an OD600 of 0.5 to 0.7, washed three times in ice-cold 10% glycerol, and concentrated approximately 250-fold. Cell-DNA suspensions were transferred to prechilled 0.1-cm electroporation cuvettes and electroporated at 1.8 kV, 25 μF, and 200 Ω. Cells were recovered in 1 ml of SOC medium (36) for 2 h at 37°C and 250 rpm. Outgrowth cultures were serially diluted, and 0.002- to 0.5-μl equivalents were plated onto selective LB agar plates. Selection of pKD46 using ampicillin was not performed following electroporation.
For ssDNA oligonucleotide recombineering, approximately 500 ng of recombinogenic (dbpA::STOP.lead or dbpA::STOP.lag) or nonrecombinogenic oligonucleotide and 100 ng of pCRISPR::dbpA or pCRISPR::ctrl were mixed with 40 μl of electrocompetent DH5α(pKD46, pCas9). Transformants capable of survival in the presence of the CRISPR/Cas9 machinery (pCRISPR::dbpA plus pCas9) were selected using kanamycin and chloramphenicol. For dsDNA gene replacements, 700 to 1,000 ng of gel-purified PCR product (recombinogenic or nonrecombinogenic) and 100 ng of pCRISPR::dbpA or pCRISPR::ctrl were mixed with 40 μl of electrocompetent DH5α(pKD46, pCas9). Transformants capable of survival in the presence of the CRISPR/Cas9 machinery (pCRISPR::dbpA plus pCas9) were selected using kanamycin, chloramphenicol, and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal).
In all CRISPR/Cas9 recombineering experiments, controls were included to assess plasmid electroporation (pCRISPR::ctrl plus nonrecombinogenic oligonucleotide [ctrl.60mer] or nonrecombinogenic PCR product), CRISPR/Cas9-mediated cell death (pCRISPR::dbpA plus ctrl.60mer or nonrecombinogenic PCR product), and recombineering in the absence of CRISPR/Cas9 selection (pCRISPR::ctrl plus recombinogenic oligonucleotide or recombinogenic PCR product). At least two replicates were utilized to assess plasmid electroporation and recombination efficiency in each CRISPR/Cas9 recombineering experiment.
Screening of ssDNA oligonucleotide recombination and dsDNA gene replacement.
Successful recombination of recombinogenic dbpA oligonucleotides was screened by colony PCR using kanamycin- and chloramphenicol-resistant transformants with primers dbpA1.Fw and dbpA1.Rv, followed by overnight digestion of the resulting PCR products with 1 U of AseI. Recombination efficiency of ssDNA recombination was assessed by the proportion of correct colonies following PCR screening and AseI digestion. Double-stranded DNA gene replacement of the dbpA locus with lacZ′ was first screened directly on chloramphenicol and kanamycin agar plates containing X-Gal. Recombination efficiency of dsDNA gene replacement was assessed by the proportion of blue colonies on selective plates containing 40 to 400 colonies. Expected gene insertion and orientation was further verified by colony PCR and Sanger sequencing across both chromosome-PCR cassette junctions using a single blue colony of each mutant type.
RESULTS
Selection of pKD46-based oligonucleotide recombination using the CRISPR/Cas9 system.
Prior to attempting chromosomal gene replacement using CRISPR/Cas9 selection, we first performed an ssDNA oligonucleotide recombination experiment using conventional pKD46-based lambda Red recombineering, as recombination of short oligonucleotides is more efficient than recombination of dsDNA PCR products (21, 22). The CRISPR/Cas9 system has recently been coupled to oligonucleotide recombineering in E. coli by utilizing a host strain possessing a chromosomal copy of the lambda bet gene expressed from a temperature-sensitive promoter (24). For metabolic and strain engineering, however, it is preferable to express the bet gene in trans, rather than carrying out recombineering in a universal recombination host and subsequently transducing the mutant allele to the desired host background. We therefore investigated the use of a three-plasmid system to couple plasmid-encoded recombineering (pKD46) to the CRISPR/Cas9 system (pCRISPR plus pCas9).
We selected the gene encoding DEAD box protein A (dbpA) in E. coli DH5α for gene disruption (10), since the gene is nonessential and deletion does not negatively affect cell growth (4). We designed recombinogenic dbpA disruption oligonucleotides targeting the leading (dbpA::STOP.lead) and lagging (dbpA::STOP.lag) strands of DNA replication whereby successful oligonucleotide recombination leads to mutation of the dbpA PAM sequence and introduces two consecutive stop codons within the dbpA ORF, in addition to a unique AseI recognition sequence for mutant screening (Fig. 1A). Cas9 was targeted to dbpA through construction of pCRISPR::dbpA, which expresses a 30-nt dbpA crRNA. To assess electroporation and oligonucleotide recombination in the absence of CRISPR/Cas9 selection, we also constructed pCRISPR::ctrl, expressing a scrambled crRNA devoid of homology to the E. coli genome. Following arabinose induction of lambda Red functions in strain DH5α(pKD46, pCas9), coelectroporation of pCRISPR::ctrl and a nonrecombinogenic control oligonucleotide (ctrl.60mer) generated approximately 1.6 × 109 CFU μg−1 of plasmid DNA, indicating efficient electroporation of pCRISPR::ctrl (Fig. 1B). Conversely, coelectroporation of pCRISPR::dbpA and ctrl.60mer led to a drastic reduction in electroporation efficiency (greater than 3 orders of magnitude), owing to CRISPR/Cas9 selection against unmodified host cells. A small proportion of pCRISPR::dbpA transformants (approximately 0.06% compared to pCRISPR::ctrl) were able to survive Cas9 endonuclease attack, presumably due to spontaneous deletion of the dbpA crRNA within pCRISPR::dbpA or mutation of the cas9 coding sequence within pCas9 (24). Having established effective CRISPR/Cas9 selection against unmodified cells, we next utilized our three-plasmid system to select for oligonucleotide recombineering mutants through mutation of the CRISPR/Cas9 PAM sequence within the dbpA coding sequence. Coelectroporation of pCRISPR::dbpA with dbpA::STOP.lead and dbpA::STOP.lag oligonucleotide led to 5- and 28-fold increases in electroporation efficiency, respectively, compared to the nonrecombinogenic control oligonucleotide (ctrl.60mer) through mutation of the dbpA PAM sequence (Fig. 1B). In line with previous studies of lambda Red-mediated oligonucleotide recombineering in E. coli (21, 37), the oligonucleotide targeting the lagging strand of DNA replication produced substantially more transformants (greater than 5-fold) than the leading-strand-targeting oligonucleotide. Using colony PCR followed by AseI digestion, we genotyped totals of 28 and 41 pCRISPR::dbpA transformant colonies generated using the dbpA::STOP.lead and dbpA::STOP.lag oligonucleotides, respectively, yielding dbpA recombination efficiencies of 50% for the leading strand oligonucleotide and 80% for the lagging strand oligonucleotide. Genotyping results from 13 representative colonies generated using the dbpA::STOP.lag oligonucleotide are shown in Fig. 1C. To further demonstrate the efficacy of CRISPR/Cas9-coupled recombineering, we set out to determine oligonucleotide recombineering efficiency in the absence of CRISPR/Cas9 selection.We coelectroporated pCRISPR::ctrl and dbpA::STOP.lag and screened the resulting transformants using colony PCR and AseI digestion. Of a total of 154 pCRISPR::ctrl transformants screened, two mutant colonies were identified, corresponding to a recombination efficiency of 1.3% (Fig. 1B). Collectively, the results presented in this section confirm that our three-plasmid system supports efficient CRISPR/Cas9 recombineering using recombinogenic oligonucleotides.
FIG 1.

Single-stranded oligonucleotide recombineering demonstration using the proposed three-plasmid system. (A) Disruption of chromosomal dbpA in E. coli. A 60-nucleotide recombinogenic oligonucleotide targeting the lagging strand of DNA replication (dbpA::STOP.lag) was utilized to disrupt dbpA by introducing six consecutive base pair changes and an AseI recognition sequence (lowercase), generating two consecutive in-frame stop codons (underlined). Sequence corresponding to the dbpA protospacer and PAM are shown in white. Rep corresponds to 36-bp repeats necessary for crRNA processing. (B) Electroporation and dbpA recombination efficiency data resulting from electroporation (pCRISPR::ctrl plus ctrl.60mer), CRISPR/Cas9 (pCRISPR::dbpA plus ctrl.60mer), and recombineering (pCRISPR::ctrl plus dbpA::STOP.lag) controls, as well as CRISPR/Cas9-coupled recombineering using leading or lagging strand oligonucleotides (pCRISPR::dbpA plus dbpA::STOP.lead or dbpA::STOP.lag). Electroporation efficiency is defined as the total number of CFU generated per microgram of plasmid DNA (pCRISPR::ctrl or pCRISPR::dbpA), and recombination efficiency was measured by determining the proportion of mutant colonies following PCR screening and AseI digestion. Recombination efficiency at the dbpA locus was not determined (ND) for electroporation of the nonrecombinogenic control oligonucleotide (ctrl.60mer). Results are averages of at least two independent experiments, and error bars depict standard deviations. (C) Colony PCR and AseI digestion screening of oligonucleotide-mediated dbpA gene disruption. A representative 13 colonies were used as the template in colony PCR with primers dbpA1.Fw and dbpA1.Rv, yielding a product of 1,002 bp. Successful oligonucleotide recombination generates products of 464 bp and 538 bp upon AseI digestion. Lane 1, marker; lanes 2, 4, 7, 8, and 13, negative, unmodified colonies; lanes 3, 5, 6, 9 to 12, and 14, positive, dbpA gene disruption colonies.
pKD46-based chromosomal gene replacement of dbpA with a markerless dsDNA PCR product using CRISPR/Cas9 selection.
We next aimed to utilize our three-plasmid CRISPR/Cas9 recombineering system to carry out chromosomal gene replacement of dbpA with a markerless dsDNA template. Recombination of dsDNA PCR products requires both lambda Exo and Beta proteins, which are coexpressed from plasmid pKD46 (3). We selected the lacZ′ subunit for gene knock-in, since the resulting PCR cassette is short (<1 kb) and recombinants can be identified directly following electroporation using agar plates supplemented with X-Gal for blue-white colony screening. We targeted the same dbpA gene region as employed in our oligonucleotide recombineering demonstration by employing pCRISPR::dbpA. We also devised two different gene replacement approaches by varying the location of Hn homology arms flanking the chromosomal DSB within the dbpA gene, generating an 8-bp (H0 plus H0) or 818-bp deletion (H1 plus H2) (Fig. 2A). To first assess plasmid electroporation efficiency and Cas9-mediated killing of unmodified host cells, we again utilized pCRISPR::ctrl and pCRISPR::dbpA, respectively, along with a 466-bp nonrecombinogenic PCR product. In a manner similar to that of the CRISPR/Cas9 oligonucleotide recombineering demonstration (Fig. 1B), coelectroporation of pCRISPR::ctrl and a 466-bp nonrecombinogenic PCR product yielded a substantially higher electroporation efficiency (more than 3 orders of magnitude) than did coelectroporation of pCRISPR::dbpA with the same control PCR product (Fig. 2B), again demonstrating efficient selection against cells possessing the dbpA PAM sequence. Compared to electroporation of pCRISPR::ctrl, approximately 0.07% of pCRISPR::dbpA transformants were able to evade Cas9 cleavage. Conversely, coelectroporation of pCRISPR::dbpA with a 560-bp recombinogenic lacZ′ PCR cassette containing 40 bp of homology generated a 1.8-fold increase in electroporation efficiency for both dbpA deletion sizes of 8 bp and 818 bp relative to that obtained with the control nonrecombinogenic PCR product (Fig. 2B). This result is in agreement with our ssDNA oligonucleotide recombineering data: high-level recombination produced a dramatic enhancement in electroporation efficiency through efficient replacement of the dbpA PAM sequence and subsequent evasion of Cas9-mediated endonuclease attack. Following electroporation of the 8-bp or 818-bp deletion PCR cassette, recombination efficiency can be determined directly by enumerating the proportion of blue transformants. Recombination efficiencies of 39% and 47% were obtained for the respective 8-bp and 818-bp deletion cassettes, validating effective selection of chromosomal gene replacement. To compare CRISPR/Cas9 recombineering to traditional lambda Red recombination of dsDNA PCR products in the absence of CRISPR/Cas9 selection, we coelectroporated pCRISPR::ctrl with the 8-bp and 818-bp dbpA deletion PCR products and obtained recombination efficiencies of 0.68% and 0.25%, respectively. Colony PCR and Sanger sequencing of one blue colony of each deletion mutant generated using CRISPR/Cas9-coupled recombineering confirmed successful replacement of the dbpA gene regions with lacZ′ (Fig. 2C).
FIG 2.

Double-stranded DNA gene replacement demonstration using the proposed three-plasmid system. (A) Replacement of 8-bp (H0 plus H0) and 818-bp (H1 plus H2) regions of dbpA with a 560-bp recombinogenic PCR product yielding lacZ′ (α). PCR primers possessing different Hn homology regions corresponding to 8-bp and 818-bp chromosomal deletions were utilized. Successful gene replacement replaces the dbpA PAM sequence and prevents Cas9 from generating a chromosomal DSB, which are both located between each set of homology regions. Genomic layout, primer binding sites, and expected PCR product sizes are shown for each dbpA gene replacement mutant, in addition to the wild-type strain. Genes corresponding to dbpA and lacZ′ are depicted to scale. “Rep” corresponds to 36-bp repeats necessary for crRNA processing. (B) Electroporation and dbpA recombination efficiency data resulting from electroporation (pCRISPR::ctrl plus control PCR product), CRISPR/Cas9 (pCRISPR::dbpA plus control PCR product), and recombineering (pCRISPR::ctrl plus 8-bp or 818-bp deletion cassette) controls, as well as an 8-bp or 818-bp chromosomal deletion using CRISPR/Cas9-coupled recombineering (pCRISPR::dbpA plus 8-bp or 818-bp deletion cassette). Electroporation efficiency is defined as the total number of CFU generated per microgram of plasmid DNA (pCRISPR::ctrl or pCRISPR::dbpA), and dbpA recombination efficiency was measured by determining the proportion of blue colonies. Recombination efficiency at the dbpA locus was not determined (ND) for electroporation of the nonrecombinogenic control PCR product. Results are averages of at least two independent experiments, and error bars depict standard deviations. (C) Colony PCR screening of dbpA gene replacement with lacZ′. Primers dbpA1.Fw and dbpA1.Rv were utilized in a colony PCR with one white and one blue colony of each gene replacement mutant type. Lane 1, marker; lane 2, wild-type E. coli colony; lanes 3 and 4, 8-bp gene replacement colonies; lanes 5 and 6, 818-bp gene replacement colonies; lanes 3 and 5, white (negative) colonies; lanes 4 and 6, blue (positive) colonies.
Challenging pKD46-based CRISPR/Cas9 gene replacement by increasing the size of chromosomal deletion.
Since dsDNA gene replacement using CRISPR/Cas9 recombineering proved to be highly efficient (39% and 47% of colonies possessed the target mutation), we sought to establish the performance of this technique with respect to the magnitude of deletion that can be efficiently introduced to the E. coli chromosome. We first examined phenotypic data from the E. coli K-12 Keio single-gene knockout mutant collection (4) to identify essential genes flanking dbpA that lead to lethality upon gene knockout. We identified a 20.3-kb nonessential chromosomal region, flanked by the essential genes insH-4 and racR (ydaR), which, theoretically, could be deleted without causing cell death. Accordingly, we varied the spacing between chromosomal homology regions (Hn) within PCR primers and held the size of our recombinogenic lacZ′ cassette constant at 560 bp to generate PCR products leading to deletions of various sizes of the E. coli genome. Specifically, we targeted chromosomal deletions of 818 bp (H1 plus H2), 2,428 bp (H3 plus H4), 5,123 bp (H5 plus H6), 9,590 bp (H7 plus H4), 11,068 bp (H8 plus H9), and 19,378 bp (H7 plus H9) (Fig. 3A). To ensure effective CRISPR/Cas9 selection in each gene replacement event, homology regions were designed to flank the dbpA PAM sequence, which is essential for Cas9 endonuclease attack. The 9,590-bp and 11,068-bp chromosomal deletions both encompass the full dbpA gene sequence, including the essential PAM sequence, and together span the entire 19,378-bp chromosomal deletion segment. An equal amount of each lacZ′ PCR cassette was electroporated with pCRISPR::dbpA to E. coli DH5α(pCas9, pKD46). Electroporation efficiency was found to vary by a factor of 2.7 between the six recombinogenic deletion PCR cassettes assayed (Fig. 3B). Notably, electroporation efficiencies corresponding to the 2,428-bp and 19,378-bp deletion PCR products were 4% and 34% lower, respectively, than that of the nonrecombinogenic control PCR product. Based on data from our preliminary ssDNA and dsDNA CRISPR/Cas9-coupled recombineering experiments (Fig. 1B and 2B), a lack of elevation in electroporation efficiency of recombinogenic DNAs relative to the nonrecombinogenic control PCR product suggests unsuccessful recombination. Surprisingly, however, target recombination events could be identified for all six chromosomal deletions. Further, recombination efficiency was found to vary dramatically between the chromosomal deletions (recombination efficiencies of 3% to 47% [Fig. 3B]). Generally, recombination efficiency decreased with increasing size of chromosomal deletion, yet not in a linear manner across the sizes of deletion assessed. A major drop in recombination efficiency (>80%) was observed by increasing the size of deletion from 818 bp to 2,428 bp. Conversely, recombination efficiency was similar across chromosomal deletions between 2,428 bp and 11,068 bp (recombination efficiencies of 8% to 11%). Deletion of a 19,378-bp region, comprising a total of 19 chromosomal genes, yielded the lowest recombination efficiency, 3%. Since replacement of 8-bp and 818-bp chromosomal regions with lacZ′ using recombineering in the absence of CRISPR/Cas9 selection could be detected with blue-white colony screening (Fig. 2B), we also assessed replacement of the 2,428-bp chromosomal region with lacZ′ using traditional lambda Red recombineering. Whereas recombination efficiency using CRISPR/Cas9 selection was 9%, we were unable to identify a blue recombinant colony among the background of unmodified cells for the 2,428-bp deletion in the absence of CRISPR/Cas9 selection (Fig. 3B). Colony PCR and Sanger sequencing using one blue colony from each of the six chromosomal deletion mutants generated using CRISPR/Cas9-coupled recombineering confirmed successful replacement of the target chromosomal gene(s) with lacZ′ (Fig. 3C).
FIG 3.

Challenging the proposed strategy of dsDNA gene replacement by varying chromosomal gene deletion size. (A) Replacement of various-size chromosomal regions with a recombinogenic PCR product encoding lacZ′ (α). PCR primers possessing different Hn homology regions corresponding to chromosomal deletions of various sizes (818 to 19,378 bp) were used to amplify a 560-bp lacZ′ product from pUC19. In each case, successful gene replacement replaces the dbpA PAM sequence and averts generation of a chromosomal DSB, which are both located between each set of homology regions. Genomic layout between the essential racR and insH genes (white) is shown for each gene replacement mutant. Chromosomal gene orientation is shown based on the dbpA coding sequence in the reverse (antisense) orientation. Primer binding sites and expected PCR product sizes are also depicted for each gene replacement mutant, in addition to the wild-type strain. All chromosomal genes and the lacZ′ PCR product are depicted to scale. (B) Electroporation and dbpA recombination efficiency data resulting from electroporation (pCRISPR::ctrl plus control PCR product), CRISPR/Cas9 (pCRISPR::dbpA plus control PCR product), and recombineering (pCRISPR::ctrl plus 818-bp or 2,428-bp PCR deletion cassette) controls, as well as six various-size chromosomal deletions using CRISPR/Cas9-coupled recombineering (pCRISPR::dbpA plus PCR deletion cassette). Electroporation efficiency is defined as the total number of CFU generated per microgram of plasmid DNA (pCRISPR::ctrl or pCRISPR::dbpA), and dbpA recombination efficiency was measured by determining the proportion of blue colonies. A recombination efficiency of 0% reflects an inability to identify blue mutant colonies on agar plates containing up to 400 transformants. Recombination efficiency at the dbpA locus was not determined (ND) for electroporation of the nonrecombinogenic control PCR product. Results shown are averages of at least two independent experiments, and error bars depict standard deviation. (C) Colony PCR screening of gene replacement mutants. Various PCR primers were used to ensure successful genomic organization by screening one white colony and one blue colony resulting from each gene replacement scheme. Lane 1, marker; lanes 2 and 3, 818-bp gene replacement colonies; lanes 4 and 5, 2,428-bp gene replacement colonies; lanes 6 and 7, 5,123-bp gene replacement colonies; lanes 8 and 9, 9,590-bp gene replacement colonies; lanes 10 and 11, 11,068-bp gene replacement colonies; lanes 12 and 13, 19,378-bp gene replacement colonies; lanes 2, 4, 6, 8, 10, and 12, white (negative) colonies; lanes 3, 5, 7, 9, 11, and 13, blue (positive) colonies. White colonies corresponding to lanes 4, 6, 8, 10, and 12 are expected to generate a PCR product that is too large to be amplified under the conditions employed.
Challenging pKD46-based CRISPR/Cas9 gene replacement by increasing the size of recombinogenic PCR products.
We next aimed to challenge our CRISPR/Cas9 recombineering approach by increasing the total size of recombinogenic PCR products. We again utilized lacZ′ as the source of recombinogenic DNA to facilitate assessment of recombination efficiency via blue-white colony screening. We held the size of dbpA homology arms (40 nt) and the distance between homology regions (818 bp) constant and varied the size of recombinogenic lacZ′ insertion DNA by PCR amplifying products of various sizes from plasmids pUC19 and pTH18cr. Specifically, we assessed recombination of lacZ′ products of 550 bp, 560 bp, 1,264 bp, 1,756 bp, 2,492 bp, and 3,000 bp (Fig. 4A). Plasmids pUC19 (2,686 bp) and pTH18cr (3,064 bp) were nearly represented in their entirety within the 2,492-bp and 3,000-bp PCR products, respectively, where portions of the sequences corresponding to plasmid origins were omitted to prevent interference with chromosomal DNA replication following recombination. An equal amount of each lacZ′ PCR cassette was electroporated along with pCRISPR::dbpA to E. coli DH5α(pCas9, pKD46), and electroporation efficiency was compared to the electroporation (pCRISPR::ctrl plus 466-bp nonrecombinogenic PCR product; 2.1 × 109 CFU μg−1 of plasmid DNA) and CRISPR/Cas9 selection (pCRISPR::dbpA plus 466-bp nonrecombinogenic PCR product; 1.4 × 106 CFU μg−1 of plasmid DNA) controls (Fig. 4B). Following coelectroporation with pCRISPR::dbpA, electroporation efficiency between the six various-sized recombinogenic PCR products varied by a factor of 3.6. In a similar manner to that of the chromosomal deletion PCR cassettes assayed (Fig. 3B), electroporation efficiencies corresponding to two of the insertion PCR cassettes (1,264 bp and 3,000 bp) were lower than that of the control electroporation. Despite this decrease in electroporation efficiency, recombinant colonies could be detected for all six recombinogenic PCR products, where recombination efficiency varied dramatically (1% to 47%) (Fig. 4B). A substantial decline in recombination efficiency (>90%) resulted from increasing the size of recombinogenic DNA from 560 bp to 1,264 bp. However, recombination efficiency remained relatively constant between PCR products of 1,264 bp to 3,000 bp (1% to 4%). We also assessed recombination of the three most recombinogenic PCR products (550 bp, 560 bp, and 2,428 bp) using lambda Red recombineering without CRISPR/Cas9 selection. Whereas PCR products of 550 bp and 560 bp yielded recombination efficiencies of 0.24% and 0.25%, respectively, mutant colonies corresponding to the 1,264-bp insertion could not be identified. Finally, colony PCR and Sanger sequencing of one blue colony from each of the six lacZ′ gene insertion mutants generated using CRISPR/Cas9-coupled recombineering confirmed successful replacement of dbpA with the lacZ′ PCR cassettes of various sizes (Fig. 4C).
FIG 4.

Challenging the proposed strategy of dsDNA gene replacement by varying dsDNA insertion size. (A) Replacement of an 818-bp region of the dbpA gene with recombinogenic PCR products of various sizes yielding lacZ′ (α). Products were PCR amplified from plasmid pUC19 (560-bp, 1,264-bp, 1,756-bp, and 2,492-bp products) or pTH18cr (550-bp and 3,000-bp products) using PCR primers possessing H1 and H2 homology regions. Regions corresponding to the β-lactamase (Apr) coding sequence, ColE1 RNA (ColE1), and repA coding sequence (pSC101) are truncated in the 1,264-bp, 2,492-bp, and 3,000-bp products, respectively. In each case, successful gene replacement replaces the dbpA PAM sequence and avoids creation of a chromosomal DSB, which are both located between the H1 and H2 homology regions. The expected genomic layout at the dbpA locus is shown for each gene replacement mutant. Primer binding sites and expected PCR product sizes are also depicted for each dbpA gene replacement mutant, in addition to the wild-type strain. Genes and plasmid regions corresponding to dbpA, lacZ′, Apr, Cmr, ColE1, and pSC101 sequences are depicted to scale. Note that size of the lacZ′ coding sequence differs between plasmids pUC19 (324 bp) and pTH18cr (237 bp). (B) Electroporation and dbpA recombination efficiency data resulting from electroporation (pCRISPR::ctrl plus control PCR product), CRISPR/Cas9 (pCRISPR::dbpA plus control PCR product), and recombineering (pCRISPR::ctrl plus 550-bp, 560-bp, or 1,264-bp PCR insertion cassette) controls, as well as six various-size chromosomal deletions using CRISPR/Cas9-coupled recombineering (pCRISPR::dbpA plus PCR insertion cassette). Electroporation efficiency is defined as the total number of CFU generated per microgram of plasmid DNA (pCRISPR::ctrl or pCRISPR::dbpA), and dbpA recombination efficiency was measured by determining the proportion of blue colonies. A recombination efficiency of 0% reflects an inability to identify blue mutant colonies on agar plates containing up to 400 transformants. Recombination efficiency at the dbpA locus was not determined (ND) for electroporation of the nonrecombinogenic control PCR product. Results shown are averages of at least two independent experiments, and error bars depict standard deviations. (C) Colony PCR screening of dbpA gene replacement mutants. Primers dbpA1.Fw and dbpA1.Rv were utilized in a colony PCR with one blue colony resulting from each gene replacement scheme. Lane 1, marker; lane 2, wild-type E. coli colony; lane 3, 550-bp insertional colony; lane 4, 560-bp insertional colony; lane 5, 1,264-bp insertional colony; lane 6, 1,756-bp insertional colony; lane 7, 2,492-bp insertional colony; lane 8, 3,000-bp insertional colony.
DISCUSSION
For more than 15 years, recombineering has served as the most powerful means of creating precise genetic changes to bacterial genomes (2, 3, 21). More recently, the CRISPR/Cas9 system has heightened the utility of recombineering by providing a method of selecting chromosomal mutations down to the single-nucleotide level (24). Still, the true potential of CRISPR/Cas9 systems for selection of bacterial genome editing has not been fully explored, as most CRISPR/Cas9 genome editing approaches to date have focused on the generation of minor chromosomal alterations using ssDNA oligonucleotides. Traditional protocols for replacing large segments of bacterial genomes with heterologous genes or genetic operons could be vastly improved by implementing the utility of CRISPR/Cas9 selection. Here we are proposing an improvement on such earlier methods (3, 9, 10) achieved by simplifying the protocol for chromosomal gene replacement and abolishing the reliance on chromosomal antibiotic markers and the use of Flp recombination for antibiotic marker recycling. We have shown that E. coli gene replacement can be greatly simplified through application of the CRISPR/Cas9 system.
To couple high-level recombineering with the powerful S. pyogenes CRISPR/Cas9 system, we devised a triple-plasmid strategy expressing the lambda Red functions, Exo, Beta, and Gam (pKD46) (3), the Cas9 protein and associated tracrRNA (pCas9), and programmable crRNA (pCRISPR) for generating DSBs at any location within the E. coli genome (24). Our gene replacement approach has three steps. First, Cas9 is directed to the target gene or chromosomal locus by reprogramming crRNA within pCRISPR (24). Next, a recombinogenic dsDNA consisting of a gene or gene operon, including associated regulatory elements, is amplified by PCR using primers possessing homology regions flanking the target chromosomal DSB region. The source of DNA for gene replacement can be of plasmid, genomic, or synthetic origin and need not encode an antibiotic resistance marker or possess FRT recombination sites. Finally, the recombinogenic PCR product is coelectroporated with retargeted pCRISPR into any E. coli host background harboring pKD46 and pCas9. A mixture of target modified cells and unmodified wild-type cells that escaped Cas9 cleavage are obtained via selection of pCas9 plus pCRISPR cotransformants and can be easily differentiated using PCR screening. In contrast to traditional recombineering protocols for chromosomal gene replacement (3, 9, 10), CRISPR/Cas9 recombineering is simpler, is less labor-intensive, and can be carried out in a substantially shorter time frame (Fig. 5). In addition, our approach is easily amendable to multiplexing through the targeting of multiple chromosomal loci (27) and circumvents the use of chromosomal antibiotic markers and Flp-mediated excision for antibiotic marker recycling. Hence, the resulting mutant genome is both markerless and “scar”-less, as no FRT “scar” sites remain following recombination.
FIG 5.

Comparison of the proposed CRISPR/Cas9-coupled recombineering methodology with previous strategies of E. coli chromosomal gene replacement in the absence of CRISPR/Cas9 (9, 10). All technical steps involved in each protocol to obtain the target markerless gene replacement mutant strain are depicted. Previous methods result in a mutant strain possessing an FRT scar site, whereas the mutant derived from the protocol proposed in this study generates a scarless mutant. ABR, antibiotic resistance marker.
Our devised three-plasmid approach was shown to allow selection of recombinant cells generated using either ssDNA oligonucleotides (Fig. 1) or heterologous dsDNA PCR products (Fig. 2). We further probed the performance of the dsDNA gene replacement protocol with respect to the magnitude of deletion and insertion that can be introduced to the host genome. We have shown that deletions up to 19.4 kb, encompassing 19 nonessential chromosomal genes (Fig. 3), and insertions up to 3 kb (Fig. 4) can be introduced with high recombination efficiency (1% to 47%). For comparison, gene replacement using the most recombinogenic substrates assayed in this study produced recombination efficiencies of 0% to 0.68% in the absence of CRISPR/Cas9 selection, as more challenging gene replacement events could not be detected without incorporation of the CRISPR/Cas9 system (Fig. 3B and 4B). When implemented, CRISPR/Cas9 selection elevated sensitivity of mutant detection up to 187-fold (Fig. 1B, 2B, 3B, and 4B). Although we utilized blue-white colony screening for detection of CRISPR/Cas9-coupled recombineering mutants, almost all gene replacements involving insertion of lacZ′ yielded recombination efficiencies enabling mutant detection via PCR screening (defined by recombination efficiency of ≥2.5% [requiring screening of ≤40 colonies]). However, attempting to replace an 818-bp region of dbpA with a 3.8-kb heterologous PCR product failed to yield the target mutant upon PCR screening of 40 colonies (data not shown), specifying a limitation on the size of DNA that can be inserted into the host chromosome or a sequence-dependent effect on gene replacement. It is also important to note that limitations on chromosomal deletions and insertions were assessed independently in this study, where chromosomal deletions were assayed using a comparatively small PCR product (560 bp) and genome insertions were evaluated using only a modest chromosomal deletion (818 bp). Recombination efficiency was found to drop significantly when the magnitude of chromosomal deletion or insertion was increased beyond these levels (Fig. 3 and 4). Interestingly, Maresca et al. (38) found similar reductions in gene insertion using substrates of ≥500 bp, yet recombination was largely unaffected by deletion of bacterial artificial chromosome (BAC) regions ranging from 0.5 kb to 50 kb. This discrepancy may arise from the use of different recipient DNAs (i.e., BACs versus the E. coli chromosome) or through incorporation of CRISPR/Cas9 selection in this study. Nevertheless, our findings imply that replacing a large chromosomal segment (>818 bp) with a large recombinogenic PCR product (>560 bp) should yield poor recombination, resulting in an inability to identify and isolate the target mutant. Therefore, gene replacements using our CRISPR/Cas9 recombineering strategy should target either large multigene deletions or chromosomal insertion of large PCR products. Owing to the high recombination efficiencies we obtained when deleting large chromosomal segments (3% to 8% for deletions of 9.6 kb to 19.4 kb), our approach could find utility in strain optimization and chromosome reduction, whereby genomes are “cleansed” through large-scale deletion of prophages, transposons, and other nonessential host elements (39). In efforts demanding both large chromosome deletion and large heterologous DNA insertion, we recommend utilizing traditional, yet more laborious, lambda Red protocols in the absence of CRISPR/Cas9 selection (3, 10), as rare recombination events can be selected directly via chromosomal antibiotic resistance. Alternatively, increasing the size of homology arms beyond the common 40-nt limit utilized in this study has been shown to enhance recombination (27), which could enable detection of low-efficiency events. It has also been reported that homologous recombination varies drastically between genomic loci (40), presumably due to secondary-structure and sequence-dependent effects. The chromosomal dbpA locus employed in this study potentially represents a recombination “hot spot,” as recombination efficiency of the lagging-strand-targeting oligonucleotide was 80%, compared to 65% at the rpsL locus reported by Jiang et al. (24). It is also worth noting that only one of the plasmids employed in our three-plasmid system, pKD46, possesses a low-copy, temperature-sensitive plasmid origin (3). Whereas previous recombineering strategies have relied exclusively on such easily curable plasmids (3, 10), pCas9 and pCRISPR possess the multicopy p15A and pBR322 origins, respectively (24), thus requiring specialized procedures for plasmid curing. Fortunately, protocols for curing multicopy plasmids from recombinant E. coli strains using a range of artificial plasmid-curing agents (e.g., acridine orange or sodium dodecyl sulfate [41]) are firmly established (42).
Overall, the approach outlined in this report demonstrates that coupling traditional lambda Red recombineering with the powerful CRISPR/Cas9 system leads to simplified construction of markerless chromosomal gene replacement mutants of E. coli. We postulate that the methodology outlined herein will pave the way to novel and more ambitious strain engineering applications through the advent of multiplexed gene replacement strategies and modifying the CRISPR/Cas9 recombineering machinery for use in a selection of biotechnologically relevant bacteria other than E. coli.
ACKNOWLEDGMENTS
This work was supported in part by grant STPGP 430106 from the Natural Sciences and Engineering Research Council of Canada (NSERC) and grant 950-211471 from the Canada Research Chairs (CRC) program.
REFERENCES
- 1.Murphy KC. 1998. Use of bacteriophage λ recombination functions to promote gene replacement in Escherichia coli. J Bacteriol 180:2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang YM, Buchholz F, Muyrers JPP, Stewart AF. 1998. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet 20:123–128. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]
- 3.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:1–11. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alper H, Fischer C, Nevoigt E, Stephanopoulos G. 2005. Tuning genetic control through promoter engineering. Proc Natl Acad Sci U S A 102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meynial-Salles I, Cervin MA, Soucaille P. 2005. New tool for metabolic pathway engineering in Escherichia coli: one-step method to modulate expression of chromosomal genes. Appl Environ Microbiol 71:2140–2144. doi: 10.1128/AEM.71.4.2140-2144.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian ZG, Xia XX, Lee SY. 2009. Metabolic engineering of Escherichia coli for the production of putrescine: a four carbon diamine. Biotechnol Bioeng 104:651–662. doi: 10.1002/bit.22502. [DOI] [PubMed] [Google Scholar]
- 8.Srirangan K, Liu X, Westbrook A, Akawi L, Pyne ME, Moo-Young M, Chou CP. 2014. Biochemical, genetic, and metabolic engineering strategies to enhance coproduction of 1-propanol and ethanol in engineered Escherichia coli. Appl Microbiol Biotechnol 98:9499–9515. doi: 10.1007/s00253-014-6093-9. [DOI] [PubMed] [Google Scholar]
- 9.Striedner G, Pfaffenzeller I, Markus L, Nemecek S, Grabherr R, Bayer K. 2010. Plasmid-free T7-based Escherichia coli expression systems. Biotechnol Bioeng 105:786–794. doi: 10.1002/bit.22598. [DOI] [PubMed] [Google Scholar]
- 10.Sukhija K, Pyne M, Ali S, Orr V, Abedi D, Moo-Young M, Chou CP. 2012. Developing an extended genomic engineering approach based on recombineering to knock-in heterologous genes to Escherichia coli genome. Mol Biotechnol 51:109–118. doi: 10.1007/s12033-011-9442-2. [DOI] [PubMed] [Google Scholar]
- 11.Yuan LZ, Rouviere PE, LaRossa RA, Suh W. 2006. Chromosomal promoter replacement of the isoprenoid pathway for enhancing carotenoid production in E. coli. Metab Eng 8:79–90. doi: 10.1016/j.ymben.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Zhang YM, Muyrers JPP, Testa G, Stewart AF. 2000. DNA cloning by homologous recombination in Escherichia coli. Nat Biotechnol 18:1314–1317. doi: 10.1038/82449. [DOI] [PubMed] [Google Scholar]
- 13.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. 2009. Programming cells by multiplex genome engineering and accelerated evolution. Nature 460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bryan A, Swanson MS. 2011. Oligonucleotides stimulate genomic alterations of Legionella pneumophila. Mol Microbiol 80:231–247. doi: 10.1111/j.1365-2958.2011.07573.x. [DOI] [PubMed] [Google Scholar]
- 15.Gerlach RG, Jaeckel D, Hoelzer SU, Hensel M. 2009. Rapid oligonucleotide-based recombineering of the chromosome of Salmonella enterica. Appl Environ Microbiol 75:1575–1580. doi: 10.1128/AEM.02509-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranallo RT, Barnoy S, Thakkar S, Urick T, Venkatesan MM. 2006. Developing live Shigella vaccines using λ Red recombineering. FEMS Immunol Med Microbiol 47:462–469. doi: 10.1111/j.1574-695X.2006.00118.x. [DOI] [PubMed] [Google Scholar]
- 17.Swingle B, Bao Z, Markel E, Chambers A, Cartinhour S. 2010. Recombineering using RecTE from Pseudomonas syringae. Appl Environ Microbiol 76:4960–4968. doi: 10.1128/AEM.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Kessel JC, Hatfull GF. 2007. Recombineering in Mycobacterium tuberculosis. Nat Methods 4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- 19.Yin J, Zhu H, Xia L, Ding X, Hoffmann T, Hoffmann M, Bian X, Müller R, Fu J, Stewart AF, Zhang Y. 2015. A new recombineering system for Photorhabdus and Xenorhabdus. Nucleic Acids Res 43:e36. doi: 10.1093/nar/gku1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosberg JA, Lajoie MJ, Church GM. 2010. Lambda Red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics 186:791–799. doi: 10.1534/genetics.110.120782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellis HM, Yu DG, DiTizio T, Court DL. 2001. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc Natl Acad Sci U S A 98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawitzke JA, Costantino N, Li X-T, Thomason LC, Bubunenko M, Court C, Court DL. 2011. Probing cellular processes with oligo-mediated recombination and using the knowledge gained to optimize recombineering. J Mol Biol 407:45–59. doi: 10.1016/j.jmb.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swaminathan S, Ellis HM, Waters LS, Yu DG, Lee EC, Court DL, Sharan SK. 2001. Rapid engineering of bacterial artificial chromosomes using oligonucleotides. Genesis 29:14–21. doi:. [DOI] [PubMed] [Google Scholar]
- 24.Jiang WY, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L, Fan X-D. 2014. CRISPR-Cas system: a powerful tool for genome engineering. Plant Mol Biol 85:209–218. doi: 10.1007/s11103-014-0188-7. [DOI] [PubMed] [Google Scholar]
- 27.Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S. 2015. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol 81:2506–2514. doi: 10.1128/AEM.04023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh J-H, van Pijkeren J-P. 2014. CRISPR-Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res 42:e131. doi: 10.1093/nar/gku623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang H, Zheng G, Jiang W, Hu H, Lu Y. 2015. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim Biophys Sin (Shanghai) 47:231–243. doi: 10.1093/abbs/gmv007. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Zhang Z-T, Seo S-O, Choi K, Lu T, Jin Y-S, Blaschek HP. 2015. Markerless chromosomal gene deletion in Clostridium beijerinckii using CRISPR/Cas9 system. J Biotechnol 200:1–5. doi: 10.1016/j.jbiotec.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Nielsen J. 1998. Metabolic engineering: techniques for analysis of targets for genetic manipulations. Biotechnol Bioeng 58:125–132. doi:. [DOI] [PubMed] [Google Scholar]
- 32.Atsumi S, Liao JC. 2008. Metabolic engineering for advanced biofuels production from Escherichia coli. Curr Opin Biotechnol 19:414–419. doi: 10.1016/j.copbio.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clomburg JM, Gonzalez R. 2010. Biofuel production in Escherichia coli: the role of metabolic engineering and synthetic biology. Appl Microbiol Biotechnol 86:419–434. doi: 10.1007/s00253-010-2446-1. [DOI] [PubMed] [Google Scholar]
- 34.Handke P, Lynch SA, Gill RT. 2011. Application and engineering of fatty acid biosynthesis in Escherichia coli for advanced fuels and chemicals. Metab Eng 13:28–37. doi: 10.1016/j.ymben.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Hashimoto-Gotoh T, Yamaguchi M, Yasojima K, Tsujimura A, Wakabayashi Y, Watanabe Y. 2000. A set of temperature sensitive-replication/-segregation and temperature resistant plasmid vectors with different copy numbers and in an isogenic background (chloramphenicol, kanamycin, lacZ, repA, par, polA). Gene 241:185–191. doi: 10.1016/S0378-1119(99)00434-5. [DOI] [PubMed] [Google Scholar]
- 36.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 37.Li XT, Costantino N, Lu LY, Liu DP, Watt RM, Cheah KSE, Court DL, Huang JD. 2003. Identification of factors influencing strand bias in oligonucleotide-mediated recombination in Escherichia coli. Nucleic Acids Res 31:6674–6687. doi: 10.1093/nar/gkg844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maresca M, Erler A, Fu J, Friedrich A, Zhang Y, Stewart AF. 2010. Single-stranded heteroduplex intermediates in λ Red homologous recombination. BMC Mol Biol 11:54. doi: 10.1186/1471-2199-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolisnychenko V, Plunkett G, Herring CD, Feher T, Posfai J, Blattner FR, Posfai G. 2002. Engineering a reduced Escherichia coli genome. Genome Res 12:640–647. doi: 10.1101/gr.217202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yahara K, Didelot X, Ansari MA, Sheppard SK, Falush D. 2014. Efficient inference of recombination hot regions in bacterial genomes. Mol Biol Evol 31:1593–1605. doi: 10.1093/molbev/msu082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keyhani J, Keyhani E, Attar F, Haddadi A. 2006. Sensitivity to detergents and plasmid curing in Enterococcus faecalis. J Ind Microbiol Biotechnol 33:238–242. doi: 10.1007/s10295-005-0261-y. [DOI] [PubMed] [Google Scholar]
- 42.Trevors JT. 1986. Plasmid curing in bacteria. FEMS Microbiol Lett 32:149–157. doi: 10.1111/j.1574-6968.1986.tb01189.x. [DOI] [Google Scholar]
- 43.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]